Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies

Abstract
:1. Introduction
1.1. Rational for Combining HDACIs with Other Anticancer Agents
1.2. Structural Requirements of Hb-HDACIs



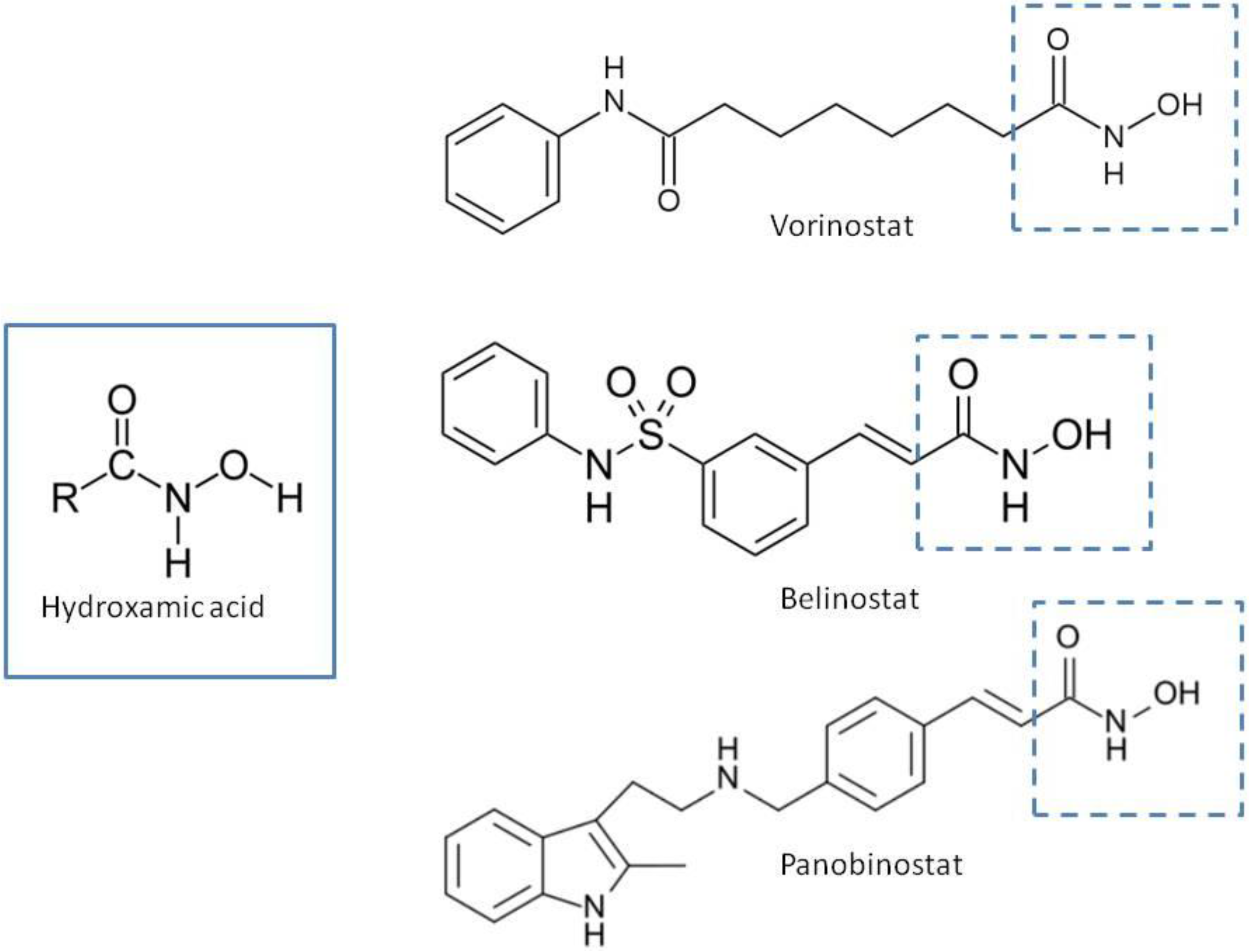{kind=link}
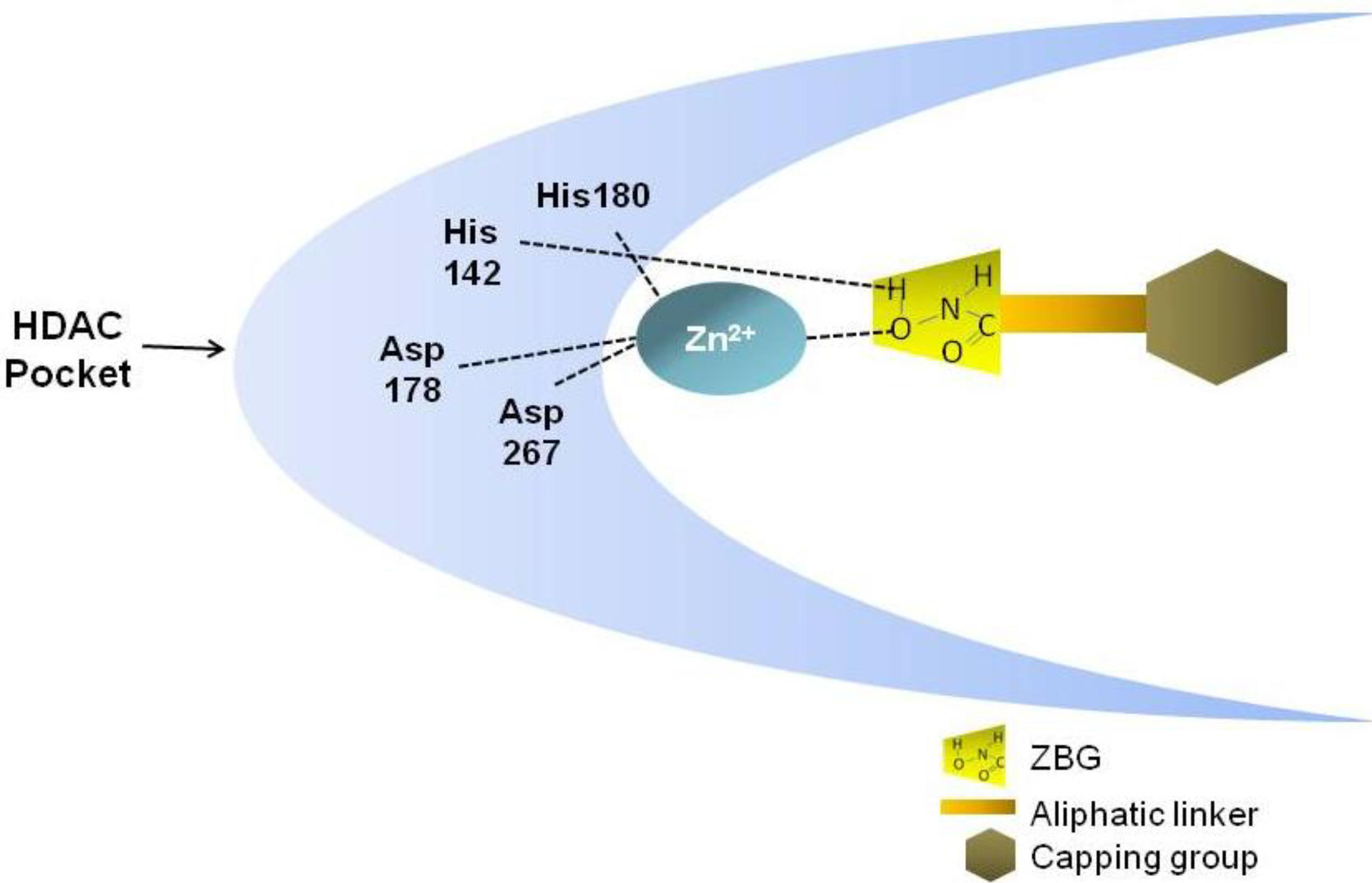{kind=link}
| Hb-HDACIs | Structure | Chemical name | Formula | Molecular mass |
|---|---|---|---|---|
| Vorinostat |  | N-hydroxy-N'-phenyl-octanediamide | C14H20N2O3 | 264.32 g/mol |
| Belinostat (PXD101) |  | (2E)-N-Hydroxy-3-[3-(phenylsulfamoyl) phenyl]prop-2-enamide | C15H14N2O4S | 318.35 g/mol |
| Panobinostat (LBH589) |  | (2E)-N-hydroxy-3-[4-({[2-(2-methyl-1H-indol-3l)ethyl]amino}methyl) phenyl]acrylamide | C21H23N3O2 | 349.42 g/mol |
| Pracinostat (SB939) |  | (E)-3-(2-butyl-1-(2-(diethylamino)ethyl)-1H-benzo[d]imidazol-5-yl)-N-hydroxyacrylamide | C20H30N4O2 | 358.48 g/mol |
| Abexinostat (PCI-24781 /CRA-024781) |  | 3-[(Dimethylamino) methyl]-N-{2-[4-(hydroxycarbamoyl) phenoxy]ethyl}-1-benzofuran-2-carboxamide | C21H23N3O5 | 397.42 g/mol |
| JNJ-26481585 |  | N-hydroxy-2-(4-(((1-methyl-1H-indol-3-yl)methylamino) methyl)piperidin-1-yl) pyrimidine-5-carboxamide | C21H26N6O2 | 394.47 g/mol |
| Dacinostat (LAQ824) |  | (E)-3-(4-(((2-(1H-indol-3-yl) ethyl)(2-hydroxyethyl)amino) methyl)phenyl)-N-hydroxyacrylamide | C22H25N3O3 | 379.45 g/mol |
| Resminostat (RAS2410/4SC-201) |  | (E)-3-(1-((4-((dimethyl amino) methyl)phenyl)sulfonyl)-1H-pyrrol-3-yl)-N-hydroxyacrylamide | C16H19N3O4S | 349.10 g/mol |
| CHR-3996 |  | 2-(6-(((6-fluoroquinolin-2-yl) methyl)amino) bicycle [3,1,0]hexan-3-yl)-N-hydroxypy rimidine-5-carboxamide | C21H20FN5O2 | 393.16 g/mol |
| HDACIs | Routes of administration | Side effects | FDA approval |
|---|---|---|---|
| Vorinostat | Oral | Anorexia, fatigue, dehydration, diarrhea, and myelosuppression | Cutaneous T-cell lymphoma |
| Belinostat (PXD101) | Oral, i.v. | Lethargy/fatigue, nausea and vomiting | Granted orphan drug and fast track designations for relapsed or refractory peripheral T-cell lymphoma |
| Panobinostat (LBH589) | Oral, i.v | Fatigue, nausea, diarrhea and myelosuppression | Not approved |
| Pracinostat (SB939) | Oral | Fatigue, nausea, vomiting, anorexia and diarrhoea | Not approved |
| Abexinostat (PCI-24781/ CRA-024781) | Oral, i.v. | Under evaluation in clinical trials | Not approved |
| JNJ-26481585 | Oral | Under evaluation in clinical trials | Not approved |
| Dacinostat (LAQ824) | i.v. | Nausea, vomiting and fatigue | Not approved |
| Resminostat (RAS2410/4SC-201) | Oral | Under evaluation in clinical trials | Granted orphan drug designation in relapsed/refractory Hodgkin’s lymphoma and hepatocellular carcinoma |
| CHR-3996 | Oral | Thrombocytopenia, fatigue, increase of plasma creatinine and atrial fibrillation | Not approved |
2. Vorinostat in Solid Tumors
2.1. Vorinostat as a Single Agent
| Disease | Regimen | No. pts. | PFS | Efficacy | Ref. |
|---|---|---|---|---|---|
| Metastatic head and neck cancer | Vorinostat: 400 mg orally daily | 13 | SD 2 pts. OR 0% | [51] | |
| Metastatic breast cancer | Vorinostat: 400 mg orally daily | 14 | SD 4 pts | [52] | |
| Relapsed NSCLC | Vorinostat: 400 mg or 300 mg once daily on days 1–14 of the 21 day cycle | 16 | Median: 2.3 m. | SD 57% | [53] |
| Metastatic prostate cancer | Vorinostat: 400 mg orally daily | 27 | Median: 2.8 m. | SD 2 pts. | [54] |
| Ovarian or primary peritoneal carcinoma | Vorinostat: 400 mg orally daily | 27 | PR 1 pt. | [55] | |
| Recurrent GBM | Vorinostat: 200 mg p.o.twice a day for 14 days, followed by a 7-day rest period | 66 | 6-m.-PFS: 9 of the first 52 patients | OR 2 pts. | [56] |
| Metastatic colorectal cancer | Vorinostat: 800 or 1,400 mg/day once a day × 3 days, every 2 weeks +5-FU: preceded by leucovorin, at 400 mg/m2 followed by a 46 h infusion at 2.400 mg/m2 on days 2–3 | 58 | PFS rate did not reach the threshold of 27 out of 43 patients | PR 1 pt. | [57] |
| Untreated stage IIIB or IV NSCLC | Vorinostat: 400 mg or Placebo on days 1 through 14 of each 3-week cycle +Carboplatin: AUC 6 + Paclitaxel: 200 mg/m2, both on day 3 of each 21-day cycle | 94 | Median: Vorinostat: 6.0 m. Placebo: 4.1 m. P = ns | RR Vorinostat: 34% Placebo: 12.5% P = 02 | [58] |
| Advanced thyroid cancer | Vorinostat: 400 mg orally daily | 19 | SD 56% OR 0 | [59] | |
