HPV DNA Integration at Actionable Cancer-Related Genes Loci in HPV-Associated Carcinomas


1
Department of Pathology, Centre Hospitalier Intercommunal de Créteil, 40, Avenue de Verdun, 94010 Créteil, France
2
Institut Curie, PSL Research University, CNRS, UMR 144, 75005 Paris, France
*
Author to whom correspondence should be addressed.
Cancers 2024, 16(8), 1584; https://doi.org/10.3390/cancers16081584
Submission received: 8 March 2024
/
Revised: 16 April 2024
/
Accepted: 17 April 2024
/
Published: 20 April 2024
(This article belongs to the Special Issue Potential Therapeutic Targets and Non-invasive Procedures for Improving the Treatment and Follow-Up of HPV-Associated Malignancies)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
In many tumor types, the development of personalized treatments has been allowed by the optimal molecular characterization of tumor cell genome alterations. In most carcinomas associated with human papillomaviruses (HPV), the integration of part of the viral genome into the tumor cell genome may lead to alterations of cancer-related genes located at the integration locus. In order to assess the overall frequency of such an event, we analyzed a large series of cases for which HPV integration sites had been determined. We found that 40% of the genes located at highly (≥3) recurrent HPV insertions corresponded to cancer-related genes. Moreover, about one-third of the genes targeted by HPV insertions correspond to actionable targets. These observations should lead to a more systematic analysis of HPV DNA integration patterns in HPV-associated carcinomas in order to identify highly specific tumor markers and develop personalized anti-tumor therapies.
Abstract
In HPV-associated carcinomas, some examples of cancer-related genes altered by viral insertion and corresponding to potential therapeutic targets have been described, but no quantitative assessment of these events, including poorly recurrent targets, has been reported to date. To document these occurrences, we built and analyzed a database comprised of 1455 cases, including HPV genotypes and tumor localizations. Host DNA sequences targeted by viral integration were classified as “non-recurrent” (one single reported case; 838 loci), “weakly recurrent” (two reported cases; 82 loci), and highly recurrent (≥3 cases; 43 loci). Whereas the overall rate of cancer-related target genes was 3.3% in the Gencode database, this rate increased to 6.5% in “non-recurrent”, 11.4% in “weakly recurrent”, and 40.1% in “highly recurrent” genes targeted by integration (p = 4.9 × 10−4). This rate was also significantly higher in tumors associated with high-risk HPV16/18/45 than other genotypes. Among the genes targeted by HPV insertion, 30.2% corresponded to direct or indirect druggable targets, a rate rising to 50% in “highly recurrent” targets. Using data from the literature and the DepMap 23Q4 release database, we found that genes targeted by viral insertion could be new candidates potentially involved in HPV-associated oncogenesis. A more systematic characterization of HPV/host fusion DNA sequences in HPV-associated cancers should provide a better knowledge of HPV-driven carcinogenesis and favor the development of personalize patient treatments.
1. Introduction
A subset of human papillomaviruses (HPV) genotypes, classified as carcinogenic to humans (group 1) (HPV16, 18), probably carcinogenic (group 2A) (HPV31, 33), and possibly carcinogenic (group 2B) (some types other than HPV16, 18, 31, 33) [1], are able to infect the mucosa of the ano-genital and oro-pharyngeal tracts and express oncogenic properties, among which HPV16 and HPV18 are the most prevalent [2,3]. Despite the development of prophylactic vaccines against potentially oncogenic HPVs, HPV-associated carcinomas remain a health burden worldwide [4,5]. HPVs are associated with most cervical [6], vaginal [7], and anal [8,9] cancers and with 25–35% of head and neck (H&N) carcinomas [4] recently estimated to be more than 50% in Western countries [10]. At the molecular level, the HPV genome consists of a 7.8Kbp double-stranded circular DNA molecule with oncogenic properties largely related to the E6 and E7 genes. These encoded proteins have the ability to inhibit the p53 and pRB host tumor suppressors, respectively, as well as many other cellular protein targets [11]. In preinvasive lesions, the viral genome commonly replicates extra-chromosomally in the cell nucleus, whereas, in most invasive carcinomas, at least a portion of the viral DNA is stably integrated into a unique or within a few sites in the tumor cell genome [12,13]. These observations led to the hypothesis that integration might represent a step in HPV-related oncogenesis [14]. Upon integration, there is often a disruption of the E1/E2 genes, which causes constitutive expression of the E6/E7 viral oncoproteins [15]. Further knowledge of the consequences of HPV integration in the cell genome progressively improved with the development of several technologies allowing both localization of the viral inserts and analysis of target sequences. Early reports based on in situ hybridization (ISH) on chromosomes obtained from cervical tumor-derived cell lines demonstrated the localization of HPV inserts at common fragile sites [16,17,18,19]. Using cloned cellular sequences adjacent to viral insertions as probes, HPV18 DNA was found 40Kbp upstream of MYC in HeLa cells, raising the hypothesis that HPV insertion can act in oncogenesis [20]. The identification of MYC as a recurrent integration site for HPV16/18 [21] and the observation of HPV insertions at translocation breakpoints [22] further supported the hypothesis of oncogene cis-activation via the insertion of viral DNA. However, the data remained scarce due to the bias introduced by the laborious cell culture steps and by the limited resolution of ISH.
Studies using PCR-based approaches, allowing the analysis of a large number of cases, confirmed that the HPV integration process was a clonal event, more frequent in invasive than in intraepithelial neoplasia, and could thus act in tumor progression [23,24,25]. A wide dispersion of mapped viral insertions throughout the genome was observed, frequently near fragile sites, but alterations of cellular genes located at the insertion sites were rarely observed [24,25]. Nevertheless, certain studies underlined the non-random distribution of HPV inserts in the genome [26], while others observed that host genome regions with high transcriptional activity were preferentially targeted by HPV insertions [27,28]. It was further shown that HPV integration frequently targeted genes [29] and was associated with structural alterations of the cell genome [30]. Some examples of gene activation [31,32] and gene inactivation [33,34] through HPV insertion were reported.
The era of NGS has profoundly modified the approach by allowing, in the same experiment, the characterization of both the entire viral sequences and the pattern of the viral/host fusion sequences. A series of cervical [35,36], H&N [37,38,39], anal [40], and vulvar [41] cancers could be analyzed using multiplex capture of HPV inserts. Improved accuracy of the rate of HPV integration in cervical tumors was obtained, ranging from 71.4% [35] to 92.2% [42] (mean 387/446 = 86.8%) and up to 100% in HPV18-associated tumors [36,43]. This rate was 60%-77% in H&N [37,38,39,44] and 54.8% in anal cancers [40]. The proportion of multiple viral insertions in individual cases could also be assessed more precisely. In the TCGA analysis [43], one single viral integration site was observed in 64% of the cases, two sites in 25%, and more than two in 11%. These studies also confirmed the integration hot spots previously reported (MYC/PVT1) and identified new recurrent insertions targeting TP63, RAD51B, MACROD2, KLF5/KLF12, PDL1/PDL2/PLGRKT, and TTC6/MIPOL1 [35,36,37,38,39].
The co-analysis of genomic and expression patterns has further reinforced the knowledge of the molecular consequences of HPV integration in tumor cells [37,39,42,43,45,46,47]. Fusion HPV genome transcripts included known or predicted genes in 70% of the integration events [43], and overexpression of the genes located near integrated viral sequences was detected in about 50% of the cases [37,45,46,47]. High transcriptional changes, preferentially observed in tumors harboring viral inserts in exonic regions [37], might be related in part to local genomic amplification upon HPV insertion, but cases of overexpression without amplification were also observed [45]. One study found silent and scattered viral inserts in tumors with multiple insertions [42]. These studies also provided some examples of gene disruption via viral insertion within RAD51B, implicated in homologous DNA repair mechanisms [39].
