1. Introduction
Obesity has been recently recognized as a new disease and one of the leading health problems in the United States [
1,
2], associated with increased risk of cardiovascular disorders, diabetes and cancer [
3]. Breast cancer, the most common cancer in women and the second leading cause of mortality from this disease in our country, is a more prevailing and aggressive disease in overweight and obese women [
4]. Obesity increases the risk of developing ER
+ postmenopausal breast cancer, although there are contradictory evidences that obesity [
5] or rather circulatory estrogen [
6] protects against premenopausal breast cancer, while there are also indications that it is associated with poor breast cancer prognosis regardless of menopausal status [
7]. However, the causes of these associations are not completely understood. Chronic inflammation is common to obesity [
8] and to many cancers [
9], and elevated numbers of macrophages colonize obese adipose tissues [
10] and also advanced cancers [
11,
12]. Macrophages, central cells in the inflammatory response, are also the most abundant immune cell type in tumor microenvironments, where they help tumor progression and are a sign of poor tumor prognosis [
13,
14,
15]. In previous studies we have shown that blood monocytes and peripheral and tumor-associated macrophages (TAMs) from mammary tumor-bearing mice are impacted to different degrees in their phenotypes and functions by the tumors and the factors they secrete [
16,
17,
18,
19,
20].
Most studies linking obesity and cancer have focused on the systemic inflammatory effects of adiposity [
21], which result in elevated circulating levels of pro-inflammatory adipokines, cytokines, chemokines, insulin, estrogen and other factors directly secreted by obese adipose tissues or indirectly induced to be secreted by other tissues. Given that the mammary gland contains a significant amount of white adipose tissue required for normal breast gland development, it is plausible that this local adipose tissue in the breast (bAT) could be inflamed in obese females as the visceral fat is, and may thus play a role in breast cancer development in conditions of obesity. The local role of bAT inflammation on the association of breast cancer with obesity has been recently analyzed in mice and humans [
22,
23], but it is still insufficiently understood and requires further examination. Breast cancer is one of the cancers not currently associated with chronic inflammation despite the association of this disease with obesity. Whether obesity also promotes breast cancer through its effect in local adipose tissue inflammation and innate immune signaling in the breast, where cancer occurs, has not received enough attention and should be more thoroughly investigated.
We propose that obesity impacts breast cancer not only systemically but also at the local level in the breast. We hypothesize that mammary paracrine factors secreted by adipocytes and tumor cells in the breast of obese/overweight females modulate macrophage recruitment to the tumor microenvironment, as well as macrophage phenotypes, activation patterns and functions, contributing to breast cancer progression. Moreover, paracrine factors resulting from the crosstalk among adipocytes, tumor cells and macrophages in the breast tumor microenvironment might contribute to tumor progression via additional mechanisms.
To analyze how adipocyte/tumor cell-derived factors in the breast affect macrophage recruitment/functions and promote breast cancer progression, we used a reductionist in vitro co-culture setting to mimic the mammary tumor microenvironment in obese mice. These in vitro studies were complemented with in vivo mouse experiments using diet-induced obese (DIO) mammary tumor-bearing female mice in which tumor progression and bAT inflammation were assessed. We examined the overarching hypothesis that bAT in obese mice exhibits inflammatory and tumor-promoting capabilities that foster breast cancer development. For the in vitro studies, we investigated the crosstalk between three critical cell types in the breast cancer microenvironment, i.e., the mammary tumor cell, the adipocyte and the macrophage. We interrogated whether adipocytes and mammary tumor cells, acting together or independently, recruit macrophages and modify their activation profiles/functions, to further tumor progression. Specifically, we analyzed whether adipocytes’ crosstalk with tumor cells and macrophages modulates their mutual production of paracrine factors such as the pro-tumor adipokine leptin, the pro-inflammatory saturated free fatty acid (FFA) lauric acid, and the chemokine CCL2, thus contributing to macrophage recruitment and tumor progression. In addition, we revealed the existence of numerous other proteins with chemotactic, proinflammatory and tumor-promoting properties as a result of the interplay between those three cell types. Furthermore we show the effects of leptin in the modulation of macrophage phenotypes and functions. Overall, our results indicate that local breast adipocytes are important players of tumor progression in the mammary cancer microenvironment of obese mice, and that their crosstalk with mammary tumor cells and macrophages is critical in the production of different paracrine factors that contribute to macrophage chemotaxis/function and tumor development. We also demonstrate that mammary tumors in obese mice are larger than those in lean ones, and that the bAT from obese tumor-bearers is inflamed, containing higher numbers of macrophages, crown-like structures (CLS) and hypertrophic adipocytes than the bAT from lean tumor-bearers. Our study also reveals that bAT distant from the tumor site is less inflamed in obese females that the bAT in the tumor microenvironment, suggesting that the contact of adipose tissue with tumor cells contributes to macrophage recruitment. Collectively, we conclude that obesity plays not only a systemic but also a local role in breast cancer development through the increased presence of inflammatory cells and molecules in the bAT.
2. Experimental Section
2.1. Materials
Recombinant (murine) and synthetic paracrine factors were from the following sources: leptin and CCL2 were from Peprotech (Rocky Hill, NJ, USA) and the saturated FFA lauric acid was from Nu-Chek Prep, Inc. (Elysian, MN, USA). Stock solutions were prepared using the following solvents: sterile water (leptin); PBS+0.1%BSA (CCL2) and 100% ethanol (lauric acid). Further dilutions were prepared in culture media to add to the cell cultures. Specific paracrine factor inhibitors were the following: CCL2 blocking antibody was from R & D System (Minneapolis, MN, USA); pegylated leptin receptor blocking peptide (LPrA2) was provided by Dr. R. R Gonzalez-Perez, whereas Eritoran, a TLR4 antagonist, was kindly supplied by Eisai Inc. (Andover, MA, USA). Matrigel was purchased from BD Biosciences (San Jose, CA, USA).