| Recurrent GBM | Vorinostat: 400 mg daily for 14 days of a 21-day cycle + Bortezomib: 1.3 mg/m2 intravenously on days 1,4, 8, and 11 of a 21-day cycle | 37 | Median: 1.5 m. | PR 1 pt. | [60] |
2.2. Vorinostat in Combination Therapy
3. Belinostat in Solid Tumors
| Disease | Regimen | No. pts. | PFS | Efficacy | Ref. |
|---|---|---|---|---|---|
| Recurrent or refractory malignant pleural mesothelioma | Belinostat: 1 g/m2 IV on days 1 to 5 of a 21-day cycle | 13 | Median: 1 m. | SD 2 pts. | [81] |
| Recurrent or refractory advanced thymic epithelial tumors | Belinostat: 1 g/m2 IV on days 1 to 5 of a 21-day cycle | 41 | 6-m.- PFS: 46% | RR 8% CB 68% | [82] |
| Unresectable hepatocellular carcinoma | Belinostat: 1,400 mg/m2 per day, on days 1–5 every 3 weeks | 54 | Median: 2.64 m. | PR 2.4% SD 45.2% | [83] |
| Platinum resistant EOC and LMP ovarian tumors | Belinostat: 1 g/m2 IV on days 1 to 5 of a 21-day cycle | 32 | EOC: Median: 2.3 m. LMP: Median: 13.4 m. | EOC: SD 9/15 pts LMP: PR 2/12 pts | [84] |
| Previously treated ovarian, fallopian tube, or primary peritoneal carcinoma | Belinostat: 1g/m² IV daily for 5 days of a 21-day cycle +Carboplatin: AUC 5 on day three of 21-day cycles | 29 | Median: 3.3 m. | RR 7.4% CR 3.7% PR 3.7% SD 44.4% | [85] |
| Previously treated ovarian cancer | Belinostat: 1g/m² IV daily for 5 days of a 21-day cycle +Carboplatin: AUC 5 +Paclitaxel: 175 mg/m² both on day 3 of each 21-day cycle | 35 | 6-m. PFS: 48% | RR 43% | [86] |
3.1. Belinostat as a Single Agent
3.2. Belinostat in Combination Therapy
4. Panobinostat in Solid Tumors
Efficacy of Panobinostat in Solid Tumor
| Disease | Regimen | No. pts. | PFS | Efficacy | Ref. |
|---|---|---|---|---|---|
| Refractory metastatic renal cell carcinoma. | Panobinostat: 45 mg orally twice a week | 20 | OR 0 | [99] | |
| Previously treated advanced pancreatic cancer | Panobinostat: 20 mg orally three times weekly +Bortezomib: 1.3 mg/m2 IV twice weekly, both during the first two weeks, followed by a 9-day rest period. | Median: 2.1 m. | OR 0 | [100] |
5. Other Hb-HDACIs in Solid Tumors
5.1. Pracinostat
5.2. Abexinostat
5.3. JNJ-26481585
5.4. Dacinostat
5.5. Resminostat
5.6. CHR-3996
6. Conclusions
| Disease | Compound | Regimen | No. Pts. | End-Point | ClinicalTrials. gov Identifier |
|---|---|---|---|---|---|
| Relapsed/refractorysarcomas-age 4–21 years | Vorinostat | Vorinostat: orally on a daily × 4 schedule +Etoposide: at a fixed dose i.v. daily × 3 days | 50 | DLT MTD RR | NCT01294670 |
| HER2-positive locally recurrent or metastatic breast cancer | Vorinostat | Vorinostat: 300 mg × 4 days on, then 3 days off +Lapatinib: 1,250 mg daily | 47 | CB | NCT01118975 |
| Metastatic RCC | Vorinostat | Vorinostat: escalating doses PO BID on days 1–14 +Bevacizumab: IV on day 1 of a 21-day cycle | 42 | MTD PFS | NCT00324870 |
| Advanced soft tissue sarcomas | Vorinostat | Vorinostat: orally once daily for 14 days + Bortezomib: IV on days 1, 4, 8, 11 of a 21-day cycle | 45 | RR | NCT00937495 |
| Metastatic NSCLC | Belinostat | Belinostat: dose escalation with starting dose 1 g/m2 i.v. on days 1-5 of a 21-day cycle +Carboplatin AUC 6 +Paclitaxel 200 mg/m2 | 35 | MTD | NCT01310244 |
| Recurrent GBM | Panobinostat | Panobinostat: orally three times per week every other week +Bevacizumab: i.v. on days 1 and 15 of a 28-day cycle | 67 | MTD PFS | NCT00859222 |
| Advanced sarcomas | Abexinostat | Abexinostat: orally on days 1–5 +Doxorubicin on day 4 of a 21-day cycle | 47 | MTD RR | NCT01027910 |
| K-Ras mutated advanced CRC | Resminostat | Resminostat: orally +OLFIRI i.v. | 80 | MTD PFS | NCT01277406 |
| HCC pretreated with sorafenib | Resminostat | orally | 60 | PFS | NCT00943449 |
Conflict of Interest
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Van Engeland, M.; Derks, S.; Smits, K.M.; Meijer, G.A.; Herman, J.G. Colorectal cancer epigenetics: Complex simplicity. J. Clin. Oncol. 2011, 29, 1382–1391. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Wade, P.A. Transcriptional control at regulatory checkpoints by histone deacetylases: Molecular connections between cancer and chromatin. Hum. Mol. Genet. 2001, 10, 693–698. [Google Scholar] [CrossRef]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (hdacs): Characterization of the classical hdac family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef]
