Upregulated Matrisomal Proteins and Extracellular Matrix Mechanosignaling Underlie Obesity-Associated Promotion of Pancreatic Ductal Adenocarcinoma

 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Model and Pancreas Homogenization
2.2. Proteomic Sample Preparation and TMT Labeling
2.3. Proteomic and Phosphoproteomic Analysis of TMT Nine-Plexes by LC-MS/MS and Systems Analysis
2.4. Pathway Analysis and Bioinformatics Tools
2.5. Histochemical and Immunocytochemical Staining of Fixed Pancreas Tissue, Spatial Analysis Using QuPath Software, and Spatial Characterization by Nanostring GeoMX
2.6. Digital Spatial Profiling of Immune Cell Subtypes by Nanostring GeoMX
3. Results
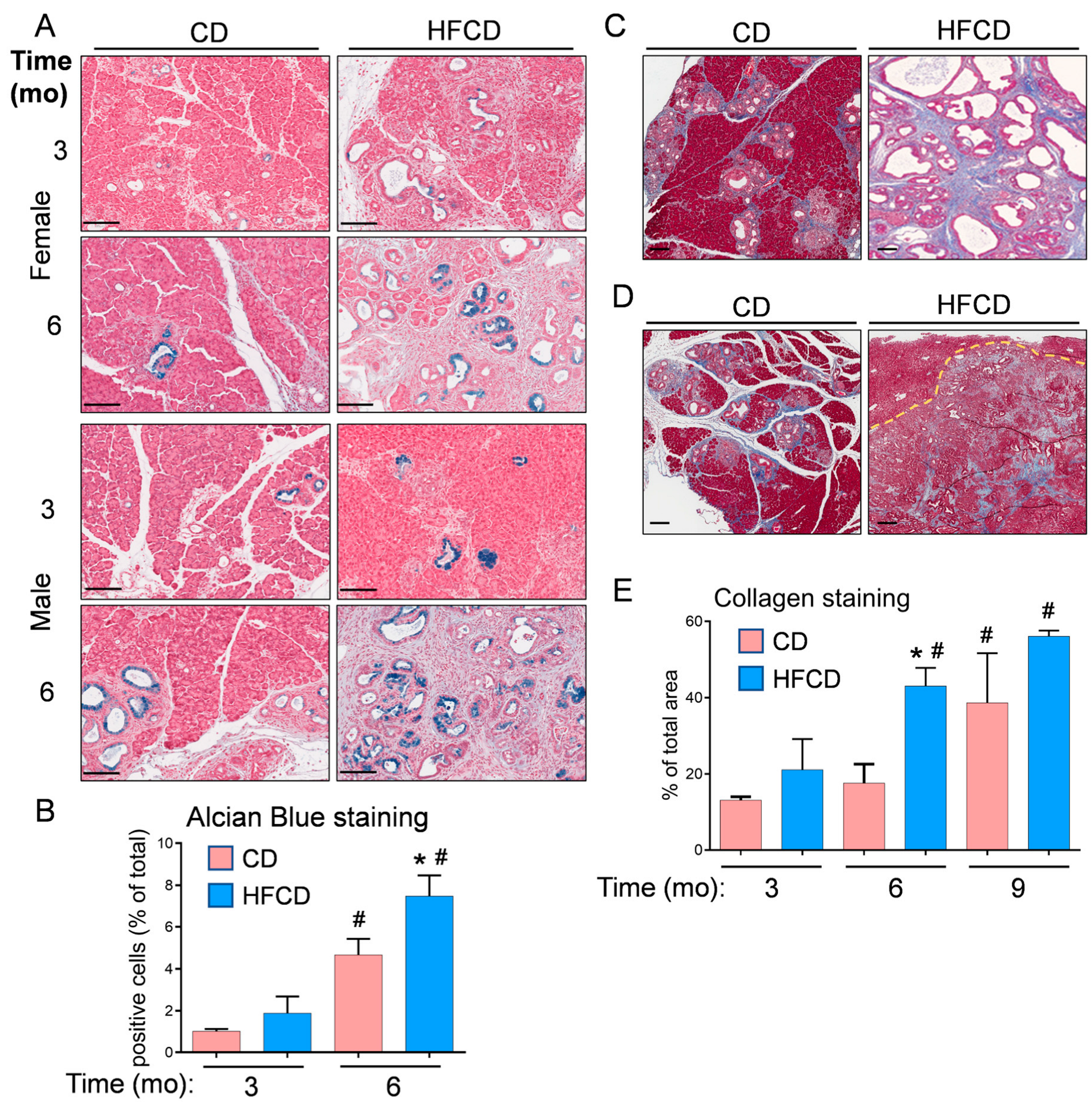
3.1. Diet-Induced Obesity Promotes Mucinous PanIN, Fibrotic Microenvironment, and Altered Cellular Phenotype Culminating in Pancreatic Tumorigenesis in KC Mice
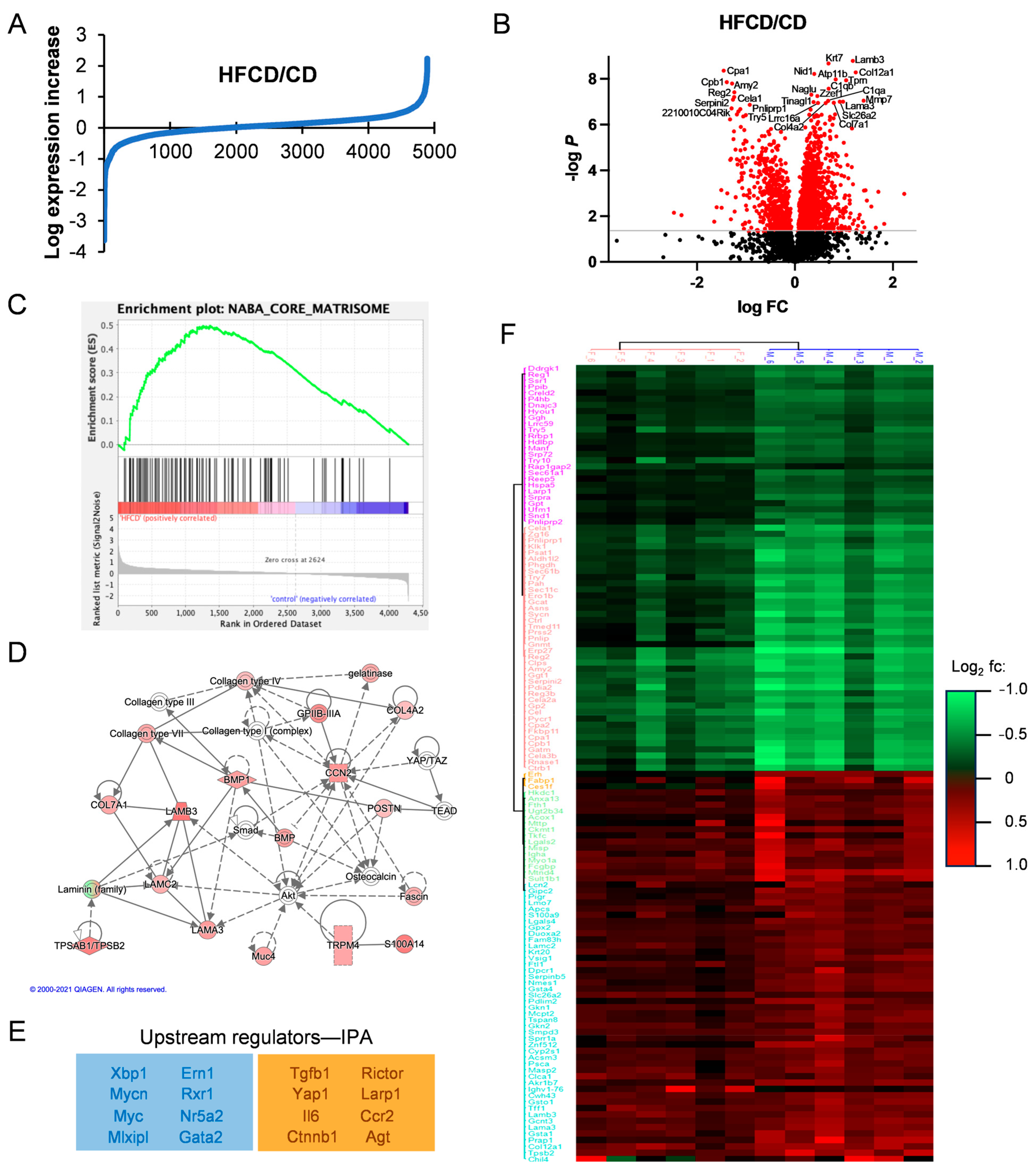
3.2. Proteomic Analysis Reveals Association of DIO-Induced PDAC with Elevated Matrisomal Proteins
3.3. Analysis of DIO-Induced Matrisomal Protein Enrichment in Subgroups of KC Mice
3.4. Phosphoproteins Associated with DIO-Induced PDAC Development in KC Mice
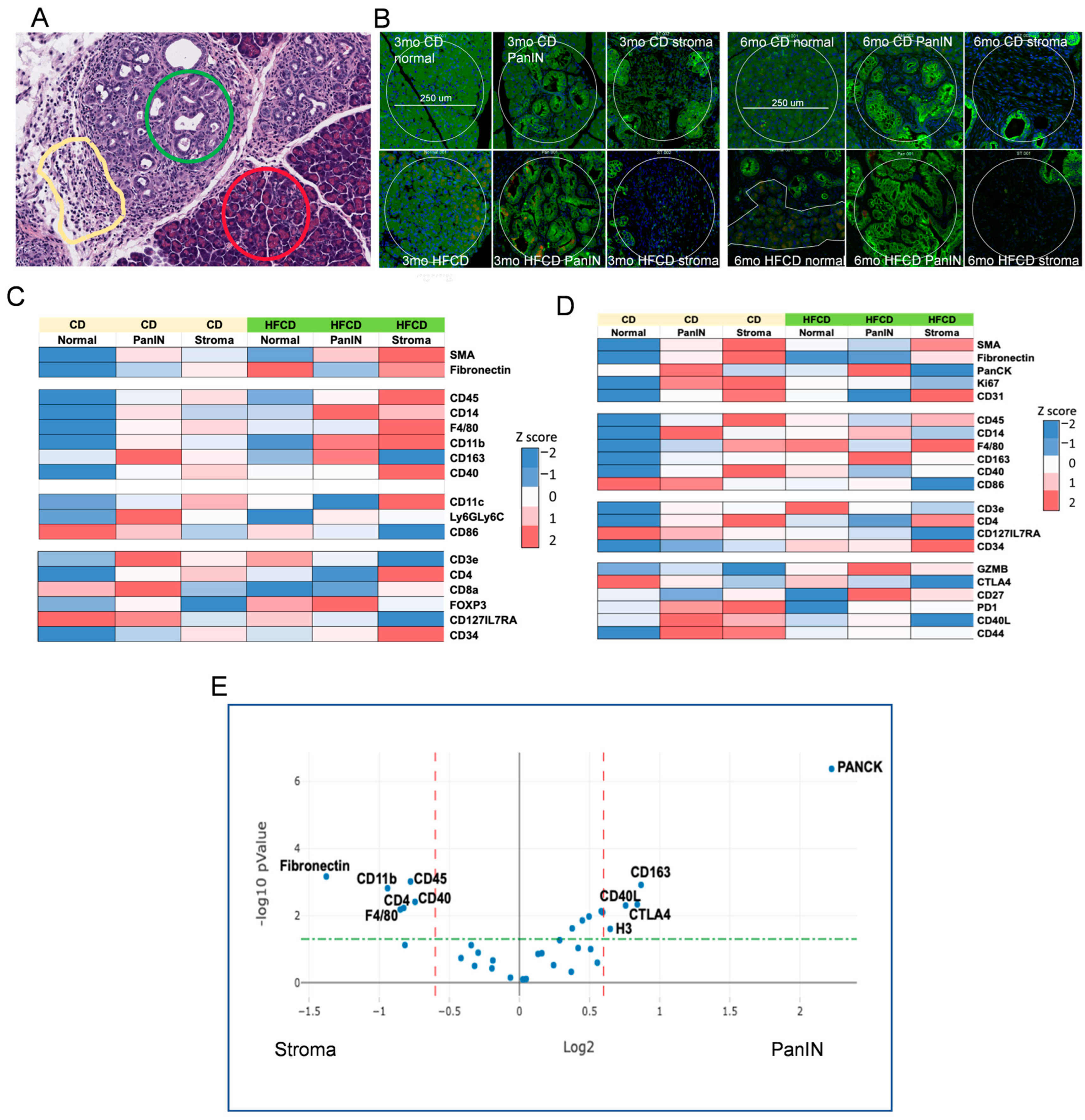
3.5. Spatial Analysis to Determine Relationships between ECM Deposition, PDAC Initiation and Progression, and Distinct Responsible Cell Types
3.6. Emergence of Atypical Flat Lesions and Substantiation of Cell Types and Marker Proteins by Immunohistochemical Staining during DIO-Induced PDAC Acceleration
3.7. Immune Cell Profiling in PanIN and Stromal Areas during DIO-Induced PDAC by Nanostring GeoMX
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Eibl, G.; Rozengurt, E. Obesity and Pancreatic Cancer: Insight into Mechanisms. Cancers 2021, 13, 5067. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W.; Hertzer, K.; Moro, A.; Donald, G.; Chang, H.H.; Go, V.L.; Pandol, S.J.; Lugea, A.; Gukovskaya, A.S.; Li, G.; et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev. Res. 2013, 6, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Moro, A.; Takakura, K.; Su, H.Y.; Mo, A.; Nakanishi, M.; Waldron, R.T.; French, S.W.; Dawson, D.W.; Hines, O.J.; et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS ONE 2017, 12, e0184455. [Google Scholar] [CrossRef] [PubMed]