2.2. Cells
E0771 mammary tumor cells (syngeneic to C57BL6 mice), a very aggressive ER
+ and estrogen-dependent breast cancer cell line that metastasizes to the peritoneal cavity and lungs and closely resembles aggressive forms of human breast cancer [
24], was generously provided by Dr. M. Kolonin (Center for Stem Cell Research, Institute of Molecular Medicine, University of Texas, Houston, TX, USA). The human acute monocytic leukemia THP-1 cell line was kindly supplied by Dr. V. Gupta (Rush University, Chicago, IL, USA). Adipocytes were either
in vitro differentiated from 3T3-L1 murine preadipocytes using the 3T3-L1 Growth and Differentiation Feeding Schedule following instructions from ZenBio Inc. (Research Triangle Park, NC, USA), or
ex vivo isolated from visceral fat of diet-induced obese (DIO) C57BL6 female mice. Peritoneal elicited macrophages (N-PEMs) and tumor-associated macrophages (TAMs) were isolated from C57BL6 normal and tumor-bearing female mice, respectively, as previously described [
17,
20].
2.3. Ex vivo Isolation of Adipocytes
Adipocytes were separated from other cell types present in the visceral white adipose tissue of diet-induced obese (DIO) female mice by enzymatic digestion of the tissue with collagenase. Briefly, approximately 200–400 mg of tissue were minced into small pieces (~1 mm) and incubated in 4 volumes of 1 mg/mL collagenase IV (Worthington Biochemical Corporation, Lakewood, NJ, USA) in PBS for 30 min at 37 °C. The sample was centrifuged at 600 × g for 2 min to obtain an adipocyte fraction that floats and the stromal vascular fraction (SVF) that pellets. Ex-vivo isolated adipocytes were then cultured in the ZenBio’s adipocyte medium AM-1-L1 (ZenBio Inc.), or they were mixed with macrophages and tumor cells in co-cultures as described below.
2.4. Pre-Treatment of Macrophages with Conditioned Medium from E0771 Cells, Adipocytes, Their Co-Cultures, and with Recombinant Paracrine Factors
Conditioned media from adipocytes (in vitro differentiated or ex vivo isolated), from E0771 mammary tumor cells and from their co-cultures without and with peritoneal elicited macrophages from normal mice (N-PEMs) were centrifuged and supernatants were frozen at −80 °C for further pre-treatment of N-PEMs and for protein analysis by ELISA and Luminex. Recombinant or synthetic paracrine factors (leptin, CCL2 and lauric acid) were prepared fresh just before use. N-PEMs were isolated and adhered to plastic tissue culture dishes, cultured in Nutridoma serum-free culture medium (Roche) and pre-treated for the referred time intervals in the different experimental conditions explained in the figures. Macrophages were lysed and Western blot analysis was performed (as described below) or supernatants were collected for ELISA or Luminex studies. Viability was assessed by trypan blue exclusion.
2.5. Cell Co-Cultures
We co-cultured mouse peritoneal elicited macrophages (N-PEMs), adipocytes (in vitro differentiated or ex vivo isolated) and E0771 mammary tumor cells for 48 h. To do this, when 3T3-L1 in vitro differentiated adipocytes were used, first 3T3-L1 fibroblasts were grown until 80%–90% confluence, at which point they started in vitro differentiation into adipocytes for 11 days following instructions from ZenBio Inc.; 5 × 105 macrophages and 5 × 105 E0771 cells per well were then plated onto those 3T3-L1 differentiated adipocytes. On the other hand, when ex vivo isolated adipocytes were used, the floating fraction of adipocytes was isolated from visceral fat as described above, and 5 × 105 adipocytes from this fraction were mixed with 5 × 105 macrophages and 5 × 105 E0771 cells and seeded altogether. Co-cultures were carried out in 6-well plates, and conditioned medium was harvested, centrifuged and supernatants were frozen at −80 °C for further analyses. By setting up the co-cultures using these cell numbers, at the end of the 48 h of co-cultures, the numbers of E0771 tumor cells, which do proliferate, will exceed the numbers of macrophages and adipocytes, as is the case in the mammary tumor microenvironment.
2.6. Migration (Chemotaxis) Assay
Assays were done in triplicate in migration chambers [24 well cell culture plate from Costar (VWR International, Radnor, PA, USA), with 8.0 μm pore size PET track-etched membrane cell culture insert (BD Falcon, Franklin Lakes, NJ, USA)]. 0.5–1 × 106 THP-1 cells were added to the upper chamber in 100 µL of serum-free medium (FBS-free adipocyte medium, AM-1-L1, ZenBio Inc.). The bottom well was filled with 600 µL cell-free supernatants from 3T3-L1-in vitro differentiated adipocytes or from ex-vivo isolated adipocytes, N-PEM, E0771 cells or their co-cultures in FBS-free AM-1-L1-SF medium or with the following recombinant paracrine factors and their antagonists: 10 and 50 ng/mL CCL2; 25 µg/mL CCL2 blocking antibody; 3, 100 and 500 ng/mL leptin; 100 nM LPrA2 (leptin receptor blocking peptide) and 2,5, 5, 10 and 100 µM lauric acid and 10 ng/mL Eritoran. After 2.5 h of incubation at 37 °C/5%CO2, the THP-1 cells that migrated to the bottom well were counted on the microscope. Assays were done in triplicate in migration chambers.
2.7. ELISA
Cell and serum-free supernatants from in vitro differentiated 3T3-L1, ex vivo isolated adipocytes, N-PEM macrophages, E0771 mammary tumor cells and their co-cultures, or from N-PEMs macrophages pre-treated with paracrine factors, were harvested after 20 h incubation and tested by ELISA for the indicated cytokines/chemokines following manufacturers’ instructions. Murine leptin and CCL2 (MCP-1) were determined by ELISA kits from R&D Systems (Minneapolis, MN, USA), and murine IL-12p70 and IL-10 were analyzed using kits from eBiosciences (San Diego, CA, USA).
2.8. Nitric Oxide
Nitric oxide was determined in cell supernatants from leptin-pretreated N-PEMs using the Griess colorimetric reaction as previously reported [
17,
25].
2.9. Free Fatty Acid (FFA) Determination
To measure the release of FFA from adipocytes, we analyzed adipocytes’ 48 h supernatants using an acyl-CoA oxidase-based enzymatic colorimetric methodology (WAKO Diagnostics, Richmond, VA, USA), following the manufacturer’s instructions and adapted for use on a Roche Cobas 6000 chemistry analyzer (Roche Diagnostics, Indianapolis, IN, USA). The intra- and inter- assay precision were <2.7% CV and 3.6%, respectively.