Several recent reviews have summarized the accumulated viral integration data in HPV-associated tumors [47,48,49,50,51]. In 2016, the studies performed by Zhang et al. [51] and Bodelon et al. [48] based on the analysis of 499 and 1.500 integration events, respectively, found that HPV DNA preferentially integrated into gene-dense regions, close to enhancers in transcriptionally active regions of the human genome and in intragenic loci. Most of the recurrent target genes in chromosomal hot spots were functionally cancer-related. In 2021, Warburton et al. [50] investigated 1.418 integration breakpoints from cervical and H&N carcinomas and observed multiple clustered HPV integration breakpoints associated with amplified regions of the host genome encompassing gene loci related to cell development and identity. Expression analyses showed that the majority of tumors had only one single transcriptionally active driver integration locus. HPV integration breakpoints were enriched at both FANCD2-associated fragile sites and enhancer-rich regions. The authors stressed that the driver of oncogenesis via HPV integrants requires a combination of events dependent on the genetic and/or epigenetic landscape of the flanking sequences.
Altogether, these studies have provided unambiguous data, indicating that HPV DNA insertion could act in oncogenesis via a direct interaction with the cell genome. However, it is not clear whether this model of cancer-related gene alteration via HPV insertion is mainly restricted to a limited number of observations revealed by their recurrency or corresponds to a more general mechanism also commonly implicated at multiple sites with low recurrency levels and may, therefore, constitute a rational basis for new strategies of therapeutic intervention. This led us to constitute and analyze a large database in order to (I) define whether the rate of multiple vs. single integration pattern within the same tumor was dependent on HPV genotypes; (II) analyze whether recurrent HPV insertions at certain host genome targets were related to a specific HPV genotype and/or tumor localization; (III) evaluate the rate of cancer-related genes targeted by viral insertion in recurrent vs. non-recurrent integration spots; and (IV) provide an assessment of insertional targets and/or pathways that might be actionable for personalized therapy development.
2. Materials and Methods
Ethical review and approval were waived for this study, which is based on literature data mining and analysis. In order to assess the rate of cancer-related gene alteration via HPV insertion in HPV-associated tumors, we first established a large database of HPV inserts, including tumor localization, tumor type, HPV genotype, and gene(s) targeted by insertion. We then separated the target genes into three groups: a group of “non-recurrent genes” (only one case of HPV insertion reported at this locus), a group of “genes with low recurrency level” (two cases of HPV insertion reported at this locus), and a third group of “genes with high recurrency level” (≥3 cases reported). In a third step, we compared the rate of tumor-related genes at HPV insertion in each of these three categories, respectively, and finally, we looked for the rate of actionable genes among HPV insertion targets in the general population of tumors and in each of the three groups of genes defined according to their recurrency level.
Our database was built using results from the literature published between 1987 and 2023. A total of 58 publications were registered [13,16,17,18,19,20,21,22,24,25,28,29,30,31,32,33,34,35,36,37,38,39,41,42,45,47,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83]. For each case, we documented the chromosomal locus of HPV insertion, HPV genotype, organ, type of lesion, sample identification, first author, date of publication, review, technique used, target gene, and coordinates of the HPV insertion when available. A list of 1455 integration loci was established. The registration of sample identification permitted us to discard cases reported several times in different publications and analyze cases with multiple integration sites according to their viral and clinical characteristics. From the analysis of the number of sites per tumor, we discarded a tumor-derived cell line that might harbor several sites developed in vitro and 9 cases associated with more than one HPV genotype that could correspond to different lesions in the same tumor diagnostic tissue sample. From the analysis of genes targeted by integration in each tumor, we excluded 34 cases explored by ISH only and 69 cases for which the sample identification was not specified. A total of 1031 cases were retained for viro-clinical analyses.
To analyze if the HPV-inserted genes were significantly enriched in cancer-related genes or in therapy targets, we compared them to a reference panel of genes without HPV inserts composed of protein-coding genes from Gencode [84] v42hg19 annotation. For the analysis, we kept only protein-coding genes with HPV insertions that were present on Gencode, removing 64 genes that were not found. Certain gene names were changed to be compatible with the Gencode v42 version, and 727 genes (1012 cases) were retained in our analysis.
The genes were classified according to their role in cancer, their role in cell proliferation in vitro using the DepMap score [85], and whether they are actionable or not. The cancer pathway for each gene was estimated according to the data from the OncoKB database [86,87]. The genes were considered to be actionable according to the Therapeutic Target Database [88] (TTD) and the Drug-Gene Interaction Database [89]. Figures and statistical tests were generated using R version 4.3.2. For the statistical analysis, we performed Fisher’s exact test, comparing the genes according to their recurrency status (0, 1, 2 ≥ 3). The contingency matrix was constructed considering the recurrency status of each gene vs. its role in cancer and vs. whether it is actionable or not. The insertion loci were also classified according to the HPV genotype (no HPV insertion; HPVs other than HPV16,18,45 (referred to as “intermediate risk HPV genotypes”); high-risk HPV16 and high-risk HPV18/45), and the contingency matrix was computed considering the role in cancer of the inserted gene. In each case, p-values were adjusted using the Holm–Bonferroni correction. For the highly recurrent genes with an unknown role in cancer, we used the DepMap CRISPR gene effect score to identify new candidate genes to be involved in cervical or H&N cancers. We excluded the genes with a very low expression value (TPM < 1) in the respective cell lines. A gene was considered to be a potential cancer gene according to the CRISPR gene effect score if more than 30% of cervical and H&N cell lines had a score below −0.3 for this gene (for the candidate oncogene) and above 0.3 (for the candidate tumor suppressor genes). Flowchart analysis in Scheme 1.
3. Results
3.1. Viral Analysis According to Tumor Localization
The 1031 tumor cases analyzed were developed in the cervix (813; 78.9%), head and neck (H&N) (164; 15,9%), anus (32; 3.1%), vulva (16; 1.5%), vagina (4; 0.4%), and penis (2; 0.2%). Most cases (970; 94%) corresponded to invasive carcinoma or carcinoma-derived cell lines (6; 0.6%). Others were high-grade intraepithelial neoplasia developed in the cervix (38), vulva (8), or vagina (2). Two low-grade intraepithelial lesions and one laryngeal papillomatosis were also recorded (four cervical lesions of undetermined histology) (Supplementary Table S1).
Virology data were available in 1.028 cases. The most frequent genotypes were HPV16 (690, 67.1%), HPV18 (184; 17.9%), HPV45 (48; 4.7%), HPV33 (30; 2.9%), HPV58 (15; 1.6%), and HPV31 (15; 1.5%). Other genotypes detected in less than 1% were HPV35 (9), HPV73 (8), HPV68 (5), HPV51 (4), HPV52 (4), HPV56 (4), HPV59 (4), HPV26 (1), HPV34 (1), HPV39 (1), HPV6 (1), HPV11 (1), HPV67 (1), HPV70 (1), and HPV82 (1) (undetermined HPV type in three cases). The comparison between HPV genotypes and tumor localizations showed that HPV18/45 were largely more prevalent in cervical than in H&N tumors (28% vs. 1%). HPV16 was highly predominant in H&N tumors (86%), together with the non-HPV16/18 types (13%), among which HPV33 and 35 were the most frequent (6.7% and 4.9%, respectively) (Figure 1). Other tumor localizations were almost exclusively (96%) associated with HPV16 (Supplementary Table S2).
3.2. In Individual Tumors, the Development of Multiple HPV DNA Inserts Is Partly Related to the Viral Genotype
The identification and HPV genotyping of samples could be unambiguously specified in 951 cases, allowing the determination of the number of viral inserts in each case. There was a single viral insert per tumor in 754 (78.5%) cases, 2 integration sites in 124 (13.7%) tumors, 3 sites in 33 (3.6%), 4 sites in 16 (1.6%), 5 sites in 11 (1.1%), 6 sites in 3 (0.3%), 7 sites in 4 (0.4%), 9 sites in 2 (0.2%), and 10, 11, 12, and 14 sites in one tumor each (0.1%) (Supplementary File SF1). We compared the number of viral inserts per case and the viral genotype. This analysis showed that HPV18 and HPV45 were less frequently associated with multiple insertion sites (8.5% and 0.6%, respectively) than HPV16 (25.0%) or the other HPV types (19.8%) (Figure 2) (p = 0.003). For instance, only 3 of the 73 cases with ≥3 insertion sites were associated with HPV18/45.