- Bertrand, P. Inside hdac with hdac inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef]
- Khabele, D.; Son, D.S.; Parl, A.K.; Goldberg, G.L.; Augenlicht, L.H.; Mariadason, J.M.; Rice, V.M. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: Implications for therapy. Cancer Biol. Ther. 2007, 6, 795–801. [Google Scholar] [CrossRef]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef]
- Bartling, B.; Hofmann, H.S.; Boettger, T.; Hansen, G.; Burdach, S.; Silber, R.E.; Simm, A. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer 2005, 49, 145–154. [Google Scholar]
- Saji, S.; Kawakami, M.; Hayashi, S.; Yoshida, N.; Hirose, M.; Horiguchi, S.; Itoh, A.; Funata, N.; Schreiber, S.L.; Yoshida, M.; et al. Significance of hdac6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 2005, 24, 4531–4539. [Google Scholar] [CrossRef]
- Tinari, N.; de Tursi, M.; Grassadonia, A.; Zilli, M.; Stuppia, L.; Iacobelli, S.; Natoli, C. An epigenetic approach to pancreatic cancer treatment: The prospective role of histone deacetylase inhibitors. Curr. Cancer Drug Targets 2012, 12, 439–452. [Google Scholar] [CrossRef]
- Mariadason, J.M. Hdacs and hdac inhibitors in colon cancer. Epigenetics 2008, 3, 28–37. [Google Scholar] [CrossRef]
- Abbas, A.; Gupta, S. The role of histone deacetylases in prostate cancer. Epigenetics 2008, 3, 300–309. [Google Scholar] [CrossRef]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of hdac inhibitors involves trail induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef]
- Atadja, P.; Gao, L.; Kwon, P.; Trogani, N.; Walker, H.; Hsu, M.; Yeleswarapu, L.; Chandramouli, N.; Perez, L.; Versace, R.; et al. Selective growth inhibition of tumor cells by a novel histone deacetylase inhibitor, nvp-laq824. Cancer Res. 2004, 64, 689–695. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.S.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Miller, C.P.; Singh, M.M.; Rivera-Del Valle, N.; Manton, C.A.; Chandra, J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011, 2011, 514261. [Google Scholar]
- Bots, M.; Johnstone, R.W. Rational combinations using hdac inhibitors. Clin. Cancer Res. 2009, 15, 3970–3977. [Google Scholar] [CrossRef]
- Stiborova, M.; Eckschlager, T.; Poljakova, J.; Hrabeta, J.; Adam, V.; Kizek, R.; Frei, E. The synergistic effects of DNA-targeted chemotherapeutics and histone deacetylase inhibitors as therapeutic strategies for cancer treatment. Curr. Med. Chem. 2012, 19, 4218–4238. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Modifying chromatin architecture during the response to DNA breakage. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 2–13. [Google Scholar] [CrossRef]
- Schneider, B.J.; Kalemkerian, G.P.; Bradley, D.; Smith, D.C.; Egorin, M.J.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, nsc 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest. New Drugs 2012, 30, 249–257. [Google Scholar] [CrossRef]
- Spratlin, J.L.; Pitts, T.M.; Kulikowski, G.N.; Morelli, M.P.; Tentler, J.J.; Serkova, N.J.; Eckhardt, S.G. Synergistic activity of histone deacetylase and proteasome inhibition against pancreatic and hepatocellular cancer cell lines. Anticancer Res. 2011, 31, 1093–1103. [Google Scholar]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase hdac6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.; Dunner, K., Jr.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef]
- Michaelis, M.; Michaelis, U.R.; Fleming, I.; Suhan, T.; Cinatl, J.; Blaheta, R.A.; Hoffmann, K.; Kotchetkov, R.; Busse, R.; Nau, H.; et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol. Pharmacol. 2004, 65, 520–527. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kato, Y.; Shabbeer, S.; Wei, Y.; Verheul, H.M.; Salumbides, B.; Sanni, T.; Atadja, P.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors: The hydroxamic acid derivative lbh589. Clin. Cancer Res. 2006, 12, 634–642. [Google Scholar] [CrossRef]
- Drappatz, J.; Lee, E.Q.; Hammond, S.; Grimm, S.A.; Norden, A.D.; Beroukhim, R.; Gerard, M.; Schiff, D.; Chi, A.S.; Batchelor, T.T.; et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J. Neurooncol. 2012, 107, 133–138. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef]