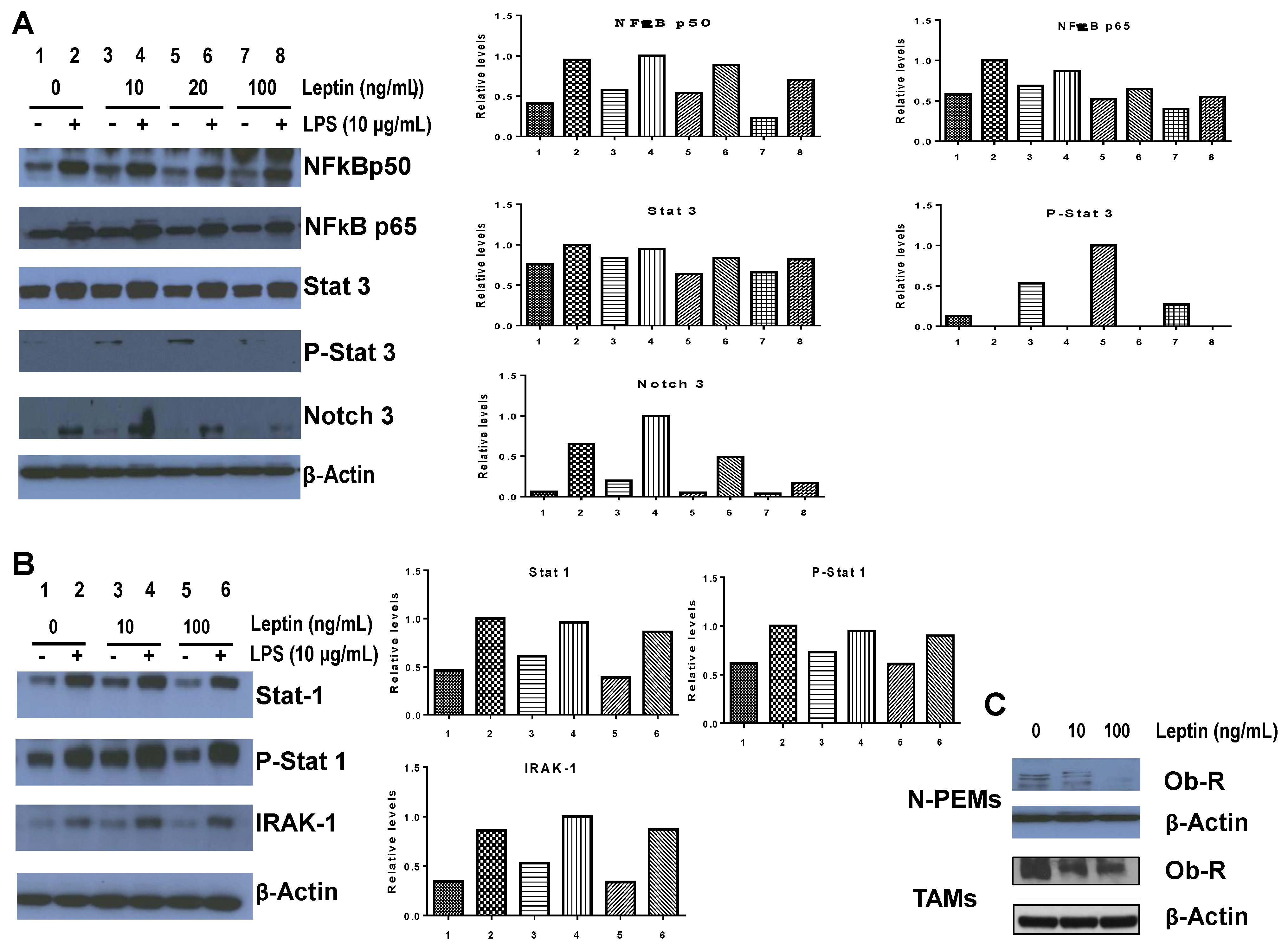
2.10. Western Blot
N-PEMs (10
7) were adhered to plastic tissue culture dishes and treated with 10, 20 and 100 ng/mL murine recombinant leptin with or without LPS (Sigma Aldrich, St. Louis, MO, USA) at 10 µg/mL for 20 h. Cells were then lysed in 100 µL RIPA lysis buffer and Western blots were performed as previously described [
18,
20]. Rabbit anti-mouse polyclonal antibodies (NFκBp50, NFκBp65), a goat anti-mouse polyclonal (Ob-R), a rabbit anti-mouse polyclonal (Notch 3) and a rabbit anti-mouse polyclonal (IRAK-1), all from Santa Cruz Biotechnologies (Santa Cruz, CA, USA), were used. Rabbit α-mouse STAT and phospho STAT (1 and 3) antibodies were from Cell Signaling (Cell Signaling Technology, Inc., Danvers, MA, USA). Rabbit α-mouse actin polyclonal antibody was obtained from Sigma-Aldrich and goat anti-rabbit IgG-HRP, bovine anti-goat IgG-HRP were both from Santa Cruz Biotechnologies. Densitometries were performed on the autoradiograms using Image J software (National Institutes of Health).
2.11. Luminex Assay
MILLIPLEXR MAP Mouse Cytokine/Chemokine kit (Millipore, Billerica, MA, USA) was used for the simultaneous quantification of 32 mouse cytokines and chemokines in cell-free/serum-free culture supernatants from in vitro differentiated 3T3-L1, ex-vivo isolated adipocytes, N-PEM and E0771 cells and their co-cultures. Cytokines and chemokines analyzed were: Eotaxin, G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7 IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-15, IL-17, IP-10, KC, LIF, LIX, MCP-1, M-CSF, MIG, MIP-1α, MIP-1β, MIP-2, RANTES, TNFα and VEGF. The procedure was performed according to the manufacturer’s protocol. Plate reading was done on the Luminex 200 platform (Luminex Corp, Austin, TX, USA).
