
Treatment with Anti-HER2 Chimeric Antigen Receptor Tumor-Infiltrating Lymphocytes (CAR-TILs) Is Safe and Associated with Antitumor Efficacy in Mice and Companion Dogs
 , , , ,
, , , ,  , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Patient Samples
2.2. Cell Experiments
2.3. Generation of TILs and CAR-TILs
2.4. CAR Detection
2.5. Cytotoxicity Experiments
2.6. Mouse Experiments
2.7. Canine CAR-TIL Expansion
2.8. First-in-Dog (FIDO) Trial
2.9. Protein Analysis
2.10. Statistical Analyses of Experimental Data
2.11. Single Cell Gene Expression Analysis
2.12. RNA-Sequencing
2.13. Transcriptomic Classification
3. Results
3.1. TILs Differentially Express Chemokine Receptors and Selectins
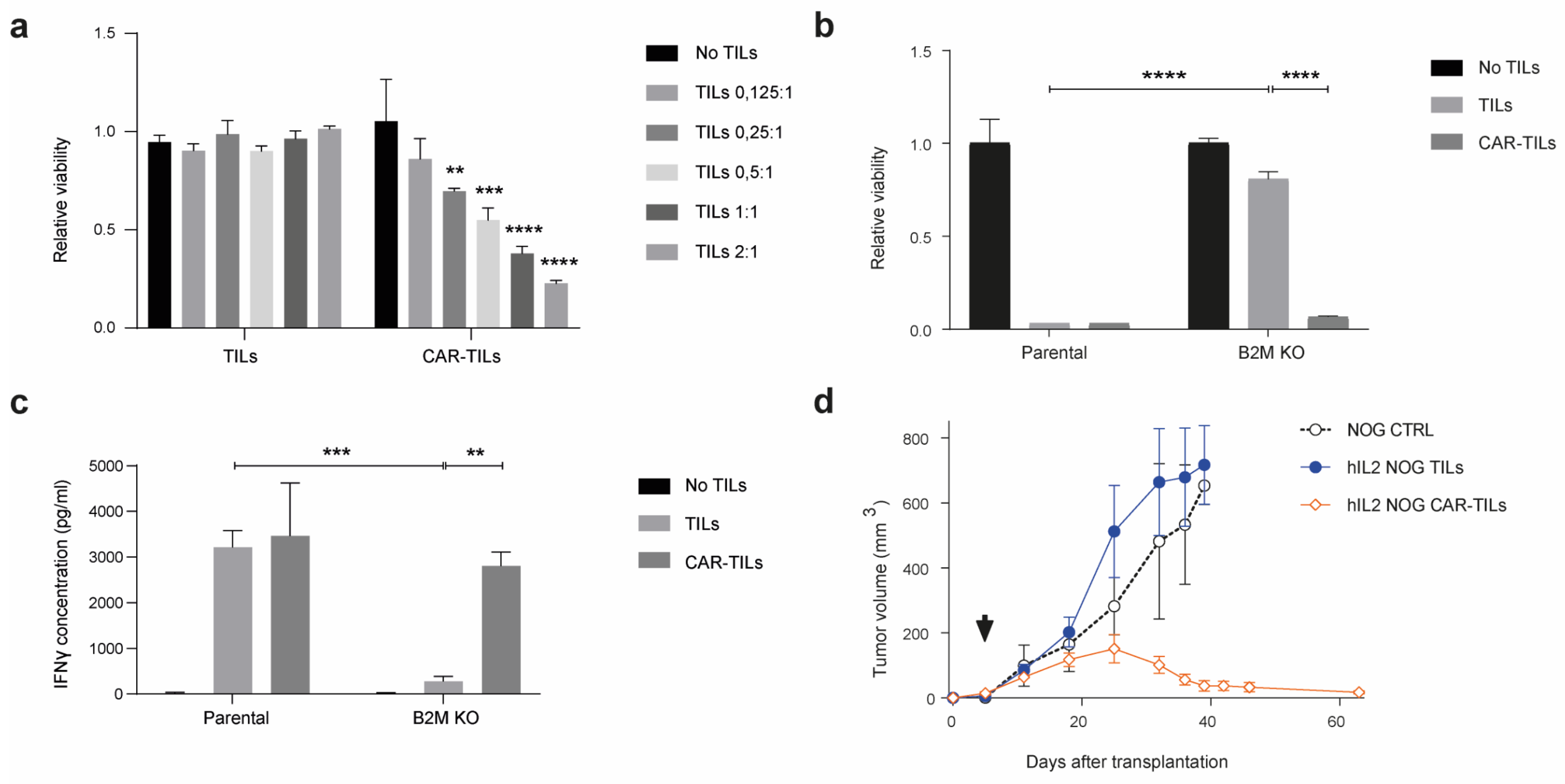
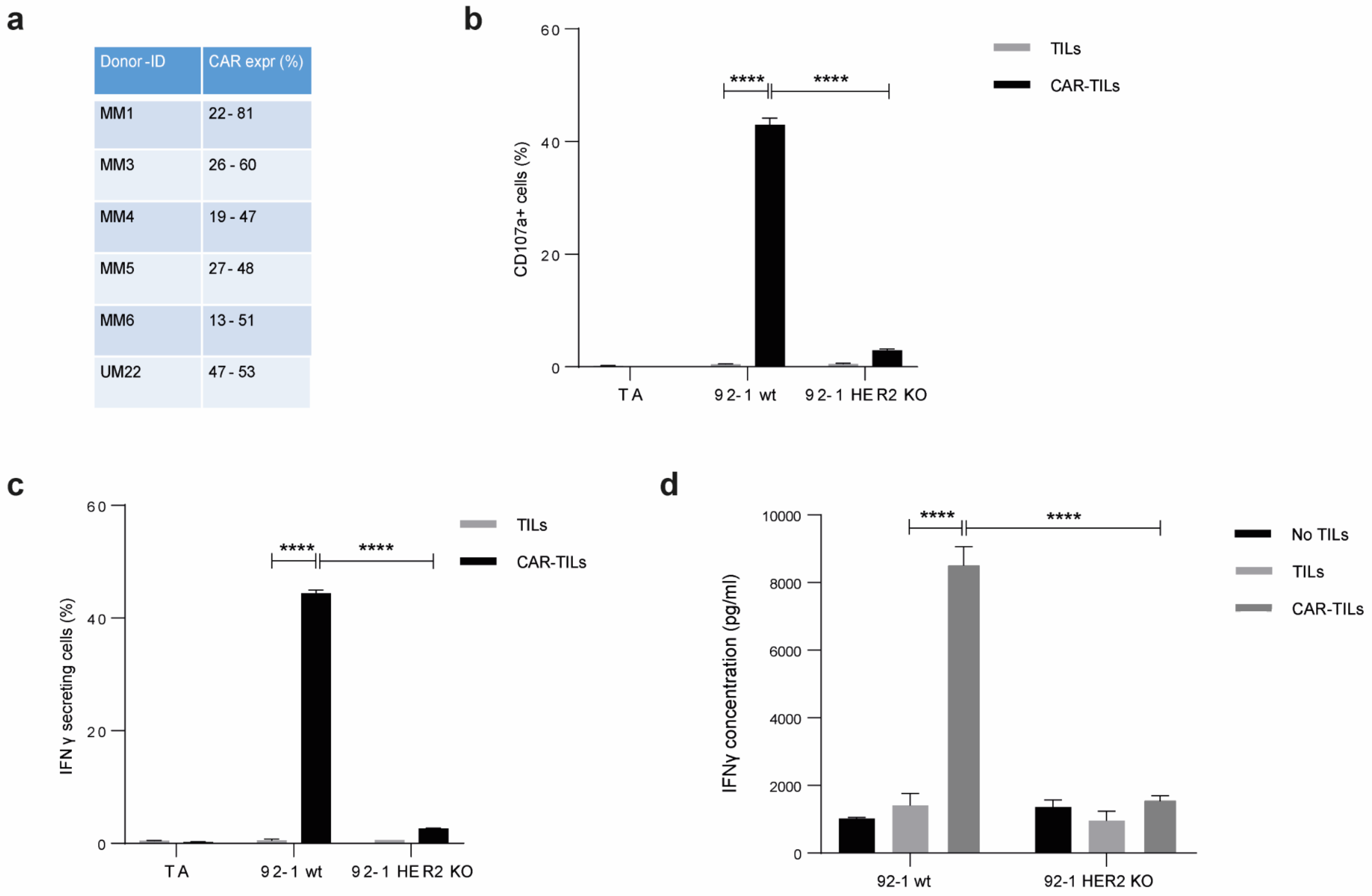
3.2. CAR-TILs Can Kill Melanoma Cells In Vitro


3.3. CAR-TILs Can Kill Melanoma Cells In Vivo
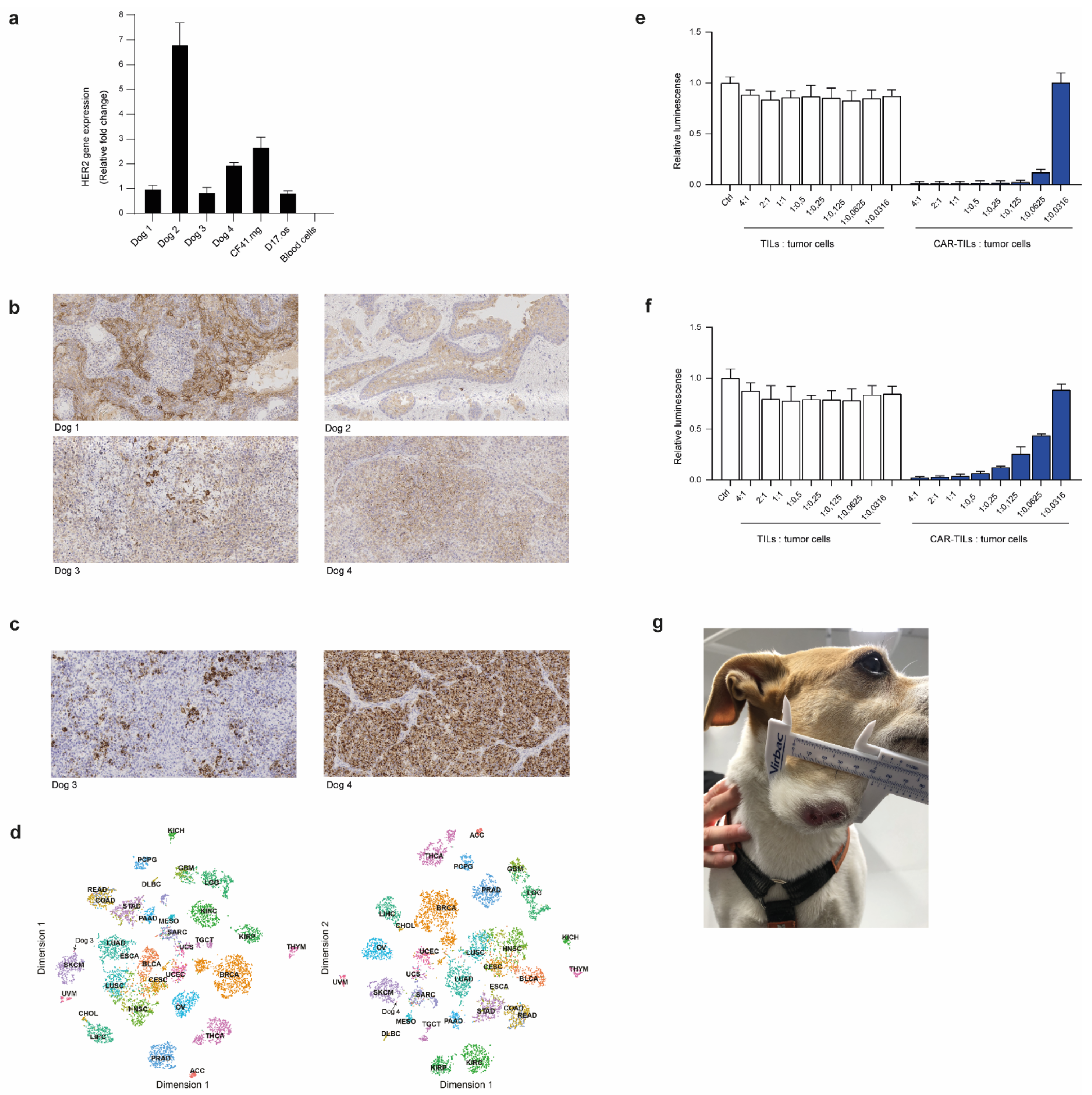
3.4. First-in-Dog (FIDO) Trial Suggests CAR-TIL Therapy Is Safe and May Have Anti-Tumoral Activity in Tumor-Bearing Companion Dogs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balch, C.M.; Gershenwald, J.E.; Soong, S.-J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final Version of 2009 AJCC Melanoma Staging and Classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller Jr., W. H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Long, G.v.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, O.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The national cancer data base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef]
- Seibel, I.; Cordini, D.; Rehak, M.; Hager, A.; Riechardt, A.I.; Böker, A.; Heufelder, J.; Weber, A.; Gollrad, J.; Besserer, A.; et al. Local Recurrence After Primary Proton Beam Therapy in Uveal Melanoma: Risk Factors, Retreatment Approaches, and Outcome. Am. J. Ophthalmol. 2015, 160, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very Long-Term Prognosis of Patients with Malignant Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [Green Version]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [PubMed]
