
Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance

1
Susan Lehman Cullman Laboratory for Cancer Research, Rutgers University, Piscataway, NJ 08854, USA
2
Graduate Program in Cellular and Molecular Pharmacology, Rutgers University, Piscataway, NJ 08854, USA
3
Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08901, USA
4
U.S. Department of Veterans Affairs, New Jersey Health Care System, East Orange, NJ 07018, USA
*
Author to whom correspondence should be addressed.
Cancers 2024, 16(8), 1571; https://doi.org/10.3390/cancers16081571
Submission received: 5 March 2024
/
Revised: 11 April 2024
/
Accepted: 16 April 2024
/
Published: 19 April 2024
(This article belongs to the Section Cancer Therapy)
Abstract
:Simple Summary
Skin cancer is the most common cancer type worldwide, and melanoma is its most aggressive and deadly form. In the past few decades, targeted therapy and immunotherapy have drastically increased survival chances for melanoma patients. However, not all patients respond to these therapies, and the majority of patients experience tumor relapse after initial response to the treatment. Continuous efforts are required to not only develop new therapeutic agents to treat this deadly disease but also to understand the underlying mechanisms leading to resistance of the tumors to treatment. In this review, we summarize data on available therapeutics for the treatment of melanoma and current knowledge on the mechanisms of resistance to melanoma therapies.
Abstract
Melanoma is the most aggressive and deadly form of skin cancer due to its high propensity to metastasize to distant organs. Significant progress has been made in the last few decades in melanoma therapeutics, most notably in targeted therapy and immunotherapy. These approaches have greatly improved treatment response outcomes; however, they remain limited in their abilities to hinder disease progression due, in part, to the onset of acquired resistance. In parallel, intrinsic resistance to therapy remains an issue to be resolved. In this review, we summarize currently available therapeutic options for melanoma treatment and focus on possible mechanisms that drive therapeutic resistance. A better understanding of therapy resistance will provide improved rational strategies to overcome these obstacles.
1. Introduction to Melanoma
Skin cancer is the most prevalent type of cancer in the United States (US) and possibly in the world. The total number of cases may be higher than current estimates, as some types of skin cancer, such as basal cell and squamous cell skin cancers, are not required to be reported to cancer registries [1]. One study estimated over 5 million annual cases in the US alone in the last decade [2]. The most common types of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), followed by melanoma. Among the rarer types are Merkel cell cancer, cutaneous T-cell lymphoma (CTCL), Kaposi sarcoma, skin adnexal tumors and sarcomas. Even though BCC and SCC are the most prevalent types of skin cancer, they are usually easily curable and not metastatic. They originate in basal and squamous skin cells, respectively. Melanoma, on the contrary, is not nearly as common—it accounts for only 1% of all skin cancers diagnosed [3]; however, it causes the majority of deaths from skin cancer. In 2024, over 100,000 new cases of invasive melanoma are predicted in the US, with approximately 8000 melanoma-related deaths, which constitutes about 90% of deaths attributed to skin cancer [1]. Melanoma originates in the pigment-forming cells of the skin, melanocytes, which produce melanin. Melanin has a photoprotective function in the skin, guarding against UV-induced DNA damage. Melanoma is categorized into cutaneous and non-cutaneous. Cutaneous melanoma (CM) comprises neoplasms that arise on most skin surfaces of the body that can be further subcategorized into chronically sun-induced melanomas and non-chronically sun-induced melanomas. The four most common molecular subtypes identified for CM have mutations in BRAF, NRAS or NF1 or a triple wild-type genotype for these genes. Non-cutaneous melanomas are rare and are classified into acral, mucosal and uveal subtypes. Acral melanoma (AM) often presents on palms, soles, fingers, toes and under the nails; mucosal melanoma (MM) occurs in mucosal membranes, such as in the mouth and nasal cavities; and uveal melanoma (UM) originates in the ocular region. These rare melanoma types are very aggressive because they are difficult to detect and therefore are not diagnosed at early stages of tumor development [4].
There are several risk factors for developing skin cancer, which include chronic exposure to UV light, having a high number and large size of moles on the skin, as well as fair skin and light hair color. Additionally, familial history of skin cancer increases the chances of developing the disease in some cases due to the association with genetic mutations, including CDKN2A, CDK4, MC1R and mutations in the nucleotide excision repair machinery pathway [5,6].
The most common melanoma type is cutaneous melanoma. BRAF mutations constitute the most common genetic mutation present in about 50% of melanoma patients, but mutated BRAF is also detected in melanocytes [7,8]. Melanomas with mutated BRAF are usually located on parts of the body not typically exposed to the sun [9]. Over 90% of BRAF mutations occur at codon 600: the most common one is the substitution of glutamic acid for valine (V600E) [7]; other minor mutations include substitution of glutamic acid for lysine (V600K), aspartic acid (V600D) and arginine (V600R) [10]. BRAF is a participant in the MAPK signaling cascade downstream of RAS (Figure 1). As such, most mutations in BRAF lead to constitutive activation of this pathway, triggering aberrant cell proliferation and inhibition of apoptosis. Both ultimately lead to tumor development and progression with additional mutations acquired, such as PTEN [11,12]. Mutations in MEK, the protein downstream of BRAF in the MAPK signaling cascade, occur less frequently than in BRAF, with 6–7% incidence that is mostly in MEK1. Upstream of BRAF, NRAS has a mutational frequency of 15–28% [7,13]. Identification of these hot-spot mutations led to the development of targeted-therapy inhibitors (Figure 1).
2. Treatment Options Available for Cutaneous Melanoma and Their Efficacy
2.1. Radiation Therapy, Topical Therapeutics and Chemotherapy
Surgical tumor removal has been the standard of care for patients with primary melanoma. Radiation therapy, which is common for many other cancer types, has not gained widespread use in melanoma, as skin tumors are usually radioresistant. Instead, therapeutic agent administration is a more likely treatment option for most patients. However, radiotherapy remains an option for patients with inoperable tumors, as well as imiquimod cream, a local immunomodulator prescribed to some patients with early-stage melanoma [14,15].
Treatment with dacarbazine, a chemotherapeutic agent that was introduced in the 1970s, was the standard of care for melanoma patients until targeted therapy was introduced in 2011 (Table 1). Dacarbazine alkylates DNA non-specifically to block DNA replication [16]. The rate of objective tumor responses in patients on dacarbazine ranged from 13 to 20%, with nearly all responses being partial [17]. In addition, dacarbazine caused severe adverse effects (AE) in patients [16].
2.2. Targeted Therapy
BRAF is the most commonly mutated gene in melanoma, and it is therefore an attractive candidate for targeted therapy. One of the first inhibitors developed against mutated BRAF was sorafenib, which is a multikinase inhibitor that targets CRAF, both wild-type and mutant BRAF, and multiple receptor tyrosine kinases (RTKs) [43]. However, the efficacy of sorafenib was limited both as a single agent and in combination with chemotherapeutics, likely due to its weak affinity for BRAF [44,45]. As a result, inhibitors that could bind specifically to mutated BRAF were developed, such as vemurafenib, dabrafenib and encorafenib (Table 1). These inhibitors bind to the ATP-binding pocket of BRAF with increased affinity for BRAF V600E mutation which enhances their selectivity [24,46,47]. They demonstrated increased efficacy over chemotherapeutic agents with a better dose-dependent tumor inhibition in preclinical studies, a higher rate of objective responses (OR) and improved overall survival (OS) in clinical trials [25,26,47,48,49,50]. Despite the improved efficacy of BRAF inhibitors (BRAFi) over chemotherapy, in most cases disease progression occurs after 6–7 months of treatment due to acquired resistance [25,51]. The molecular mechanism underlying this induced resistance is often the reactivation of MAPK signaling through initiation of the MEK cascade [43]; this will be discussed in detail later in the review. The first MEK1/2 inhibitor (MEKi)—trametinib—was developed along with BRAF inhibitors and was shown to also have improved treatment outcomes over chemotherapeutic agents. Unfortunately, patients also developed resistance to the inhibitor shortly after the beginning of treatment [51,52]. Therefore, reactivation of the MAPK cascade after treatment with BRAFi led to the use of a combination of BRAFi and MEKi to overcome therapy resistance and improve treatment outcomes. Three different combinations of BRAFi and MEKi were approved for melanoma patients: dabrafenib/trametinib, vemurafenib/cobimetinib and encorafenib/binimetinib [43]. These approaches increased overall response and survival rates for patients with advanced melanoma to 50–70% [53,54,55,56,57,58,59,60]. The combination therapy, both as a standalone therapy and as an adjunctive therapy, demonstrated strong efficacy in patients [35]. Although effective, BRAFi/MEKi combinations had their shortcomings, mostly in the form of adverse effects, which ranged from mild to severe and occurred in 45–70% of patients who participated in the COMBI-D, COMBI-V, coBRIM and COLUMBUS clinical trials [61].
2.3. Immunotherapy
The development of immunotherapeutic agents, such as inhibitors of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), was of immense significance, as their modes of action are independent of the genetic profiles of any cancers, including melanoma. One of the first immunotherapy options available for the treatment of melanoma was interferon alpha (IFNα) as an adjuvant treatment (Table 1). It modulates the tumor immune environment by increasing the number of tumor-infiltrating cells, decreasing circulating T-regulatory cells, altering cytokine levels and activating transcription 1 (STAT1) and transcription 3 (STAT3) signal transducers [20]. The OS rate was shown to improve in patients treated with IFNα; however, more effective therapies have since replaced IFNα as a melanoma treatment option. Another early immunotherapy option was a high-dose interleukin-2 (IL-2) regimen that modulated immune signaling (Table 1). With response rates (RR) at about 16%, IL-2 therapy did not differ much in efficacy from chemotherapy; however, it offered an alternative to patients who were not responding to dacarbazine, and the response seemed to be more durable [22]. The first immune checkpoint inhibitor introduced was ipilimumab, an antibody against the CTLA-4 receptor present on the surface of T-cells. It was approved for use in melanoma patients in 2011 (Table 1) [27]. CTLA-4 is a co-receptor on the surface of T-cells that is responsible for their activation. When blocked by CTLA-4 specific antibodies, immune responses against tumor cells were observed through an increase in various immune cells (tumor-specific T-cells and non-tumor-specific immune cells) that promoted an increase in anti-tumor activity but also toxicity [62]. Ipilimumab was shown to be effective in patients previously treated with dacarbazine or IL-2, with a median OS of 10.1 months, an overall response rate of 10.9% and an objective response observed in 60.0% of patients that was maintained for at least 2 years in a phase III clinical trial. Grade 3 or 4 adverse effects that could be life-threatening and/or treatment-limiting were reported to occur in 10–15% of patients [28]. Another study, using a combination of ipilimumab and dacarbazine in previously untreated metastatic melanoma patients, confirmed the effectiveness of ipilimumab over the traditional chemotherapy regimen. The OS rates on the combinational ipilimumab/dacarbazine treatment were 11.2 months vs. 9.1 months on dacarbazine alone. However, the rate of adverse effects was increased in the combinational therapy group to 56.3% compared to 27.5% in a single-treatment arm using dacarbazine or placebo [63].
Another promising immunotherapy axis is programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) [27]. PD-1 is a receptor on T-cells that is responsible for maintaining self-tolerance and the protection of host cells against autoimmunity by suppressing the activity of host T-cells. PD-L1 is the endogenous ligand for PD-1 that is expressed in many normal and immune cells. Binding of PD-L1 to PD-1 activates the downstream immune signaling cascade that deactivates T-cells. Overexpression of PD-L1 has been found in several cancer types and is thought to promote evasion from immune detection in tumor cells [64,65,66]. Antibodies specific to PD-1 and PD-L1, respectively, were found to inhibit solid tumor growth in several studies [67,68] and in clinical trials with melanoma patients [29,31,69,70,71]. Two PD-1 antibodies, pembrolizumab and nivolumab, were approved by the FDA for melanoma patients in 2014 (Table 1). Pembrolizumab proved to be more effective than ipilimumab, with an estimated 6-month progression-free-survival (PFS) rate of 46.5% compared to that of 26.5% for ipilimumab. The response rate (RR) was also significantly improved for pembrolizumab, with 32.5% compared to an RR of 11.9% for ipilimumab. Additionally, the incidence of adverse effects was also lower for pembrolizumab (10–13%) than for ipilimumab (19.9%) [31]. Nivolumab demonstrated effectiveness in patients with ipilimumab-refractory tumors in a phase III study when compared to a chemotherapy treatment, with objective responses of 32% vs. 17% [30]. Another phase III study that involved patients with previously untreated melanoma tumors showed an overall survival of 72.9% after 1 year on nivolumab versus 42.1% after 1 year on dacarbazine. The objective response rate (ORR) was 40.0% for nivolumab versus 13.9% for dacarbazine, establishing survival benefits with nivolumab over dacarbazine. Grade 3 or 4 adverse effects rates were also slightly lower in patients on nivolumab (11.7%) than in those on dacarbazine (17.6%) [29]. Thus, immunotherapy approaches paved the way to a new era of treatment for melanoma patients.
Given the success in preclinical models testing combinations of immune checkpoint inhibitors in the treatment of melanoma, clinical trials were held to assess the benefits of using such combinations in patients [34,72]. The combination of nivolumab and ipilimumab proved to be a successful therapeutic strategy in a phase III clinical trial, with an improved median OS rate in comparison with nivolumab monotherapy. This approach was approved by the FDA in 2015 (Table 1) [34]. However, despite showing improved immediate efficacy of the combination, including increased responses, the long-term survival data demonstrated a 6.5-year OS in 50% of participants in the CheckMate 067 phase III trial [34]. Unfortunately, there was a higher risk of severe adverse effects in patients on the combination compared to those on the monotherapy. This sparked an interest in identifying prognostic biomarkers that could single out patients who are likely to benefit from immunotherapy treatment [73].
Antibodies to block PD-L1 were also explored. An in vitro study revealed a higher potency of PD-L1 compared to PD-1 antibodies based on a T-cell reporter platform using EC50 values as an indicator [74]. To this end, atezolizumab, a PD-L1 antibody, was tested in a phase III clinical trial in combination with BRAFi or MEKi, or with vemurafenib or cobimetinib, respectively. The overall survival rates between the combination group and the control group (which received a combination of vemurafenib and cobimetinib as standard of care at the time) were similar. However, PFS rates for the combination group were significantly better than those for the control group at rates of 21.0 months vs. 12.6 months, suggesting a prolonged duration of efficacy when using the PD-L1 antibody atezolizumab with either vemurafenib or cobimetinib. Therefore, it was established that the combination of PD-1/PD-L1 inhibitors with BRAF and MEK inhibitors demonstrated a similar response rate to the combination of PD-1 blockers but that it provided longer-lasting benefit [75,76,77,78,79]. In 2020, the FDA approved the use of atezolizumab in combination with cobimetinib and vemurafenib for patients with advanced unresectable melanoma harboring the BRAF V600E mutation (Table 1) [37].
In 2022, the FDA approved the first lymphocyte-activation gene 3 (LAG-3) immune checkpoint inhibitor, relatlimab, in combination with nivolumab for the treatment of advanced melanoma (Table 1). LAG-3 is a receptor on the surface of T-cells that regulates the activity of T-cell mediated cell proliferation and function. The combination of relatlimab and nivolumab was tested in patients with resectable and previously untreated/unresectable melanoma [39,80]. When used in the neoadjuvant setting, the 1- and 2-year recurrence-free survival rates for patients with a pathological response were 100% and 92%, respectively, compared to 88% and 55% for patients without a pathologic response. The absence of grade 3 or 4 adverse effects was also noted [80]. The trial involved patients with either previously untreated or unresectable melanoma tumors who demonstrated progression-free-survival rates of 47.7% at the 1-year timepoint with relatimab/nivolumab versus 36.0% for nivolumab monotherapy. Grade 3 or 4 adverse effects occurred in 18.9% of patients in the combination therapy group compared to 9.7% of patients in the monotherapy group [39]. As seen in these trials, LAG-3 inhibitor was unable to reduce therapy toxicity; however, it provides an alternative treatment for patients with refractory tumors [40].