2.12. Flow Cytometry
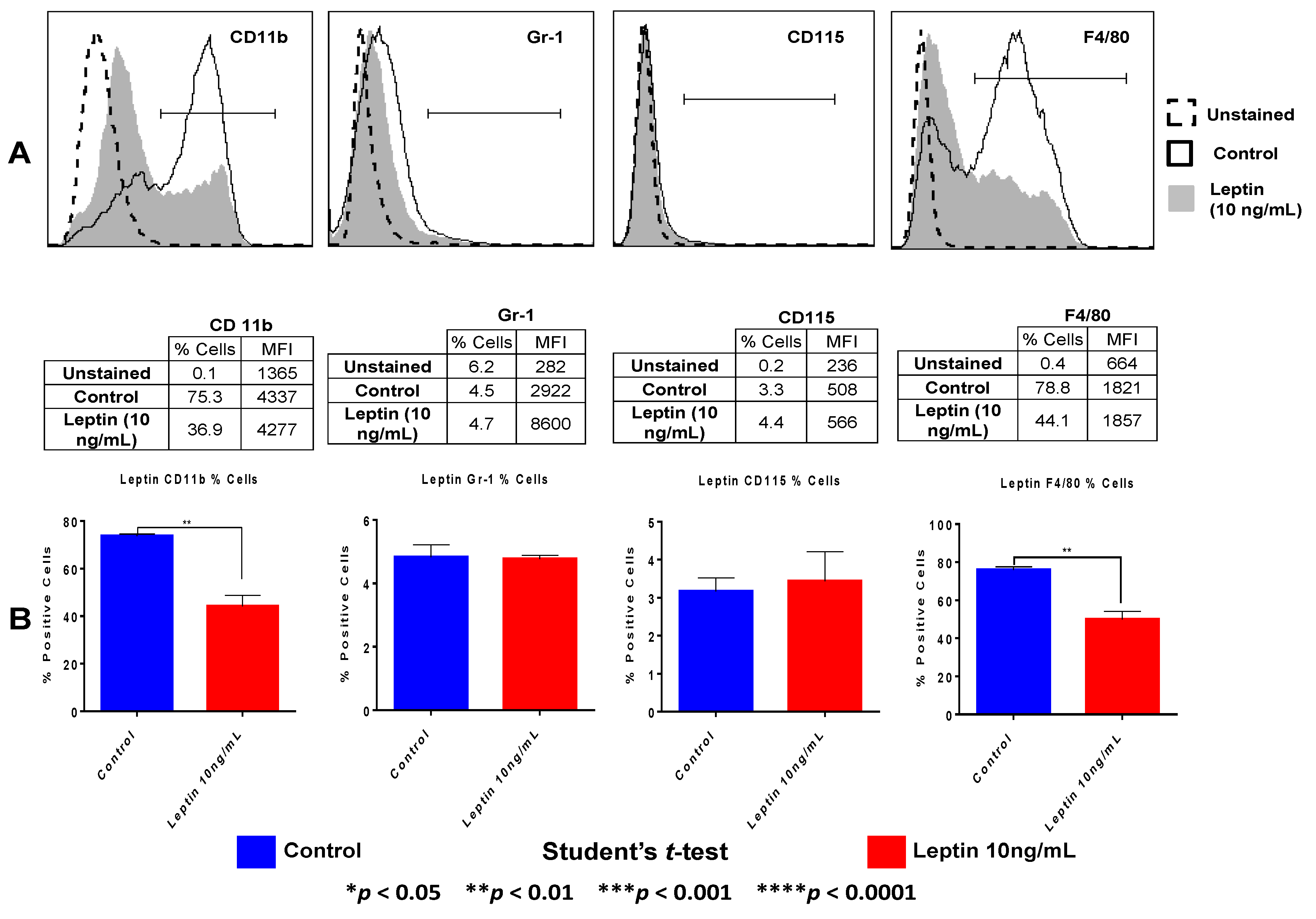
N-PEMs were gently scraped from tissue culture plates, washed and counted after 20 h treatment with 10 ng/mL leptin. Fc receptors were blocked for 5 min with a Fc receptor antibody (eBioscience), and cells were stained with CD11b-FITC, CD115-PE, Gr-1-APC antibodies, all from eBioscience, and F4/80-PE Cy7 (BioLegend). Samples were acquired in a FACS Canto-II Analyzer and analyzed using FlowJo vX.0.6 (BD Biosciences, San Jose, CA, USA). Percentage of cells expressing the antibody and Mean Fluorescent Intensity (MFI) were determined.
2.13. Mice, Diets, Tumors and Treatments
C57BL6 female mice 6 weeks of age (NCI Frederick, MD) were used. Institutional animal care and use committee (IACUC) approved all the animal experiments.
Diet-induced-obesity: To induce obesity, we used a high fat diet (HFD) containing 33%Kcal from fat (TD.03438, Harlan Laboratories, Inc., Madison, WI, USA), whereas a control low fat diet (LFD) containing 10%Kcal/fat was employed to generate lean control mice (TD.94048, Harlan Laboratories, Inc.). Five mice were kept per cage for both types of diets and body weight was evaluated weekly. Obese (and overweight) status was assessed by body weight (BW) increase: overweight was defined as a 15%–20% increase and obesity ≥25% BW higher than normal BW group (lean). Obesity-resistant mice were mice fed the 33% HFD where BW increased less than 15% weight.
Tumor cell inoculation: Eight weeks after the diets were introduced, E0771 breast tumor cells, syngeneic to C57BL6 mice, were resuspended in PBS:Matrigel 1:1 and subcutaneously (s.c) injected in the fat pad of the fourth mammary gland in the lower abdomen at 2.5 × 10
5 cells/50 µL/mouse. Primary tumor growth was measured at weekly intervals using calipers, and mice were kept in their respective diets until they were euthanized five weeks after tumor cell injection.
Treatment with the leptin receptor antagonist peptide PEG-LPrA: A week after tumor cell inoculation, mice started treatments (50 uL/100 uM i.v. in tail vein) once a week with the peptide (PEG-LPrA2) or vehicle for 4 weeks, at the end of which they were euthanized. The peptide (PEG-LPrA2, pegylated leptin receptor antagonist peptide 2) was synthesized, purified, pegylated and provided by Dr. R.R Gonzalez-Perez. The mice were stratified into two major groups: (1) treated (PEG-LPrA2) and (2) treated with vehicle (designated either as “untreated” of “control” in
Figure 9). Within each major group, there were 4 subgroups: lean, obese-resistant, overweight and obese, each with
n = 10 per subgroup. Right before euthanasia, serum samples were obtained and frozen at −80 °C for leptin, CCL2, glucose, and insulin determinations.
