PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis?


{kind=link}
{kind=link}
Abstract
:1. Introduction
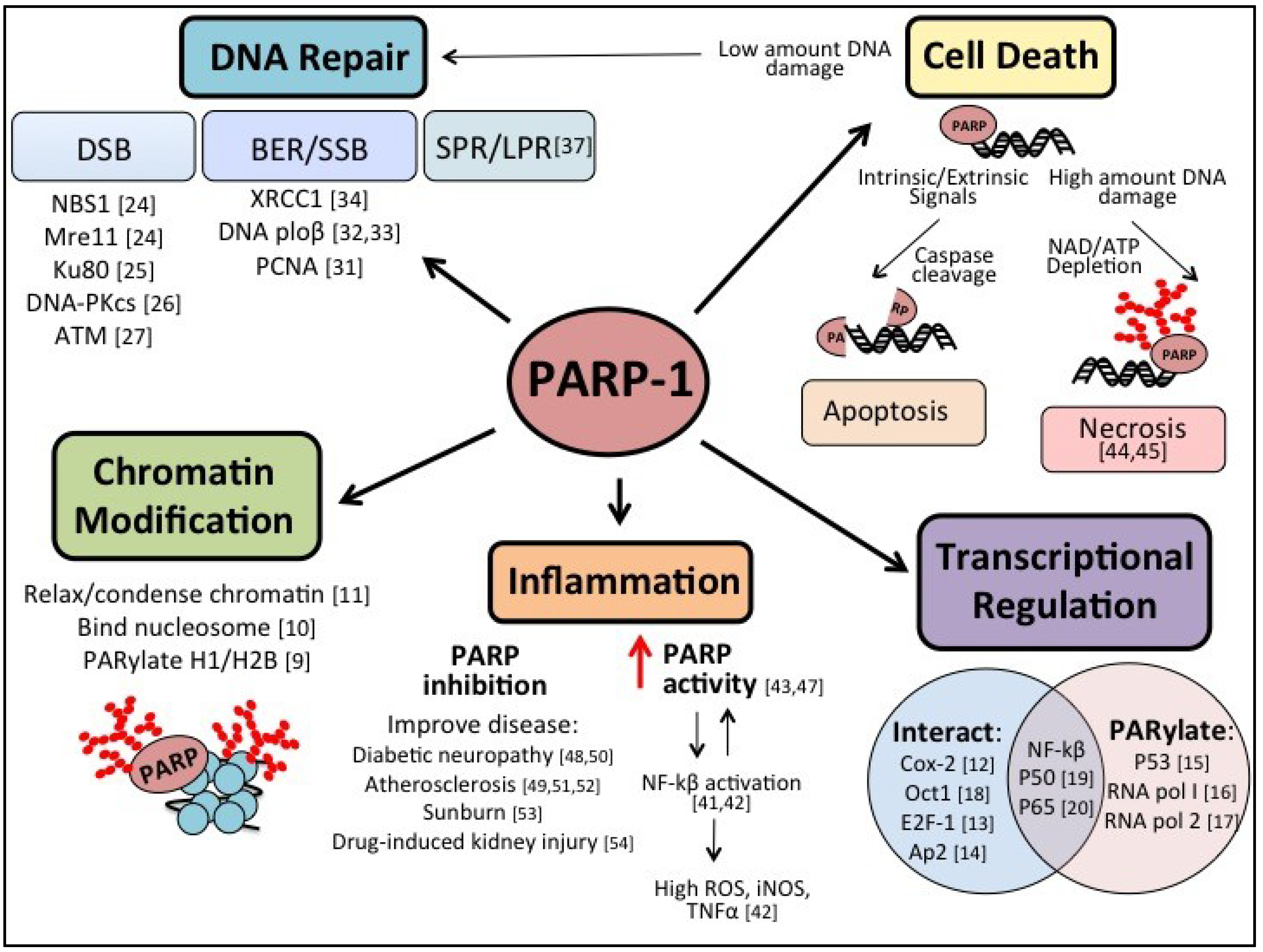
2. PARP-1
2.1. PARP-1 in Inflammation