- Chen, P.C.; Patil, V.; Guerrant, W.; Green, P.; Oyelere, A.K. Synthesis and structure–activity relationship of histone deacetylase (hdac) inhibitors with triazole-linked cap group. Bioorg. Med. Chem. 2008, 16, 4839–4853. [Google Scholar] [CrossRef]
- Witter, D.J.; Belvedere, S.; Chen, L.; Secrist, J.P.; Mosley, R.T.; Miller, T.A. Benzo[b]thiophene-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4562–4567. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Howman, R.A.; Prince, H.M. New drug therapies in peripheral T-cell lymphoma. Expert Rev. Anticancer Ther. 2011, 11, 457–472. [Google Scholar] [CrossRef]
- Lemoine, M.; Younes, A. Histone deacetylase inhibitors in the treatment of lymphoma. Discov. Med. 2010, 10, 462–470. [Google Scholar]
- Prince, H.M.; Bishton, M.J.; Harrison, S.J. Clinical studies of histone deacetylase inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. [Google Scholar] [CrossRef]
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug Discov. 2007, 6, 21–22. [Google Scholar] [CrossRef]
- Federico, M.; Bagella, L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J. Biomed. Biotechnol. 2011, 2011, 475641. [Google Scholar]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (hdacis): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar]
- Andreeff, M.; Stone, R.; Michaeli, J.; Young, C.W.; Tong, W.P.; Sogoloff, H.; Ervin, T.; Kufe, D.; Rifkind, R.A.; Marks, P.A. Hexamethylene bisacetamide in myelodysplastic syndrome and acute myelogenous leukemia: A phase II clinical trial with a differentiation-inducing agent. Blood 1992, 80, 2604–2609. [Google Scholar]
- Richon, V.M.; Webb, Y.; Merger, R.; Sheppard, T.; Jursic, B.; Ngo, L.; Civoli, F.; Breslow, R.; Rifkind, R.A.; Marks, P.A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 5705–5708. [Google Scholar] [CrossRef]
- Kelly, W.K.; Richon, V.M.; O’Connor, O.; Curley, T.; MacGregor-Curtelli, B.; Tong, W.; Klang, M.; Schwartz, L.; Richardson, S.; Rosa, E.; et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003, 9, 3578–3588. [Google Scholar]
- Ramalingam, S.S.; Kummar, S.; Sarantopoulos, J.; Shibata, S.; LoRusso, P.; Yerk, M.; Holleran, J.; Lin, Y.; Beumer, J.H.; Harvey, R.D.; et al. Phase i study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: A national cancer institute organ dysfunction working group study. J. Clin. Oncol. 2010, 28, 4507–4512. [Google Scholar]
- Vansteenkiste, J.; van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.L.; Randolph, S.S.; Schoffski, P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest. New Drugs 2008, 26, 483–488. [Google Scholar] [CrossRef]
- Blumenschein, G.R., Jr.; Kies, M.S.; Papadimitrakopoulou, V.A.; Lu, C.; Kumar, A.J.; Ricker, J.L.; Chiao, J.H.; Chen, C.; Frankel, S.R. Phase ii trial of the histone deacetylase inhibitor vorinostat (zolinza, suberoylanilide hydroxamic acid, saha) in patients with recurrent and/or metastatic head and neck cancer. Invest. New Drugs 2008, 26, 81–87. [Google Scholar] [CrossRef]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Wong, C.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A california cancer consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef]
- Traynor, A.M.; Dubey, S.; Eickhoff, J.C.; Kolesar, J.M.; Schell, K.; Huie, M.S.; Groteluschen, D.L.; Marcotte, S.M.; Hallahan, C.M.; Weeks, H.R.; et al. Vorinostat (nsc# 701852) in patients with relapsed non-small cell lung cancer: A wisconsin oncology network phase II study. J. Thorac. Oncol. 2009, 4, 522–526. [Google Scholar] [CrossRef]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (national cancer institute trial 6862): Trial results and interleukin-6 analysis: A study by the department of defense prostate cancer clinical trial consortium and university of chicago phase 2 consortium. Cancer 2009, 115, 5541–5549. [Google Scholar]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef]
- Fakih, M.G.; Groman, A.; McMahon, J.; Wilding, G.; Muindi, J.R. A randomized phase II study of two doses of vorinostat in combination with 5-fu/lv in patients with refractory colorectal cancer. Cancer Chemother. Pharmacol. 2012, 69, 743–751. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef]