3.3. HPV DNA Preferentially Integrates near or into Genes and Recurrent Targets Are Identified
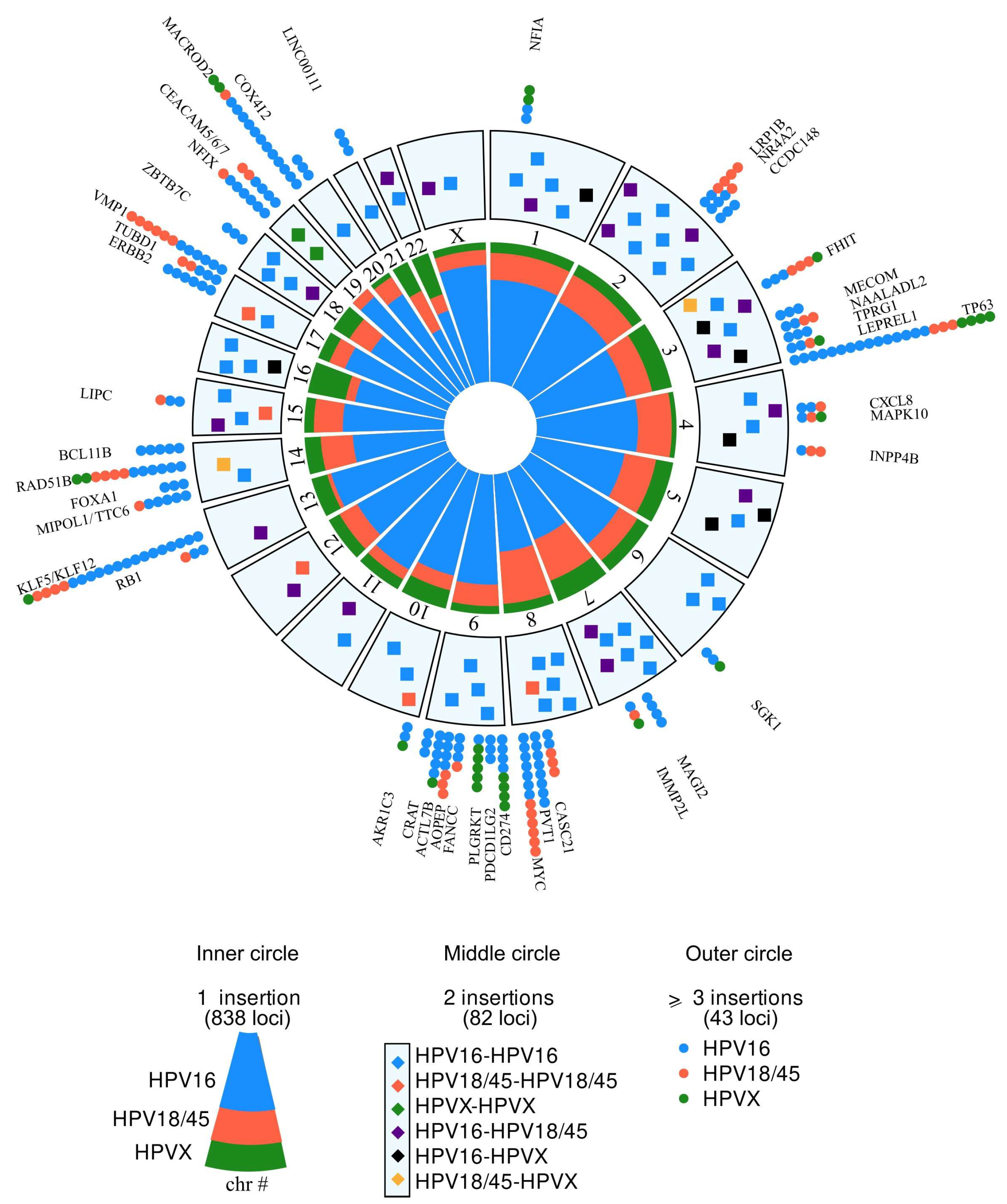
A total of 1455 insertions were recorded, from lesions developed in the cervix (1145 insertions), H&N (233), anus (53), vulva (18), vagina (4), and penis (2). In 1.186 of these cases (81.5%), a target sequence was identified at the insertion locus. Among these, 72 did not contain known or expressed sequences (Alu, pseudogenes, or sequences of unknown functional significance). Sequences with potential functional relevance were observed in 1104/1455 (75.8%) of the cases. Among these, 125 genes were recurrently targeted by viral insertion. The most frequent were TP63 (22 cases), KLF5/12 (21 cases), MACROD2 (15), MYC (13), VMP1 (12), RAD51B (12), CEACAM (10), PVT1 (8), CD174 (8), FHIT (7), NFIX (7), C9orf3 (7), MIPOL1/TTC, LRP1B, PLGRKT, ACTL7B, ERBB2 (6 cases each), CASC21/CASC8, TUBD1, and BCL11B (5 cases each) (Figure 3). In addition, 23 genes were targeted in three or four cases each and 82 genes in two cases (Figure 3 and Supplementary Table S3) (all data on Supplementary File SF2).
The comparison between recurrent HPV insertional targets and tumor localizations showed that all of the insertions at MYC (13), FHIT (7), and VMP1 (12) were found in genital tumors and were preferentially associated with HPV18/45, whereas the insertions on ACTL7B (6/6), CD274 (7/8), and PLGRKT (4/6) were mainly found in HNCC cases (one CD274 insertion in anal cancer) and preferentially associated with HPV16. Finally, NFIX was the preferential target for HPV16 in anal carcinoma (5/7). Comparing genes at viral integration and HPV genotypes, we found a limited number of hot spots that were preferentially targeted by specific HPV genotypes: VMP1, MYC, and LRP1B were preferential targets for HPV18/45, PVT1 for HPV16, and PLGRKT for HPV33. Other targets (TP63, KLF5/12…) showed a balanced repartition between the different viral genotypes and tumor localizations (Supplementary File SF3).
3.4. The Proportion of Target Genes Implicated in Oncogenesis Increases Proportionally with Increasing HPV Insertion Recurrency Levels and with the Oncogenicity of the HPV Genotypes
In order to further assess the targeting frequency of cellular cancer-related genes via HPV DNA insertion, we analyzed separately the proportion of this event in groups of non-recurrent vs. recurrent genes for HPV integration. Of the 792 target genes identified in our series, 727 matched with genes from Gencode. Using the OncoKB Cancer Gene List, these 727 genes were classified into four functional categories: genes with pro-tumorigenic or oncogenic (OG) or with tumor suppressive (TSG) effects, genes expressing both OG and TSG (OG/TSG) effects, and genes with no recognized effect in tumorigenesis (N). We compared the proportion of these functional categories in a panel of 19,402 genes with that observed among genes found targeted in one single tumor case (604 genes), in two cases (79 genes), or in more than two cases (44 genes). The proportion of cancer-related genes was 3.3% in the reference panel of genes. It was 6.5% among the group of genes with a single integration event, 11.4% among genes reported twice as HPV insertion targets, and 40.9% among genes reported more than two times as viral insertion targets (Figure 4A) (p = 4.9 × 10−4).
We next examined whether the proportion of cancer-related genes targeted by viral integration might be influenced by the genotype of the HPV inserts. For this analysis, we focused on cervical cancers in which the viral heterogeneity was the highest. Viral genotypes were separated into three categories: high-risk HPV16 (514 cases), high-risk HPV18/45 (167 cases), and intermediate-risk non-HPV16/18/45 (82 cases). As compared with cases in the reference data set, the rate of cancer-related genes targeted by integration was higher in tumors associated with intermediate-risk HPVs and significantly increased in tumors with high-risk HPV genotypes that were highest in the group of HPV18/45-associated carcinomas (Figure 4B).
Taken together, the observations that the rate of cancer-related genes increases proportionally to both HPV insertion recurrency level and viral genotype oncogenicity constitute a strong argument for a direct role of viral insertion as a common mechanism in HPV-associated oncogenesis and should lead to the identification of new genes also implied in tumor processes that may constitute attractive therapeutic targets.
3.5. The Proportion of Genes Targeted through HPV Inserts and Corresponding to Therapeutic Targets Increases According to Viral Integration Recurrency
We used the DGIdb database to look for the proportion of genes targeted through viral integration that correspond to therapeutic onco-targets or are implied in targetable molecular pathways. About 19% of the 19,402 genes in the database were recorded as direct or indirect therapeutic targets. This proportion was 27.6% among the genes reported as HPV insertion targets in a single case, 40.5% among the genes reported as HPV targets two times, and 50% in the genes reported as targets more than two times (Figure 5) (overall rate 30.2%). The proportion was the same for cervical or H&N tumors. Since several HPV integration sites may be observed in the same tumor, we assessed the proportion of patients for whom a potential therapeutic target could be identified by an HPV insertion pattern as about 26%.