Tumor detection: Mammary glands were palpated weekly for tumor formation. Tumor growth was determined every week in live animals by caliper measurement (as an estimated measure of growth, ╥/6 × width
2 × length, where the width is the smaller of the two measurements) and upon euthanasia. Tumors were fixed in 10% formaldehyde for further histological and immunohistochemistry studies.
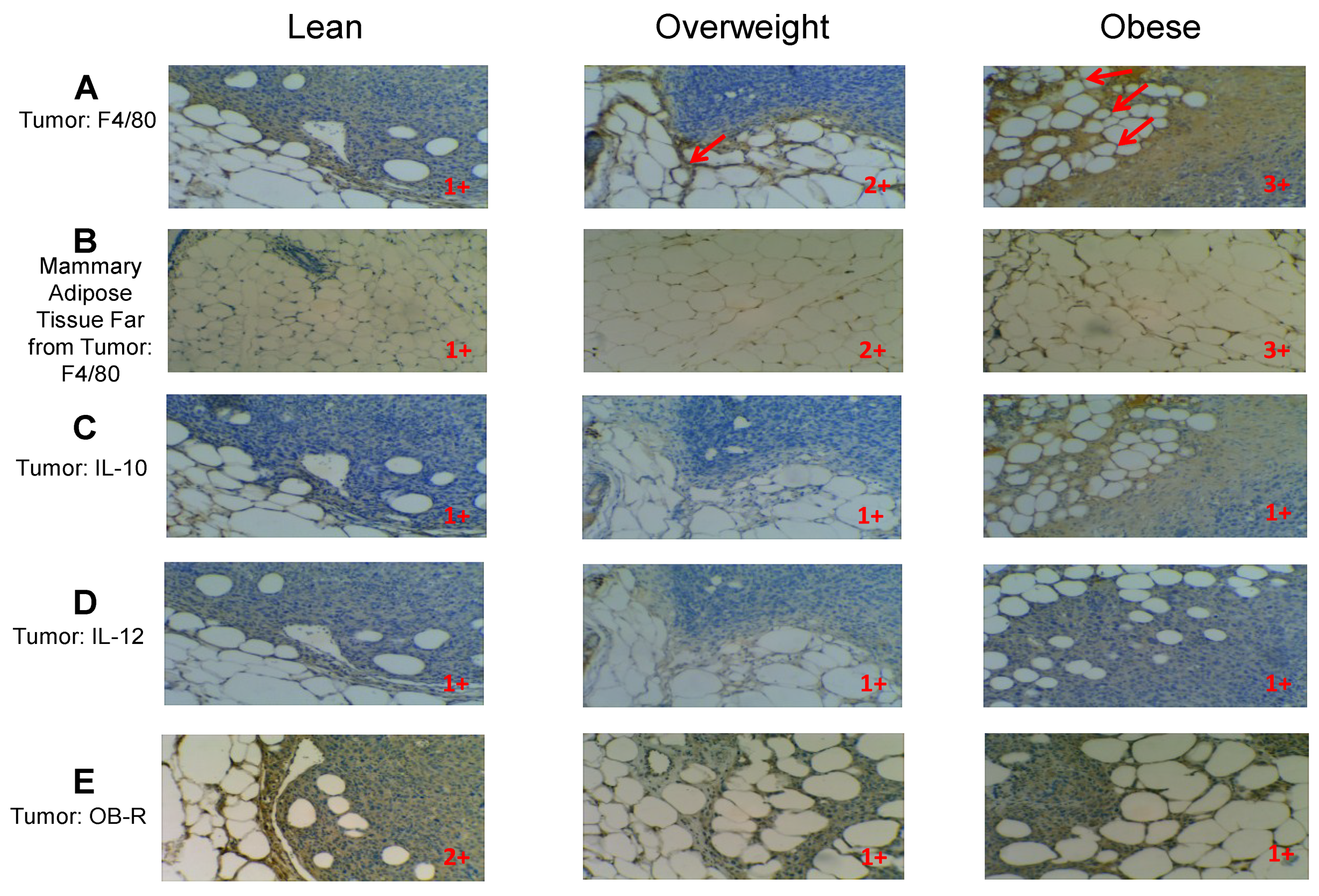
2.14. Histology and Immunohistochemistry
Tissue sections were stained with Hematoxylin and Eosin (H&E) to reveal the histology of the tumors. Immunohistochemistry (IHC) was performed as previously reported [
20] using the VECTASTAIN ABC Kit as described by the manufacturer (Vectors Laboratories, Burlingame, CA, USA). The following were the primary antibodies used for IHC: rat monoclonal anti-mouse F4/80 (Abcam, Cambridge, MA, USA), rat monoclonal anti-mouse IL-10 (Santa Cruz), rat monoclonal anti-mouse IL-12 (Santa Cruz), and rabbit polyclonal anti-Ob-R (H300, Santa Cruz, which recognizes the long and short forms of the leptin receptor). Antigen retrieval was performed using the following conditions: for F4/80, IL-10 and IL-12 it was made in Citrate Buffer (Antigen Unmasking Solution, Vector Laboratories), whereas for leptin receptor (Ob-R), it was carried out in High pH Buffer (Antigen Unmasking Solution High pH, Vector Laboratories). First antibodies were incubated overnight at 4 °C.
2.15. Determination of Glucose, Leptin, CCL2 and Insulin in Sera
The serum levels of glucose (Colorimetric Assay Kit, Cayman Chemical Company, Ann Arbor, MI, USA), insulin (Mouse Insulin ELISA, Mercodia, Uppsala, Sweden), leptin and CCL2 (both by ELISAS from R & D System) were determined following the manufacturer’s instructions.
2.16. Statistical Analysis
Prism software was used to statistically analyze our results. One or two-way ANOVAs were used when comparing more than two experimental groups; a significant ANOVA test shows that the different groups are statistically different among themselves as a group. However, to compare pairs of groups, ANOVA was then followed by Tukey’s Multiple Comparisons Test (TMCT) or by Sidak’s Multiple Comparison Test (SMCT), which allow comparing the means between pairs of groups among the larger group analyzed by ANOVA. Levels of significance were provided by Prism analysis (
p ≤ 0.05 *,
p ≤ 0.01 **,
p ≤ 0.001 *** and
p ≤ 0.0001 ****. These
p values apply to all presented figures using the different statistical tests. The Multiple Comparisons derived from significant ANOVAs, Tukey’s or Sidak’s (whether significant or not) are shown for each figure in
Supplemental Data, and only the significant values of biological relevance are depicted in each figure. Paired Student’s
t-test was used when means between two groups were initially compared to analyze their statistical difference. Error bars represent standard error of the mean (SEM) and alpha was set at 5%.