- Woyach, J.A.; Kloos, R.T.; Ringel, M.D.; Arbogast, D.; Collamore, M.; Zwiebel, J.A.; Grever, M.; Villalona-Calero, M.; Shah, M.H. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J. Clin. Endocrinol. Metab. 2009, 94, 164–170. [Google Scholar]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro Oncol. 2012, 14, 215–221. [Google Scholar] [CrossRef]
- Wilson, P.M.; El-Khoueiry, A.; Iqbal, S.; Fazzone, W.; LaBonte, M.J.; Groshen, S.; Yang, D.; Danenberg, K.D.; Cole, S.; Kornacki, M.; et al. A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-fu-based chemotherapy. Cancer Chemother. Pharmacol. 2010, 65, 979–988. [Google Scholar] [CrossRef]
- Fakih, M.G.; Fetterly, G.; Egorin, M.J.; Muindi, J.R.; Espinoza-Delgado, I.; Zwiebel, J.A.; Litwin, A.; Holleran, J.L.; Wang, K.; Diasio, R.B. A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin. Cancer Res. 2010, 16, 3786–3794. [Google Scholar] [CrossRef]
- Fakih, M.G.; Pendyala, L.; Fetterly, G.; Toth, K.; Zwiebel, J.A.; Espinoza-Delgado, I.; Litwin, A.; Rustum, Y.M.; Ross, M.E.; Holleran, J.L.; et al. A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin. Cancer Res. 2009, 15, 3189–3195. [Google Scholar] [CrossRef]
- Fazzone, W.; Wilson, P.M.; Labonte, M.J.; Lenz, H.J.; Ladner, R.D. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int. J. Cancer 2009, 125, 463–473. [Google Scholar] [CrossRef]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar] [CrossRef]
- Gandia, P.; Arellano, C.; Chalret du Rieu, Q.; Lochon, I.; Campone, M.; Pierga, J.Y.; Poublanc, M.; Hennebelle, I.; Filleron, T.; Chatelut, E.; et al. Unexpected high levels of vorinostat when combined with vinorelbine in patients with advanced cancer. Curr. Clin. Pharmacol. 2011, 6, 274–279. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Parise, R.A.; Ramanathan, R.K.; Lagattuta, T.F.; Musguire, L.A.; Stoller, R.G.; Potter, D.M.; Argiris, A.E.; Zwiebel, J.A.; Egorin, M.J.; et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin. Cancer Res. 2007, 13, 3605–3610. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. Hdac6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Amit, L.; Ben-Aharon, I.; Vidal, L.; Leibovici, L.; Stemmer, S. The impact of bevacizumab (avastin) on survival in metastatic solid tumors- a meta-analysis and systematic review. PLoS One 2013, 8, e51780. [Google Scholar]
- Ramaswamy, B.; Fiskus, W.; Cohen, B.; Pellegrino, C.; Hershman, D.L.; Chuang, E.; Luu, T.; Somlo, G.; Goetz, M.; Swaby, R.; et al. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: Evidence for vorinostat-induced tubulin acetylation and hsp90 inhibition in vivo. Breast Cancer Res. Treat. 2012, 132, 1063–1072. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Chowdhary, S.; Potthast, L.; Prabhu, A.; Tsai, Y.Y.; Sarcar, B.; Kahali, S.; Brem, S.; Yu, H.M.; Rojiani, A.; et al. Phase I trial of vorinostat combined with bevacizumab and cpt-11 in recurrent glioblastoma. Neuro Oncol. 2012, 14, 93–100. [Google Scholar] [CrossRef]
- Du, X.L.; Chen, Q. Recent advancements of bortezomib in acute lymphocytic leukemia treatment. Acta Haematol. 2013, 129, 207–214. [Google Scholar] [CrossRef]
- Kaur, G.; Stetler-Stevenson, M.; Sebers, S.; Worland, P.; Sedlacek, H.; Myers, C.; Czech, J.; Naik, R.; Sausville, E. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone l86–8275. J. Natl. Cancer Inst. 1992, 84, 1736–1740. [Google Scholar] [CrossRef]
- Dickson, M.A.; Rathkopf, D.E.; Carvajal, R.D.; Grant, S.; Roberts, J.D.; Reid, J.M.; Ames, M.M.; McGovern, R.M.; Lefkowitz, R.A.; Gonen, M.; et al. A phase I pharmacokinetic study of pulse-dose vorinostat with flavopiridol in solid tumors. Invest. New Drugs 2011, 29, 1004–1012. [Google Scholar] [CrossRef]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase I clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; la Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor pxd101. Mol. Cancer Ther 2003, 2, 721–728. [Google Scholar]
- Qian, X.; LaRochelle, W.J.; Ara, G.; Wu, F.; Petersen, K.D.; Thougaard, A.; Sehested, M.; Lichenstein, H.S.; Jeffers, M. Activity of pxd101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol. Cancer Ther. 2006, 5, 2086–2095. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II study of belinostat (pxd101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef]