A therapeutic option of a different class is talimogene laherparepevec (T-VEC), an oncolytic virus that is used for the treatment of unresectable and advanced melanoma. It was approved by the FDA in 2015 (Table 1). T-VEC induces the expression of cytokine granulocyte–macrophage colony-stimulating factor (GM-CSF) that recruits and activates antigen-presenting cells that mobilize the host’s immune system to the tumor [81,82]. T-VEC was demonstrated to increase survival rates by approximately 4 months when compared to GM-CSF monotherapy. It was proposed for use as a component of combinational therapy with other therapeutic agents based on its mechanism of action [81,82,83,84].
In 2024, the first cell therapy approach was approved by the FDA for the treatment of melanoma patients with unresectable or metastatic melanoma that has been previously treated with anti-PD-1 and BRAFi [42]. Lifileucel is a tumor-derived autologous T-cell immunotherapy based on in vitro T-cell expansion. This line of treatment demonstrated OR rates of 31.5% in a phase II clinical trial [85]. A phase III clinical trial is currently underway, with results expected in 2028–2030 (NCT05727904).
Another direction that has potential in the treatment of melanoma is photodynamic therapy (PDT). PDT has had some success in treating several cancers, such as head and neck, breast, lung, non-melanoma skin cancer and esophageal cancer [86,87,88,89,90,91]. It typically involves using photosensitizer molecules that target the diseased tissue. Lasers with certain light wavelengths activate photosensitizers to yield singlet oxygen, which is oxygen in an excited state, that will act by the same principle as reactive oxygen species (ROS) to cause further damage to cancer cells. Such damage includes, but is not limited to, increased immune responses and DNA damage, which lead to cell death. Through its mechanism of action, PDT limits damage to the normal tissue and demonstrates increased selectivity and low AEs [92,93,94]. In melanoma specifically, PDT has been shown to cause apoptosis, tumor vasculature damage and anti-tumor immune responses [92]. Additionally, the combination of PDT with immunotherapy is a promising axis with favorable results demonstrated in vivo and in situ [92,95,96,97]. Approaches combining PDT with nanoparticles and phytochemical products are also being explored for their efficacy against melanoma [98,99]. PDT has not yet gained widespread application, in part due to its relatively low efficiency. However, it is a promising direction in the field of melanoma therapy.
3. Available Treatment for Acral, Mucosal and Uveal Melanoma
Acral melanoma (AM) and mucosal melanoma (MM) have been noted to be biologically and clinically distinct from cutaneous melanoma (CM) [4]. Low incidence rates of these melanoma subtypes have made it very difficult to determine the effectiveness of treatment due to problems with patient recruitment and sample size. AM constitutes 2–3% of melanoma cases, and MM accounts for approximately 1.2% of all melanoma cases [100,101]. The rarity of AM and MM greatly hinders the identification of critical genes involved in the onset and progression of AM and MM, impeding the development of appropriate treatment options. Because of that, retrospective studies and subgroup analysis of clinical trials not specific to patients with AM or MM subtypes of melanoma had to be conducted to obtain data on available therapeutic options for these patients.
For AM, current standard of care involves surgical removal of the tumor [4]. Given the specificity of this subtype, depending on the location of the tumor, plastic surgery is often required, and amputation could be an additional treatment option in the case of subungual melanomas. Some retrospective studies have demonstrated that a non-amputation treatment option coupled with a full-thickness skin graft is an effective alternative to amputation [102,103]. However, at this time, amputation remains the recommended strategy for subungual melanoma [4]. Interferon alpha has been used as an adjuvant therapy for AM patients with a high risk of relapse and it was demonstrated to reduce the risk of melanoma recurrence; however, its efficacy remains controversial, with a lack of consistency in overall survival improvement for AM patients [104,105,106].
Activating mutations in BRAF are detected only in 15–20% of AM and 3–11% of MM tumors, rendering BRAF/MEK inhibitors effective in only a small population of patients [52,107]. In a retrospective analysis of a clinical trial involving BRAF inhibitors, the ORRs were 38.1% for AM patients and 20% for MM patients [108]. Another study suggested that these inhibitors are just as effective for AM and MM patients with a BRAF V600 mutation as for CM patients with the same mutation [109]. Activating mutations in the receptor tyrosine kinase KIT are more common than those in BRAF and have incidence rates of 36% for AM and 39% for MM patients [110]. Several clinical trials using imatinib, the most well-studied KIT inhibitor, were conducted and the responses to the inhibitor were found to be variable [111,112,113]. Even though some patients benefited from the KIT inhibitor therapy, in most cases the disease ultimately progressed within 1–2 years [4]. Interestingly, KIT was found to have ambiguous functions in cancer. There are multiple indications suggesting that KIT has tumor-suppressing functions in melanoma. Its low expression was associated with higher metastatic potential, and its expression has also been lost in the majority of melanoma cell lines [114,115,116,117]. On the other hand, in other cancer types such as acute myeloid leukemia, gastrointestinal stromal tumors and small-cell lung carcinomas, KIT was associated with tumor progression, with activating mutations in 70–80% of cases. Unlike BRAF and NRAS, KIT mutations were reported not to occur at the same codon in the majority of cases. Rather, they might be spread more heterogeneously throughout the gene, making the development of inhibitors for mutated KIT a challenge [117,118]. This heterogeneity could account for a lack of response to a specific KIT inhibitor in some patients [117]. Other KIT inhibitors, such as nilotinib, were explored for their efficacy in AM and MM, but they ultimately showed similar outcomes to imatinib [119,120].
Surgical excision of the tumor remains standard of care for MM patients. However, given the anatomic sites of the malignancy, morbidity rates for such procedures are higher for MM patients than those for AM and CM. Additionally, neoadjuvant therapy is gaining importance and wider clinical application for MM patients; however, to date, there is no systematic data on its use in clinical settings [4].
Currently, immunotherapy is the most conventional option for adjuvant therapy in AM and MM patients. However, due to the low incidence rates of these melanoma subtypes, data on the safety and efficacy of such immunotherapy is very limited. In a study comparing efficacy between nivolumab and ipilimumab for AM and MM patients, at the 4-year follow-up there was no significant improvement with nivolumab versus ipilimumab treatment; this is likely due to the low number of patient participants [121]. Retrospective analyses of several studies revealed that anti-PD-1 treatment is more effective than a high-dose of interferon α-2b in the adjuvant setting in CM patients but not in AM patients, which could be due to a lower tumor mutational burden in AM patients than in CM patients [122,123]. Despite the lack of sufficient data on the efficacy of immunotherapy in AM and MM, patients with a high risk of relapse will be considered for immunotherapy in an adjuvant setting. In the case of aggressive metastatic AM, pembrolizumab and nivolumab were shown to be effective for disease management to a degree similar to that in CM patients [124]. Several other studies reported conflicting outcomes on the efficacy of anti-PD-1 therapy in AM patients, with shorter OS and ORR rates observed compared to non-AM patients [125,126]. A study using a combination of nivolumab and ipilimumab demonstrated an improved ORR comparable with non-AM patients [127]. Retrospective analyses of several trials testing the efficacy of anti-PD-1 in advanced MM patients revealed lower responses, which is in line with other studies [124,126,128,129,130]. Similar to what was seen in CM, increased efficacy in combinational immune therapies over single therapy regimens was demonstrated in AM and MM, albeit to lower degrees than for CM patients [129].
Uveal melanoma (UM) is a form of ocular cancer. There are approximately 5 cases per 1 million people in the US registered annually [131,132], but the distal metastasis rate in UM is approximately 50% [133]. The management of UM is highly individualized. If the disease is localized, then laser therapy, radiation therapy and surgery are the primary treatment options depending on the size of the tumor and other accompanying circumstances. Chemotherapy is surprisingly ineffective for uveal melanoma, with a response rate of less than 1% [134]. However, in 2023, the FDA approved the use of the chemotherapeutic agent melphalan for UM patients with liver metastasis using a special hepatic delivery system which limits systemic toxicity of the drug (Table 1) [41]. A targeted approach to treating UM is hindered by the genetic distinction of UM from CM, and the rarity of UM greatly impedes the development of novel therapeutic options. Most clinical trial outcomes available for UM are retrospective analyses of larger trials. So far, no significant responses in immunotherapy or targeted-therapy clinical trials have been reported for metastatic uveal melanoma patients. Liver is the most common site of metastasis for UM. Apart from melphalan, direct intervention by means of surgery, hepatic artery embolization and radiofrequency ablation can be used [134]. A recent breakthrough is the development of tebentafusp, a T-cell receptor-bispecific molecule that targets both glycoprotein 100 and CD3. It was approved by the FDA in 2022 for the treatment of unresectable or metastatic UM in patients who are positive for HLA-A*02:01, which is the most prevalent and polymorphic major histocompatibility complex (MHC)-allele family in humans (Table 1) [38,135]. A recently concluded phase III trial found that tebentafusp treatment was effective for UM patients, with a 3-year OS of 22% in the treatment group compared to 17% in the control group [136]. Carvajal et al. described in detail the current state of clinical management of UM and potential therapeutic strategies for UM patients [135]. More studies are also available on UM, and this review is not an exhaustive source of information on the subject.
Despite the significant progress made in the past two decades much still needs to be done to advance available therapeutic options for melanoma patients, especially those with rare types of melanoma. As demonstrated in some cases with immune checkpoint inhibitors, targeted agents not specific to the tumor (sub)type can be successful and in fact preferential over targeted therapy to achieve better results in a variety of melanoma types. Therefore, it is of great importance to investigate and develop ‘multifaceted’ therapeutics capable of treating various subtypes of the disease.
4. mGluR1-Driven Melanoma
4.1. Glutamate in Cancer Development and Progression
Glutamate is a non-essential amino acid that has been largely implicated in both the central nervous system (CNS) and peripheral nervous system as a major excitatory neurotransmitter and a key regulator in learning and memory. Its link to neuropathology has been firmly established. However, glutamate is also an evolutionarily conserved signaling molecule found in various other non-neuronal tissues of the body that contributes to metabolism and structure, as well as to numerous signaling pathways [78,79,137,138,139]. Therefore, it is not surprising that aberrant glutamatergic signaling can lead to the pathogenesis of other diseases such as cancer. The main characteristic of cancer cells is their ability to proliferate uncontrollably. To sustain such intense expansion, tumor cells rewire metabolic pathways, including glutamate metabolism, to meet the increased demand by amplifying the supply of energy and nutrient material necessary for the synthesis of new biomolecules for dividing cells [140]. Glutamate is linked to the production of the major cellular energy source, adenosine triphosphate (ATP), through the tricarboxylic acid cycle (TCA). In cells, glutamate is produced from glutamine by the enzyme glutaminase (GLS) in a hydrolytic reaction with the formation of ammonia that takes place in the mitochondria. It is then converted to α-ketoglutarate by glutamate dehydrogenase; subsequently, α-ketoglutarate enters the TCA cycle to contribute to ATP production [141].
Cancer cells proliferate rapidly, causing a constant need for energy to support their viability and expansion. However, oxygen supply inside a tumor is often limited, leading to hypoxia and hindering the use of aerobic oxidative phosphorylation, which is the main energy source for non-neoplastic cells. Cancer cells then switch over to using anaerobic glycolysis as their major energy source, a phenomenon also known as the Warburg effect [142,143]. It has been reported for many different cancer types that tumors rewire cells to use glutamine/glutamate as the major metabolite, serving as an alternative energy source. This is known as ‘glutamine addiction’ [144,145]. One of the proposed mechanisms for such rewiring is the upregulation of GLS to increase available glutamate levels inside the cell [146,147,148,149].
Taken together, the notion of targeting glutamine/glutamate metabolism is a promising therapeutic strategy for cancer therapy. Additionally, glutamate is a ligand for multiple cell surface receptors that initiate downstream signaling cascades, including metabotropic glutamate receptors (mGluRs) and ionotropic glutamate receptors (iGluR). mGluRs have been specifically implicated in development and progression of several cancer types [140,150]. Our group described the involvement of an aberrant expression of metabotropic glutamate receptor 1 (mGluR1) in melanomagenesis [151].
4.2. mGluR1-Driven Melanoma
mGluR1 expression and activation were found to induce the transformation of normal melanocytes into melanoma cells. This was discovered through establishing transgenic mice where one out of five founder mice developed pigmented lesions. These lesions were subsequently identified histologically to be melanoma [152,153]. The transgenic mice were established to study cell differentiation by the use of a small genomic DNA fragment that showed the ability to induce adipocyte differentiation in vitro, as confirmed by biochemical and histological characterization [154,155]. Molecular analysis revealed that an insertional mutagenesis had occurred in the transgenic mouse (TG-3) that presented with pigmented lesions. The insertion of the small genomic DNA fragment, termed clone B, into the intron 3 region of the Grm1 gene coding for mGluR1 was concurrent with the deletion of a 70 kB host DNA fragment, which led to the aberrant expression of mGluR1 in melanocytes. Normal melanocytes do not express mGluR1 [151]. To confirm the role of the ectopically expressed mGluR1 in the transformation, a second transgenic mouse line, named Tg(Grm1)EPv, was generated using the cloned murine Grm1 cDNA expression that was driven by a melanocyte-specific promoter dopachrome tautomerase. These mice presented with pigmented lesions that were similar in onset and progression to the tumors of TG-3 mice, confirming the role of aberrant mGluR1 expression in the transformation of normal melanocytes into melanoma cells [151]. Analysis of human melanoma cell lines and biopsy samples revealed mGluR1 expression in 92% (no. = 25) of human melanoma cell lines, 65% (no. = 175) of primary and metastatic human melanoma biopsy samples and 33% of human dysplastic nevus samples. In contrast, in human melanocytes mGluR1 expression was not detected [151,156,157]. Further studies by several other groups demonstrated that functional mGluR1 was necessary for not only the development but also the progression of melanoma tumors in vitro and in vivo [157,158,159,160]. In mGluR1-positive tumors, levels of extracellular glutamate were shown to be elevated, suggesting the establishment of an autocrine/paracrine loop that promotes increased glutamatergic signaling through mGluR1 (Figure 2) [150].
The activation of mGluR1 initiates downstream signaling through the MAPK pathway. The PI3K/AKT cascade is thought to be activated by mGluR1 in an indirect manner via the activation of Src and the subsequent launch of insulin-like growth factor receptor 1 (IGF-R1) signaling cascade. Stimulation of these pathways provides signals for cell proliferation, resistance to cell death, angiogenesis, metastasis, dysregulation of cellular metabolism and immune evasion [157,158,159,161,162,163]. Another prominent feature of cancer cells, neoangiogenesis, was reported to be promoted by mGluR1 activity through the induction of vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) via the PI3K/AKT/mTOR/HIF-1 pathway (Figure 2) [164]. mGluR1 was also suspected to be involved in the Deubiquitinase cylindromatosis (CYLD)–NF-κB axis. CYLD is a tumor suppressor, and downregulation of CYLD leads to increased levels of cyclin D and N-cadherin that promote cell proliferation, migration and invasion (Figure 2) [165]. A connection between CYLD expression and the onset of melanoma was demonstrated with Tg(Grm1)EPv. The homozygous loss of CYLD resulted in quicker cancer development and progression as well as increased neoangiogenesis [166]. Another study demonstrated that NF-κB, a transcription factor known for its involvement in cancer development and progression, is regulated by CYLD [167,168]. Earlier studies from our lab showed constitutive activation of NF-κB in mGluR1-positive melanoma cells, further suggesting a link between mGluR1 and CYLD/NF-κB [169]. We hypothesize that mGluR1 stimulates NF-κB through downregulation of CYLD to promote tumor growth; however, this theory requires further investigation and validation.