- Leyvraz, S.; Konietschke, F.; Peuker, C.; Schütte, M.; Kessler, T.; Ochsenreither, S.; Ditzhaus, M.; Sprünken, E.D.; Dörpholz, G.; Lamping, M.; et al. Biomarker-driven therapies for metastatic uveal melanoma: A prospective precision oncology feasibility study. Eur. J. Cancer 2022, 169, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Piulats, J.M.; Espinosa, E.; de la Cruz, M.L.; Varela, M.; Alonso, C.L.; Martin-Algarra, S.; Lopez Castro, R.; Curiel, T.; Rodriguez-Abreu, D.; Redrado, M.; et al. Nivolumab Plus Ipilimumab for Treatment-Naive Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J. Clin. Oncol. 2021, 39, 586–598. [Google Scholar] [CrossRef]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results from a Single-Arm Phase II Study. J. Clin. Oncol. 2021, 39, 599–607. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Khoja, L.; Atenafu, E.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.; Piperno-Neumann, S.; et al. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: An international rare cancers initiative (IRCI) ocular melanoma study. Ann. Oncol. 2019, 30, 1370–1380. [Google Scholar] [CrossRef]
- Olofsson, B.R.; Nelson, A.; Shafazand, A.; All-Ericsson, C.; Cahlin, C.; Elander, N.; Helgadottir, H.; Kiilgaard, J.F.; Kinhult, S.; Ljuslinder, I.; et al. Isolated hepatic perfusion as a treatment for uveal melanoma liver metastases, first results from a phase III randomized controlled multicenter trial (the SCANDIUM trial). J. Clin. Oncol. 2022, LBA9509. [Google Scholar] [CrossRef]