- Giaccone, G.; Rajan, A.; Berman, A.; Kelly, R.J.; Szabo, E.; Lopez-Chavez, A.; Trepel, J.; Lee, M.J.; Cao, L.; Espinoza-Delgado, I.; et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J. Clin. Oncol. 2011, 29, 2052–2059. [Google Scholar] [CrossRef]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y.; et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the mayo phase II consortium and the cancer therapeutics research group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [CrossRef]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.A.; Tsao, M.S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (lmp) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; Disilvestro, P.A.; Fader, A.N. A phase ii evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef]
- Dizon, D.S.; Damstrup, L.; Finkler, N.J.; Lassen, U.; Celano, P.; Glasspool, R.; Crowley, E.; Lichenstein, H.S.; Knoblach, P.; Penson, R.T. Phase II activity of belinostat (pxd-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 979–986. [Google Scholar] [CrossRef]
- Steele, N.L.; Plumb, J.A.; Vidal, L.; Tjornelund, J.; Knoblauch, P.; Rasmussen, A.; Ooi, C.E.; Buhl-Jensen, P.; Brown, R.; Evans, T.R.; et al. A phase 1 pharmacokinetic and harmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 804–810. [Google Scholar] [CrossRef]
- Takada, M.; Kataoka, A.; Toi, M.; Bando, H.; Toyama, K.; Horiguchi, S.; Ueno, T.; Linder, S.; Saji, S.; Hayashi, Y.; et al. A close association between alteration in growth kinetics by neoadjuvant chemotherapy and survival outcome in primary breast cancer. Int. J. Oncol. 2004, 25, 397–405. [Google Scholar]
- Lassen, U.; Molife, L.R.; Sorensen, M.; Engelholm, S.A.; Vidal, L.; Sinha, R.; Penson, R.T.; Buhl-Jensen, P.; Crowley, E.; Tjornelund, J.; et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br. J. Cancer 2010, 103, 12–17. [Google Scholar] [CrossRef]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor lbh589 and the hsp90 inhibitor 17-aag is highly active against human cml-bc cells and aml cells with activating mutation of flt-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Breslow, R.; Rifkind, R.A. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 2001, 13, 477–483. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Hamberg, P.; Woo, M.M.; Chen, L.C.; Verweij, J.; Porro, M.G.; Zhao, L.; Li, W.; van der Biessen, D.; Sharma, S.; Hengelage, T.; et al. Effect of ketoconazole-mediated cyp3a4 inhibition on clinical pharmacokinetics of panobinostat (lbh589), an orally active histone deacetylase inhibitor. Cancer Chemother. Pharmacol. 2011, 68, 805–813. [Google Scholar] [CrossRef]
- Morita, S.; Oizumi, S.; Minami, H.; Kitagawa, K.; Komatsu, Y.; Fujiwara, Y.; Inada, M.; Yuki, S.; Kiyota, N.; Mitsuma, A.; et al. Phase I dose-escalating study of panobinostat (lbh589) administered intravenously to japanese patients with advanced solid tumors. Invest. New Drugs 2012, 30, 1950–1957. [Google Scholar] [CrossRef]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer. Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef]
- Jones, S.F.; Infante, J.R.; Thompson, D.S.; Mohyuddin, A.; Bendell, J.C.; Yardley, D.A.; Burris, H.A., 3rd. A phase I trial of oral administration of panobinostat in combination with paclitaxel and carboplatin in patients with solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 471–475. [Google Scholar] [CrossRef]
- Strickler, J.H.; Starodub, A.N.; Jia, J.; Meadows, K.L.; Nixon, A.B.; Dellinger, A.; Morse, M.A.; Uronis, H.E.; Marcom, P.K.; Zafar, S.Y.; et al. Phase I study of bevacizumab, everolimus, and panobinostat (lbh-589) in advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 251–258. [Google Scholar] [CrossRef]
- Jones, S.F.; Bendell, J.C.; Infante, J.R.; Spigel, D.R.; Thompson, D.S.; Yardley, D.A.; Greco, F.A.; Murphy, P.B.; Burris, H.A., 3rd. A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin. Adv. Hematol. Oncol. 2011, 9, 225–230. [Google Scholar]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Invest. 2011, 29, 451–455. [Google Scholar]
- Wang, H.; Cao, Q.; Dudek, A.Z. Phase II study of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy. Anticancer Res. 2012, 32, 1027–1031. [Google Scholar]