- Cascetta, P.; Cavaliere, A.; Piro, G.; Torroni, L.; Santoro, R.; Tortora, G.; Melisi, D.; Carbone, C. Pancreatic Cancer and Obesity: Molecular Mechanisms of Cell Transformation and Chemoresistance. Int. J. Mol. Sci. 2018, 19, 3331. [Google Scholar] [CrossRef] [PubMed]
- Eibl, G.; Cruz-Monserrate, Z.; Korc, M.; Petrov, M.S.; Goodarzi, M.O.; Fisher, W.E.; Habtezion, A.; Lugea, A.; Pandol, S.J.; Hart, P.A.; et al. Diabetes Mellitus and Obesity as Risk Factors for Pancreatic Cancer. J. Acad. Nutr. Diet. 2018, 118, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Eibl, G. Obesity-Induced Adipose Tissue Inflammation as a Strong Promotional Factor for Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.S.; Chang, H.H.; Hauer, M.; Lagishetty, V.; Katzka, W.; Rozengurt, E.; Jacobs, J.P.; Eibl, G. Metformin alters the duodenal microbiome and decreases the incidence of pancreatic ductal adenocarcinoma promoted by diet-induced obesity. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G763–G772. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Recio, C.; Aranda-Tavio, H.; Guerra-Rodriguez, M.; Garcia-Castellano, J.M.; Fernandez-Perez, L. The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef] [PubMed]
- Oni, T.E.; Biffi, G.; Baker, L.A.; Hao, Y.; Tonelli, C.; Somerville, T.D.D.; Deschenes, A.; Belleau, P.; Hwang, C.I.; Sanchez-Rivera, F.J.; et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J. Exp. Med. 2020, 217, 20192389. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R. Filter-Aided Sample Preparation: The Versatile and Efficient Method for Proteomic Analysis. Methods Enzymol. 2017, 585, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; Kadiyala, V.; Lee, L.S.; Banks, P.A.; Conwell, D.L.; Steen, H. Proteomic analysis (GeLC-MS/MS) of ePFT-collected pancreatic fluid in chronic pancreatitis. J. Proteome Res. 2012, 11, 1897–1912. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Teper, Y.; Ye, L.; Waldron, R.T.; Lugea, A.; Sun, X.; Sinnett-Smith, J.; Hines, O.J.; Pandol, S.J.; Rozengurt, E.; Eibl, G. Low dosage combination treatment with metformin and simvastatin inhibits obesity-promoted pancreatic cancer development in male KrasG12D mice. Sci. Rep. 2023, 13, 16144. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.t.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Krah, N.M.; De La, O.J.; Swift, G.H.; Hoang, C.Q.; Willet, S.G.; Chen Pan, F.; Cash, G.M.; Bronner, M.P.; Wright, C.V.; MacDonald, R.J.; et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015, 4, e07125. [Google Scholar] [CrossRef] [PubMed]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and repair of the exocrine pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef]