- Zager, J.S.; Orloff, M.M.; Ferrucci, P.F.; Glazer, E.S.; Ejaz, A.; Richtig, E.; Ochsenreither, S.; Lowe, M.C.; Reddy, S.A.; Beasley, G.; et al. FOCUS phase 3 trial results: Percutaneous hepatic perfusion (PHP) with melphalan for patients with ocular melanoma liver metastases (PHP-OCM-301/301A). J. Clin. Oncol. 2022, 9510. [Google Scholar] [CrossRef]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, a TCR/anti-CD3 bispecific fusion protein targeting gp100, potently activated anti-tumor immune responses in patients with metastatic melanoma. Clin. Cancer Res. 2020, 26, 5869–5878. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Olivier, T.; Prasad, V. Tebentafusp in first-line melanoma trials: An outperforming outlier. Transl. Oncol. 2022, 20, 101408. [Google Scholar] [CrossRef]
- Slingluff, C.L.; Colella, T.A.; Thompson, L.; Graham, D.D.; Skipper, J.C.; Caldwell, J.; Brinckerhoff, L.; Kittlesen, D.J.; Deacon, D.H.; Oei, C.; et al. Melanomas with concordant loss of multiple melanocytic differentiation proteins: Immune escape that may be overcome by targeting unique or undefined antigens. Cancer Immunol. Immunother. 2000, 48, 661–672. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of Immune Evasion by Tumors. Adv Immunol. 2006, 90, 51–81. [Google Scholar]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Aparicio, S.; Hidalgo, M.; Kung, A. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 2015, 15, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef]
- Bonnett, B.N.; Egenvall, A.; Hedhammar, A.; Olson, P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: I. Breed-, gender-, age- and cause-specific rates. Acta Veter Scand. 2005, 46, 105–120. [Google Scholar] [CrossRef]
- Gordon, I.; Paoloni, M.; Mazcko, C.; Khanna, C. The Comparative Oncology Trials Consortium: Using Spontaneously Occurring Cancers in Dogs to Inform the Cancer Drug Development Pathway. PLOS Med. 2009, 6, e1000161. [Google Scholar] [CrossRef]
- Pazzi, P.; Steenkamp, G.; Rixon, A.J. Treatment of Canine Oral Melanomas: A Critical Review of the Literature. Vet. Sci. 2022, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; van der Weyden, L.; Schott, C.R.; Foote, A.; Constantino-Casas, F.; Smith, S.; Dobson, J.M.; Murchison, E.P.; Wu, H.; Yeh, I.; et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat. Commun. 2019, 10, 353. [Google Scholar] [CrossRef]