5. Therapeutic Potential to Treat mGluR1-Driven Melanoma and Other Cancers with Riluzole
5.1. Use of Riluzole in Melanoma
The encouraging data suggesting the contribution of mGluR1 to the onset and progression of melanoma delivers a promising target to explore for therapeutic purposes. Our group has assessed a small-molecule drug named riluzole (Rilutek®) that has been approved by the FDA for treatment of Amyotrophic Lateral Sclerosis (ALS). One of its known functions is the inhibition of glutamate export to the extracellular space. This reduces the amount of the ligand glutamate that is available to activate glutamate receptors and glutamatergic signaling. Riluzole was shown to reduce levels of extracellular glutamate in mGluR1-positive melanoma cells, decrease cell proliferation and increase cell apoptosis in vitro and in vivo. Furthermore, mGluR1-positive melanoma cells were more sensitive to riluzole than were normal melanocytes or mGluR1-negative melanoma cells, supporting the notion of riluzole being a selective agent in targeted therapy [158,170]. These findings were first translated in a phase 0 clinical trial for patients with advanced melanoma (stage III/IV) that used riluzole at the dose approved for ALS patients. mGluR1 expression was not a requirement for participation in this clinical trial; however, it was detected in all participants in the trial. This trial revealed a significant reduction in MAPK and PI3K/AKT signaling, as well as a decrease in FDG-PET intensity in 34% of paired pre- and post-treatment patient samples [171]. Based on these observations, a phase II clinical trial using riluzole monotherapy was conducted in patients with unresectable stage III/IV melanoma. A stable disease span of 23–56 weeks was observed in 46% of the patients. Downregulation of MAPK and PI3K/AKT pathways was also seen in this phase II trial in 33% of paired pre- and post-treatment tumor samples [172]. As previously reported for ALS patients, this phase II trial also revealed high variability in the levels of bioavailable riluzole among patients [172]. This observed variability may in part account for the lack of response in some patients. It was shown in several studies that the difference in the metabolic clearance of riluzole is likely to be contributed to by the inconsistent expression of the cytochrome P450 isoform CYP1A2, a key metabolic enzyme for riluzole [173]. On account of such variability, the prodrug troriluzole was developed with the goal of giving more consistent effects in patients by bypassing liver metabolism and increasing riluzole bioavailability. Troriluzole is proven to have equal or higher efficacy than riluzole in preclinical studies (Patent Publication US20210228549) [174]. Based on the results of the monotherapy clinical trials, riluzole has the potential to be beneficial to some melanoma patients; however, the option of using it as part of a combinational therapy presents as a more promising one.
The exact molecular mechanism by which riluzole inhibits the export of glutamate has not been elucidated. Our group demonstrated earlier that only mGluR1-expressing cells were arrested at the G2/M phase at 24 h in the presence of riluzole with subsequent apoptosis, as indicated by the accumulation of cells in subG1 by 48 h post-treatment. mGluR1-negative cells did not show such alterations in cell cycle profiles [158]. A high proportion of cells in G2/M phase indicates the repair of DNA damage. This was confirmed by increased levels of ROS and γH2AX, a marker of DNA double-stranded breaks, in riluzole-treated mGluR1-positive melanoma cells. It was further supported by elevated γH2AX levels in post-treatment melanoma specimens from patients with stable disease who participated in the phase II single-agent riluzole trial [175]. We further determined that riluzole specifically produces double-stranded DNA breaks and induces nonhomologous end joining (NHEJ) repair pathways [176]. Based on these results, we propose that riluzole may act by mediating the export of glutamate via the xCT antiporter. xCT facilitates the export of one molecule of glutamate in exchange for the import of one molecule of cystine. Cystine is then intracellularly reduced to cysteine that participates in the synthesis of glutathione (GSH) [177]. The decrease in the export of glutamate in the presence of riluzole in mGluR1-positive cells leads to the reduction in the import of cystine, diminished cysteine levels, and less GSH being synthesized for the clearance of ROS. Increased ROS accumulation within the cell ultimately leads to DNA damage, enhanced mutational burden and cell death (Figure 2). We showed that ROS levels are elevated only in riluzole-treated mGluR1-expressing melanoma cells with decreased levels of GSH [175]. Additionally, cells with higher expression of xCT were shown to be more sensitive to riluzole treatment, which corroborates our working hypothesis [178].
This presumed mode of action for riluzole suggests that combining riluzole with an anti-PD-1 inhibitor would be beneficial. Riluzole will promote tumor mutational burden via increased DNA damage and enhanced neoantigen production and mobilize anti-tumor immune cells to the tumor site. In line with this concept, a phase II riluzole monotherapy clinical trial demonstrated that patients with stable disease had increased immune cell infiltration compared to patients with progressive disease [172]. Taken together, these results suggest that the combination of anti-PD-1, an agent that enhances the ability of cytotoxic immune cells to identify and eliminate cancer cells, with troriluzole, the prodrug of riluzole, will enhance the cytotoxic effects of infiltrating immune cells. This hypothesis was tested in our lab by conducting a preclinical study using traditional graft models (allograft and xenograft) as well as the melanoma-prone transgenic mouse models described above. In order to enable easier monitoring of tumor progression, we crossed a TG-3 mouse with a hairless SKH mouse and derived TGS mice. TGS mice exhibit similar onset and progression of melanoma to those described for TG-3 and Tg(Grm1)EPv mice, and they mimic human melanoma development and progression [174].
Many in vivo preclinical therapeutic studies are short-term (4–6 weeks) and do not accurately recapitulate patients’ circumstances. Our unique TGS mouse model provided an opportunity to assess the longitudinal treatment response (up to 15 months in the studies of troriluzole combined with anti-PD-1) without obvious toxicity [174,179]. A sex-dichotomy in treatment response was observed in which male mice benefited from a lower dose of troriluzole than did female mice. When we increased the troriluzole dosage, we detected a rescued response rate in some female mice. We also observed a decrease in response across treatment modalities in both sexes, suggesting the emergence of resistant cell populations and/or a reduction in immune cell infiltrates [179]. Results from these in vivo studies suggest that the TGS model is a responsive and tractable system for evaluating therapeutic regimens for melanoma in a long-term immunocompetent setting. Troriluzole and nivolumab were shown to be safe and tolerable in patients with advanced and refractory solid tumors in a phase I clinical trial [180]. A phase II trial using this combination for melanoma patients with brain metastasis was initiated (NCT04899921) but had to be terminated due to poor enrollment during the COVID pandemic. However, an ongoing clinical trial of troriluzole and anti-PD-1 treatment for glioma patients shows promising results for the use of this combination as a therapeutic strategy [181].
5.2. Use of Riluzole in Cancers Other Than Melanoma
In the last decade, riluzole has been tested in various other cancers with some success. Beneficial effects were seen in breast, pancreas, colon, liver, bone, brain, lung, prostate, thyroid, blood-related and nasopharynx tumors [182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198]. Interestingly, not all of these cancers express mGluR1 or other members of the glutamate receptor family. Despite this, some of these cancers respond to riluzole treatment [185]. Furthermore, results obtained from some studies, mostly in breast cancer, demonstrate that the therapeutic effects of riluzole could be disconnected from its role as an inhibitor of glutamatergic signaling [185,188].
As a single-agent therapy in pancreatic cancer, riluzole was used in both in vitro and in vivo studies, where it showed efficacy by inhibiting the Wnt-β-catenin/TCF-LEF pathway. This pathway is involved in the expression of glutamate transporter, glutamine synthetase and glucose transporter 2, which modulate apoptosis, cell migration and autophagy among other markers and pathways [184,186]. In prostate cancer, riluzole was reported to have anti-invasive, as well as anti-proliferative, activity [183,189,191]. One of the potential mechanisms underlying its mode of action in prostate cancer is the downregulation of androgen receptor expression through the endoplasmic reticulum stress pathway and selective autophagy [191]. Riluzole also inhibited the growth of both estrogen receptor-positive (ER+) and triple negative (TNBC) breast cancer cells [185]. In liver cancer, riluzole demonstrated anti-proliferative activity in vitro and in vivo via the same mechanism as in melanoma by inducing cell cycle arrest, apoptosis and modulating levels of GSH and ROS [190]. In bone cancer, riluzole showed treatment efficacy both in vitro and in vivo, especially when it was delivered via iron oxide nanocage [192,193,195]. In osteosarcomas, the presence of riluzole was shown to alter the phosphorylation status of AKT/P70 S6 kinase, ERK1/2 and JNK1/2 signaling cascades involved in cell proliferation and cell survival [195].
Given the prominent role of glutamate signaling in the CNS, riluzole has been extensively used in brain cancer therapy [194,196,197,198]. It inhibited tumor cell growth in tumor stem-like-cell-enriched cultures isolated from two human glioblastomas (GBM) through the suppression of glucose transporter 3 expression and inhibition of the p-AKT/HIF1α pathway. Additionally, downregulation of DNA (cytosine-5)-methyltransferase 1 (DNMT1), which is involved in the hypermethylation of various tumor-suppressor genes, and poor prognosis in GBM were reported [197]. In glioma U87 cells, the inclusion of riluzole in in vitro cultures and in vivo xenografts led to the suppression of mGluR1 signaling and significant decreases in tumor cell proliferation, potentially through reduced phosphorylation of PI3K, AKT, mTOR and P70S6K, and this has been confirmed in several studies [196]. In addition to its efficacy in brain tumors, riluzole’s therapeutic effectiveness has also been investigated in metastasis to the brain [199,200]. Combining riluzole with other agents, including radiation therapy, seems to create a promising approach to battle melanoma metastasis to the brain [199].
In general, combinational therapy has proven to be an advantageous strategy for many diseases in the last decade. Riluzole has also been tested in combination with other therapeutics to achieve improved synergistic outcomes. In liver cancer, riluzole was coupled with sorafenib, a protein kinase inhibitor, and the combination therapy showed stronger inhibition of cell proliferation than did either compound alone [190]. A phase I clinical trial using the combination of riluzole and sorafenib in patients with advanced solid tumors was completed. Stable disease was reached in 36% of patients, and partial response was observed in 2.9% of patients. This combination was also found to be safe and tolerable [201]. In TNBC, synergistic effects were observed with riluzole combined with paclitaxel, a chemotherapeutic agent, in vitro in cell lines that were resistant to paclitaxel and in vivo in a xenograft model [202]. In colorectal cancer, together with cisplatin, a conventional chemotherapeutic, riluzole proved to be effective in inhibiting growth of cisplatin-resistant cell lines [203]. In another study, Li et al. developed a platinum (IV)–riluzole conjugate compound that displayed IC50 values significantly exceeding those of cisplatin. It increased DNA damage and cell apoptosis and also suppressed invasion in the HCT-116 colon cancer cell line. In GBM, it was found that, together with an mTORC1/2 inhibitor PP242, riluzole synergistically inhibited the proliferation of GBM cell lines and a patient-derived cell line, as well as tumor growth in xenograft models. This synergistic result seems to derive from reduced levels of cyclin D1, c-MYC, G1 arrest and induced apoptosis [204]. In another study, riluzole was combined with temozolomide, a chemotherapeutic agent, and tested on GBM cell lines as well as in an orthotopic mouse allograft model of O6-methylguanine DNA methyltransferase (MGMT)-positive GBM. Riluzole was found to enhance the anti-tumor activities of temozolomide in MGMT-positive but not MGMT-negative GBM cell lines [205]. Furthermore, riluzole was also found to sensitize some tumor types to ionizing radiation [194,200,206]. These investigations of riluzole using in vitro and in vivo assessments further point to the promise of using riluzole or the prodrug troriluzole in improved therapeutic strategies for various cancer types [207].
However, despite the investigation of riluzole as a therapeutic option in numerous other cancers, the most comprehensive studies with riluzole or troriluzole were performed in melanoma. Our recently completed preclinical longitudinal studies in TGS mice treated with troriluzole and/or anti-PD-1 are the first preclinical studies to show a loss of response across treatment modalities, suggesting that intrinsic and/or acquired resistance must be involved in the reduced treatment efficacy as seems to be the case in many human patients [174,179]. This also suggests that the TGS model is suitable to use for not only tractable treatment responses but also for gradual resistance development over a prolonged period, which provides an opportunity to examine the early steps in the onset of treatment resistance. To date, little is known about the origins of resistance to riluzole in ALS or different cancer types. One possibility is that the acquired resistance to riluzole may be related to the upregulation of the multidrug resistance protein 1 (MDR1) reported in ALS mouse models and patient samples [208,209,210,211,212,213]. MDR1 is a cell membrane protein that serves as an efflux pump for many small-molecule compounds. Riluzole was shown to be a substrate for MDR1, supporting the notion that MDR1 is likely to be in part responsible for the resistance to riluzole [208,214]. Other evidence suggests that riluzole may be a substrate for breast cancer resistance protein (BCRP) at the blood–brain barrier (BBB) as well. Therefore, administration of riluzole together with MDR1/BCRP inhibitors may improve its therapeutic efficacy [215,216]. To our knowledge, there are currently no reports on the basis for resistance to riluzole in melanoma or other cancers.
6. Resistance to Different Treatment Options in Melanoma
Cancer therapies have advanced significantly in the last decade with increasing success in treatment responses. However, despite this improvement, the majority of therapeutic agents are effective only for a limited time period before the responses decrease and resistance develops. Treatment resistance has been postulated to derive from intrinsic and/or acquired genetic and/or functional changes from the status quo in cells. Intrinsic resistance comprises the inherent factors that allow tumors to tolerate treatment modalities from the beginning of treatment and continue to progress. Acquired resistance encompasses the adaptation of tumors to treatments via mutations, rewiring of signaling and/or metabolic pathways, epigenetic changes and modulation of the tumor/stromal microenvironment.
Tumors are heterogeneous entities that consist of cancer cells with different genetic profiles. Therefore, multiple cell populations exist within the same tumor. Both acquired and/or intrinsic resistance are likely to occur in one or more of such cell populations that will progress to form tumors that are non-responsive to the treatment (Figure 3). In this review, we discuss the current knowledge of the underlying mechanisms of resistance to targeted therapies and immunotherapy in melanoma. We do not claim this review to be an exhaustive source of information, but we are summarizing the most common and well-studied types of acquired resistance and strive to provide a general overview of the topic.
7. Resistance to Targeted Therapies
7.1. MAPK Proteins
The most prevalent mutation in melanoma patients is the BRAF V600E mutation. As such, resistance to BRAF-targeted therapy is one of the best studied topics in the therapeutic resistance of melanomas. For BRAF/MEK inhibitors, it is the specific reactivation of the MAPK pathway that is the most prominent mechanism of resistance [217,218,219,220,221] (Figure 4). The development of MEK inhibitors was initiated to overcome resistance to small molecules targeting mutated BRAF. RAS protein can activate several forms of RAF, including ARAF, BRAF and CRAF, which mediate phosphorylation of MEK, followed by phosphorylation of ERK and its downstream effectors that promote cell proliferation and survival signals. RAS activates RAF by relocating RAF to the plasma membrane and causing dimerization. BRAF inhibitors only bind to a mutated version of the protein, specifically BRAF V600E. Melanoma cells that harbor this mutation exhibit low levels of activated RAS that are less likely to mediate BRAF V600E dimerization and direct downstream signaling. However, increased RAS activity will lead to BRAF signaling even in the presence of BRAF inhibitor. The BRAF in the dimer that is not bound to the inhibitor will initiate downstream signaling desensitizing tumor cells to the targeted therapy (Figure 4) [222]. Another resistance mechanism involves a secondary mutation in BRAF that can alter its function and cause a loss of response to treatment. One such mutation was characterized by Poulikakos and colleagues: p61BRAF(V600E) protein, which enhanced dimerization in a RAS-independent manner [223]. Therefore, mutations in RAS or other upstream proteins that increase its signaling and drive RAF-dimer formation can contribute to the acquired resistance to targeted BRAF inhibitor treatments. Examples of such mutations are gain-of-function (GOF) mutations in RAS and loss-of-function (LOF) mutations in NF1 that have been described by several groups and demonstrated to drive resistance to BRAF inhibitors [219,220]. Some other possibilities include mutation-independent structural changes in BRAF itself that are mediated by alternative splicing, dimerization of BRAF and the constitutive activation of the MAPK pathway regardless of the status of upstream proteins [223]. The significance of BRAF dimerization in driving resistance was confirmed in a study using patient-derived xenograft models (PDX) established from metastasized human melanoma tumors. Duplications of the BRAF kinase domain led to resistance in approximately 10% of PDXs, which was verified in a patient cohort [224]. Some other means to drive BRAF-dimer formation include upregulated CRAF expression and increased copy numbers of BRAF [217].
Activation of the MAPK pathway downstream of BRAF has been noted as another way to induce resistance to BRAFi, including somatic mutations in MAP2K1 coding for MEK1 protein or indirect activation of MEK through MAP3K8, a MAPK substrate [225]. Further downstream in the MAPK signaling cascade, ERK has been implicated in acquired resistance to BRAFi through the loss of stromal antigen 2 or 3 (STAG2 or STAG3), which encodes subunits of the cohesin complex that is important for DNA replication. Reactivation of ERK signaling is mediated by deregulation of the expression of dual specificity phosphatase 6 (DUSP6), an inhibitor and regulator of ERK activity in melanoma, which is mediated by CCCTC-binding factor (CTCF) and by the loss of STAG2. LOF in dual specificity phosphatase 4 (DUSP4) may also be implicated in this process [226].