3.6. The Localization of the Viral Inserts at the Molecular Level Regarding the Structure of the Targeted Gene Does Not Differ Significantly between Pro-Tumorigenic and Tumor Suppressive Genes
In order to determine whether the integration pattern differed between pro-tumorigenic and tumor suppressor genes, we compared the position of the viral inserts in a few bona fide examples of genes in these two categories. Depmap plots were used to verify the functional properties of these genes in cervical cancer- and H&N tumor-derived cells. Most of the viral inserts were found located in introns or upstream of the genes, regardless of their respective pro- or anti-tumorigenic effect (Figure 6). In particular, no preferential occurrence of viral DNA integration within gene exons, suggestive of disruptive gene alteration, was observed in tumor suppressors.
3.7. Identification of New Genes Involved in HPV-Associated Cancers
A total of 78% of the genes recurrently targeted by viral insertion were not classified as oncogene or tumor suppressor genes in OncoKB. At least three explanations could account for this observation: (I) genes not involved in oncogenesis may be targeted by viral insertion regarding their proximity to fragile sites; (II) genes not classified as oncogene or tumor suppressor may nevertheless, upon activation or inactivation, provide an advantage in vivo to cancer cells; for instance, overexpression of FOXA1 associated with viral insertion (three cases) may induce tumor cells protection from the host immune system; (III) oncogene or tumor suppressor in HPV-associated cancer may not be yet identified as such in OncoKB. To identify the genes belonging to this latter category, we used two strategies: a literature search and the analysis of gene effects using a large CRISPR screen including 17 cervical and 56 H&N cancer cell lines (https://depmap.org/portal/ accessed on 6 December 2023). Among the 43 genes corresponding to hot spots (≥3) targets for HPV insertion, 26 were not referred as cancer-related in OncoKB. The literature search and/or the CRISPR screen allowed us to further classify some of these genes as oncogenes (AKR1C3, TUBD1, and VMP1), tumor suppressors (LEPREL1), or genes with dual pro- and anti-tumor effects (FAM110B, KLF12, and MAGI2), while others were found to act in the tumor process as transcription factors (NAALADL2, NFIA, NFIX, and NR4A2), cell adhesion (CEACAM5/6), cell proliferation (TPRG1), matrix modeling molecules (PLGRKT), or immunoevasion (MAPK10 and CXCL8). Only seven genes (ACTL7B, CCDC148, COX4I2, CRAT, IMMP2L, LIPC, and ZBTB7C) were not found to be associated with tumor processes (Supplementary Table S4). Altogether, these observations indicate that genes recurrently targeted by viral insertion are a suitable source of candidate genes to be involved in HPV-associated oncogenesis.
4. Discussion
The observation that HPV insertion in integration hot spots may target cancer-related genes has been well documented [27,38,43,45]. We report here that the frequency of this event is observed in 5–10% of the non-recurrent integration sites dispersed throughout the genome and progressively increases proportionally with increasing HPV insertion recurrency levels. We show, in addition, that high-risk HPV16/18/45 more frequently target cancer-related genes than the other viral genotypes. Finally, we report that HPV18/45, exhibiting the highest frequency of targeting cancer-related genes, were rarely integrated at multiple chromosome sites in individual tumors. This correlates with the fact that HPV18/45 DNA is always found in the integrated form in carcinoma cells [36,46] and found in patients, on average, younger than those with other HPV-associated tumors [90]. These data suggest that the oncogenicity of HPV18/45 is stronger than that of other genotypes, for which a longer preinvasive step may allow the development of multiple integration events that are less critical for tumor progression. In accordance, Brant et al. [54] also observed HPV18 more frequently inserted into genes than HPV16. However, the nature of the targets does not seem to be different between the viral genotypes; although certain preferential hot spots for HPV18/45 were found at the VMP1, MYC, and LRP1B loci, no specific gene targets for these high-risk genotypes were identified.
Altogether, these data indicate that in addition to its impact leading to the constitutive expression of the E6/E7 viral oncoproteins, HPV integration acts in oncogenesis via direct interactions between the viral and host cell genomes. Our results provide significant features sustaining the dynamic process of HPV-driven carcinogenesis acting via the selection of driver integration clones that harbor HPV integrants at gene loci susceptible to tumor progression. Importantly, this constitutes a rational basis for the development of potential innovative therapies aiming to counteract the underlying processes via specific molecular interventions. We found that about 30% of the genes targeted by HPV insertion correspond to direct or indirect actionable targets.
Different processes may lead to viral DNA insertion into the genome and account for distinct integration patterns with various impacts on the HPV/host gene expression. There is growing evidence that HPV DNA integration takes place in part through microhomology-mediated repair (MHMR) mechanisms of DNA breaks [34,35,39,91,92,93,94], and this may account for the two main mechanisms of “direct” and “looping” integration processes identified thus far for viral DNA insertions [35,91,92]. HPV18/45 genotypes more frequently integrate following the “looping” or amplification processes, and this may account for the specificities observed for the integration pattern of these genotypes. Whether this is linked to the strong HPV18 E7 impact on cell proliferation [95] or has consequences on disease outcome remains to be determined. Besides genomic amplification, various mechanisms for gene overexpression through HPV DNA insertion have been described, such as viral enhancer insertions, inter-chromosomal translocation hijacking of host enhancers, and long-range cis-activation or chromatin remodeling [39,50,92,96]. Different processes leading to gene silencing have also been reported [33,34,39]. It is also mentioned that an indirect influence of HPV DNA inserts may favor the tumor process: for instance, in a large review of sequencing data from various types of cancers [47], an increase in mutations number within viral integration sites was observed, which might reflect chromatin features facilitating integration into regions with limited access to DNA repair or direct influence of integrated viral sequences on the proximal host genome.
The development of innovative therapeutic approaches using HPV as a target for molecular- [97] or immuno-therapy [98] has already provided encouraging results. It has also been suggested that host genes deregulated via HPV integration could be attractive targets for personalized therapy, such as the functional ERBB2 and RAD51 genes or CD274, PDCD1LG2, and BRCA4, which may provide a rational basis for immunotherapy [43].
The optimal characterization of the viral physical status in HPV-associated carcinomas constitutes information that can be clinically relevant. A negative outcome of HPV-associated tumors harboring integrated vs. exclusively episomal viral sequences has been observed [37,58,99]. More importantly, hybrid viral/host sequences represent a highly specific tumor marker, detectable in the blood [35,76,100,101], that may be useful in patient follow-up care [60]. Recently, the value of using circulating HPV DNA as a tumor marker predictive of tumor relapse following treatment of HPV-associated tumors has been clinically validated [102]. It is important to underline that NGS-based analyses provide, in a single experiment, a full characterization of the viral pattern, including the exact genotyping via the complete DNA sequence, the viral physical status, and the localization of the viral inserts into the host genome. The possibility of performing such analyses from a standard blood sample should facilitate its use in clinical practice.
5. Conclusions
In conclusion, the analysis of our database permitted us to show that (I) HPV18/45 DNA is rarely found at multiple sites in individual tumors, (II) recurrent insertions were not specific to HPV genotypes or tumor localizations, (III) the proportion of target genes implicated in oncogenesis increases proportionally with increasing HPV insertion recurrency levels and with the oncogenicity of the HPV genotypes, and (IV) 30% of the genes at HPV insertion loci correspond to potential therapeutic targets. The characterization of the full viral pattern in HPV-associated cancers provides data that can be used for diagnosis, follow-up, and personalized therapy. The systematic and prospective biobanking and informatic analyses of these data should help to improve the knowledge of HPV-associated carcinogenesis and favor the evolution of the traditional histological classification of HPV tumors toward a more relevant histo-molecular approach, as in other tumor types.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers16081584/s1, Table S1: Histology and tumor localizations; Table S2: HPV types and tumor localizations; Table S3: Genes recurrently targeted by HPV insertions; Table S4: Functions of gene targets; File SF1: HPV inserts per tumor; File SF2: All data; File SF3: Recurrent targets per HPVs and localizations.