- Prouteau, A. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, E.; Lindberg, M.F.; Jespersen, H.; Alsén, S.; Bagge, R.O.; Donia, M.; Svane, I.M.; Nilsson, O.; Ny, L.; Nilsson, L.M.; et al. HER2 CAR-T Cells Eradicate Uveal Melanoma and T-cell Therapy-Resistant Human Melanoma in IL2 Transgenic NOD/SCID IL2 Receptor Knockout Mice. Cancer Res. 2019, 79, 899–904. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Söderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8, 707. [Google Scholar] [CrossRef]
- Einarsdottir, B.O.; Bagge, R.O.; Bhadury, J.; Jespersen, H.; Mattsson, J.; Nilsson, L.M.; Truvé, K.; López, M.D.; Naredi, P.; Nilsson, O.; et al. Melanoma patient-derived xenografts accurately model the disease and develop fast enough to guide treatment decisions. Oncotarget 2014, 5, 9609–9618. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Nilsson, L.M.; Mitra, S.; Alsén, S.; Shelke, G.V.; Sah, V.R.; Forsberg, E.M.V.; Stierner, U.; All-Eriksson, C.; Einarsdottir, B.; et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, S.M.; Thamm, D.; Vail, D.M.; London, C.A. Response evaluation criteria for solid tumours in dogs (v1.0): A Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet. Comp. Oncol. 2013, 13, 176–183. [Google Scholar] [CrossRef]
- LeBlanc, A.K.; Atherton, M.; Bentley, R.T.; Boudreau, C.E.; Burton, J.H.; Curran, K.M.; Dow, S.; Giuffrida, M.A.; Kellihan, H.B.; Mason, N.J.; et al. Veterinary Cooperative Oncology Group—Common Terminology Criteria for Adverse Events (VCOG-CTCAE v2) following investigational therapy in dogs and cats. Vet. Comp. Oncol. 2021, 19, 311–352. [Google Scholar]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Bagge, R.O.; Demir, A.; Karlsson, J.; Alaei-Mahabadi, B.; Einarsdottir, B.O.; Jespersen, H.; Lindberg, M.F.; Muth, A.; Nilsson, L.M.; Persson, M.; et al. Mutational Signature and Transcriptomic Classification Analyses as the Decisive Diagnostic Tools for a Cancer of Unknown Primary. JCO Precis. Oncol. 2018, 2, 1–25. [Google Scholar] [CrossRef]