7.2. Receptor Tyrosine Kinases
The upregulation and activation of other receptor tyrosine kinases (RTK) are other ways for melanoma cells to circumvent the activities of BRAF/MEK inhibitors [221]. The intricate network of cellular signaling pathways is highly interconnected, which means that changes in the activity of pathways other than MAPK can also influence the MAPK pathway indirectly. Increases in RTK ligands and growth factors also contribute to alterations in cellular signaling, reducing innate drug sensitivity and driving acquired resistance [217]. Key RTKs that were noted to be implicated in driving acquired resistance to BRAFi are MET, insulin-like growth factor 1 receptor (IFG-1R), epidermal growth factor receptor (EGFR), platelet derived growth factor receptor ß (PDGFRß) and hepatocyte growth factor (HGF) [227,228].
7.3. MITF and Phenotypic Switching
Microphthalmia-associated transcription factor (MITF) plays a critical role in melanocyte lineage survival by regulating cell survival and anti-apoptotic gene expression; it has also been implicated in therapeutic resistance [229]. Depending on its level in the cell, MITF can have different functions: high levels of MITF contribute to differentiation, moderate levels contribute to proliferation, and low levels contribute to invasiveness [229]. With such multifaceted functions, overexpression of MITF has been reported to reduce the therapeutic effects of BRAFi, MEKi and their combination therapy by increasing cAMP signaling pathway activity [217,230,231,232,233,234,235]. Low MITF levels were shown to contribute to drug resistance by upregulation of the AXL receptor tyrosine kinase (AXL-RTK). Melanoma cell lines harboring BRAF or NRAS mutations frequently showed low MITF levels and high AXL-RTK levels. MITF and AXL-RTL levels have been used as a predictor of early resistance to multiple targeted therapy drugs [236,237]. Changes associated with MITF and AXL expression patterns in melanoma cells were proposed as means for cells to withstand therapeutic effects by transitioning from one differentiation state to another, which is often referred to as phenotypic switching [238]. For example, migrating melanoma cells were shown to exhibit low pigmentation and reduced proliferation rates; however, once they settled in a secondary site in the organism, they switched back to a more typical melanoma phenotype with increased pigmentation and proliferation [239]. Frequently, slow-cycling cells can withstand treatment and re-emerge as tumors at a later time as most therapies target fast dividing cells [240]. Such decreases in melanoma cell proliferation have been linked not only to MITF levels but also to regulation by histone demethylase-mediated chromatin remodeling factors. An example of these chromatin remodeling factors is Jumonji/ARID domain-containing protein 1B (JARID1B). These factors are regulated by hypoxia and cytokine signaling; their activity also leads to an increase in PI3K/AKT signaling [241,242]. Although several studies have proposed the involvement of MITF in phenotype switching, the precise role of MITF in all of these processes remains to be elucidated [236,243,244].
7.4. NF-κB Signaling and Tumor Microenvironment
Another study identified NF-κB signaling downstream of AXL as being responsible for incurred resistance to BRAFi [237]. Tirosh et al. proposed distinct tumor-microenvironmental patterns, including cell-to-cell interactions, for melanoma cells resistant to BRAFi with MITFlow/AXLhigh profiles [243]. Apart from the significance of the MITF/AXL ratio in the development of resistance to BRAFi, MITFlow/JARID1Bhigh and MITFhigh/PGC1αhigh (PPARG coactivator 1 alpha) ratios also have been linked to drug resistance in melanoma [245,246].
Interestingly, a transcriptome and methylome analysis study of matched paired pre- and post-treatment melanoma samples showed that DNA mutations were not recurrently present in samples resistant to the treatment. In contrast, transcriptome alterations were very common, with high abundances of Yes-associated protein 1 (YAP1) and MET and low levels of lymphoid enhancer-binding factor-1 (LEF1). Additionally, altered methylation mediated by MAPK inhibition was observed in key regulatory genes and the transcriptome in the resistant samples in a time-dependent manner during treatment [217,247]. Enhanced activation of the NF-κB pathway, together with increased numbers of monocytes and M2 macrophages, suggest that MAPK inhibition may lead to changes in the tumor microenvironment and contribute to therapy resistance together with alterations within the tumor itself [247].
7.5. Parallel Signaling Cascades
An alternative path for the onset of resistance is the activation of parallel signaling cascades, such as PI3K/AKT/mTOR, through changes in the expression of negative regulators of the pathway: PTEN and/or RB1 (Figure 4) [248]. Identifying such pathways will provide options for overcoming acquired resistance. This strategy was utilized in a study by Greger and co-workers where they demonstrated in vitro synergistic effects of combining dabrafenib with trametinib [249]. Subsequently, the FDA granted approval for the use of this combination in patients with unresectable or metastatic melanoma and later extended its use to other tumor types harboring the same mutations. Another study identified ligand-independent ephrin type-A receptor 2 (EPHA2) signaling as a result of BRAFi resistance was associated with increased invasion and metastasis in an AKT-dependent manner. Based on this observation, inhibition of EPHA2 signaling was proposed to be a promising strategy to prevent the onset of metastasis in BRAFi-treated patients [250].
7.6. Autophagy
Autophagy occupies an ambiguous position in cancer therapy as both a tumor-promoting and tumor-inhibiting component [251]. Autophagy can promote tumor resistance to BRAFi by enhancing ATP secretion. Excessive extracellular ATP contributes to tumor cell migration and invasion, possibly through the purinergic receptor P2RX7, which is a player in the autocrine–paracrine pathway that promotes an invasive tumor cell phenotype [252]. Another mechanism that may be involved in autophagy-mediated BRAFi resistance is the induction of endoplasmic reticulum (ER) stress and activation of the TAM (TYRO3, AXL, MER) receptor pathway [238,253]. Targeting pathways that can increase ER stress could be a viable anti-cancer strategy. Cerezo et al. developed a compound that induces ER stress by targeting BiP/GRP78/HSPA5 and triggering the concomitant induction of autophagy and apoptosis in melanoma cells. They also confirmed that this compound inhibits tumor growth in vivo in a xenograft model using both BRAF-sensitive and -resistant cells [254]. In brain cancer, targeting autophagy was shown to inhibit tumor growth and increase cell death in BRAFi-resistant cells, which has been confirmed in a patient population [255].
7.7. MicroRNAs
MicroRNAs (miRNAs) are involved in the transcriptional regulation of numerous genes. Specifically, they were found to mediate the protein expression of genes involved in signaling cascades linked to BRAFi-associated resistance [256]. For example, miR-204 and miR-211 were shown to confer BRAFi resistance in vitro [257]. miR-204 is regulated by STAT3 in amelanotic melanoma cells, where it targets AP-1 complex subunit sigma-2 (AP1S2) and inhibits motility. miR-211 is a transcriptional target of MITF in melanotic melanoma cells that targets ER degradation-enhancing alpha-mannosidase-like 1 (EDEM1) of tyrosinase through the ER-associated degradation (ERAD) pathway. miR-211 also promotes pigmentation, which was shown to be an adaptive response to BRAFi treatment and to contribute to therapy resistance [258].
Results from these studies clearly point out the complexity associated with resistance to BRAF/MEK inhibitors. It is not surprising that no compounds have been developed and incorporated into existing therapeutic regimens to increase the effectiveness of therapy upon a loss of response to BRAFi or MEKi. Therefore, much work remains to be completed in this area.
8. Resistance to Immunotherapies
8.1. Tumor Mutational Burden
The rationale and principle of immunotherapy are based on sensitization of the host’s immune system. Therefore, reducing immune system responses to cancer cells is a logical approach for tumors to use to evade destruction. One way to do that is to diminish T-cell activity. Depending on the subtype, T-cells can have either immune-stimulating or immune-suppressive activity. Effector T-cells include several T-cell subtypes that respond to stimuli and mediate an appropriate immune response. T-cells become activated by interacting with a major histocompatibility complex (MHC) coupled with an antigen to initiate a T-cell immune response. Interruptions and errors in antigen presentation can lead to reduced immune responses; as such, antigen presentation is a process commonly hijacked by tumors to evade immune recognition. Antigens that are presented for T-cell recognition can be in different forms; frequently, mutated or broken-down proteins can serve as antigens in cancer cells. The number of such newly generated antigens in tumor cells is commonly described as the tumor mutational burden (TMB). Higher TMB usually denotes a larger number of mutations that increase antigenicity and promote immune system recognition of tumor cells. In general, melanoma patients with higher TMB showed better responses to immunotherapy and improved OS [259,260,261,262,263]. Evidence exists that TMB can even be used to predict therapy outcomes and the onset of immunotherapy resistance [264]. Immunotherapeutic agents naturally target cells with higher TMB and establish selective pressure for the survival of cancer cells with lower TMB that do not respond well to therapy but will eventually become the source of a resistant tumor [265]. Using therapeutic approaches that target ‘cold’ unresponsive tumors together with immunotherapy will help to overcome this hurdle.
8.2. Antigen Presentation: LAG-3 and B2M
Another way in which cancer cells modulate antigen presentation is through the upregulation of LAG-3, an immune checkpoint receptor often expressed on activated immune cells [266]. LAG-3 has a dual role, being both an activator of T-cells when bound to MHC-II in the process of antigen presentation, and a T-cell suppressor when it is located on T-cells. Because of this ambiguity, LAG-3 has been used as a target for therapeutic intervention and a marker to predict poor therapy responses [267,268,269]. Elevated levels of soluble LAG-3 in serum from patients receiving anti-PD-1 therapy have been linked to poorer responses. Increased tumor infiltration of LAG-3-positive T-cells, T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3) were also associated with shorter PFS. TIM3 is a co-inhibitory receptor expressed on IFN-γ-producing T-cells involved in immune tolerance [268,270]. As such, patients who participated in a combination clinical trial using PD-1 antibody, nivolumab, and a monoclonal antibody targeting LAG-3, namely relatlimab, showed improved PFS [39]. Beta-2-microglobulin (B2M) is another protein involved in antigen presentation that was implicated in the onset of resistance to immunotherapy in melanoma patients. In a paired-patient-sample study, a truncating mutation in the gene coding for B2M was identified as being responsible for the loss of MHC-I on the cell surface [271].
8.3. IFN Signaling and Other Cellular Regulators
Once T-cells are activated, IFN-γ serves to amplify immune responses by recruiting leukocytes, macrophages and natural killer T-cells. Over time, IFN-γ induces the expression of several factors, including indolamine-2,3-dioxygenase (IDO), to reduce this immune response [272,273]. Additionally, IFN-γ has been shown to induce PD-L1 expression and limit anti-PD-1-related immune responses [274,275]. PD-L1 upregulation is a major mechanism involved in acquired resistance to immunotherapy. Several other regulators, including PTEN, nucleophosmin (NPM)/anaplastic lymphoma kinase (ALK), STAT3, EGFR and nuclear factor erythroid 2-related factor 2 (NRF2), were also reported to be involved in this process [276,277,278,279,280]. Carcinoembryonic antigen cell adhesion molecule-1 (CEACAM1) is another target in IFN-γ signaling that heterodimerizes with TIM-3 and promotes T-cell exhaustion to suppress the immune system [281]. Both IFN-α and IFN-γ receptors regulate Janus kinase 1 (JAK1) and/or Janus kinase 2 (JAK2) activity that have been implicated in the development of resistance to anti-PD-1 therapy in several studies, including one using paired patient samples [271,282]. LOF mutations in JAK1 and JAK2 that were concurrent with the deletion of their wild-type alleles were found to be responsible for the onset of resistance in several patients [271].
Stimulator of interferon genes (STING) is an ER protein that is responsible for the regulation of IFN and chemokine signaling [283]. STING expression promotes an increase in MHC-I expression, antigenicity and lysis by tumor-infiltrating T-cells (TILs) via the upregulation of type I IFN signaling and the activity of the CXCL10 chemokine [284]. In one study, the induction of STING signaling improved anti-PD-1 and anti-CTLA-4 therapeutic responses by increasing IFN signaling [285]. However, defects in STING signaling promote tumor progression by protecting melanoma cells from increased immune recognition by TILs [284]. Based on these results, several studies reported on the development of drugs that induce STING signaling in melanoma cells [286,287].
High expression of sphingosine kinase 1 (SK1), a protein that catalyzes phosphorylation of sphingosine to give sphingosine-1-phosphate, is reported to be associated with immunotherapy resistance in melanoma patients. SK1 was proposed to regulate the process of lymphocyte trafficking and differentiation. The exact mechanism of resistance involving SK1 has not been established. However, its inhibition enhanced the efficacy of anti-CTLA-4 and anti-PD-1 therapy in vitro [288,289].
Another protein implicated in immunotherapy resistance is nucleotide-binding domain, leucine-rich containing family, pyrin domain-containing-3 (NLRP3). It induces an inflammasome signaling cascade in response to anti-PD-1 therapy, resulting in the recruitment of granulocytic myeloid-derived suppressor cells (MDSCs), immune cells known for their ability to suppress immune response, to the tumor site and the decreased efficacy of anti-PD-1 therapy [290].
8.4. Regulatory T-Cells
Regulatory T-cells are another type of immunosuppressive cell whose main function is to decrease proliferation and downregulate the activity of effector T-cells in order to maintain appropriate immune responses and avoid excessive immune reactions or autoimmunity. Within resistance to immunotherapy in melanoma, levels of regulatory T-cells expressing forkhead box P3 (FOXP3) are elevated. Their mobilization is dependent on the presence of CD8+ T-cells and is driven by the production of CCR4-binding chemokines and increased proliferation [274].
8.5. Phenotypic Switching
Similar to acquired resistance to targeted therapy, phenotypic switching occurs in cancer cells as a mechanism of resistance to immunotherapy. It involves tumor necrosis factor α (TNFα) signaling in tumor-specific T-cells during the T-cell-mediated immune response to cancer cells. TNFα is a cytokine responsible for modulating inflammatory responses. Its signaling has been associated with the reduced expression of genes that are specific to melanoma cells and an increase in the expression of genes that trigger de-differentiation, resulting in a profile more similar to neural crest-derived cells/immature melanocytes [291].
8.6. Epigenetic Regulation
Finally, epigenetic change is another way to influence cellular proteomic profiles, via processes of the methylation, demethylation and acetylation of DNA or histone proteins. In immunotherapy resistance, epigenetic changes can influence the expression of proteins that promote resistance [292]. DNA hypermethylation reduces expression of 4-1BB (CD137), which helps to stimulate T-cell and B-cell proliferation as well as dendritic cell maturation. Upregulated IL-10 and PD-L1 expression mediated through histone deacetylase 6 (HDAC6) decreases immune response to tumors, as discussed above. Mutations in enzymes that regulate epigenetic functions also influence immune responses to tumors. An example involves mutations in the histone methyltransferase EZH2, whose activity causes reduced expression of RAS association domain-containing protein 5 (RASSF5) and integrin beta chain-2 (ITGB2); these are proteins that are involved in anti-tumor response [293]. KDM5B, another histone demethylase, has also been implicated in the onset of resistance to immunotherapy by recruiting SETDB1, an H3K9 methyltransferase whose overexpression has been linked to reduced anti-tumor activities in several cancers [293,294,295,296]. However, to our knowledge there are no reports regarding their role(s) in resistance to immunotherapy in melanoma.
The loss of response to immunotherapy treatment has been extensively studied, and this review does not claim to be an exhaustive source on the topic. For a more detailed review on the mechanisms of immune evasion in melanomas, please refer to Eddy et al., 2020 [84].
9. Conclusions and Future Directions
Significant progress has been made in melanoma therapy in the past two decades with the development of targeted therapeutics against mutated BRAF/MEK and the introduction of immunotherapy. BRAF/MEK inhibitors significantly improved patient responses and outcomes in comparison to previously available options of chemotherapy and radiation therapy. However, their efficacy and ability to overcome resistance remains to be improved. Based on the outcomes from clinical trials conducted in the last ten years, immunotherapy seems to achieve better efficacy for overall patient survival. But despite the advantages that immune checkpoint inhibitors offer, their efficacy as single agents remains at approximately 50%, and this is often accompanied by AEs [34,39,73]. Using combinations of different therapeutical agents (combining different molecular-tumor-targeted inhibitors with immune checkpoint inhibitors against different immune-related receptors) has been efficacious for improving patient responses. However, such increased effectiveness often leads to more severe AEs [34]. Therefore, further exploration of new targets and development of new therapeutic agents are still needed to maximize the benefits of treatment and minimize side effects.