2.2. PARP-1 in Cancer
3. Oxidative Clustered DNA Lesions (OCDLs)
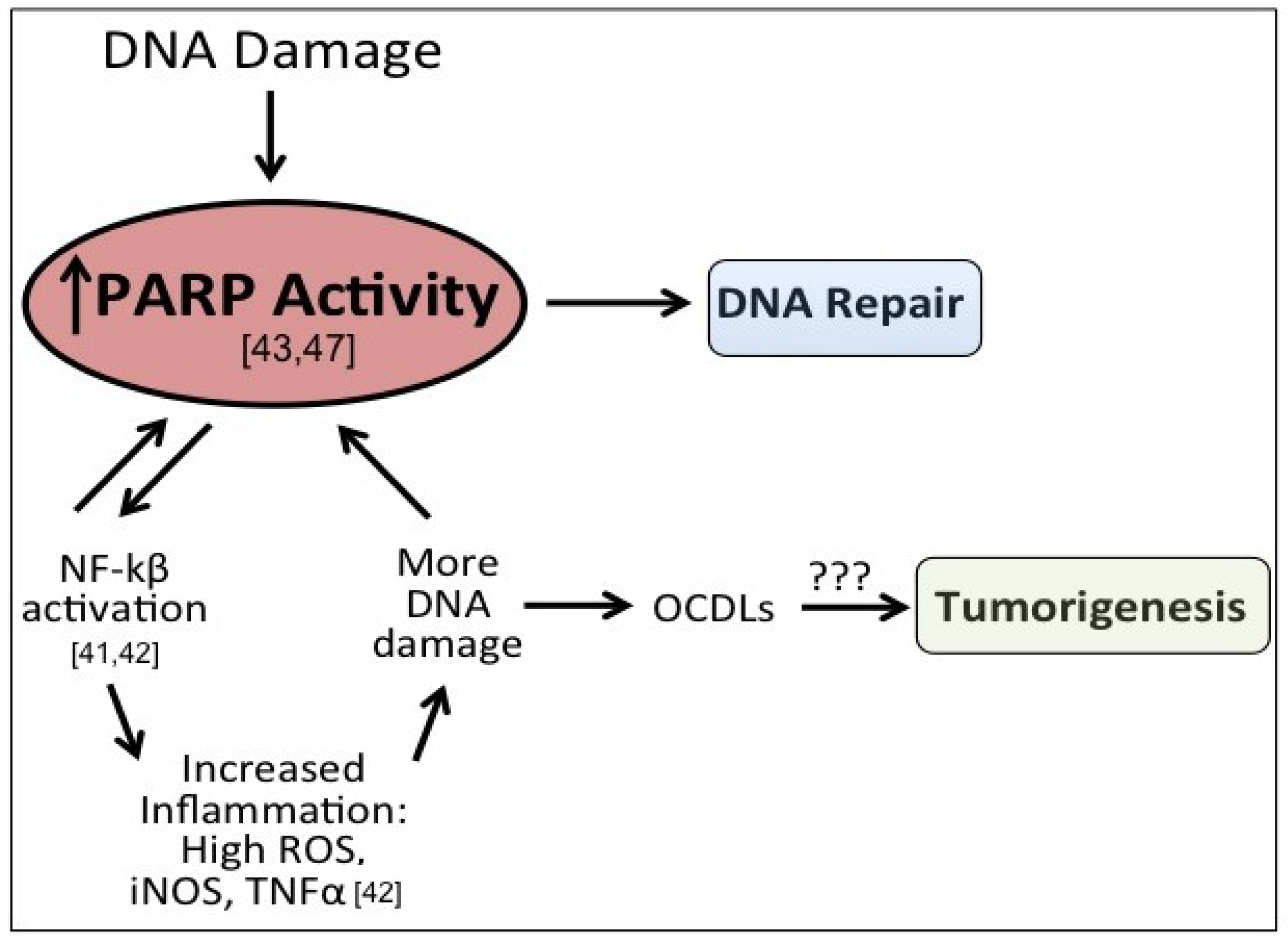
3.1. OCLDs and Cancer
3.2. PARP-1-Friend or Foe of OCDLs?

4. Conclusions
Acknowledgments
Conflict of Interest
References
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer. 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Kameshita, I.; Matsuda, Z.; Taniguchi, T.; Shizuta, Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 1984, 259, 4770–4776. [Google Scholar]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Kraus, W.L.; Lis, J.T. PARP goes transcription. Cell 2003, 113, 677–683. [Google Scholar] [CrossRef]
- Huletsky, A.; de Murcia, G.; Muller, S.; Hengartner, M.; Menard, L.; Lamarre, D.; Poirier, G.G. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J. Biol. Chem. 1989, 264, 8878–8886. [Google Scholar]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef]
- Lin, Y.; Tang, X.; Zhu, Y.; Shu, T.; Han, X. Identification of PARP-1 as one of the transcription factors binding to the repressor element in the promoter region of COX-2. Arch. Biochem. Biophys. 2011, 505, 123–129. [Google Scholar] [CrossRef]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Samara, R.; Espinoza, L.A.; Hassa, P.O.; Hottiger, M.O.; Smulson, M.E. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene 2003, 22, 8460–8471. [Google Scholar] [CrossRef]
- Kannan, P.; Yu, Y.; Wankhade, S.; Tainsky, M.A. PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Res. 1999, 27, 866–874. [Google Scholar] [CrossRef]
- Wesierska-Gadek, J.; Schmid, G. Poly(ADP-ribose) polymerase-1 regulates the stability of the wild-type p53 protein. Cell. Mol. Biol. Lett. 2001, 6, 117–140. [Google Scholar]
- Muller, W.E.; Zahn, R.K. Poly ADP-ribosylation of DNA-dependent RNA polymerase I from quail oviduct. Dependence on progesterone stimulation. Mol. Cell. Biochem. 1976, 12, 147–159. [Google Scholar] [CrossRef]
- Taniguchi, T.; Suzuki, S.; Shizuta, Y. Poly (ADP-ribosyl)ation of RNA polymerase II from wheat germ. Biochem. Biophys. Res. Commun. 1985, 127, 526–532. [Google Scholar] [CrossRef]
- Nie, J.; Sakamoto, S.; Song, D.; Qu, Z.; Ota, K.; Taniguchi, T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998, 424, 27–32. [Google Scholar] [CrossRef]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar]
- Kameoka, M.; Ota, K.; Tetsuka, T.; Tanaka, Y.; Itaya, A.; Okamoto, T.; Yoshihara, K. Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. Biochem. J. 2000, 346, 641–649. [Google Scholar] [CrossRef]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef]
- Shrivastav, M.; de Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–208. [Google Scholar]
- Galande, S.; Kohwi-Shigematsu, T. Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem. 1999, 274, 20521–20528. [Google Scholar] [CrossRef]