Author Contributions
Conceptualization: X.S.-G. and F.R.; Methodology: X.S.-G., L.E.-V. and F.R.; Data curation: X.S.-G. and L.E.-V.; Writing original draft preparation: X.S.-G.; Supervision: F.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study is based on literature data analysis and did not require ethical approval.
Informed Consent Statement
Patient consent was waived since this study is based on literature data analysis.
Data Availability Statement
All of the data collected and used for this study are provided in the Supplementary Materials Section.
Acknowledgments
We are indebted to Allyson Holmes and Jérôme Couturier for their critical review of our manuscript. This work is dedicated to Gérard Orth.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- IARC. Monographs on the Evaluation of Carcinogenic Risks to Humans–Volume 90: Human Papillomaviruses; IARC: Lyon, France, 2007. [Google Scholar]
- de Sanjose, S.; Alemany, L.; Ordi, J.; Tous, S.; Alejo, M.; Bigby, S.M.; Joura, E.A.; Maldonado, P.; Laco, J.; Bravo, I.G.; et al. Worldwide human papillomavirus genotype attribution in over 2000 cases of intraepithelial and invasive lesions of the vulva. Eur. J. Cancer 2013, 49, 3450–3461. [Google Scholar] [CrossRef] [PubMed]
- Haeggblom, L.; Ramqvist, T.; Tommasino, M.; Dalianis, T.; Nasman, A. Time to change perspectives on HPV in oropharyngeal cancer. A systematic review of HPV prevalence per oropharyngeal sub-site the last 3 years. Papillomavirus Res. 2017, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.I.; Francoeur, A.A.; Kapp, D.S.; Caesar, M.A.P.; Huh, W.K.; Chan, J.K. Trends in Human Papillomavirus-Associated Cancers, Demographic Characteristics, and Vaccinations in the US, 2001–2017. JAMA Netw. Open 2022, 5, e222530. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosh, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Preti, M.; Boldorini, R.; Gallio, N.; Cavagnetto, C.; Borella, F.; Pisapia, E.; Ribaldone, R.; Bovio, E.; Bertero, L.; Airoldi, C.; et al. Human papillomavirus genotyping in high-grade vaginal intraepithelial neoplasia: A multicentric Italian study. J. Med. Virol. 2024, 96, e29474. [Google Scholar] [CrossRef] [PubMed]
- Alemany, L.; Saunier, M.; Alvarado-Cabrero, I.; Quiros, B.; Salmeron, J.; Shin, H.R.; Pirog, E.C.; Guimera, N.; Hernandez-Suarez, C.; Felix, A.; et al. Human papillomavirus DNA prevalence and type distribution in anal carcinomas worldwide. Int. J. Cancer 2014, 136, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Baricevic, I.; He, X.; Chakrabarty, B.; Oliver, A.W.; Bailey, C.; Hampson, L.; Hampson, I.; Gilbert, D.C.; Renehan, A.G. High-sensitivity human papilloma virus genotyping reveals near universal positivity in anal squamous cell carcinoma: Different implications for vaccine prevention and prognosis. Eur. J. Cancer 2015, 51, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Liu, J.; Masterson, L.; Fenton, T.R. HPV-associated oropharyngeal cancer: Epidemiology, molecular biology and clinical management. Nat. Rev. Clin. Oncol. 2022, 19, 306–327. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Cullen, A.P.; Reid, R.; Campion, M.; Lorincz, A.T. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, M.; Blennow, E.; Hagmar, B.; Johansson, B. Physical state of HPV16 and chromosomal mapping of the integrated form in cervical carcinomas. Diagn. Mol. Pathol. 2001, 10, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Hopman, A.H.; Smedts, F.; Dignef, W.; Ummelen, M.; Sonke, G.; Mravunac, M.; Vooijs, G.P.; Speel, E.J.; Ramaekers, F.C. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J. Pathol. 2004, 202, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Cannizzaro, L.; Dürst, M.; Mendez, M.; Hecht, B.; Hecht, F. Regional chromosome localization of human papillomavirus integration sites near fragile sites, oncogenes, and cancer chromosome breakpoints. Cancer Genet. Cytogenet. 1988, 33, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Mincheva, A.; Gissmann, L.; zur Hausen, H. Chromosomal integration sites of human papillomavirus DNA in three cervical cancer cell lines mapped by in situ hybridization. Med. Microbiol. Immunol. 1987, 176, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Popescu, N.; Amsbaugh, S.; DiPaolo, J. Human papillomavirus type 18 DNA is integrated at a single chromosome site in cervical carcinoma cell line SW756. J. Virol. 1987, 61, 1682–1685. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.; Myers, S.; Persing, D.; Sakar, G.; McGovern, R.; Gostout, B.; Smith, D. Human papillomavirus type 16 integration in cervical tumors frequently occur in common fragile sites. Cancer Res. 2000, 60, 5916–5921. [Google Scholar] [PubMed]
- Dürst, M.; Croce, C.M.; Gissmann, L.; Schwarz, E.; Huebner, K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc. Natl. Acad. Sci. USA 1987, 84, 1070–1074. [Google Scholar] [CrossRef]
- Couturier, J.; Sastre-Garau, X.; Schneider-Maunoury, S.; Labib, A.; Orth, G. Integration of papillomavirus DNA near myc genes in genital carcinomas and its consequences for proto-oncogene expression. J. Virol. 1991, 65, 4534–4538. [Google Scholar] [CrossRef]
- Koopman, L.; Szuhai, K.; van Eedenburg, J.; Bezrookove, V.; Kenter, G.; Schuuring, E.; Tanke, H.; Fleuren, G. Recurrent integration of human papillomaviruses 16, 45, and 67 near translocation breakpoints in new cervical cancer cell lines. Cancer Res. 1999, 59, 5615–5624. [Google Scholar]
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high risk cervical intra-epithelial neoplasia and cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136. [Google Scholar] [PubMed]
- Luft, F.; Klaes, R.; Nees, M.; Dürst, M.; Heilmann, V.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Int. J. Cancer 2001, 92, 9–17. [Google Scholar] [CrossRef]
- Wentzensen, N.; Ridder, R.; Klaes, R.; Vinokurova, S.; Schaefer, U.; Doeberitz, M.K. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002, 21, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Driesch, C.; Jansen, L.; Runnebaum, I.B.; Durst, M. Non-random integration of the HPV genome in cervical cancer. PLoS ONE 2012, 7, e39632. [Google Scholar] [CrossRef]
- Christiansen, I.K.; Sandve, G.K.; Schmitz, M.; Durst, M.; Hovig, E. Transcriptionally active regions are the preferred targets for chromosomal HPV integration in cervical carcinogenesis. PLoS ONE 2015, 10, e0119566. [Google Scholar] [CrossRef] [PubMed]
- Klimov, E.; Vinokourova, S.; Moisjak, E.; Rakhmanaliev, E.; Kobseva, V.; Laimins, L.; Kisseljov, F.; Sulimova, G. Human papilloma viruses and cervical tumours: Mapping of integration sites and analysis of adjacent cellular sequences. BMC Cancer 2002, 2, 24. [Google Scholar] [CrossRef]
- Kraus, I.; Driesch, C.; Vinokurova, S.; Hovig, E.; Schneider, A.; von Knebel Doeberitz, M.; Dürst, M. The majority of viral-cellular fusion transcripts in cervical carcinomas cotranscribe cellular sequences of known or predicted genes. Cancer Res. 2008, 68, 2514–2522. [Google Scholar] [CrossRef]
- Peter, M.; Stransky, N.; Couturier, J.; Huppe, P.; Barillot, E.; de Cremoux, P.; Cottu, P.; Radvanyi, F.; Sastre-Garau, X. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J. Pathol. 2010, 221, 320–330. [Google Scholar] [CrossRef]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar] [CrossRef]