An additional challenge for cancer therapy is the onset of resistance to treatment for nearly every type of drug. Investigations are ongoing to understand the underlying mechanisms in intrinsic and acquired resistance to therapeutics. Many of them are associated with the regulation of genes in redundant pathways and/or epigenetic regulation [217,238,292]. Understanding the loss of response to therapy at the molecular level will allow the design of novel therapeutic options based on rational strategies. Apart from that, the current paradigm for preclinical investigation of potential drug candidates—the ‘in vitro to in vivo’ model—is limited; the use of 3D culture approaches and suitable in vivo experimental models are expected to contribute significantly to assessment and the better prediction of long-term outcomes for many candidate therapeutics.
Author Contributions
Conceptualization, A.F. and S.C.; writing—original draft preparation, A.F. and K.E.; writing—review and editing, A.F., K.E. and S.C.; supervision, S.C.; funding acquisition, S.C. All authors have read and agreed to the published version of the manuscript.
Funding
We wish to acknowledge the support of the National Cancer Institute Small Business Innovation Research (R44CA156781), the VA Merit Award (101BX003742) and the VA RCS Award (1lK6Bx006319).
Acknowledgments
We would also like to thank Christina Marinaro for her help with editing the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures 2023; American Cancer Society: Atlanta, GA, USA, 2023. [Google Scholar]
- American Society of Clinical Oncology Cancer. Net Melanoma Statistics. Available online: https://www.cancer.net/cancer-types/melanoma/statistics (accessed on 15 May 2023).
- Jung, S.; Johnson, D.B. Management of Acral and Mucosal Melanoma: Medical Oncology Perspective. Oncologist 2022, 27, 703–710. [Google Scholar] [CrossRef]
- American Cancer Society Risk Factors for Melanoma Skin Cancer. Available online: https://www.cancer.org/cancer/types/melanoma-skin-cancer/causes-risks-prevention/risk-factors.html (accessed on 15 May 2023).
- American Cancer Society Skin Cancer (Non-Melanoma): Risk Factors and Prevention. Available online: https://www.cancer.net/cancer-types/skin-cancer-non-melanoma/risk-factors-and-prevention (accessed on 15 May 2023).
- Massand, S.; Neves, R.I. Emerging Therapies in the Treatment of Advanced Melanoma. Clin. Plast. Surg. 2021, 48, 713–733. [Google Scholar] [CrossRef]
- Tétu, P.; Baroudjian, B.; Lebbe, C. Targeting BRAF and MEK inhibitors in melanoma in the metastatic, neoadjuvant and adjuvant setting. Curr. Opin. Oncol. 2020, 32, 85–90. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF Heterogeneity in Melanoma. Curr. Treat. Options Oncol. 2021, 22, 20. [Google Scholar] [CrossRef]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Milagre, C.; Dhomen, N.; Geyer, F.C.; Hayward, R.; Lambros, M.; Reis-Filho, J.S.; Marais, R. A mouse model of melanoma driven by oncogenic KRAS. Cancer Res. 2010, 70, 5549–5557. [Google Scholar] [CrossRef]
- Shi, W. Role for radiation therapy in melanoma. Surg. Oncol. Clin. N. Am. 2015, 24, 323–335. [Google Scholar] [CrossRef]
- Powell, A.M.; Russell-Jones, R.; Barlow, R.J. Topical imiquimod immunotherapy in the management of lentigo maligna. Clin. Exp. Dermatol. 2004, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Teimouri, F.; Nikfar, S.; Abdollahi, M. Efficacy and side effects of dacarbazine in comparison with temozolomide in the treatment of malignant melanoma: A meta-analysis consisting of 1314 patients. Melanoma Res. 2013, 23, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Kirkwood, J.M. Re-evaluating the role of dacarbazine in metastatic melanoma: What have we learned in 30 years? Eur. J. Cancer 2004, 40, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Drugs Approved for Melanoma. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/melanoma (accessed on 27 March 2023).
- Velho, T.R. Metastatic melanoma-a review of current and future drugs. Drugs Context 2012, 2012, 212242. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M. Adjuvant therapy of melanoma with interferon: Lessons of the past decade. J. Transl. Med. 2008, 6, 62. [Google Scholar] [CrossRef]
- Cook, J.; Zitelli, J.A. Treating patients with melanoma with interferon. Arch. Dermatol. 1997, 133, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Search Orphan Drug Designations and Approvals. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=100696 (accessed on 27 March 2023).
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Weber, J.S.; Minor, D.R.; D’Angelo, S.P.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Grob, J.J.; et al. A phase 3 randomized, open-label study of nivolumab (anti-PD-1; BMS-936558; ONO-4538) versus investigator’s choice chemotherapy (ICC) in patients with advanced melanoma with prior anti-CTLA-4 therapy. In Proceedings of the Presented at the European Society for Medical Oncology 2014 Congress, Madrid, Spain, 26–30 September 2014. [Google Scholar]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- National Cancer Institute. FDA Approves Cobimetinib as Part of Drug Combination for Advanced Melanoma. 2015. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2015/cobimetinib-melanoma (accessed on 27 March 2023).
- Wang, C.; Lu, N.; Yan, L.; Li, Y. The efficacy and safety assessment of oncolytic virotherapies in the treatment of advanced melanoma: A systematic review and meta-analysis. Virol. J. 2023, 20, 252. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Approves Dabrafenib Plus Trametinib for Adjuvant Treatment of Melanoma with BRAF V600E or V600K Mutations. 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-plus-trametinib-adjuvant-treatment-melanoma-braf-v600e-or-v600k-mutations (accessed on 27 March 2023).
- U.S. Food & Drug Administration. FDA Approves Atezolizumab for BRAF V600 Unresectable or Metastatic Melanoma. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-braf-v600-unresectable-or-metastatic-melanoma (accessed on 27 March 2023).
- Song, S.; Han, M.; Zhang, H.; Wang, Y.; Jiang, H. Full screening and accurate subtyping of HLA-A*02 alleles through group-specific amplification and mono-allelic sequencing. Cell Mol. Immunol. 2013, 10, 490–496. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Au, L.; Larkin, J.; Turajlic, S. Relatlimab and nivolumab in the treatment of melanoma. Cell 2022, 185, 4866–4869. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Approves Melphalan as a Liver-Directed Treatment for Uveal Melanoma. 2023. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-melphalan-liver-directed-treatment-uveal-melanoma (accessed on 27 March 2023).
- FDA Press Announcement. FDA Approves First Cellular Therapy to Treat Patients with Unresectable or Metastatic Melanoma; U.S. Food and Drug Administration: Washington, DC, USA, 2024. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cellular-therapy-treat-patients-unresectable-or-metastatic-melanoma (accessed on 22 February 2024).
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Eisen, T.; Ahmad, T.; Flaherty, K.T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L.M.; et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Agarwala, S.S.; Trefzer, U.; Hogg, D.; Robert, C.; Hersey, P.; Eggermont, A.; Grabbe, S.; Gonzalez, R.; Gille, J.; et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J. Clin. Oncol. 2009, 27, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Koelblinger, P.; Thuerigen, O.; Dummer, R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 2018, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A., Jr.; Laquerre, S.G.; King, A.J.; et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies-update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsová, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients With BRAF V600-Mutant Melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Heinzerling, L.; Eigentler, T.K.; Fluck, M.; Hassel, J.C.; Heller-Schenck, D.; Leipe, J.; Pauschinger, M.; Vogel, A.; Zimmer, L.; Gutzmer, R. Tolerability of BRAF/MEK inhibitor combinations: Adverse event evaluation and management. ESMO Open 2019, 4, e000491. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef]
- Gandini, S.; Massi, D.; Mandalà, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98. [Google Scholar] [CrossRef]
- Strome, S.E.; Dong, H.; Tamura, H.; Voss, S.G.; Flies, D.B.; Tamada, K.; Salomao, D.; Cheville, J.; Hirano, F.; Lin, W.; et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003, 63, 6501–6505. [Google Scholar]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Ascierto, P.A.; Maio, M. Updated survival in patients (pts) with BRAF mutant melanoma administered pembrolizumab (pembro), dabrafenib (D), and trametinib. In Proceedings of the International Congress of the Society for Melanoma Research, Salt Lake City, UT, USA, 20–23 November 2019. [Google Scholar]
- Melchiorri, D.; Cappuccio, I.; Ciceroni, C.; Spinsanti, P.; Mosillo, P.; Sarichelou, I.; Sale, P.; Nicoletti, F. Metabotropic glutamate receptors in stem/progenitor cells. Neuropharmacology 2007, 53, 473–480. [Google Scholar] [CrossRef]
- Tong, Q.; Ouedraogo, R.; Kirchgessner, A.L. Localization and function of group III metabotropic glutamate receptors in rat pancreatic islets. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1324–E1333. [Google Scholar] [CrossRef]
- Amaria, R.N.; Postow, M.; Burton, E.M.; Tetzlaff, M.T.; Ross, M.I.; Torres-Cabala, C.; Glitza, I.C.; Duan, F.; Milton, D.R.; Busam, K.; et al. Neoadjuvant relatlimab and nivolumab in resectable melanoma. Nature 2022, 611, 155–160. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Achard, C.; Surendran, A.; Wedge, M.E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. eBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Lu, Y.G.; Wang, Y.Y.; Yang, Y.D.; Zhang, X.C.; Gao, Y.; Yang, Y.; Zhang, J.B.; Li, G.L. Efficacy of topical ALA-PDT combined with excision in the treatment of skin malignant tumor. Photodiagnosis Photodyn. Ther. 2014, 11, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Muto, M.; Minashi, K.; Iwasaki, J.; Kojima, T.; Fuse, N.; Doi, T.; Kaneko, K.; Ohtsu, A. Photodynamic therapy as salvage treatment for local failure after chemoradiotherapy in patients with esophageal squamous cell carcinoma: A phase II study. Int. J. Cancer 2012, 131, 1228–1234. [Google Scholar] [CrossRef]
- Lou, P.J.; Jäger, H.R.; Jones, L.; Theodossy, T.; Bown, S.G.; Hopper, C. Interstitial photodynamic therapy as salvage treatment for recurrent head and neck cancer. Br. J. Cancer 2004, 91, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Isola, V.; Pece, A.; Pierro, L. Photodynamic therapy with verteporfin of choroidal malignancy from breast cancer. Am. J. Ophthalmol. 2006, 142, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.B., 2nd; Cengel, K.A. Photodynamic therapy for lung cancer and malignant pleural mesothelioma. Semin. Oncol. 2014, 41, 820–830. [Google Scholar] [CrossRef]
- Salvio, A.G.; Stringasci, M.D.; Requena, M.B.; Fregolenti, B.A.; Medeiro, M.; Santos, R.G.; Bagnato, V.S. Long-term follow-up results of a pilot study for nodular basal cell carcinoma with PDT using partial home treatment protocol. Photodiagnosis Photodyn. Ther. 2024, 45, 103930. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Tan, L.C.; Dong, L.W.; Zhang, W.Q.; Shen, X.X.; Lu, X.; Zheng, H.; Lu, Y.G. Susceptibility and Resistance Mechanisms During Photodynamic Therapy of Melanoma. Front. Oncol. 2020, 10, 597. [Google Scholar] [CrossRef]
- Kim, M.M.; Ghogare, A.A.; Greer, A.; Zhu, T.C. On the in vivo photochemical rate parameters for PDT reactive oxygen species modeling. Phys. Med. Biol. 2017, 62, R1–R48. [Google Scholar] [CrossRef]
- Lim, M.E.; Lee, Y.L.; Zhang, Y.; Chu, J.J. Photodynamic inactivation of viruses using upconversion nanoparticles. Biomaterials 2012, 33, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Saji, H.; Song, W.; Furumoto, K.; Kato, H.; Engleman, E.G. Systemic antitumor effect of intratumoral injection of dendritic cells in combination with local photodynamic therapy. Clin. Cancer Res. 2006, 12, 2568–2574. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.F.; Chen, W.R.; Teague, T.K.; Perry, L.A.; Nordquist, R.E. In situ photoimmunotherapy: A tumour-directed treatment for melanoma. Br. J. Dermatol. 2006, 155, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Naylor, M.F.; Le, H.; Nordquist, R.E.; Teague, T.K.; Howard, C.A.; Murray, C.; Chen, W.R. Clinical effects of in situ photoimmunotherapy on late-stage melanoma patients: A preliminary study. Cancer Biol. Ther. 2010, 10, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.H.; Youness, R.A.; Sebak, A.A.; Handoussa, H. Phytochemical-derived tumor-associated macrophage remodeling strategy using Phoenix dactylifera L. boosted photodynamic therapy in melanoma via H19/iNOS/PD-L1 axis. Photodiagnosis Photodyn. Ther. 2023, 44, 103792. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Photodynamic Therapy for Metastatic Melanoma Treatment: A Review. Technol. Cancer Res. Treat. 2018, 17, 1533033818791795. [Google Scholar] [CrossRef]
- Carbó-Bagué, A.; Rubió-Casadevall, J.; Puigdemont, M.; Sanvisens, A.; Oliveras, G.; Coll, M.; Del Olmo, B.; Perez-Bueno, F.; Marcos-Gragera, R. Epidemiology and Molecular Profile of Mucosal Melanoma: A Population-Based Study in Southern Europe. Cancers 2022, 14, 780. [Google Scholar] [CrossRef] [PubMed]
- Kolla, A.M.; Vitiello, G.A.; Friedman, E.B.; Sun, J.; Potdar, A.; Daou, H.; Farrow, N.E.; Farley, C.R.; Vetto, J.T.; Han, D.; et al. Acral Lentiginous Melanoma: A United States Multi-Center Substage Survival Analysis. Cancer Control 2021, 28, 10732748211053567. [Google Scholar] [CrossRef] [PubMed]
- Sureda, N.; Phan, A.; Poulalhon, N.; Balme, B.; Dalle, S.; Thomas, L. Conservative surgical management of subungual (matrix derived) melanoma: Report of seven cases and literature review. Br. J. Dermatol. 2011, 165, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.E.; Han, G.; Gabrick, K.; Kanouzi, J.; Lo, Y.C.; Galan, A.; Clune, J.; Narayan, D.; Ariyan, S.; Han, D. Acral Lentiginous Melanoma: Do Surgical Approach and Sentinel Lymph Node Biopsy Matter? Plast. Reconstr. Surg. Glob. Open 2020, 8, e2698. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Suciu, S.; Testori, A.; Santinami, M.; Kruit, W.H.; Marsden, J.; Punt, C.J.; Salès, F.; Dummer, R.; Robert, C.; et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J. Clin. Oncol. 2012, 30, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Si, L.; Chi, Z.; Cui, C.; Sheng, X.; Li, S.; Tang, B.; Guo, J. A randomised phase II trial of 1 month versus 1 year of adjuvant high-dose interferon α-2b in high-risk acral melanoma patients. Eur. J. Cancer 2011, 47, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Lerner, B.A.; Stewart, L.A.; Horowitz, D.P.; Carvajal, R.D. Mucosal Melanoma: New Insights and Therapeutic Options for a Unique and Aggressive Disease. Oncology 2017, 31, e23–e32. [Google Scholar]