- Ariumi, Y.; Masutani, M.; Copeland, T.D.; Mimori, T.; Sugimura, T.; Shimotohno, K.; Ueda, K.; Hatanaka, M.; Noda, M. Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 1999, 18, 4616–4625. [Google Scholar] [CrossRef]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Menisser-de Murcia, J.; Mark, M.; Wendling, O.; Wynshaw-Boris, A.; de Murcia, G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol. Cell. Biol. 2001, 21, 1828–1832. [Google Scholar] [CrossRef]
- Henrie, M.S.; Kurimasa, A.; Burma, S.; Menissier-de Murcia, J.; de Murcia, G.; Li, G.C.; Chen, D.J. Lethality in PARP-1/Ku80 double mutant mice reveals physiological synergy during early embryogenesis. DNA Repair 2003, 2, 151–158. [Google Scholar] [CrossRef]
- Srivastava, D.K.; Berg, B.J.; Prasad, R.; Molina, J.T.; Beard, W.A.; Tomkinson, A.E.; Wilson, S.H. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 1998, 273, 21203–21209. [Google Scholar]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702. [Google Scholar]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998, 18, 3563–3571. [Google Scholar]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef]
- Trucco, C.; Oliver, F.J.; de Murcia, G.; Menissier-de Murcia, J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998, 26, 2644–2649. [Google Scholar] [CrossRef]
- Dantzer, F.; de La Rubia, G.; Menissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Giansanti, V.; Dona, F.; Tillhon, M.; Scovassi, A.I. PARP inhibitors: New tools to protect from inflammation. Biochem. Pharmacol. 2010, 80, 1869–1877. [Google Scholar] [CrossRef]
- Bai, P.; Virag, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Ke, Y.; Jiang, X.; He, F.; Pan, L.; Xu, L.; Zeng, X.; Ba, X. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-kappaB transcription in murine macrophages. Hum. Immunol. 2012, 73, 439–447. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol. Chem. 1999, 380, 953–959. [Google Scholar]
- Oliver, F.J.; Menissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef]
- Czapski, G.A.; Adamczyk, A.; Strosznajder, R.P.; Strosznajder, J.B. Expression and activity of PARP family members in the hippocampus during systemic inflammation: Their role in the regulation of prooxidative genes. Neurochem. Int. 2013, 62, 664–673. [Google Scholar] [CrossRef]
- Filipovic, D.M.; Meng, X.; Reeves, W.B. Inhibition of PARP prevents oxidant-induced necrosis but not apoptosis in LLC-PK1 cells. Am. J. Physiol. 1999, 277, F428–F436. [Google Scholar]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef]
- Tentori, L.; Balduzzi, A.; Portarena, I.; Levati, L.; Vernole, P.; Gold, B.; Bonmassar, E.; Graziani, G. Poly (ADP-ribose) polymerase inhibitor increases apoptosis and reduces necrosis induced by a DNA minor groove binding methyl sulfonate ester. Cell Death Differ. 2001, 8, 817–828. [Google Scholar] [CrossRef]