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-Garau, X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 2006, 25, 5985–5993. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Bartelmann, M.; Vogt, M.; Geisen, C.; Napierski, I.; Kahn, T.; Delius, H.; Lichter, P.; Weitz, S.; Korn, B.; et al. APM-1, a novel human gene, identified by aberrant co-transcription with papillomavirus oncogenes in a cervical carcinoma cell line, encodes a BTB/POZ-zinc finger protein with growth inhibitory activity. EMBO J. 1998, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Driesch, C.; Beer-Grondke, K.; Jansen, L.; Runnebaum, I.B.; Durst, M. Loss of gene function as a consequence of human papillomavirus DNA integration. Int. J. Cancer 2012, 131, E593–E602. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Lameiras, S.; Deloger, M.; Morel, A.; Vacher, S.; Lecerf, C.; Dupain, C.; Jeannot, E.; Girard, E.; Baulande, S.; et al. Human papilloma virus (HPV) integration signature in Cervical Cancer: Identification of MACROD2 gene as HPV hot spot integration site. Br. J. Cancer 2021, 124, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Koneva, L.A.; Zhang, Y.; Virani, S.; Hall, P.B.; McHugh, J.B.; Chepeha, D.B.; Wolf, G.T.; Carey, T.E.; Rozek, L.S.; Sartor, M.A. HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol. Cancer Res. 2018, 16, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Mainguene, J.; Vacher, S.; Kamal, M.; Hamza, A.; Masliah-Planchon, J.; Baulande, S.; Ibadioune, S.; Borcoman, E.; Cacheux, W.; Calugaru, V.; et al. Human papilloma virus integration sites and genomic signatures in head and neck squamous cell carcinoma. Mol. Oncol. 2022, 16, 3001–3016. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.; Neuzillet, C.; Wack, M.; Lameiras, S.; Vacher, S.; Deloger, M.; Servant, N.; Veyer, D.; Péré, H.; Mariani, O.; et al. Mechanistic Signatures of Human Papillomavirus Insertions in Anal Squamous Cell Carcinomas. Cancers 2019, 11, 1846. [Google Scholar] [CrossRef]
- Thomas, J.; Leufflen, L.; Chesnais, V.; Diry, S.; Demange, J.; Depardieu, C.; Bani, M.A.; Marchal, F.; Charra-Brunaud, C.; Merlin, J.L.; et al. Identification of Specific Tumor Markers in Vulvar Carcinoma Through Extensive Human Papillomavirus DNA Characterization Using Next Generation Sequencing Method. J. Low Genit. Tract. Dis. 2020, 24, 53–60. [Google Scholar] [CrossRef]
- Fan, J.; Fu, Y.; Peng, W.; Li, X.; Shen, Y.; Guo, E.; Lu, F.; Zhou, S.; Liu, S.; Yang, B.; et al. Multi-omics characterization of silent and productive HPV integration in cervical cancer. Cell Genom. 2023, 3, 100211. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center &Research Institute at Christiana Care Health Services; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Symer, D.E.; Akagi, K.; Geiger, H.M.; Song, Y.; Li, G.; Emde, A.K.; Xiao, W.; Jiang, B.; Corvelo, A.; Toussaint, N.C.; et al. Diverse tumorigenic consequences of human papillomavirus integration in primary oropharyngeal cancers. Genome Res. 2022, 32, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4, 2513. [Google Scholar] [CrossRef] [PubMed]
- Zapatka, M.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sultmann, H.; Moch, H.; Pathogens, P.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 52, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Rusan, M.; Li, Y.Y.; Hammerman, P.S. Genomic landscape of human papillomavirus-associated cancers. Clin. Cancer Res. 2015, 21, 2009–2019. [Google Scholar] [CrossRef]
- Warburton, A.; Markowitz, T.E.; Katz, J.P.; Pipas, J.M.; McBride, A.A. Recurrent integration of human papillomavirus genomes at transcriptional regulatory hubs. NPJ Genom. Med. 2021, 6, m101. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, C.; Zhao, L.; Wang, J.; McCrae, M.; Chen, X.; Lu, F. Dysregulation of host cellular genes targeted by human papillomavirus (HPV) integration contributes to HPV-related cervical carcinogenesis. Int. J. Cancer 2016, 138, 1163–1174. [Google Scholar] [CrossRef]
- Annunziata, C.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Characterization of the human papillomavirus (HPV) integration sites into genital cancers. Pathol. Oncol. Res. 2012, 18, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Vinokurova, S.; Sampson, J.N.; den Boon, J.A.; Walker, J.L.; Horswill, M.A.; Korthauer, K.; Schiffman, M.; Sherman, M.E.; Zuna, R.E.; et al. Chromosomal copy number alterations and HPV integration in cervical precancer and invasive cancer. Carcinogenesis 2016, 37, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Brant, A.C.; Menezes, A.N.; Felix, S.P.; de Almeida, L.M.; Sammeth, M.; Moreira, M.A.M. Characterization of HPV integration, viral gene expression and E6E7 alternative transcripts by RNA-Seq: A descriptive study in invasive cervical cancer. Genomics 2019, 111, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Brink, A.A.; Wiegant, J.C.; Szuhai, K.; Tanke, H.J.; Kenter, G.G.; Fleuren, G.J.; Schuuring, E.; Raap, A.K. Simultaneous mapping of human papillomavirus integration sites and molecular karyotyping in short-term cultures of cervical carcinomas by using 49-color combined binary ratio labeling fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2002, 134, 145–150. [Google Scholar] [CrossRef]
- Bryant, D.; Onions, T.; Raybould, R.; Flynn, A.; Tristram, A.; Meyrick, S.; Giles, P.; Ashelford, K.; Hibbitts, S.; Fiander, A.; et al. mRNA sequencing of novel cell lines from human papillomavirus type-16 related vulval intraepithelial neoplasia: Consequences of expression of HPV16 E4 and E5. J. Med. Virol. 2014, 86, 1534–1541. [Google Scholar] [CrossRef]
- Chaiwongkot, A.; Pientong, C.; Ekalaksananan, T.; Vinokurova, S.; Kongyingyoes, B.; Chumworathayi, B. Patarapadungkit, N, Siriaunkgul, S.; von Knebel Doeberitz, M. Detection of the human papillomavirus 58 physical state using the amplification of papillomavirus oncogene transcripts assay. J. Virol. Methods 2013, 189, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Thomas, A.; Mahantshetty, U.; Shrivastava, S.K.; Deodhar, K.; Mulherkar, R. HPV genotyping and site of viral integration in cervical cancers in Indian women. PLoS ONE 2012, 7, e41012. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Johnson, S.H.; Kasperbauer, J.L.; Eckloff, B.W.; Tombers, N.M.; Vasmatzis, G.; Smith, D.I. Mate pair sequencing of oropharyngeal squamous cell carcinomas reveals that HPV integration occurs much less frequently than in cervical cancer. J. Clin. Virol. 2014, 59, 195–200. [Google Scholar] [CrossRef]
- Harle, A.; Guillet, J.; Thomas, J.; Demange, J.; Dolivet, G.; Peiffert, D.; Leroux, A.; Sastre-Garau, X. HPV insertional pattern as a personalized tumor marker for the optimized tumor diagnosis and follow-up of patients with HPV-associated carcinomas: A case report. BMC Cancer 2019, 19, 277. [Google Scholar] [CrossRef]
- Hori, T.; Ichimura, H.; Minamihisamatsu, M.; Takahashi, E.I.; Yamauchi, M.; Hama, Y.; Kurimura, O.; Yamasaki, M.; Kurimura, T. Chromosomal insertion and amplification of human papillomavirus 16 DNA sequences in a cell line of Argyrophil small cell carcinoma of the uterine cervix. Jpn. J. Cancer Res. 1991, 82, 371–375. [Google Scholar] [CrossRef]
- Huebbers, C.U.; Preuss, S.F.; Kolligs, J.; Vent, J.; Stenner, M.; Wieland, U.; Silling, S.; Drebber, U.; Speel, E.J.; Klussmann, J.P. Integration of HPV6 and downregulation of AKR1C3 expression mark malignant transformation in a patient with juvenile-onset laryngeal papillomatosis. PLoS ONE 2013, 8, e57207. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, E.; Harle, A.; Holmes, A.; Sastre-Garau, X. Nuclear factor I X is a recurrent target for HPV16 insertions in anal carcinomas. Genes Chromosomes Cancer 2018, 57, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; Tcga Network; Meric-Bernstam, F.; Medeiros, L.J.; et al. Landscape of DNA virus associations across human malignant cancers: Analysis of 3,775 cases using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed]
- Lace, M.J.; Anson, J.R.; Klussmann, J.P.; Wang, D.H.; Smith, E.M.; Haugen, T.H.; Turek, L.P. Human papillomavirus type 16 (HPV-16) genomes integrated in head and neck cancers and in HPV-16-immortalized human keratinocyte clones express chimeric virus-cell mRNAs similar to those found in cervical cancers. J. Virol. 2011, 85, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Lagstrom, S.; Umu, S.U.; Lepisto, M.; Ellonen, P.; Meisal, R.; Christiansen, I.K.; Ambur, O.H.; Rounge, T.B. TaME-seq: An efficient sequencing approach for characterisation of HPV genomic variability and chromosomal integration. Sci. Rep. 2019, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, R.; Cai, Y.; Li, Y.; Cheng, X.; Yang, Y.; Xiang, Y. Determination of integrated HPV58 sequences in cervical lesions. Int. J. Gynecol. Cancer 2012, 22, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.S.; Aldrich, J.; Nasser, S.; Kurdoglu, A.; Phillips, L.; Reiman, R.; McDonald, J.; Izatt, T.; Christoforides, A.; Baker, A.; et al. Simultaneous characterization of somatic events and HPV-18 integration in a metastatic cervical carcinoma patient using DNA and RNA sequencing. Int. J. Gynecol Cancer 2014, 24, 329–338. [Google Scholar] [CrossRef]