- Li, Q.; Li, B.; Miao, X.; Ramgattie, C.; Gao, Z.H.; Liu, J.L. Reg2 Expression Is Required for Pancreatic Islet Compensation in Response to Aging and High-Fat Diet-Induced Obesity. Endocrinology 2017, 158, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Loftus, S.K.; Cannons, J.L.; Incao, A.; Pak, E.; Chen, A.; Zerfas, P.M.; Bryant, M.A.; Biesecker, L.G.; Schwartzberg, P.L.; Pavan, W.J. Acinar cell apoptosis in Serpini2-deficient mice models pancreatic insufficiency. PLoS Genet. 2005, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Hwang, S.; Seong, R.H. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem. Biophys. Res. Commun. 2016, 471, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yin, X.; Liu, Z.; Wang, J. Emerging Potential Mechanism and Therapeutic Target of Ferroptosis in PDAC: A Promising Future. Int. J. Mol. Sci. 2022, 23, 15031. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 2015, 161, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Dumartin, L.; Alrawashdeh, W.; Trabulo, S.M.; Radon, T.P.; Steiger, K.; Feakins, R.M.; di Magliano, M.P.; Heeschen, C.; Esposito, I.; Lemoine, N.R.; et al. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene 2017, 36, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Shi, G.; Schmidt, C.M.; Hruban, R.H.; Konieczny, S.F. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am. J. Pathol. 2007, 171, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Eibl, G.; Rozengurt, E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin. Cancer Biol. 2019, 54, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yang, J.; Su, H.Y.; Waldron, R.T.; Zhi, M.; Li, L.; Xia, Q.; Pandol, S.J.; Lugea, A. Yes-Associated Protein 1 Plays Major Roles in Pancreatic Stellate Cell Activation and Fibroinflammatory Responses. Front. Physiol. 2019, 10, 1467. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, L.; Shi, J.; He, R.; Yang, W.; Habtezion, A.; Niu, N.; Lu, P.; Xue, J. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. eBioMedicine 2020, 58, 102920. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Washington, M.K.; Chaturvedi, R.; Drosos, Y.; Revetta, F.L.; Weaver, C.J.; Buzhardt, E.; Yull, F.E.; Blackwell, T.S.; Sosa-Pineda, B.; et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab. Investig. 2014, 94, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Konukiewitz, B.; Schlitter, A.M.; Kloppel, G. Pathology of pancreatic ductal adenocarcinoma: Facts, challenges and future developments. World J. Gastroenterol. 2014, 20, 13833–13841. [Google Scholar] [CrossRef] [PubMed]
- von Figura, G.; Fahrenkrog-Petersen, L.; Hidalgo-Sastre, A.; Hartmann, D.; Huser, N.; Schmid, R.M.; Hebrok, M.; Roy, N.; Esposito, I. Atypical flat lesions derive from pancreatic acinar cells. Pancreatology 2017, 17, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.S.; Maller, O.; Pickup, M.W.; Weaver, V.M.; Hansen, K.C. Compartment resolved proteomics reveals a dynamic matrisome in a biomechanically driven model of pancreatic ductal adenocarcinoma. J. Immunol. Regen. Med. 2018, 1, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Clauser, K.R.; Ohlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, J.S.; Court, C.M.; Hou, S.; Li, Q.; Song, M.; Wu, D.; Chen, J.F.; Lee, T.; Lin, M.; Sho, S.; et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br. J. Cancer 2016, 114, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Doppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Prakash, H.; Nadella, V.; Singh, S.; Schmitz-Winnenthal, H. CD14/TLR4 priming potentially recalibrates and exerts anti-tumor efficacy in tumor associated macrophages in a mouse model of pancreatic carcinoma. Sci. Rep. 2016, 6, 31490. [Google Scholar] [CrossRef] [PubMed]
- Honselmann, K.C.; Finetti, P.; Birnbaum, D.J.; Monsalve, C.S.; Wellner, U.F.; Begg, S.K.S.; Nakagawa, A.; Hank, T.; Li, A.; Goldsworthy, M.A.; et al. Neoplastic-Stromal Cell Cross-talk Regulates Matrisome Expression in Pancreatic Cancer. Mol. Cancer Res. 2020, 18, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Penzel, R.; Chaib-Harrireche, M.; Barcena, U.; Bergmann, F.; Riedl, S.; Kayed, H.; Giese, N.; Kleeff, J.; Friess, H.; et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J. Pathol. 2006, 208, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Ishiwata, T.; Kumbasar, A.; Friess, H.; Buchler, M.W.; Lander, A.D.; Korc, M. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J. Clin. Investig. 