- De Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Lupachyk, S.; Shevalye, H.; Maksimchyk, Y.; Drel, V.R.; Obrosova, I.G. PARP inhibition alleviates diabetes-induced systemic oxidative stress and neural tissue 4-hydroxynonenal adduct accumulation: Correlation with peripheral nerve function. Free Radic. Biol. Med. 2011, 50, 1400–1409. [Google Scholar] [CrossRef]
- Oumouna-Benachour, K.; Hans, C.P.; Suzuki, Y.; Naura, A.; Datta, R.; Belmadani, S.; Fallon, K.; Woods, C.; Boulares, A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: Effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 2007, 115, 2442–2450. [Google Scholar] [CrossRef]
- Szabo, C.; Biser, A.; Benko, R.; Bottinger, E.; Susztak, K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes 2006, 55, 3004–3012. [Google Scholar] [CrossRef]
- Benko, R.; Pacher, P.; Vaslin, A.; Kollai, M.; Szabo, C. Restoration of the endothelial function in the aortic rings of apolipoprotein E deficient mice by pharmacological inhibition of the nuclear enzyme poly(ADP-ribose) polymerase. Life Sci. 2004, 75, 1255–1261. [Google Scholar] [CrossRef]
- Hans, C.P.; Zerfaoui, M.; Naura, A.S.; Troxclair, D.; Strong, J.P.; Matrougui, K.; Boulares, A.H. Thieno[2,3-c]isoquinolin-5-one, a potent poly(ADP-ribose) polymerase inhibitor, promotes atherosclerotic plaque regression in high-fat diet-fed apolipoprotein E-deficient mice: Effects on inflammatory markers and lipid content. J. Pharmacol. Exp. Ther. 2009, 329, 150–158. [Google Scholar] [CrossRef]
- Farkas, B.; Magyarlaki, M.; Csete, B.; Nemeth, J.; Rabloczky, G.; Bernath, S.; Literáti Nagy, P.; Sümegi, B. Reduction of acute photodamage in skin by topical application of a novel PARP inhibitor. Biochem. Pharmacol. 2002, 63, 921–932. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horvath, B.; Kechrid, M.; Tanchian, G.; Rajesh, M.; Naura, A.S.; Boulares, A.H.; Pacher, P. Poly(ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic. Biol. Med. 2011, 51, 1774–1788. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Weil, M.K.; Chen, A.P. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer 2011, 35, 7–50. [Google Scholar] [CrossRef]
- Martin-Oliva, D.; O’Valle, F.; Munoz-Gamez, J.A.; Valenzuela, M.T.; Nunez, M.I.; Aguilar, M.; Ruiz de Almodóvar, J.M.; Garcia del Moral, R.; Oliver, F.J.; et al. Crosstalk between PARP-1 and NF-kappaB modulates the promotion of skin neoplasia. Oncogene 2004, 23, 5275–5283. [Google Scholar] [CrossRef]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, J.L.; Karin, M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Bonner, J.A.; LoBuglio, A.F.; Yang, E.S. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res. 2012, 72, 4796–4806. [Google Scholar] [CrossRef]