- Matovina, M.; Sabol, I.; Grubisic, G.; Gasperov, N.M.; Grce, M. Identification of human papillomavirus type 16 integration sites in high-grade precancerous cervical lesions. Gynecol. Oncol. 2009, 113, 120–127. [Google Scholar] [CrossRef]
- Nakanishi, G.; Fujii, K.; Asagoe, K.; Tanaka, T.; Iwatsuki, K. Human papillomavirus genome integration in multifocal vulvar Bowen’s disease and squamous cell carcinoma. Clin. Exp. Dermatol. 2009, 34, e965–e967. [Google Scholar] [CrossRef]
- Nambaru, L.; Meenakumari, B.; Swaminathan, R.; Rajkumar, T. Prognostic significance of HPV physical status and integration sites in cervical cancer. Asian Pac. J. Cancer Prev. 2009, 10, 355–360. [Google Scholar]
- Olthof, N.C.; Speel, E.J.; Kolligs, J.; Haesevoets, A.; Henfling, M.; Ramaekers, F.C.; Preuss, S.F.; Drebber, U.; Wieland, U.; Silling, S.; et al. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PLoS ONE 2014, 9, e88718. [Google Scholar] [CrossRef] [PubMed]
- Peitsaro, P.; Johansson, B.; Syrjanen, S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J. Clin. Microbiol. 2002, 40, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Ragin, C.C.; Reshmi, S.C.; Gollin, S.M. Mapping and analysis of HPV16 integration sites in a head and neck cancer cell line. Int. J. Cancer 2004, 110, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garau, X.; Couturier, J.; Favre, M.; Orth, G. A recurrent human papillomavirus integration site at chromosome region 12q14-q15 in SW756 and SK-v cell lines derived from genital tumors. Comptes Rendus L’académie Sci. 1995, 318, 475–478. [Google Scholar]
- Sastre-Garau, X.; Diop, M.; Martin, F.; Dolivet, G.; Marchal, F.; Charra-Brunaud, C.; Peiffert, D.; Leufflen, L.; Dembele, B.; Demange, J.; et al. A NGS-based Blood Test For the Diagnosis of Invasive HPV-associated Carcinomas with Extensive Viral Genomic Characterization. Clin. Cancer Res. 2021, 27, 5307–5316. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garau, X.; Favre, M.; Couturier, J.; Orth, G. Distinct patterns of alteration of myc genes associated with integration of human papillomavirus type 16 or type 45 DNA in two genital tumours. J. Gen. Virol. 2000, 81, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garau, X.; Schneider-Maunoury, S.; Couturier, J.; Orth, G. Human papillomavirus type 16 DNA is integrated in chromosome region 12q14-q15 in a cell line derived from a vulvar intraepithelial neoplasia. Cancer Genet. Cytogenet. 1990, 44, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Myers, S.L.; Gostout, B.S.; Smith, D.I. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 2003, 22, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Walline, H.M.; Komarck, C.M.; McHugh, J.B.; Bellile, E.L.; Brenner, J.C.; Prince, M.E.; McKean, E.L.; Chepeha, D.B.; Wolf, G.T.; Worden, F.P.; et al. Genomic Integration of High-Risk HPV Alters Gene Expression in Oropharyngeal Squamous Cell Carcinoma. Mol. Cancer Res. 2016, 14, 941–952. [Google Scholar] [CrossRef]
- Wilke, C.; Hall, B.; Hoge, A.; Paradee, W.; Smith, D.; Glover, T. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: Direct evidence for the coincidence of viral integration site and fragile site. Hum. Mol. Genet. 1996, 5, 187–195. [Google Scholar] [CrossRef]
- Zhou, L.; Qiu, Q.; Zhou, Q.; Li, J.; Yu, M.; Li, K.; Xu, L.; Ke, X.; Xu, H.; Lu, B.; et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat. Commun. 2022, 13, 2563. [Google Scholar] [CrossRef] [PubMed]
- Popescu, N.; DiPaolo, J. Preferential sites for viral integration on mammalian genome. Cancer Genet. Cytogenet. 1989, 42, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Amstrong, J.; Barnes, I.; et al. Gencode 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Broad, D.; Mustafa, K. Repurposing Public 23Q2. figshare. Dataset 2023. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumlar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Wang, S.; Qiu, Y.; Chen, Y.; Zhu, F. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024, 52, D1465–D1477. [Google Scholar] [CrossRef] [PubMed]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef] [PubMed]
- de Cremoux, P.; de la Rochefordiere, A.; Savignoni, A.; Kirova, Y.; Alran, S.; Fourchotte, V.; Plancher, C.; Thioux, M.; Salmon, R.J.; Cottu, P.; et al. Different outcome of invasive cervical cancer associated with high-risk versus intermediate-risk HPV genotype. Int. J. Cancer 2009, 124, 778–782. [Google Scholar] [CrossRef]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, C.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef]
- Groves, I.J.; Drane, E.L.A.; Michalski, M.; Monahan, J.M.; Scarpini, C.G.; Smith, S.P.; Bussoti, G.; Varnai, C.; Schoenfelder, S.; Fraser, P.; et al. Short- and long-range cis interactions between integrated HPV genomes and cellular chromatin dysregulate host gene expression in early cervical carcinogenesis. PLoS Pathog. 2021, 17, e1009875. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, C.; Gao, W.; Wang, L.; Pan, Y.; Gao, Y.; Lu, Z.; Ke, Y. Genome-wide profiling of the human papillomavirus DNA integration in cervical intraepithelial neoplasia and normal cervical epithelium by HPV capture technology. Sci. Rep. 2016, 6, 35427. [Google Scholar] [CrossRef]
- Rosty, C.; Sheffer, M.; Tsafrir, D.; Stransky, N.; Tsafir, I.; Peter, M.; de Cremoux, P.; de la Rochefordière, A.; Salmon, R.J.; Dorval, T.; et al. Identification of a proliferation gene cluster associated with HPV E6/E7 expression level and viral DNA load in invasive cervical carcinoma. Oncogene 2005, 24, 7094–7104. [Google Scholar] [CrossRef] [PubMed]
- Karimzadeh, M.; Arlidge, C.; Rostami, A.; Lupien, M.; Bratman, S.V.; Hoffman, M.M. Human papillomavirus integration transforms chromatin to drive oncogenesis. Genome Biol. 2023, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Jubair, L.; Fallaha, S.; McMillan, N.A.J. Systemic Delivery of CRISPR/Cas9 Targeting HPV Oncogenes Is Effective at Eliminating Established Tumors. Mol. Ther. 2019, 27, 2091–2099. [Google Scholar] [CrossRef]
- Doran, S.L.; Stevanovic, S.; Adhikary, S.; Gartner, J.J.; Jia, L.; Kwong, M.L.M.; Faquin, W.C.; Hewitt, S.M.; Sherry, R.M.; Yang, J.C.; et al. T-Cell Receptor Gene Therapy for Human Papillomavirus-Associated Epithelial Cancers: A First-in-Human, Phase I/II Study. J. Clin. Oncol. 2019, 37, 2759–2768. [Google Scholar] [CrossRef]
- Shin, H.J.; Joo, J.; Yoon, J.H.; Yoo, C.W.; Kim, J.Y. Physical status of human papillomavirus integration in cervical cancer is associated with treatment outcome of the patients treated with radiotherapy. PLoS ONE 2014, 9, e78995. [Google Scholar] [CrossRef]
- Caballero-Mellado, J.; Martinez-Aguilar, L.; Paredes-Valdez, G.; Santos, P.E. Burkholderia unamae sp. nov., an N2-fixing rhizospheric and endophytic species. Int. J. Syst. Evol. Microbiol. 2004, 54, 1165–1172. [Google Scholar] [CrossRef]
- Campitelli, M.; Jeannot, E.; Peter, M.; Lappartient, E.; Saada, S.; de la Rochefordière, A.; Fourchotte, V.; Alran, S.; Petrow, P.; Cottu, P.; et al. Human papillomavirus mutational insertion: Specific marker of circulating tumor DNA in cervical cancer patients. PLoS ONE 2012, 7, e43393. [Google Scholar] [CrossRef]
- Han, K.; Zou, J.; Zhao, Z.; Baskurt, Z.; Zheng, Y.; Barnes, E.; Croke, J.; Ferguson, S.E.; Fyles, A.; Gien, L.; et al. Clinical Validation of Human Papilloma Virus Circulating Tumor DNA for Early Detection of Residual Disease After Chemoradiation in Cervical Cancer. J. Clin. Oncol. 2024, 42, 431–440. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Flowchart of the analysis of HPV integration sites.