1998, 102, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Paron, I.; Berchtold, S.; Voros, J.; Shamarla, M.; Erkan, M.; Hofler, H.; Esposito, I. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PLoS ONE 2011, 6, e21684. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Y.; Bai, B. The Expression, Regulation, and Biomarker Potential of Glypican-1 in Cancer. Front. Oncol. 2019, 9, 614. [Google Scholar] [CrossRef] [PubMed]
- Yokosaki, Y.; Monis, H.; Chen, J.; Sheppard, D. Differential effects of the integrins α9β1, αvβ3, and αvβ6 on cell proliferative responses to tenascin. Roles of the β subunit extracellular and cytoplasmic domains. J. Biol. Chem. 1996, 271, 24144–24150. [Google Scholar] [CrossRef] [PubMed]
- Turaga, R.C.; Yin, L.; Yang, J.J.; Lee, H.; Ivanov, I.; Yan, C.; Yang, H.; Grossniklaus, H.E.; Wang, S.; Ma, C.; et al. Rational design of a protein that binds integrin αvβ3 outside the ligand binding site. Nat. Commun. 2016, 7, 11675. [Google Scholar] [CrossRef] [PubMed]
- Turaga, R.C.; Sharma, M.; Mishra, F.; Krasinskas, A.; Yuan, Y.; Yang, J.J.; Wang, S.; Liu, C.; Li, S.; Liu, Z.R. Modulation of Cancer-Associated Fibrotic Stroma by An Integrin α(v)β(3) Targeting Protein for Pancreatic Cancer Treatment. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Diaz Brinton, R. Minireview: Translational animal models of human menopause: Challenges and emerging opportunities. Endocrinology 2012, 153, 3571–3578. [Google Scholar] [CrossRef]
- Gomez-Chou, S.B.; Swidnicka-Siergiejko, A.K.; Badi, N.; Chavez-Tomar, M.; Lesinski, G.B.; Bekaii-Saab, T.; Farren, M.R.; Mace, T.A.; Schmidt, C.; Liu, Y.; et al. Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res. 2017, 77, 2647–2660. [Google Scholar] [CrossRef]
- Liu, X.; Yuan, H.; Zhou, J.; Wang, Q.; Qi, X.; Bernal, C.; Avella, D.; Kaifi, J.T.; Kimchi, E.T.; Timothy, P.; et al. LMO7 as an Unrecognized Factor Promoting Pancreatic Cancer Progression and Metastasis. Front. Cell Dev. Biol. 2021, 9, 647387. [Google Scholar] [CrossRef] [PubMed]
- Deal, B.; Phillips, K.; Crelli, C.; Janjic, J.M.; Pollock, J.A. RNA-Seq Reveals Sex Differences in Gene Expression during Peripheral Neuropathic Inflammation and in Pain Relief from a COX-2 Inhibiting Theranostic Nanoemulsion. Int. J. Mol. Sci. 2023, 24, 9163. [Google Scholar] [CrossRef] [PubMed]