- Shimizu, S.; Nomura, F.; Tomonaga, T.; Sunaga, M.; Noda, M.; Ebara, M.; Saisho, H. Expression of poly(ADP-ribose) polymerase in human hepatocellular carcinoma and analysis of biopsy specimens obtained under sonographic guidance. Oncol. Rep. 2004, 12, 821–825. [Google Scholar]
- Rojo, F.; Garcia-Parra, J.; Zazo, S.; Tusquets, I.; Ferrer-Lozano, J.; Menendez, S.; Eroles, P.; Chamizo, C.; Servitja, S.; Ramírez-Merino, N.; et al. Nuclear PARP-1 protein overexpression is associated with poor overall survival in early breast cancer. Ann. Oncol. 2012, 23, 1156–1164. [Google Scholar] [CrossRef]
- Domagala, P.; Huzarski, T.; Lubinski, J.; Gugala, K.; Domagala, W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: Possible implications for PARP-1 inhibitor therapy. Breast Cancer Res. Treat. 2011, 127, 861–869. [Google Scholar] [CrossRef]
- Michels, J.; Vitale, I.; Galluzzi, L.; Adam, J.; Olaussen, K.A.; Kepp, O.; Senovilla, L.; Talhaoui, I.; Guegan, J.; Enot, D.P.; et al. Cisplatin Resistance Associated with PARP Hyperactivation. Cancer Res. 2013, 73, 2271–2280. [Google Scholar] [CrossRef]
- Cazzalini, O.; Dona, F.; Savio, M.; Tillhon, M.; Maccario, C.; Perucca, P.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p21CDKN1A participates in base excision repair by regulating the activity of poly(ADP-ribose) polymerase-1. DNA Repair 2010, 9, 627–635. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Georgakilas, A.G. Processing of DNA damage clusters in human cells: Current status of knowledge. Mol. Biosyst. 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Ward, J.F. Some biochemical consequences of the spatial distribution of ionizing radiation-produced free radicals. Radiat. Res. 1981, 86, 185–195. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef]
- Gulston, M.; de Lara, C.; Jenner, T.; Davis, E.; O’Neill, P. Processing of clustered DNA damage generates additional double-strand breaks in mammalian cells post-irradiation. Nucleic Acids Res. 2004, 32, 1602–1609. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutation Res. 2011, 711, 123–133. [Google Scholar] [CrossRef]
- Malyarchuk, S.; Youngblood, R.; Landry, A.M.; Quillin, E.; Harrison, L. The mutation frequency of 8-oxo-7,8-dihydroguanine (8-oxodG) situated in a multiply damaged site: Comparison of a single and two closely opposed 8-oxodG in Escherichia coli. DNA Repair 2003, 2, 695–705. [Google Scholar] [CrossRef]
- Shikazono, N.; Pearson, C.; O’Neill, P.; Thacker, J. The roles of specific glycosylases in determining the mutagenic consequences of clustered DNA base damage. Nucleic Acids Res. 2006, 34, 3722–3730. [Google Scholar] [CrossRef]
- Malyarchuk, S.; Brame, K.L.; Youngblood, R.; Shi, R.; Harrison, L. Two clustered 8-oxo-7,8-dihydroguanine (8-oxodG) lesions increase the point mutation frequency of 8-oxodG, but do not result in double strand breaks or deletions in Escherichia coli. Nucleic Acids Res. 2004, 32, 5721–5731. [Google Scholar] [CrossRef]