Figure 1.
Distribution of the different HPV genotypes (outer circle) according to tumor localization (inner circle). HPV18/45 are more prevalent in cervical tumors than in other tumors. Non-HPV16/18/45 genotypes (HPVX) are present in 10–13% of cervical and head and neck (H&N) tumors, whereas HPV16 is highly prevalent in other tumor localizations.
Figure 1.
Distribution of the different HPV genotypes (outer circle) according to tumor localization (inner circle). HPV18/45 are more prevalent in cervical tumors than in other tumors. Non-HPV16/18/45 genotypes (HPVX) are present in 10–13% of cervical and head and neck (H&N) tumors, whereas HPV16 is highly prevalent in other tumor localizations.

Figure 2.
Proportion of unique and multiple viral inserts per individual tumor according to HPV genotypes. Multiple inserts at different chromosomal sites are found in 25% of HPV16 or HPVX (non-HPV16, 18, 45) tumor cases, whereas multiple viral inserts are found in only 8% of the HPV18/45-associated tumors (p = 0.003).
Figure 2.
Proportion of unique and multiple viral inserts per individual tumor according to HPV genotypes. Multiple inserts at different chromosomal sites are found in 25% of HPV16 or HPVX (non-HPV16, 18, 45) tumor cases, whereas multiple viral inserts are found in only 8% of the HPV18/45-associated tumors (p = 0.003).

Figure 3.
Circosplot of the distribution of 963 HPV insertion targets on the cell genome according to viral genotypes. In the external ring, each point corresponds to a tumor case harboring a viral insert at the indicated gene locus with its genotype color code. In the intermediate ring, every square corresponds to the insertion at the same gene locus in two different tumor cases, with the corresponding color code for each of the two genotypes. The central circle shows the respective frequency of the cases with a single insert according to viral genotypes.
Figure 3.
Circosplot of the distribution of 963 HPV insertion targets on the cell genome according to viral genotypes. In the external ring, each point corresponds to a tumor case harboring a viral insert at the indicated gene locus with its genotype color code. In the intermediate ring, every square corresponds to the insertion at the same gene locus in two different tumor cases, with the corresponding color code for each of the two genotypes. The central circle shows the respective frequency of the cases with a single insert according to viral genotypes.

Figure 4.
Cancer-related genes rate according to HPV insertion recurrency (A) and HPV genotypes (B). (A) As compared with the rate in the reference data base (no HPV insert; column 1), there is an increasing proportion of cancer-related genes from the group of non-recurrent gene targets (a single insertion case at each site; column 2) (p = 4.9 × 10−3) from those with two cases (column 3) (p = 4.9 × 10−3) and more than two cases (column 4) of HPV insertion at each site. (B) As compared with the rate in the reference data base (no HPV insert; column 1), there is a higher rate of cancer-related genes at HPV insertion for the high-risk HPV16/18/45 than for the other HPV genotypes (p = 0.03). ns p ≥ 0.05; * p < 0.05; ** p < 0.01.
Figure 4.
Cancer-related genes rate according to HPV insertion recurrency (A) and HPV genotypes (B). (A) As compared with the rate in the reference data base (no HPV insert; column 1), there is an increasing proportion of cancer-related genes from the group of non-recurrent gene targets (a single insertion case at each site; column 2) (p = 4.9 × 10−3) from those with two cases (column 3) (p = 4.9 × 10−3) and more than two cases (column 4) of HPV insertion at each site. (B) As compared with the rate in the reference data base (no HPV insert; column 1), there is a higher rate of cancer-related genes at HPV insertion for the high-risk HPV16/18/45 than for the other HPV genotypes (p = 0.03). ns p ≥ 0.05; * p < 0.05; ** p < 0.01.

Figure 5.
Rate of actionable target genes found in the DGIdb according to HPV insertion recurrency. As compared with the rate found in the reference database (no HPV insert; column 1), there is an increasing proportion of actionable genes from the group of non-recurrent gene targets (a single insertion case at each site; column 2) to those of two cases (column 3) or more than two cases (column 4) of HPV insertion at each site (p = 4.9 × 10−4). ns p ≥ 0.05; * p < 0.05; ** p <0.01; *** p < 0.001.
Figure 5.
Rate of actionable target genes found in the DGIdb according to HPV insertion recurrency. As compared with the rate found in the reference database (no HPV insert; column 1), there is an increasing proportion of actionable genes from the group of non-recurrent gene targets (a single insertion case at each site; column 2) to those of two cases (column 3) or more than two cases (column 4) of HPV insertion at each site (p = 4.9 × 10−4). ns p ≥ 0.05; * p < 0.05; ** p <0.01; *** p < 0.001.

Figure 6.
Comparison of the localization of HPV inserts on the physical maps of oncogenes (upper panel) and tumor suppressor genes (lower panel). Depmap patterns were used to differentiate the two groups of genes. Cervical cancer-derived cell lines as yellow circle, H&N cancer cells as brown triangles, and other cell lines as gray squares. Under each Depmap plot, the physical map is represented for each respective gene, with exons depicted as vertical bars and HPV inserts as vertical arrows. Arrows in blue when located in exon.
Figure 6.
Comparison of the localization of HPV inserts on the physical maps of oncogenes (upper panel) and tumor suppressor genes (lower panel). Depmap patterns were used to differentiate the two groups of genes. Cervical cancer-derived cell lines as yellow circle, H&N cancer cells as brown triangles, and other cell lines as gray squares. Under each Depmap plot, the physical map is represented for each respective gene, with exons depicted as vertical bars and HPV inserts as vertical arrows. Arrows in blue when located in exon.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sastre-Garau, X.; Estrada-Virrueta, L.; Radvanyi, F. HPV DNA Integration at Actionable Cancer-Related Genes Loci in HPV-Associated Carcinomas. Cancers 2024, 16, 1584. https://doi.org/10.3390/cancers16081584
AMA Style
Sastre-Garau X, Estrada-Virrueta L, Radvanyi F. HPV DNA Integration at Actionable Cancer-Related Genes Loci in HPV-Associated Carcinomas. Cancers. 2024; 16(8):1584. https://doi.org/10.3390/cancers16081584
Chicago/Turabian StyleSastre-Garau, Xavier, Lilia Estrada-Virrueta, and François Radvanyi. 2024. "HPV DNA Integration at Actionable Cancer-Related Genes Loci in HPV-Associated Carcinomas" Cancers 16, no. 8: 1584. https://doi.org/10.3390/cancers16081584
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.