- Bai, X.; Mao, L.L.; Chi, Z.H.; Sheng, X.N.; Cui, C.L.; Kong, Y.; Dai, J.; Wang, X.; Li, S.M.; Tang, B.X.; et al. BRAF inhibitors: Efficacious and tolerable in BRAF-mutant acral and mucosal melanoma. Neoplasma 2017, 64, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Ito, T.; Kato, H.; Irie, H.; Kaji, T.; Maekawa, T.; Asai, J.; Yamamoto, Y.; Fujimura, T.; Nakai, Y.; et al. Outcome of combination therapy using BRAF and MEK inhibitors among Asian patients with advanced melanoma: An analysis of 112 cases. Eur. J. Cancer 2021, 145, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Montone, K.T.; van Belle, P.; Elenitsas, R.; Elder, D.E. Proto-oncogene c-kit expression in malignant melanoma: Protein loss with tumor progression. Mod. Pathol. 1997, 10, 939–944. [Google Scholar] [PubMed]
- Isabel Zhu, Y.; Fitzpatrick, J.E. Expression of c-kit (CD117) in Spitz nevus and malignant melanoma. J. Cutan. Pathol. 2006, 33, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Luca, M.; Gutman, M.; McConkey, D.J.; Langley, K.E.; Lyman, S.D.; Bar-Eli, M. Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene 1996, 13, 2339–2347. [Google Scholar] [PubMed]
- Slipicevic, A.; Herlyn, M. KIT in melanoma: Many shades of gray. J. Investig. Dermatol. 2015, 135, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Eton, O.; Davis, D.W.; Frazier, M.L.; McConkey, D.J.; Diwan, A.H.; Papadopoulos, N.E.; Bedikian, A.Y.; Camacho, L.H.; Ross, M.I.; et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br. J. Cancer 2008, 99, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Lawrence, D.P.; Weber, J.S.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase II Study of Nilotinib in Melanoma Harboring KIT Alterations Following Progression to Prior KIT Inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Li, D.; Wen, X.; Ding, Y.; Liu, X.; Jiang, H.; Huang, F.; Zhang, X. Adjuvant PD-1 inhibitor versus high-dose interferon α-2b for Chinese patients with cutaneous and acral melanoma: A retrospective cohort analysis. Dermatol. Ther. 2021, 34, e15067. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Wang, J.; Nadeem, Z.; Patel, N.; Postow, M.A.; Shoushtari, A.N.; Plitas, G.; Balachandran, V.P.; et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 2021, 53, 11–15. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Munhoz, R.R.; Kuk, D.; Ott, P.A.; Johnson, D.B.; Tsai, K.K.; Rapisuwon, S.; Eroglu, Z.; Sullivan, R.J.; Luke, J.J.; et al. The efficacy of anti-PD-1 agents in acral and mucosal melanoma. Cancer 2016, 122, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Klemen, N.D.; Wang, M.; Rubinstein, J.C.; Olino, K.; Clune, J.; Ariyan, S.; Cha, C.; Weiss, S.A.; Kluger, H.M.; Sznol, M. Survival after checkpoint inhibitors for metastatic acral, mucosal and uveal melanoma. J. Immunother. Cancer 2020, 8, e000341. [Google Scholar] [CrossRef] [PubMed]
- Ogata, D.; Haydu, L.E.; Glitza, I.C.; Patel, S.P.; Tawbi, H.A.; McQuade, J.L.; Diab, A.; Ekmekcioglu, S.; Wong, M.K.; Davies, M.A.; et al. The efficacy of anti-programmed cell death protein 1 therapy among patients with metastatic acral and metastatic mucosal melanoma. Cancer Med. 2021, 10, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, K.; Kiyohara, Y.; Takenouchi, T.; Uhara, H.; Uchi, H.; Yoshikawa, S.; Takatsuka, S.; Koga, H.; Wada, N.; Minami, H.; et al. Efficacy and safety of nivolumab in combination with ipilimumab in Japanese patients with advanced melanoma: An open-label, single-arm, multicentre phase II study. Eur. J. Cancer 2018, 105, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Ascierto, P.A.; Haanen, J.; Espinosa, E.; Demidov, L.; Garbe, C.; Guida, M.; Lorigan, P.; Chiarion-Sileni, V.; Gogas, H.; et al. Safety and efficacy of nivolumab in patients with rare melanoma subtypes who progressed on or after ipilimumab treatment: A single-arm, open-label, phase II study (CheckMate 172). Eur. J. Cancer 2019, 119, 168–178. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Larkin, J.; Sosman, J.A.; Lebbé, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J.C.; Hodi, F.S.; Lorigan, P.; et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J. Clin. Oncol. 2017, 35, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Zhang, X.; Shu, Y.; Pan, H.; Wu, D.; Liu, J.; Lou, F.; Mao, L.; Wang, X.; Wen, X.; et al. A Phase Ib Study of Pembrolizumab as Second-Line Therapy for Chinese Patients With Advanced or Metastatic Melanoma (KEYNOTE-151). Transl. Oncol. 2019, 12, 828–835. [Google Scholar] [CrossRef]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Carvajal, R.D. Treatment of Uveal Melanoma. Cancer Treat. Res. 2016, 167, 281–293. [Google Scholar] [CrossRef]
- Chattopadhyay, C.; Kim, D.W.; Gombos, D.S.; Oba, J.; Qin, Y.; Williams, M.D.; Esmaeli, B.; Grimm, E.A.; Wargo, J.A.; Woodman, S.E.; et al. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Sacco, J.J.; Jager, M.J.; Eschelman, D.J.; Olofsson Bagge, R.; Harbour, J.W.; Chieng, N.D.; Patel, S.P.; Joshua, A.M.; Piperno-Neumann, S. Advances in the clinical management of uveal melanoma. Nat. Rev. Clin. Oncol. 2023, 20, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Piperno-Neumann, S.; Rutkowski, P.; Baurain, J.F.; Schlaak, M.; Butler, M.O.; Sullivan, R.J.; Dummer, R.; Kirkwood, J.M.; Orloff, M.; et al. Three-Year Overall Survival with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2023, 389, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Arcella, A.; Iacovelli, L.; Battaglia, G.; Giangaspero, F.; Melchiorri, D. Metabotropic glutamate receptors: New targets for the control of tumor growth? Trends Pharmacol. Sci. 2007, 28, 206–213. [Google Scholar] [CrossRef]
- Storto, M.; Capobianco, L.; Battaglia, G.; Molinaro, G.; Gradini, R.; Riozzi, B.; Di Mambro, A.; Mitchell, K.J.; Bruno, V.; Vairetti, M.P.; et al. Insulin secretion is controlled by mGlu5 metabotropic glutamate receptors. Mol. Pharmacol. 2006, 69, 1234–1241. [Google Scholar] [CrossRef]
- Morimoto, R.; Uehara, S.; Yatsushiro, S.; Juge, N.; Hua, Z.; Senoh, S.; Echigo, N.; Hayashi, M.; Mizoguchi, T.; Ninomiya, T.; et al. Secretion of L-glutamate from osteoclasts through transcytosis. EMBO J. 2006, 25, 4175–4186. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate signaling in benign and malignant disorders: Current status, future perspectives, and therapeutic implications. Int. J. Biol. Sci. 2013, 9, 728–742. [Google Scholar] [CrossRef]
- Fazzari, J.; Linher-Melville, K.; Singh, G. Tumour-Derived Glutamate: Linking Aberrant Cancer Cell Metabolism to Peripheral Sensory Pain Pathways. Curr. Neuropharmacol. 2017, 15, 620–636. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, microRNAs and glutamine addiction in cancers. Cell Cycle 2009, 8, 3243–3245. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, A.P.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Eddin, M.N.; Fateeva, A.; Pompili, S.V.B.; Shah, R.; Doshi, S.; Chen, S. Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression. Cells 2022, 11, 2857. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.S.; et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, H.; Wetzel, W.J.; Philbert, M.A. Spontaneous melanocytosis in transgenic mice. J. Investig. Dermatol. 1996, 106, 1145–1151. [Google Scholar] [CrossRef]
- Zhu, H.; Reuhl, K.; Zhang, X.; Botha, R.; Ryan, K.; Wei, J.; Chen, S. Development of heritable melanoma in transgenic mice. J. Investig. Dermatol. 1998, 110, 247–252. [Google Scholar] [CrossRef]
- Chen, S.; Teicher, L.C.; Kazim, D.; Pollack, R.E.; Wise, L.S. Commitment of mouse fibroblasts to adipocyte differentiation by DNA transfection. Science 1989, 244, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Colón-Teicher, L.; Wise, L.S.; Martino, J.J.; Baskin, L.; Sakoulas, G.; Pollack, R.E.; Chen, S. Genomic sequences capable of committing mouse and rat fibroblasts to adipogenesis. Nucleic Acids Res. 1993, 21, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- Funusaka, Y.H.; Harada, T.; Aiba, A.; Nishigori, C. Expression of metabotropic glutamate receptor 1 and phosphorylated extracellular signal-regulated kinase 1/2 proteins in human melanocytic lesions. Pigm. Cell Res. 2006, 19, 256. [Google Scholar]
- Wangari-Talbot, J.; Wall, B.A.; Goydos, J.S.; Chen, S. Functional effects of GRM1 suppression in human melanoma cells. Mol. Cancer Res. 2012, 10, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, J.; Shin, S.S.; Lee, H.J.; Marín, Y.E.; Wall, B.A.; Goydos, J.S.; Chen, S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007, 67, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.S.; Namkoong, J.; Wall, B.A.; Gleason, R.; Lee, H.J.; Chen, S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment Cell Melanoma Res. 2008, 21, 368–378. [Google Scholar] [CrossRef]
- Ohtani, Y.; Harada, T.; Funasaka, Y.; Nakao, K.; Takahara, C.; Abdel-Daim, M.; Sakai, N.; Saito, N.; Nishigori, C.; Aiba, A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene 2008, 27, 7162–7170. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Glutamatergic Signaling a Therapeutic Vulnerability in Melanoma. Cancers 2021, 13, 3874. [Google Scholar] [CrossRef]
- Shin, S.S.; Wall, B.A.; Goydos, J.S.; Chen, S. AKT2 is a downstream target of metabotropic glutamate receptor 1 (Grm1). Pigment Cell Melanoma Res. 2010, 23, 103–111. [Google Scholar] [CrossRef]
- Marín, Y.E.; Namkoong, J.; Cohen-Solal, K.; Shin, S.S.; Martino, J.J.; Oka, M.; Chen, S. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell Signal. 2006, 18, 1279–1286. [Google Scholar] [CrossRef]
- Wen, Y.; Li, J.; Koo, J.; Shin, S.S.; Lin, Y.; Jeong, B.S.; Mehnert, J.M.; Chen, S.; Cohen-Sola, K.A.; Goydos, J.S. Activation of the glutamate receptor GRM1 enhances angiogenic signaling to drive melanoma progression. Cancer Res. 2014, 74, 2499–2509. [Google Scholar] [CrossRef]
- Massoumi, R.; Kuphal, S.; Hellerbrand, C.; Haas, B.; Wild, P.; Spruss, T.; Pfeifer, A.; Fässler, R.; Bosserhoff, A.K. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 2009, 206, 221–232. [Google Scholar] [CrossRef]
- de Jel, M.M.; Schott, M.; Lamm, S.; Neuhuber, W.; Kuphal, S.; Bosserhoff, A.K. Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model. Oncogenesis 2019, 8, 56. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. Onco Targets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef]
- Marín, Y.E.; Wall, B.A.; Wang, S.; Namkoong, J.; Martino, J.J.; Suh, J.; Lee, H.J.; Rabson, A.B.; Yang, C.S.; Chen, S.; et al. Curcumin downregulates the constitutive activity of NF-kappaB and induces apoptosis in novel mouse melanoma cells. Melanoma Res. 2007, 17, 274–283. [Google Scholar] [CrossRef]
- Shah, R.; Singh, S.J.; Eddy, K.; Filipp, F.V.; Chen, S. Concurrent Targeting of Glutaminolysis and Metabotropic Glutamate Receptor 1 (GRM1) Reduces Glutamate Bioavailability in GRM1(+) Melanoma. Cancer Res. 2019, 79, 1799–1809. [Google Scholar] [CrossRef]
- Yip, D.; Le, M.N.; Chan, J.L.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin. Cancer Res. 2009, 15, 3896–3902. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Silk, A.W.; Lee, J.H.; Dudek, L.; Jeong, B.S.; Li, J.; Schenkel, J.M.; Sadimin, E.; Kane, M.; Lin, H.; et al. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (GRM1) signaling, in patients with advanced melanoma. Pigment Cell Melanoma Res. 2018, 31, 534–540. [Google Scholar] [CrossRef] [PubMed]
- van Kan, H.J.; Groeneveld, G.J.; Kalmijn, S.; Spieksma, M.; van den Berg, L.H.; Guchelaar, H.J. Association between CYP1A2 activity and riluzole clearance in patients with amyotrophic lateral sclerosis. Br. J. Clin. Pharmacol. 2005, 59, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Gupta, K.; Pelletier, J.C.; Isola, A.L.; Marinaro, C.; Rasheed, M.A.; Campagnolo, J.; Eddin, M.N.; Rossi, M.; Fateeva, A.; et al. A Spontaneous Melanoma Mouse Model Applicable for a longitudinal Chemotherapy and Immunotherapy Study. J. Investig. Dermatol. 2023, 143, 2007–2018.e6. [Google Scholar] [CrossRef]
- Wall, B.A.; Wangari-Talbot, J.; Shin, S.S.; Schiff, D.; Sierra, J.; Yu, L.J.; Khan, A.; Haffty, B.; Goydos, J.S.; Chen, S. Disruption of GRM1-mediated signalling using riluzole results in DNA damage in melanoma cells. Pigment Cell Melanoma Res. 2014, 27, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Cerchio, R., Jr.; Marinaro, C.; Foo, T.K.; Xia, B.; Chen, S. Nonhomologous end-joining repair is likely involved in the repair of double-stranded DNA breaks induced by riluzole in melanoma cells. Melanoma Res. 2020, 30, 303–308. [Google Scholar] [CrossRef]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef]
- Eddy, K.; Gupta, K.; Eddin, M.N.; Marinaro, C.; Putta, S.; Sauer, J., Jr.; Chaly, A.; Freeman, K.B.; Pelletier, J.C.; Fateeva, A.; et al. Assessing Longitudinal Treatment Efficacies and Alterations in Molecular Markers Associated with Glutamatergic Signaling and Immune Checkpoint Inhibitors in a Spontaneous Melanoma Mouse Model. JID Innov. 2024, 4, 100262. [Google Scholar] [CrossRef]
- Silk, A.W.; Saraiya, B.; Groisberg, R.; Chan, N.; Spencer, K.; Girda, E.; Shih, W.; Palmeri, M.; Saunders, T.; Berman, R.M.; et al. A phase Ib dose-escalation study of troriluzole (BHV-4157), an oral glutamatergic signaling modulator, in combination with nivolumab in patients with advanced solid tumors. Eur. J. Med. Res. 2022, 27, 107. [Google Scholar] [CrossRef]
- Medikonda, R.; Choi, J.; Pant, A.; Saleh, L.; Routkevitch, D.; Tong, L.; Belcaid, Z.; Kim, Y.H.; Jackson, C.M.; Jackson, C.; et al. Synergy between glutamate modulation and anti-programmed cell death protein 1 immunotherapy for glioblastoma. J. Neurosu. 2022, 136, 379–388. [Google Scholar] [CrossRef]
- Blyufer, A.; Lhamo, S.; Tam, C.; Tariq, I.; Thavornwatanayong, T.; Mahajan, S.S. Riluzole: A neuroprotective drug with potential as a novel anticancer agent (Review). Int. J. Oncol. 2021, 59, 95. [Google Scholar] [CrossRef] [PubMed]
- Rizaner, N.; Uzun, S.; Fraser, S.P.; Djamgoz, M.B.A.; Altun, S. Riluzole: Anti-invasive effects on rat prostate cancer cells under normoxic and hypoxic conditions. Basic Clin. Pharmacol. Toxicol. 2020, 127, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Ma, Y.; Lam, B.Q.; Shrivastava, A.; Srivastav, S.; Shankar, S.; Srivastava, R.K. Riluzole regulates pancreatic cancer cell metabolism by suppressing the Wnt-β-catenin pathway. Sci. Rep. 2022, 12, 11062. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.L.; Nassar, M.A.; Hachem, A.H.; Bukhsh, M.A.; Jafry, W.S.; Khansa, R.M.; Gorski, D.H. Riluzole mediates anti-tumor properties in breast cancer cells independent of metabotropic glutamate receptor-1. Breast Cancer Res. Treat. 2016, 157, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; He, X.; Jiang, X.; Tao, H. The new role of riluzole in the treatment of pancreatic cancer through the apoptosis and autophagy pathways. J. Cell Biochem. 2021, 122, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Lemieszek, M.K.; Stepulak, A.; Sawa-Wejksza, K.; Czerwonka, A.; Ikonomidou, C.; Rzeski, W. Riluzole Inhibits Proliferation, Migration and Cell Cycle Progression and Induces Apoptosis in Tumor Cells of Various Origins. Anticancer. Agents Med. Chem. 2018, 18, 565–572. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Medina, D.J.; Kareddula, A.; Paratala, B.; Rose, A.; Dhami, J.; Chen, S.; Ganesan, S.; Mackay, G.; Vazquez, A.; et al. Riluzole exerts distinct antitumor effects from a metabotropic glutamate receptor 1-specific inhibitor on breast cancer cells. Oncotarget 2017, 8, 44639–44653. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, K.; Shibata, M.A.; Ito, Y.; Sohma, Y.; Azuma, H.; Otsuki, Y. Riluzole induces apoptotic cell death in human prostate cancer cells via endoplasmic reticulum stress. Anticancer Res. 2009, 29, 2195–2204. [Google Scholar]