4. Discussion
White adipose tissue occurs basically in two different anatomical locations, visceral and subcutaneous, which differ in their capacities to become inflamed in obesity. Normally, visceral adipose tissue—but not subcutaneous—shows signs of chronic inflammation in obese individuals, characterized by elevated numbers of M1 macrophages, CLS and enlarged adipocytes [
35]. The normal adult female breast is comprised largely of adipocytes and epithelial cells [
36]. Interestingly bAT is considered subcutaneous fat due to its anatomical location in the breast. However, given the significant amount of adipose tissue that proportionally exists in the breast and its potential crosstalk with other cells of the parenchyma, it will be important to elucidate whether intermediate inflammatory changes do occur in the bAT of obese females, and particularly, of obese females with breast cancer. This obesity-induced bAT inflammation may have important implications in defining the tumor- promoting role of locally generated breast inflammation in breast cancer development.
Until very recently, breast cancer was considered among the cancers not associated with chronic inflammation. Although probably still true in lean females who eat a healthy non inflammatory diet, this is not valid for obese females. In obese females, it is known that visceral obesity triggers systemic inflammation, enhancing cancer risk in general, including increasing risk of breast cancer and poor tumor prognosis. However, we reason that obesity may also locally impact bAT, generating local inflammation within the breast. Compared to other subcutaneous adipose tissues as the one underneath the skin, bAT encompasses a larger volume. This may allow adipocytes to expand their cellular size and their numbers, becoming hypertrophic and hyperplastic. The existence of different cell types within the breast cancer microenvironment—adipocytes included—facilitates crosstalk between them. We have shown that this interaction is capable of recruiting inflammatory cells and producing pro-inflammatory, tumor-promoting molecules in the DIO mouse.
Macrophages, central cells in the inflammatory response, are the most frequent immune cell type in tumor microenvironments. TAMs assist tumor progression via different mechanisms and are considered a sign of poor tumor prognosis. These cells contribute to tumor initiation while they are pro-inflammatory and pro-mutagenic in tissues (M1) via their expression of ROI/RNI. They also participate in tumor progression when they acquire immunosuppressive and tissue repairing properties (M2), contributing to ECM remodeling, angiogenesis, invasion and metastasis. On the other hand, resident macrophages in lean adipose tissues are immunosuppressive M2, whereas they are pro-inflammatory M1 when they colonize obese AT, providing its inflammatory signature.
We hypothesize that bAT from overweight/obese subjects is inflamed and promotes breast cancer development. This is due in large extent to the recruitment of macrophages to the bAT and tumor. For our studies, we used the E0771 mouse mammary tumor model, syngeneic to C57BL6 mice, which is the inbred mouse strain that is most sensitive to diet-induced obesity (DIO). In addition, E0771 is the only mammary tumor cell line syngeneic to C57BL6 mice; thus, the model used is the indicated one for our studies in obesity and mammary cancer in the mouse, given that genetic models of obesity in mice, such as the ob/ob or db/db mice, deficient in leptin or its receptor respectively, are resistant to mammary cancer [
37]. To address our hypothesis we first carried out a series of
in vitro studies focused on three main cell types of the mammary tumor microenvironment: adipocytes, mammary tumor cells and macrophages, and we studied the impact of their mutual interactions on macrophage recruitment and on tumor progression using
in vitro co-cultures, and then conducted further studies in
in vivo settings.
In the
in vitro studies, we centered our attention on three factors that could be produced in a paracrine fashion by these cells or their interactions, exerting monocyte chemotaxis by themselves or their combinations, and resulting in macrophage recruitment to the mammary tumor microenvironment. These paracrine factors are the adipokine leptin, the saturated fatty acid lauric acid and the chemokine CCL2. Adipocytes from obese fat tissues are known to produce high amounts of leptin and undergo lipolysis, releasing saturated fatty acids. Obese adipocytes express several pro-inflammatory chemokines, such as CCL2 (MCP-1). Mammary adipocytes from obese females may express all these molecules locally in the mammary gland, some of which could also be expressed by tumor cells. Co-culturing murine and human breast tumor cells with mature adipocytes, Dirat
et al. [
38] have shown that invasive cancer cells impact surrounding adipocytes, modifying their phenotype and biological features to become cancer-associated adipocytes, which alter the cancer cell’s characteristics leading to a more aggressive behavior. Their results strongly support the concept that adipocytes in the breast participate in breast cancer progression in a process orchestrated by tumor cells which may be amplified in obese women.
Leptin—the central regulator of satiety in the body—has been characterized as a pro-inflammatory, pro-angiogenic and proliferation-inducing adipokine essential to breast cancer [
39]. The role of leptin signaling in enhancing the tumorigenic potential of tumor cells is well known [
40], however whether leptin’s signaling in macrophages induces their recruitment to the breast tumor microenvironment or whether it activates in other ways macrophage’s pro-tumor functions, has not been thoroughly examined. Lauric acid is a saturated fatty acid released by adipocytes, especially upon obesity-induced lipolysis, and a ligand of TLR4, highly expressed by macrophages; the possible macrophage chemotactic capacity of this fatty acid had not been analyzed either. Chemokine CCL2 (MCP-1), one of the main chemo attractants for tissue monocytes/macrophages, might synergize with the other paracrine factors, further increasing macrophage recruitment to the tumor. Chronic inflammation is a potent inducer of many cancers, and expression of cytokines/chemokines within tumors has been correlated with poor prognosis [
41]. Production of pro-inflammatory adipokines, cytokines, chemokines and saturated fatty acids by the different cell types within the breast tumor microenvironment could be exacerbated in obesity, resulting in an inflammatory situation in the breast of obese females.