- Novotny-Diermayr, V.; Sangthongpitag, K.; Hu, C.Y.; Wu, X.; Sausgruber, N.; Yeo, P.; Greicius, G.; Pettersson, S.; Liang, A.L.; Loh, Y.K.; et al. Sb939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol. Cancer Ther. 2010, 9, 642–652. [Google Scholar] [CrossRef]
- Yong, W.P.; Goh, B.C.; Soo, R.A.; Toh, H.C.; Ethirajulu, K.; Wood, J.; Novotny-Diermayr, V.; Lee, S.C.; Yeo, W.L.; Chan, D.; et al. Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, sb939, in patients with refractory solid malignancies. Ann. Oncol. 2011, 22, 2516–2522. [Google Scholar] [CrossRef]
- Razak, A.R.; Hotte, S.J.; Siu, L.L.; Chen, E.X.; Hirte, H.W.; Powers, J.; Walsh, W.; Stayner, L.A.; Laughlin, A.; Novotny-Diermayr, V.; et al. Phase I clinical, pharmacokinetic and pharmacodynamicstudy of sb939, an oral histone deacetylase (hdac) inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2011, 104, 756–762. [Google Scholar] [CrossRef]
- Buggy, J.J.; Cao, Z.A.; Bass, K.E.; Verner, E.; Balasubramanian, S.; Liu, L.; Schultz, B.E.; Young, P.R.; Dalrymple, S.A. Cra-024781: A novel synthetic inhibitor of histone deacetylase enzymes with antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2006, 5, 1309–1317. [Google Scholar] [CrossRef]
- Lopez, G.; Liu, J.; Ren, W.; Wei, W.; Wang, S.; Lahat, G.; Zhu, Q.S.; Bornmann, W.G.; McConkey, D.J.; Pollock, R.E.; et al. Combining pci-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin. Cancer Res. 2009, 15, 3472–3483. [Google Scholar] [CrossRef]
- Yang, C.; Choy, E.; Hornicek, F.J.; Wood, K.B.; Schwab, J.H.; Liu, X.; Mankin, H.; Duan, Z. Histone deacetylase inhibitor (hdaci) pci-24781 potentiates cytotoxic effects of doxorubicin in bone sarcoma cells. Cancer Chemother. Pharmacol. 2011, 67, 439–446. [Google Scholar] [CrossRef]
- Yang, C.; Choy, E.; Hornicek, F.J.; Wood, K.B.; Schwab, J.H.; Liu, X.; Mankin, H.; Duan, Z. Histone deacetylase inhibitor pci-24781 enhances chemotherapy-induced apoptosis in multidrug-resistant sarcoma cell lines. Anticancer Res. 2011, 31, 1115–1123. [Google Scholar]
- Lopez, G.; Torres, K.; Liu, J.; Hernandez, B.; Young, E.; Belousov, R.; Bolshakov, S.; Lazar, A.J.; Slopis, J.M.; McCutcheon, I.E.; et al. Autophagic survival in resistance to histone deacetylase inhibitors: Novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011, 71, 185–196. [Google Scholar] [CrossRef]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of lsd1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011, 13, 894–903. [Google Scholar] [CrossRef]
- Kitamura, T.; Connolly, K.; Ruffino, L.; Ajiki, T.; Lueckgen, A.; DiGiovanni, J.; Kiguchi, K. The therapeutic effect of histone deacetylase inhibitor pci-24781 on gallbladder carcinoma in bk5. Erbb2 mice. J. Hepatol. 2012, 57, 84–91. [Google Scholar] [CrossRef]
- Arts, J.; King, P.; Marien, A.; Floren, W.; Belien, A.; Janssen, L.; Pilatte, I.; Roux, B.; Decrane, L.; Gilissen, R.; et al. Jnj-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin. Cancer Res. 2009, 15, 6841–6851. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Tai, Y.T.; Atadja, P.; Remiszewski, S.; Hideshima, T.; Mitsiades, N.; Shringarpure, R.; LeBlanc, R.; Chauhan, D.; et al. Nvp-laq824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 2003, 102, 2615–2622. [Google Scholar] [CrossRef]
- Fuino, L.; Bali, P.; Wittmann, S.; Donapaty, S.; Guo, F.; Yamaguchi, H.; Wang, H.G.; Atadja, P.; Bhalla, K. Histone deacetylase inhibitor laq824 down-regulates her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone b. Mol. Cancer Ther. 2003, 2, 971–984. [Google Scholar]
- Remiszewski, S.W.; Sambucetti, L.C.; Bair, K.W.; Bontempo, J.; Cesarz, D.; Chandramouli, N.; Chen, R.; Cheung, M.; Cornell-Kennon, S.; Dean, K.; et al. N-hydroxy-3-phenyl-2-propenamides as novel inhibitors of human histone deacetylase with in vivo antitumor activity: Discovery of (2e)-n-hydroxy-3-[4-[[(2-hydroxyethyl)[2-(1h-indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide (nvp-laq824). J. Med. Chem. 2003, 46, 4609–4624. [Google Scholar] [CrossRef]
- Chen, L.; Meng, S.; Wang, H.; Bali, P.; Bai, W.; Li, B.; Atadja, P.; Bhalla, K.N.; Wu, J. Chemical ablation of androgen receptor in prostate cancer cells by the histone deacetylase inhibitor laq824. Mol. Cancer Ther. 2005, 4, 1311–1319. [Google Scholar]