- Seol, H.S.; Lee, S.E.; Song, J.S.; Lee, H.Y.; Park, S.; Kim, I.; Singh, S.R.; Chang, S.; Jang, S.J. Glutamate release inhibitor, Riluzole, inhibited proliferation of human hepatocellular carcinoma cells by elevated ROS production. Cancer Lett. 2016, 382, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Shourideh, M.; Goodrich, D.W.; Koochekpour, S. Riluzole induces AR degradation via endoplasmic reticulum stress pathway in androgen-dependent and castration-resistant prostate cancer cells. Prostate 2019, 79, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Raghubir, M.; Rahman, C.N.; Fang, J.; Matsui, H.; Mahajan, S.S. Osteosarcoma growth suppression by riluzole delivery via iron oxide nanocage in nude mice. Oncol. Rep. 2020, 43, 169–176. [Google Scholar] [CrossRef]
- Raghubir, M.; Azeem, S.M.; Hasnat, R.; Rahman, C.N.; Wong, L.; Yan, S.; Huang, Y.Q.; Zhagui, R.; Blyufer, A.; Tariq, I.; et al. Riluzole-induced apoptosis in osteosarcoma is mediated through Yes-associated protein upon phosphorylation by c-Abl Kinase. Sci. Rep. 2021, 11, 20974. [Google Scholar] [CrossRef]
- Khan, A.J.; LaCava, S.; Mehta, M.; Schiff, D.; Thandoni, A.; Jhawar, S.; Danish, S.; Haffty, B.G.; Chen, S. The glutamate release inhibitor riluzole increases DNA damage and enhances cytotoxicity in human glioma cells, in vitro and in vivo. Oncotarget 2019, 10, 2824–2834. [Google Scholar] [CrossRef]
- Liao, S.; Ruiz, Y.; Gulzar, H.; Yelskaya, Z.; Ait Taouit, L.; Houssou, M.; Jaikaran, T.; Schvarts, Y.; Kozlitina, K.; Basu-Roy, U.; et al. Osteosarcoma cell proliferation and survival requires mGluR5 receptor activity and is blocked by Riluzole. PLoS ONE 2017, 12, e0171256. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, X.R.; Li, H.Y.; Zhao, Z.J.; Liao, Y.W.; Wang, X.Y.; Su, J.; Sang, S.S.; Liu, Q. Anti-cancer effect of metabotropic glutamate receptor 1 inhibition in human glioma U87 cells: Involvement of PI3K/Akt/mTOR pathway. Cell Physiol. Biochem. 2015, 35, 419–432. [Google Scholar] [CrossRef]
- Sperling, S.; Aung, T.; Martin, S.; Rohde, V.; Ninkovic, M. Riluzole: A potential therapeutic intervention in human brain tumor stem-like cells. Oncotarget 2017, 8, 96697–96709. [Google Scholar] [CrossRef]
- Yelskaya, Z.; Carrillo, V.; Dubisz, E.; Gulzar, H.; Morgan, D.; Mahajan, S.S. Synergistic inhibition of survival, proliferation, and migration of U87 cells with a combination of LY341495 and Iressa. PLoS ONE 2013, 8, e64588. [Google Scholar] [CrossRef]
- Yu, L.J.; Wall, B.A.; Chen, S. The current management of brain metastasis in melanoma: A focus on riluzole. Expert Rev. Neurother. 2015, 15, 779–792. [Google Scholar] [CrossRef]
- Wall, B.A.; Yu, L.J.; Khan, A.; Haffty, B.; Goydos, J.S.; Chen, S. Riluzole is a radio-sensitizing agent in an in vivo model of brain metastasis derived from GRM1 expressing human melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 105–109. [Google Scholar] [CrossRef]
- Spencer, K.R.; Portal, D.E.; Aisner, J.; Stein, M.N.; Malhotra, J.; Shih, W.; Chan, N.; Silk, A.W.; Ganesan, S.; Goodin, S.; et al. A phase I trial of riluzole and sorafenib in patients with advanced solid tumors: CTEP #8850. Oncotarget 2023, 14, 302–315. [Google Scholar] [CrossRef]
- Speyer, C.L.; Bukhsh, M.A.; Jafry, W.S.; Sexton, R.E.; Bandyopadhyay, S.; Gorski, D.H. Riluzole synergizes with paclitaxel to inhibit cell growth and induce apoptosis in triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 166, 407–419. [Google Scholar] [CrossRef]
- Fortunato, A. The role of hERG1 ion channels in epithelial-mesenchymal transition and the capacity of riluzole to reduce cisplatin resistance in colorectal cancer cells. Cell Oncol. 2017, 40, 367–378. [Google Scholar] [CrossRef]
- Benavides-Serrato, A.; Saunders, J.T.; Holmes, B.; Nishimura, R.N.; Lichtenstein, A.; Gera, J. Repurposing Potential of Riluzole as an ITAF Inhibitor in mTOR Therapy Resistant Glioblastoma. Int. J. Mol. Sci. 2020, 21, 344. [Google Scholar] [CrossRef]
- Yamada, T.; Tsuji, S.; Nakamura, S.; Egashira, Y.; Shimazawa, M.; Nakayama, N.; Yano, H.; Iwama, T.; Hara, H. Riluzole enhances the antitumor effects of temozolomide via suppression of MGMT expression in glioblastoma. J. Neurosurg. 2020, 134, 701–710. [Google Scholar] [CrossRef]
- Sun, L.; Wu, C.; Ming, J.; Nie, X.; Guo, E.; Zhang, W.; Hu, G. Riluzole Enhances the Response of Human Nasopharyngeal Carcinoma Cells to Ionizing Radiation via ATM/P53 Signalling Pathway. J. Cancer 2020, 11, 3089–3098. [Google Scholar] [CrossRef]
- Li, Z.; Qiao, X.; Liu, X.M.; Shi, S.H.; Qiao, X.; Xu, J.Y. Blocking xCT and PI3K/Akt pathway synergized with DNA damage of Riluzole-Pt(IV) prodrugs for cancer treatment. Eur. J. Med. Chem. 2023, 250, 115233. [Google Scholar] [CrossRef]
- Pasinelli, P.; Dena, J. Rethinking Drug Treatment Approaches in ALS by Targeting ABC Efflux Transporters; Annual Rept. Accession Number: ADA594930. Defenst Technical Information Center: Fort Belvoir, VA, USA, 2012.
- Mohamed, L.A.; Markandaiah, S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Blood-Brain Barrier Driven Pharmacoresistance in Amyotrophic Lateral Sclerosis and Challenges for Effective Drug Therapies. AAPS J. 2017, 19, 1600–1614. [Google Scholar] [CrossRef]
- Jablonski, M.; Miller, D.S.; Pasinelli, P.; Trotti, D. ABC transporter-driven pharmacoresistance in Amyotrophic Lateral Sclerosis. Brain Res. 2015, 1607, 1–14. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Markandaiah, S.S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Excess glutamate secreted from astrocytes drives upregulation of P-glycoprotein in endothelial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2019, 316, 27–38. [Google Scholar] [CrossRef]
- Chan, G.N.; Evans, R.A.; Banks, D.B.; Mesev, E.V.; Miller, D.S.; Cannon, R.E. Selective induction of P-glycoprotein at the CNS barriers during symptomatic stage of an ALS animal model. Neurosci. Lett. 2017, 639, 103–113. [Google Scholar] [CrossRef]
- Milane, A.; Fernandez, C.; Dupuis, L.; Buyse, M.; Loeffler, J.P.; Farinotti, R.; Meininger, V.; Bensimon, G. P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 472, 166–170. [Google Scholar] [CrossRef]
- Milane, A.; Fernandez, C.; Vautier, S.; Bensimon, G.; Meininger, V.; Farinotti, R. Minocycline and riluzole brain disposition: Interactions with p-glycoprotein at the blood-brain barrier. J. Neurochem. 2007, 103, 164–173. [Google Scholar] [CrossRef]
- Milane, A.; Vautier, S.; Chacun, H.; Meininger, V.; Bensimon, G.; Farinotti, R.; Fernandez, C. Interactions between riluzole and ABCG2/BCRP transporter. Neurosci. Lett. 2009, 452, 12–16. [Google Scholar] [CrossRef]
- Jablonski, M.R.; Markandaiah, S.S.; Jacob, D.; Meng, N.J.; Li, K.; Gennaro, V.; Lepore, A.C.; Trotti, D.; Pasinelli, P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann. Clin. Transl. Neurol. 2014, 1, 996–1005. [Google Scholar] [CrossRef]
- Mandalà, M.; Romano, E. Mechanisms of Drug Resistance in Cancer Therapy; Springer: Paris, France, 2018. [Google Scholar]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef]
- Whittaker, S.R.; Theurillat, J.P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef]
- Solit, D.B.; Rosen, N. Resistance to BRAF inhibition in melanomas. N. Engl. J. Med. 2011, 364, 772–774. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Kemper, K.; Krijgsman, O.; Kong, X.; Cornelissen-Steijger, P.; Shahrabi, A.; Weeber, F.; van der Velden, D.L.; Bleijerveld, O.B.; Kuilman, T.; Kluin, R.J.C.; et al. BRAF(V600E) Kinase Domain Duplication Identified in Therapy-Refractory Melanoma Patient-Derived Xenografts. Cell Rep. 2016, 16, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.H.; Kim, S.H.; Trousil, S.; Frederick, D.T.; Piris, A.; Yuan, P.; Cai, L.; Gu, L.; Li, M.; Lee, J.H.; et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat. Med. 2016, 22, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef]
- Haq, R.; Yokoyama, S.; Hawryluk, E.B.; Jönsson, G.B.; Frederick, D.T.; McHenry, K.; Porter, D.; Tran, T.N.; Love, K.T.; Langer, R.; et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. USA 2013, 110, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Smith, M.P.; Ferguson, J.; Arozarena, I.; Hayward, R.; Marais, R.; Chapman, A.; Hurlstone, A.; Wellbrock, C. Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J. Natl. Cancer Inst. 2013, 105, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Pinner, S.; Jordan, P.; Sharrock, K.; Bazley, L.; Collinson, L.; Marais, R.; Bonvin, E.; Goding, C.; Sahai, E. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009, 69, 7969–7977. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.; Chatterjee, A.; Eccles, M.R. The Slow Cycling Phenotype: A Growing Problem for Treatment Resistance in Melanoma. Mol. Cancer Ther. 2017, 16, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Vachtenheim, J.; Réda, J.; Horák, P.; Ondrušová, L. Inducibly decreased MITF levels do not affect proliferation and phenotype switching but reduce differentiation of melanoma cells. J. Cell Mol. Med. 2018, 22, 2240–2251. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Bristot, I.J.; Kehl Dias, C.; Chapola, H.; Parsons, R.B.; Klamt, F. Metabolic rewiring in melanoma drug-resistant cells. Crit. Rev. Oncol. Hematol. 2020, 153, 102995. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Das Thakur, M.; Fang, B.; Koomen, J.M.; Fedorenko, I.V.; John, J.K.; Tsao, H.; Flaherty, K.T.; Sondak, V.K.; Messina, J.L.; et al. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 2015, 5, 264–273. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An autophagy-driven pathway of ATP secretion supports the aggressive phenotype of BRAF(V600E) inhibitor-resistant metastatic melanoma cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef]
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Prêtre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. mTORC1/autophagy-regulated MerTK in mutant BRAFV600 melanoma with acquired resistance to BRAF inhibition. Oncotarget 2017, 8, 69204–69218. [Google Scholar] [CrossRef]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Pisanu, M.E.; Liguoro, D.; Cerri, A.; Costantini, S.; Capone, F.; Acunzo, M.; Romano, G.; Nigita, G.; et al. Reprogramming miRNAs global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ. 2019, 26, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Martínez, M.; Benito-Jardón, L.; Alonso, L.; Koetz-Ploch, L.; Hernando, E.; Teixidó, J. miR-204-5p and miR-211-5p Contribute to BRAF Inhibitor Resistance in Melanoma. Cancer Res. 2018, 78, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, M.; Tuccoli, A.; D’Aurizio, R.; Sarti, S.; Giannecchini, L.; Lubrano, S.; Marranci, A.; Evangelista, M.; Peppicelli, S.; Ippolito, C.; et al. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget 2017, 8, 25395–25417. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Rajczykowski, M.; Widłak, W. Biological Factors behind Melanoma Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4071. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Wolchok, J.D.; Schadendorf, D.; Larkin, J.; Long, G.V.; Qian, X.; Saci, A.; Young, T.C.; Srinivasan, S.; Chang, H.; et al. TMB and Inflammatory Gene Expression Associated with Clinical Outcomes following Immunotherapy in Advanced Melanoma. Cancer Immunol. Res. 2021, 9, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, X.; Zhang, H.; Liang, Y.; Li, L.; Wei, H.; Sun, W.; Wang, Y. Prognostic Role of Tumor Mutational Burden in Cancer Patients Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 706652. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef]
- Wang, L.; Chen, F.; Liu, R.; Shi, L.; Zhao, G.; Yan, Z. Gene expression and immune infiltration in melanoma patients with different mutation burden. BMC Cancer 2021, 21, 379. [Google Scholar] [CrossRef]
- Abbott, C.W.; Boyle, S.M.; Pyke, R.M.; McDaniel, L.D.; Levy, E.; Navarro, F.C.P.; Mellacheruvu, D.; Zhang, S.V.; Tan, M.; Santiago, R.; et al. Prediction of Immunotherapy Response in Melanoma through Combined Modeling of Neoantigen Burden and Immune-Related Resistance Mechanisms. Clin. Cancer Res. 2021, 27, 4265–4276. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef]
- Huo, J.L.; Wang, Y.T.; Fu, W.J.; Lu, N.; Liu, Z.S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. LAG3 inhibition improves outcomes. Nat. Rev. Clin. Oncol. 2022, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Postow, M.A.; Adamow, M.; Arora, A.; Hannum, M.; Maher, C.; Wong, P.; Curran, M.A.; Hollmann, T.J.; Jia, L.; et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabf5107. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Machiraju, D.; Schäfer, S.; Hassel, J.C. Potential Reasons for Unresponsiveness to Anti-PD1 Immunotherapy in Young Patients with Advanced Melanoma. Life 2021, 11, 1318. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Bald, T.; Landsberg, J.; Lopez-Ramos, D.; Renn, M.; Glodde, N.; Jansen, P.; Gaffal, E.; Steitz, J.; Tolba, R.; Kalinke, U.; et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014, 4, 674–687. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Kent, M.S.; Grossenbacher, S.K.; Mall, C.; Zamora, A.E.; Mirsoian, A.; Chen, M.; Kol, A.; Shiao, S.L.; Reddy, A.; et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clin. Cancer Res. 2016, 22, 4328–4340. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [PubMed]
- Falahat, R.; Perez-Villarroel, P.; Mailloux, A.W.; Zhu, G.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. STING Signaling in Melanoma Cells Shapes Antigenicity and Can Promote Antitumor T-cell Activity. Cancer Immunol. Res. 2019, 7, 1837–1848. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, T.; Endo, R.; Sasaki, S.; Takahashi, N.; Sato, Y.; Hyodo, M.; Hayakawa, Y.; Harashima, H. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J. Immunother. Cancer 2021, 9, e002852. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Dai, J.; Tang, L.; Yang, L.; Si, L.; Sheng, X.; Cui, C.; Chi, Z.; Kong, Y.; Guo, J. EZH2 Inhibitor Enhances the STING Agonist—Induced Antitumor Immunity in Melanoma. J. Investig. Dermatol. 2022, 142, 1158–1170.e1158. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, 323. [Google Scholar] [CrossRef] [PubMed]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Tumeh, P.C. Cancer therapy: Tumours switch to resist. Nature 2012, 490, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Shirley, C.A.; Ahmad, N. Mechanisms of Immunotherapy Resistance in Cutaneous Melanoma: Recognizing a Shapeshifter. Front. Oncol. 2022, 12, 880876. [Google Scholar] [CrossRef]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef]
- Lazaro-Camp, V.J.; Salari, K.; Meng, X.; Yang, S. SETDB1 in cancer: Overexpression and its therapeutic implications. Am. J. Cancer Res. 2021, 11, 1803–1827. [Google Scholar]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Melanoma therapies currently approved by the FDA. Some elements of the figure were created with BioRender.