- Mills, J.K.; Henderson, M.A.; Giuffrida, L.; Petrone, P.; Westwood, J.A.; Darcy, P.K.; Neeson, P.J.; Kershaw, M.H.; Gyorki, D.E. Generating CAR T cells from tumor-infiltrating lymphocytes. Ther. Adv. Vaccines Immunother. 2021, 9, 25151355211017119. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; DePinho, R. The mighty mouse: Genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov. 2006, 5, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; E Dudley, M.; Laurencot, C.M. A Rosenberg. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, D.; Gordon, A.A.; Kajitani, H.; Enokihara, H.; Barrett, A.J. Interleukin-2 treatment-associated eosinophilia is mediated by interleukin-5 production. Br. J. Haematol. 1990, 76, 168–173. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Days after Treatment | Treatment | Number of Cells/kg | CAR Expression | Tumor Size (mm) | CRP (mg/L) | WBC (109/L) | Eosinophils (109/L) | Side Effects |
|---|---|---|---|---|---|---|---|---|
| 0 | Dose 1: CAR-TILs | 0.1 million | 70% (mRNA) | <7 | 5 | 0.3 | ||
| 14 | Dose 2: CAR-TILs | 10 million | 75% (mRNA) | 15 × 13 | <7 | 8.4 | 0.4 | Diarrhea that resolved after one day |
| 35 | 27 × 14 | <7 | 7.9 | 0.5 | ||||
| 63 | 35 × 29 | <7 | 7.4 | 0.4 | ||||
| 78 | Dose 3: CAR-TILs | 10 million | 1–7% (virus) | 42 × 24 | <7 | 7.2 | 0.6 | |
| 78 | IL-2 injections 30,000 IU/kg q12h | Diarrhea and vomiting, decreased appetite all grade 1 | ||||||
| 84 | IL-2 injections 30,000 IU/kg q12h | 36 × 22 | 58 | 10.5 | 1.3 | Eosinophilia grade 1 | ||
| 89 | IL-2 injections 30,000 IU/kg q12h | 34 × 24 | 51 | 23.2 | 9.7 | Eosinophilia grade 1 | ||
| 112 | 52 × 44 | <7 | 11.2 | |||||