- Pearson, C.G.; Shikazono, N.; Thacker, J.; O’Neill, P. Enhanced mutagenic potential of 8-oxo-7,8-dihydroguanine when present within a clustered DNA damage site. Nucleic Acids Res. 2004, 32, 263–270. [Google Scholar] [CrossRef]
- Gollapalle, E.; Wang, R.; Adetolu, R.; Tsao, D.; Francisco, D.; Sigounas, G.; Georgakilas, A.G. Detection of oxidative clustered DNA lesions in X-irradiated mouse skin tissues and human MCF-7 breast cancer cells. Radiat. Res. 2007, 167, 207–216. [Google Scholar] [CrossRef]
- Bennett, P.V.; Cuomo, N.L.; Paul, S.; Tafrov, S.T.; Sutherland, B.M. Endogenous DNA damage clusters in human skin, 3-D model, and cultured skin cells. Free Radic. Biol. Med. 2005, 39, 832–839. [Google Scholar] [CrossRef]
- Chastain, P.D., 2nd; Nakamura, J.; Swenberg, J.; Kaufman, D. Nonrandom AP site distribution in highly proliferative cells. FASEB J. 2006, 20, 2612–2614. [Google Scholar] [CrossRef]
- Nowsheen, S.; Wukovich, R.L.; Aziz, K.; Kalogerinis, P.T.; Richardson, C.C.; Panayiotidis, M.I.; Bonner, W.M.; Sedelnikova, O.A.; Georgakilas, A.G. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat. Res. 2009, 674, 131–136. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- El-Bayoumy, K. The protective role of selenium on genetic damage and on cancer. Mutat. Res. 2001, 475, 123–139. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Bennett, P.V.; Wilson, D.M., 3rd; Sutherland, B.M. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res. 2004, 32, 5609–5620. [Google Scholar] [CrossRef]
- Singleton, B.K.; Griffin, C.S.; Thacker, J. Clustered DNA damage leads to complex genetic changes in irradiated human cells. Cancer Res. 2002, 62, 6263–6269. [Google Scholar]
- Bock, F.J.; Krumschnabel, G.; Manzl, C.; Peintner, L.; Tanzer, M.C.; Hermann-Kleiter, N.; Baier, G.; Llacuna, L.; Yelamos, J.; Villunger, A. Loss of PIDD limits NF-kappaB activation and cytokine production but not cell survival or transformation after DNA damage. Cell Death Differ. 2013, 20, 546–557. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Swindall, A.F.; Stanley, J.A.; Yang, E.S. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers 2013, 5, 943-958. https://doi.org/10.3390/cancers5030943
Swindall AF, Stanley JA, Yang ES. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers. 2013; 5(3):943-958. https://doi.org/10.3390/cancers5030943
Chicago/Turabian StyleSwindall, Amanda F., Jennifer A. Stanley, and Eddy S. Yang. 2013. "PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis?" Cancers 5, no. 3: 943-958. https://doi.org/10.3390/cancers5030943
APA StyleSwindall, A. F., Stanley, J. A., & Yang, E. S. (2013). PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers, 5(3), 943-958. https://doi.org/10.3390/cancers5030943