Figure 1.
Melanoma therapies currently approved by the FDA. Some elements of the figure were created with BioRender.

Figure 2.
Molecular signaling pathways in mGluR1-positive melanoma cells. Some elements of the figure were created with BioRender.
Figure 2.
Molecular signaling pathways in mGluR1-positive melanoma cells. Some elements of the figure were created with BioRender.

Figure 3.
Process of resistance development and molecules likely to participate in the emergence of resistance. Some elements of the figure were created with BioRender.
Figure 3.
Process of resistance development and molecules likely to participate in the emergence of resistance. Some elements of the figure were created with BioRender.

Figure 4.
Some of the possible mechanisms of resistance to BRAF and MEK inhibitors. (A) Silencing of MAPK signaling pathway using BRAF/MEK inhibitors can lead to the activation of PI3K/AKT signaling pathway, resulting in circumvention of the inhibitors’ effect. (B–D) Mechanisms of BRAF signaling activation in the presence of a BRAF inhibitor and different levels of RAS. Some elements of the figure were created with BioRender.
Figure 4.
Some of the possible mechanisms of resistance to BRAF and MEK inhibitors. (A) Silencing of MAPK signaling pathway using BRAF/MEK inhibitors can lead to the activation of PI3K/AKT signaling pathway, resulting in circumvention of the inhibitors’ effect. (B–D) Mechanisms of BRAF signaling activation in the presence of a BRAF inhibitor and different levels of RAS. Some elements of the figure were created with BioRender.


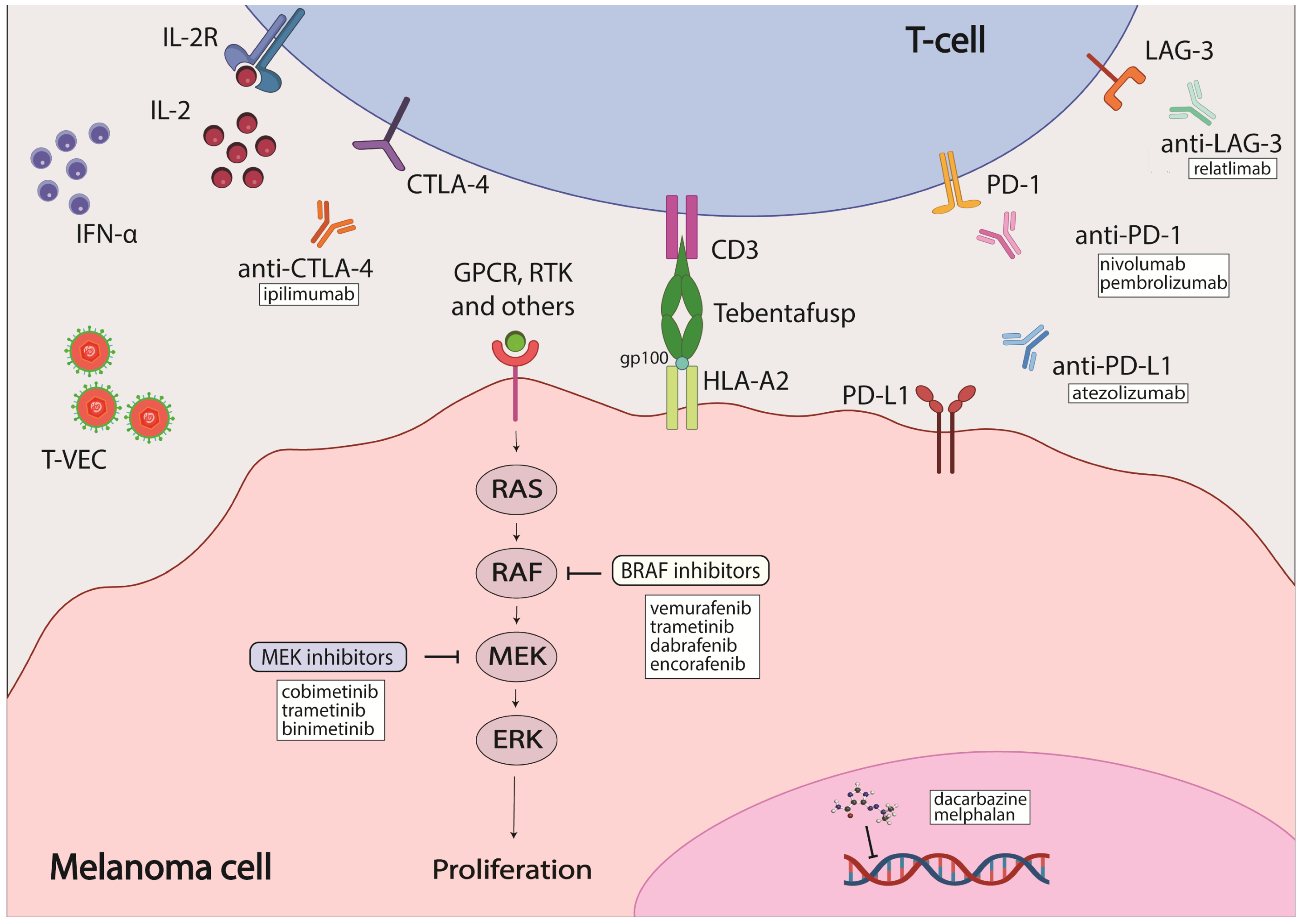{kind=link}
{kind=link}
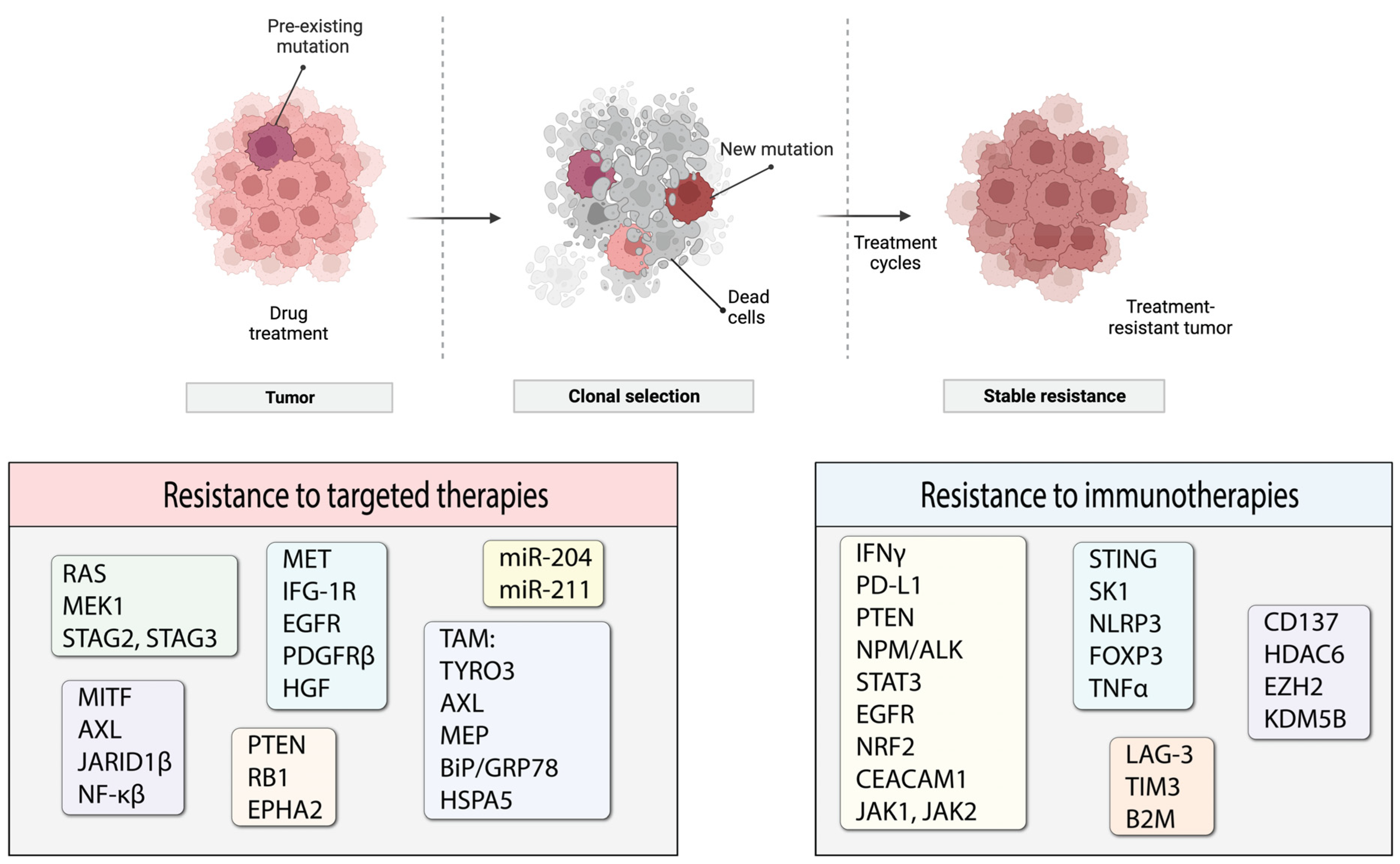{kind=link}
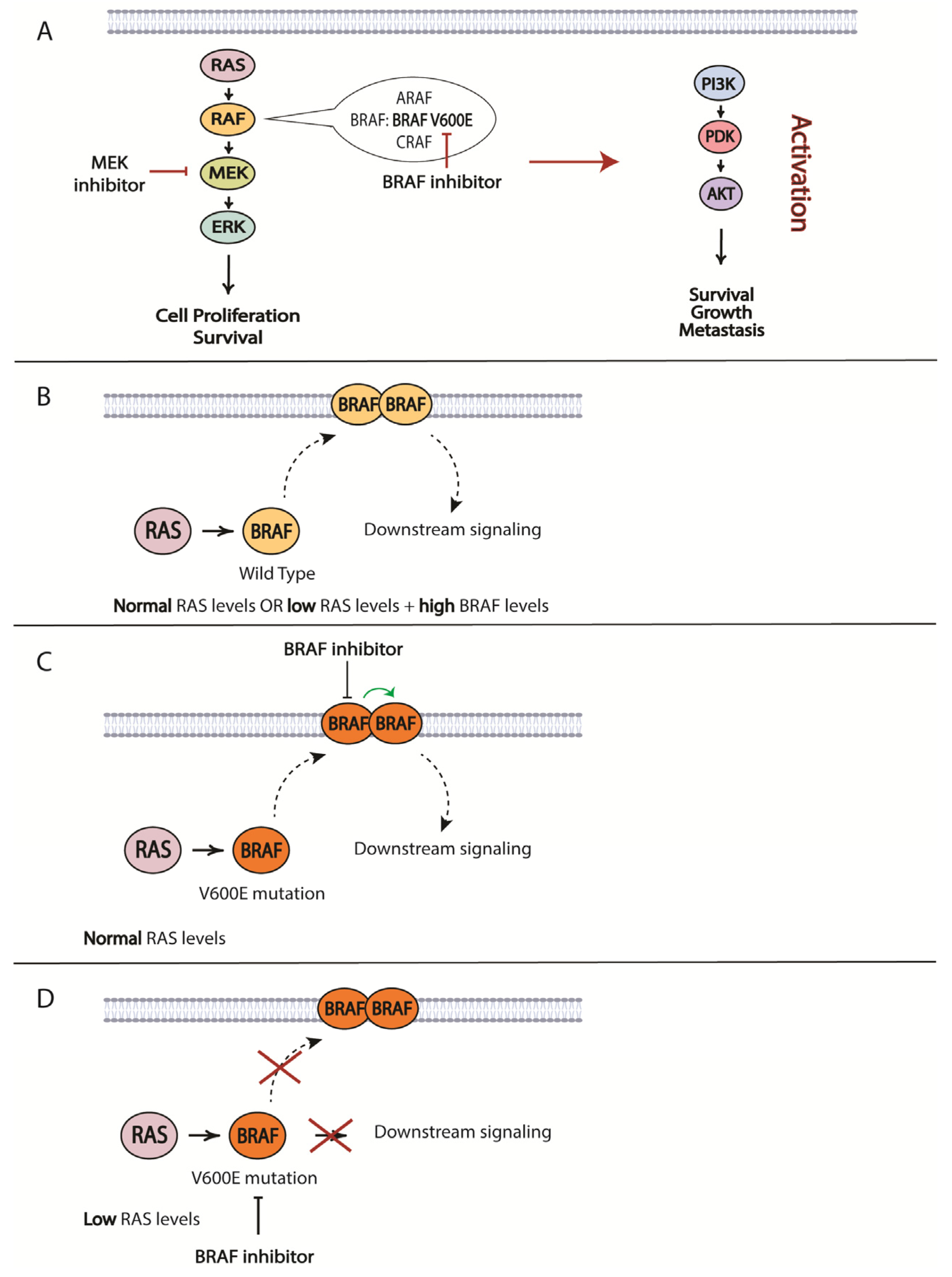{kind=link}
Table 1.
Therapeutics currently approved by the FDA for treatment of patients with melanoma tumors [18].
Table 1.
Therapeutics currently approved by the FDA for treatment of patients with melanoma tumors [18].
| Name | Therapy Type | Disease Conditions | Year of Approval | Details | Limitations | References |
|---|---|---|---|---|---|---|
| Dacarbazine | Chemotherapy |
| 1975 | DNA-alkylating agent |
| [16,19] |
| Recombinant Interferon Alfa-2b | Immunotherapy |
| 1995 | Recombinant form of the protein interferon alpha-2 |
| [20,21] |
| Aldesleukin | Immunotherapy |
| 1998 | Human recombinant interleukin-2 |
| [22,23] |
| Vemurafenib | Targeted therapy |
| 2011 | BRAF inhibitor |
| [24,25,26] |
| Ipilimumab | Immunotherapy |
| 2011 | Antibody against CTLA-4 |
| [27,28] |
| Nivolumab | Immunotherapy |
| 2014 | Antibody against PD-1 |
| [29,30] |
| Pembrolizumab | Immunotherapy |
| 2014 | Antibody against PD-1 |
| [31] |
| Cobimetinib + vemurafenib | Targeted therapy |
| 2015 | MEK inhibitor + BRAF inhibitor |
| [32] |
| Talimogene Laherparepvec (T-VEC) | Immunotherapy |
| 2015 | Oncolytic virus |
| [33] |
| Ipilimumab + nivolumab | Immunotherapy |
| 2015 | Antibody against CTLA-4 + antibody against PD-1 |
| [34] |
| Binimetinib + Encorafenib | Targeted therapy |
| 2018 | MEK inhibitor + BRAF inhibitor |
| [23] |
| Dabrafenib + trametinib | Targeted therapy |
| 2018 | BRAF inhibitor + MEK inhibitor |
| [35,36] |
| Atezolizumab + cobimetinib + vemurafenib | Immunotherapy + targeted therapy |
| 2020 | Antibody against PD-L1 + MEK inhibitor + BRAF inhibitor |
| [37] |
| Tebentafusp-tebn | Immunotherapy |
| 2022 | T-cell receptor-bispecific molecule that targets both glycoprotein 100 and CD3 |
| [38] |
| Nivolumab + relatlimab | Immunotherapy |
| 2022 | Antibody against PD-1 + antibody against LAG-3 |
| [39,40] |
| Melphalan | Chemotherapy |
| 2023 | DNA-alkylating agent |
| [41] |
| Lifileucel | Cellular therapy |
| 2024 | Tumor-derived autologous T-cell immunotherapy |
| [42] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fateeva, A.; Eddy, K.; Chen, S. Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance. Cancers 2024, 16, 1571. https://doi.org/10.3390/cancers16081571
AMA Style
Fateeva A, Eddy K, Chen S. Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance. Cancers. 2024; 16(8):1571. https://doi.org/10.3390/cancers16081571
Chicago/Turabian StyleFateeva, Anna, Kevinn Eddy, and Suzie Chen. 2024. "Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance" Cancers 16, no. 8: 1571. https://doi.org/10.3390/cancers16081571
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.