| 133 | Euthanized due to tumor progression |
| Days after Treatment | Treatment | Number of Cells/kg | CAR Expression | CRP (mg/L) | WBC (109/L) | Eosinophils (109/L) | Side Effects |
|---|---|---|---|---|---|---|---|
| 0 | Dose 1: CAR-TILs | 0.1 million | 79% (mRNA) | <7 | 6.6 | 0.5 | |
| 20 | Dose 2: CAR-TILs | 10 million | 9% (virus) | <7 | 4.4 | 0.3 | |
| 21–24 | IL-2 injections 30,000 IU/kg q12h | Altered appetite, vomiting and diarreha all grade 1 | |||||
| 27–32 | IL-2 injections 30,000 IU/kg q24h | 43 | 12.9 | 2.5 | Eosinophilia, altered appetite, vomiting, diarreha and lethargy all grade 1 | ||
| 34 | 12 | 22.1 | 10.9 | Eosinophilia grade 1 | |||
| 241 | Revisit, in CR | ||||||
| 290 | Revisit, still in CR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forsberg, E.M.V.; Riise, R.; Saellström, S.; Karlsson, J.; Alsén, S.; Bucher, V.; Hemminki, A.E.; Olofsson Bagge, R.; Ny, L.; Nilsson, L.M.; et al. Treatment with Anti-HER2 Chimeric Antigen Receptor Tumor-Infiltrating Lymphocytes (CAR-TILs) Is Safe and Associated with Antitumor Efficacy in Mice and Companion Dogs. Cancers 2023, 15, 648. https://doi.org/10.3390/cancers15030648
Forsberg EMV, Riise R, Saellström S, Karlsson J, Alsén S, Bucher V, Hemminki AE, Olofsson Bagge R, Ny L, Nilsson LM, et al. Treatment with Anti-HER2 Chimeric Antigen Receptor Tumor-Infiltrating Lymphocytes (CAR-TILs) Is Safe and Associated with Antitumor Efficacy in Mice and Companion Dogs. Cancers. 2023; 15(3):648. https://doi.org/10.3390/cancers15030648
Chicago/Turabian StyleForsberg, Elin M. V., Rebecca Riise, Sara Saellström, Joakim Karlsson, Samuel Alsén, Valentina Bucher, Akseli E. Hemminki, Roger Olofsson Bagge, Lars Ny, Lisa M. Nilsson, and et al. 2023. "Treatment with Anti-HER2 Chimeric Antigen Receptor Tumor-Infiltrating Lymphocytes (CAR-TILs) Is Safe and Associated with Antitumor Efficacy in Mice and Companion Dogs" Cancers 15, no. 3: 648. https://doi.org/10.3390/cancers15030648