- Bluethner, T.; Niederhagen, M.; Caca, K.; Serr, F.; Witzigmann, H.; Moebius, C.; Mossner, J.; Wiedmann, M. Inhibition of histone deacetylase for the treatment of biliary tract cancer: A new effective pharmacological approach. World J. Gastroenterol. 2007, 13, 4761–4770. [Google Scholar]
- Egler, V.; Korur, S.; Failly, M.; Boulay, J.L.; Imber, R.; Lino, M.M.; Merlo, A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin. Cancer Res. 2008, 14, 3132–3140. [Google Scholar] [CrossRef]
- Haefner, M.; Bluethner, T.; Niederhagen, M.; Moebius, C.; Wittekind, C.; Mossner, J.; Caca, K.; Wiedmann, M. Experimental treatment of pancreatic cancer with two novel histone deacetylase inhibitors. World J. Gastroenterol. 2008, 14, 3681–3692. [Google Scholar] [CrossRef]
- Hurtubise, A.; Bernstein, M.L.; Momparler, R.L. Preclinical evaluation of the antineoplastic action of 5-aza-2'-deoxycytidine and different histone deacetylase inhibitors on human ewing’s sarcoma cells. Cancer Cell Int. 2008, 8, 16. [Google Scholar] [CrossRef]
- Kato, Y.; Salumbides, B.C.; Wang, X.F.; Qian, D.Z.; Williams, S.; Wei, Y.; Sanni, T.B.; Atadja, P.; Pili, R. Antitumor effect of the histone deacetylase inhibitor laq824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol. Cancer Ther. 2007, 6, 70–81. [Google Scholar]
- De Bono, J.S.; Kristeleit, R.; Tolcher, A.; Fong, P.; Pacey, S.; Karavasilis, V.; Mita, M.; Shaw, H.; Workman, P.; Kaye, S.; et al. Phase I pharmacokinetic and pharmacodynamic study of laq824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 6663–6673. [Google Scholar] [CrossRef]
- Mandl-Weber, S.; Meinel, F.G.; Jankowsky, R.; Oduncu, F.; Schmidmaier, R.; Baumann, P. The novel inhibitor of histone deacetylase resminostat (ras2410) inhibits proliferation and induces apoptosis in multiple myeloma (mm) cells. Br. J. Haematol. 2010, 149, 518–528. [Google Scholar] [CrossRef]
- Brunetto, A.T.; Ang, J.E.; Lal, R.; Olmos, D.; Frentzas, S.; Mais, A.; Hauns, B.; Mollenhauer, M.; Lahu, G.; de Bono, J.S. A first-in-human phase I study of 4sc-201, an oral histone deacetylase (hdac) inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2009, 27, 3530. [Google Scholar]
- Bitzer, M.; Horger, M.; Ganten, T.M.; Lauer, U.M.; Woerns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Wege, H.; Giannini, E.G.; et al. Efficacy, safety, tolerability, and pk of the hdac inhibitor resminostat in sorafenib-refractory hepatocellular carcinoma (hcc): Phase II shelter study. J. Clin. Oncol. 2012, 30, Abstract 4115. [Google Scholar]
- Moffat, D.; Patel, S.; Day, F.; Belfield, A.; Donald, A.; Rowlands, M.; Wibawa, J.; Brotherton, D.; Stimson, L.; Clark, V.; et al. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo [3.1.0]hex-3-yl)-n-hydroxypyrim idine-5-carboxamide (chr-3996), a class I selective orally active histone deacetylase inhibitor. J. Med. Chem. 2010, 53, 8663–8678. [Google Scholar] [CrossRef]
- Banerji, U.; van Doorn, L.; Papadatos-Pastos, D.; Kristeleit, R.; Debnam, P.; Tall, M.; Stewart, A.; Raynaud, F.; Garrett, M.D.; Toal, M.; et al. A phase I pharmacokinetic and pharmacodynamic study of chr-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin. Cancer Res. 2012, 18, 2687–2694. [Google Scholar] [CrossRef]
- Clinicaltrials.Gov, U.S. National institutes of health. Available online: http://www.clinicaltrial.gov/ (accessed on 15 April 2013).
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Invest. 2002, 109, 1139–1142. [Google Scholar]
- Fantin, V.R.; Loboda, A.; Paweletz, C.P.; Hendrickson, R.C.; Pierce, J.W.; Roth, J.A.; Li, L.; Gooden, F.; Korenchuk, S.; Hou, X.S.; et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous t-cell lymphoma. Cancer Res. 2008, 68, 3785–3794. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grassadonia, A.; Cioffi, P.; Simiele, F.; Iezzi, L.; Zilli, M.; Natoli, C. Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies. Cancers 2013, 5, 919-942. https://doi.org/10.3390/cancers5030919
Grassadonia A, Cioffi P, Simiele F, Iezzi L, Zilli M, Natoli C. Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies. Cancers. 2013; 5(3):919-942. https://doi.org/10.3390/cancers5030919
Chicago/Turabian StyleGrassadonia, Antonino, Pasquale Cioffi, Felice Simiele, Laura Iezzi, Marinella Zilli, and Clara Natoli. 2013. "Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies" Cancers 5, no. 3: 919-942. https://doi.org/10.3390/cancers5030919