- Kessel, J.C.; Weiskirchen, R.; Schroder, S.K. Expression Analysis of Lipocalin 2 (LCN2) in Reproductive and Non-Reproductive Tissues of Esr1-Deficient Mice. Int. J. Mol. Sci. 2023, 24, 9280. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Ludescher, M.; Poschmann, G.; Stuhler, K.; Wyrich, M.; Oles, J.; Franken, A.; Rivandi, M.; Abramova, A.; Reinhardt, F.; et al. PGRMC1 Promotes Progestin-Dependent Proliferation of Breast Cancer Cells by Binding Prohibitins Resulting in Activation of ERα Signaling. Cancers 2021, 13, 5635. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Esposito, K.; Vietri, M.T.; Gazzerro, P.; D’Auria, A.; Ardovino, I.; Puca, G.A.; Molinari, A.M. Cytokine pattern in postmenopause. Maturitas 2002, 41, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef]
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Xu, Q.; Wang, J.; Yu, S.; Chang, H.H.; Sinnett-Smith, J.; Eibl, G.; Rozengurt, E. Lipophilic statins inhibit YAP nuclear localization, co-activator activity and colony formation in pancreatic cancer cells and prevent the initial stages of pancreatic ductal adenocarcinoma in KrasG12D mice. PLoS ONE 2019, 14, e0216603. [Google Scholar] [CrossRef] [PubMed]
- Papalazarou, V.; Drew, J.; Juin, A.; Spence, H.J.; Whitelaw, J.; Nixon, C.; Salmeron-Sanchez, M.; Machesky, L.M. Collagen VI expression is negatively mechanosensitive in pancreatic cancer cells and supports the metastatic niche. J. Cell Sci. 2022, 135, 259978. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Zhang, Q.; Wu, L.S.; Salaria, S.N.; Winter, J.W.; Hruban, R.H.; Goggins, M.S.; Abbruzzese, J.L.; Maitra, A.; Ho, L. Prognostic significance of maspin in pancreatic ductal adenocarcinoma: Tissue microarray analysis of 223 surgically resected cases. Mod. Pathol. 2007, 20, 570–578. [Google Scholar] [CrossRef]
- Gautam, S.K.; Kumar, S.; Cannon, A.; Hall, B.; Bhatia, R.; Nasser, M.W.; Mahapatra, S.; Batra, S.K.; Jain, M. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert. Opin. Ther. Targets 2017, 21, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Gumpper, K.; Dangel, A.W.; Pita-Grisanti, V.; Krishna, S.G.; Lara, L.F.; Mace, T.; Papachristou, G.I.; Conwell, D.L.; Hart, P.A.; Cruz-Monserrate, Z. Lipocalin-2 expression and function in pancreatic diseases. Pancreatology 2020, 20, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Liu, X.; Wang, F.; Ma, Y.; Zhang, S.; Shao, Z.; Yang, Y.; Tian, X. Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma. Genes 2022, 13, 1760. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, Z.; Ren, X.; Chen, Z.; Bao, X.; Zheng, J.; Zuo, Z. The Crosstalk Between Malignant Cells and Tumor-Promoting Immune Cells Relevant to Immunotherapy in Pancreatic Ductal Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 821232. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Norberg, K.J.; Mezheyeuski, A.; Schnittert, J.; Kuninty, P.R.; Moro, C.F.; Paulsson, J.; Schultz, N.A.; Calatayud, D.; Lohr, J.M.; et al. Stroma-regulated HMGA2 is an independent prognostic marker in PDAC and AAC. Br. J. Cancer 2017, 117, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Jagadeesh, K.A.; Guo, J.A.; Hoffman, H.I.; Yadollahpour, P.; Reeves, J.W.; Mohan, R.; Drokhlyansky, E.; Van Wittenberghe, N.; Ashenberg, O.; et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 2022, 54, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Gukovskaya, A.; Edderkaoui, M.; Dawson, D.; Eibl, G.; Lugea, A. Epidemiology, risk factors, and the promotion of pancreatic cancer: Role of the stellate cell. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 127–134. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldron, R.T.; Lugea, A.; Chang, H.-H.; Su, H.-Y.; Quiros, C.; Lewis, M.S.; Che, M.; Ramanujan, V.K.; Rozengurt, E.; Eibl, G.; et al. Upregulated Matrisomal Proteins and Extracellular Matrix Mechanosignaling Underlie Obesity-Associated Promotion of Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 1593. https://doi.org/10.3390/cancers16081593
Waldron RT, Lugea A, Chang H-H, Su H-Y, Quiros C, Lewis MS, Che M, Ramanujan VK, Rozengurt E, Eibl G, et al. Upregulated Matrisomal Proteins and Extracellular Matrix Mechanosignaling Underlie Obesity-Associated Promotion of Pancreatic Ductal Adenocarcinoma. Cancers. 2024; 16(8):1593. https://doi.org/10.3390/cancers16081593
Chicago/Turabian StyleWaldron, Richard T., Aurelia Lugea, Hui-Hua Chang, Hsin-Yuan Su, Crystal Quiros, Michael S. Lewis, Mingtian Che, V. Krishnan Ramanujan, Enrique Rozengurt, Guido Eibl, and et al. 2024. "Upregulated Matrisomal Proteins and Extracellular Matrix Mechanosignaling Underlie Obesity-Associated Promotion of Pancreatic Ductal Adenocarcinoma" Cancers 16, no. 8: 1593. https://doi.org/10.3390/cancers16081593