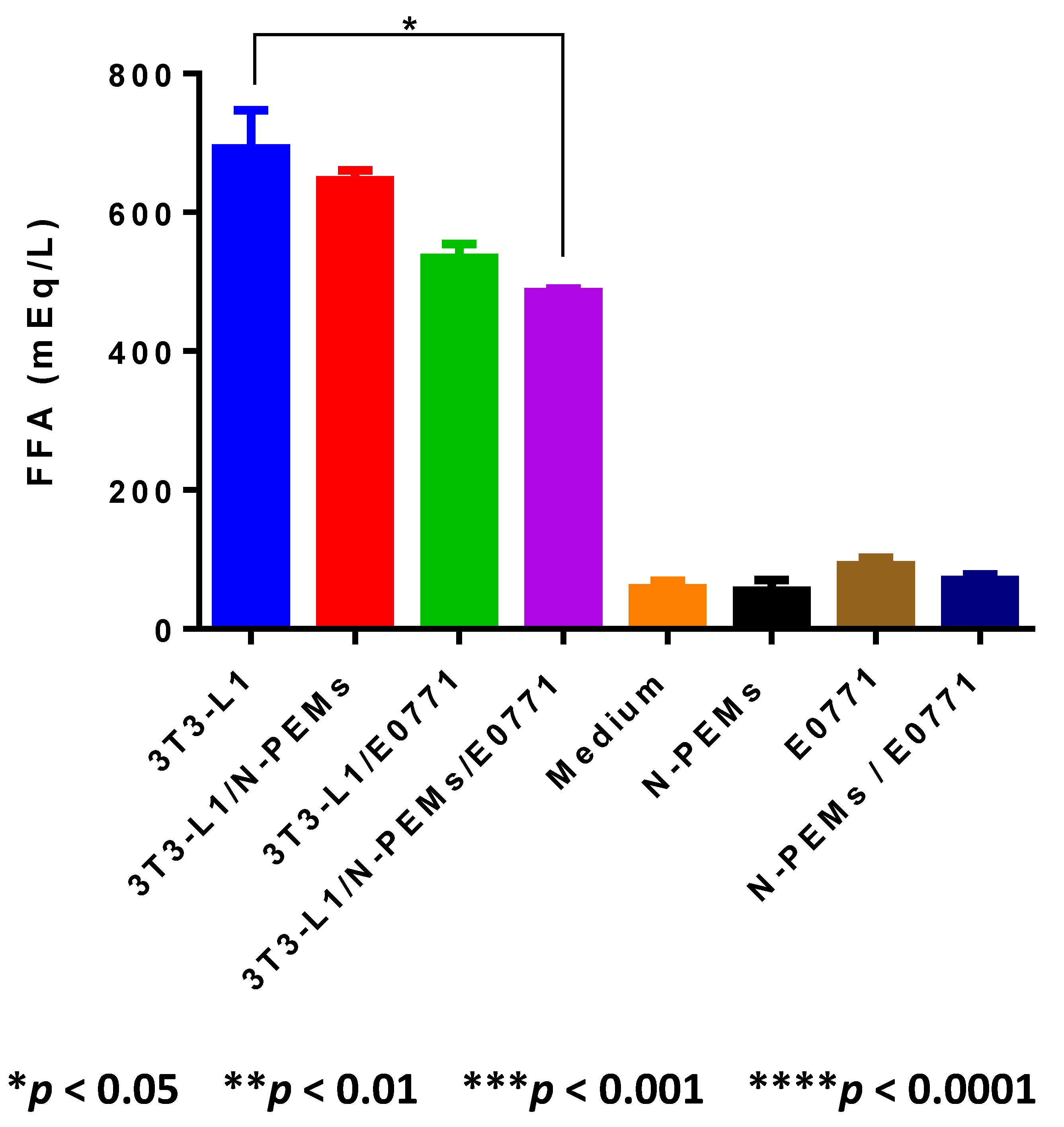
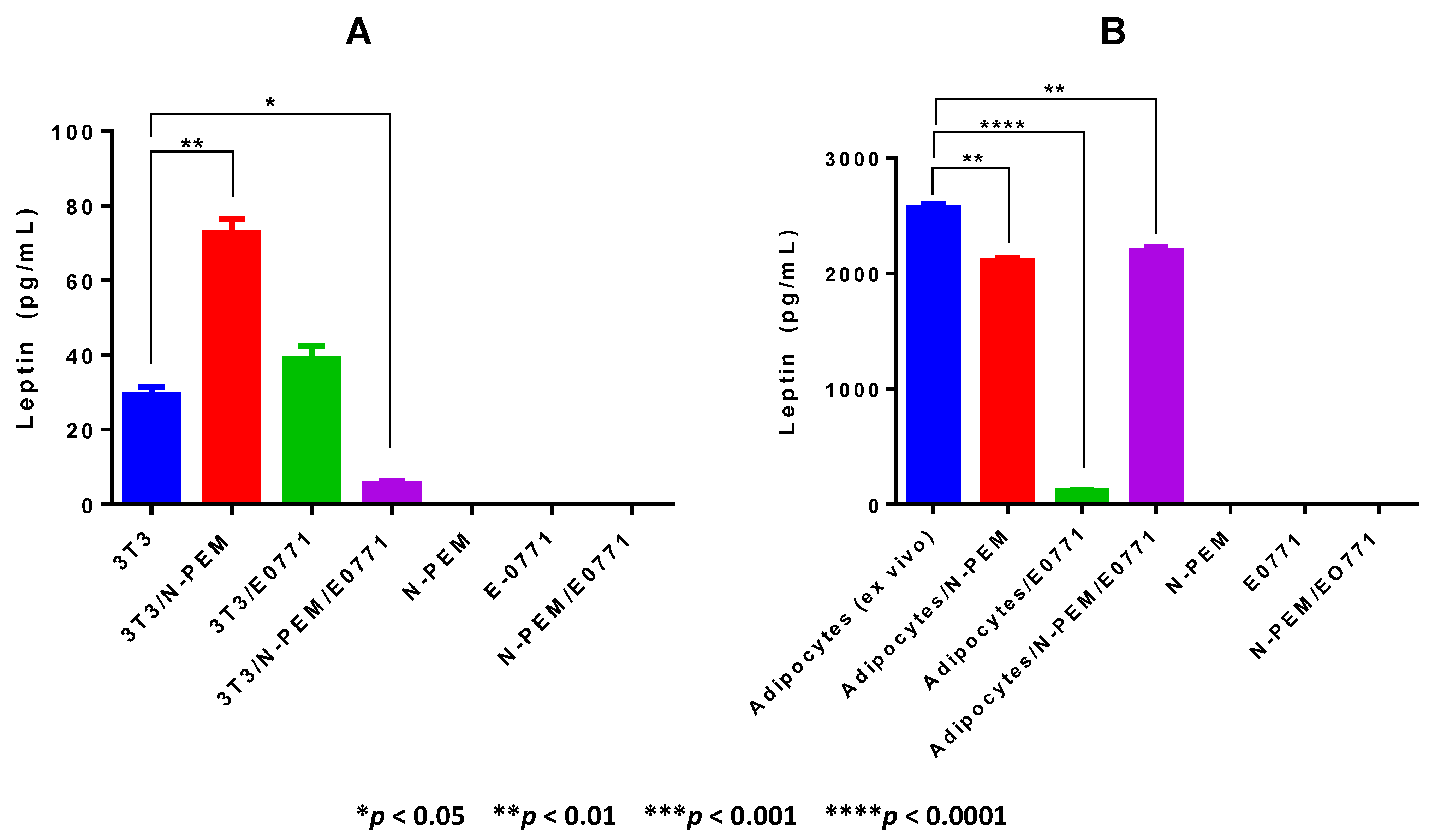
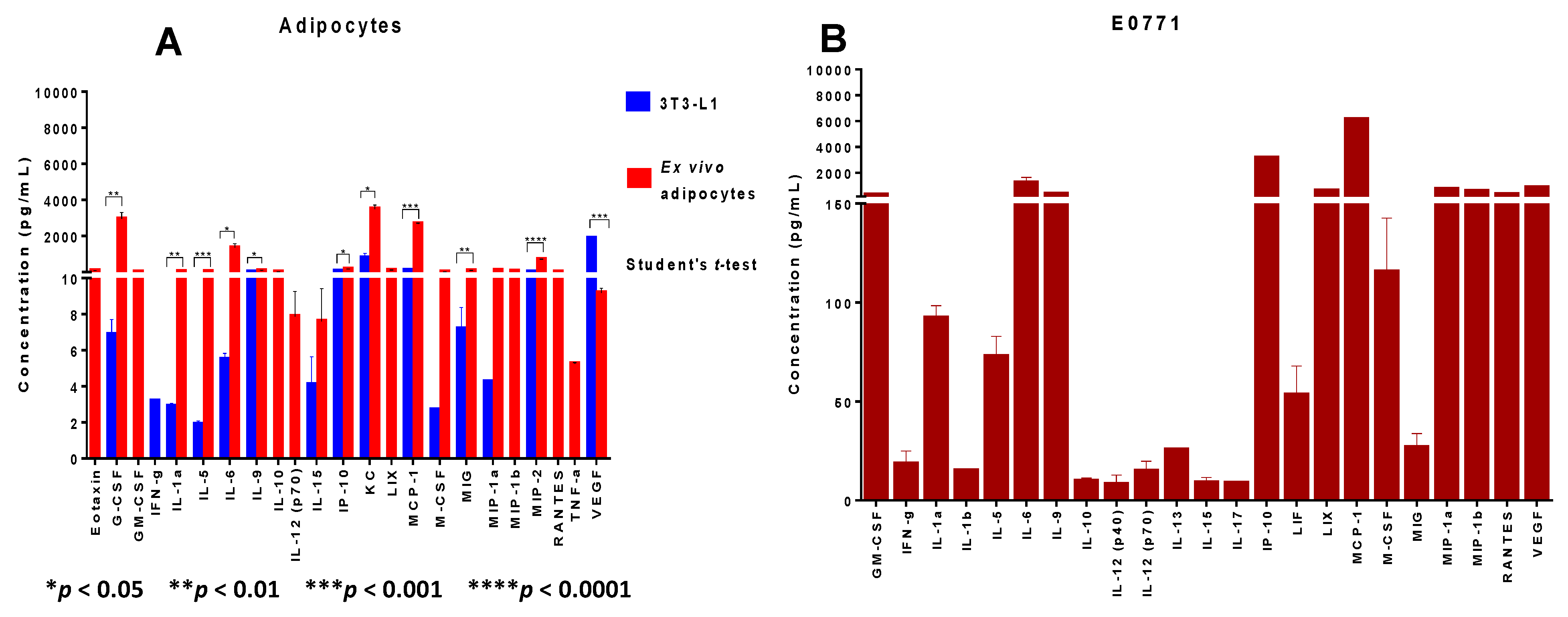
Our work demonstrates that adipocytes isolated ex vivo from visceral fat of obese female mice undergo lipolysis and release fatty acids. We observed that in the crosstalk of adipocytes with mammary tumor cells and macrophages—a setting that mimics the mammary/breast tumor microenvironment—lipolysis may be decreased, favoring inflammation. The decrease in FFA observed in the supernatants of the cell co-cultures may be explained mainly in two different ways: (1) an actual downregulation of lipolysis within adipocytes, which may result in increased accumulation of triglycerides and therefore in larger, hypertrophic adipocytes, characteristic of inflamed adipose tissue seen in the visceral fat of obese subjects; or (2) an increase in the uptake of FFA produced by adipocytes through lipolysis by tumor cells or macrophages. Either possibility, or the occurrence of both, is consistent with the reduced amount of FFA detected. Indeed, decreased lipolysis results in increased accumulation of triglycerides, and therefore in larger, hypertrophic adipocytes, characteristic of the inflamed obese adipose tissues of obese subjects. Alternatively, tumor cells and/or macrophages may increase uptake of fatty acids, as alternative sources of energy. We showed that ex vivo isolated adipocytes from obese adipose tissue are the major source of leptin production in our tumor model: we demonstrated that E0771 mammary tumor cells, in contrast to the majority of mammary/breast tumor cell lines, do not express leptin. Our results also demonstrate that co-culture with the tumor cells and/or macrophages decreases leptin production by adipocytes, suggesting that in the obese mammary tumor microenvironment of our E0771 DIO tumor model, leptin amounts may be lowered. Our results suggest that leptin is a marker of obesity rather than of adipocyte differentiation, since obese ex vivo isolated adipocytes but not differentiated 3T3-L1 adipocytes were the main producers of leptin in the system. Further studies with ex vivo adipocytes isolated from visceral adipose tissue of lean and obese mice will enable us to substantiate a broader generalization of this conclusion.
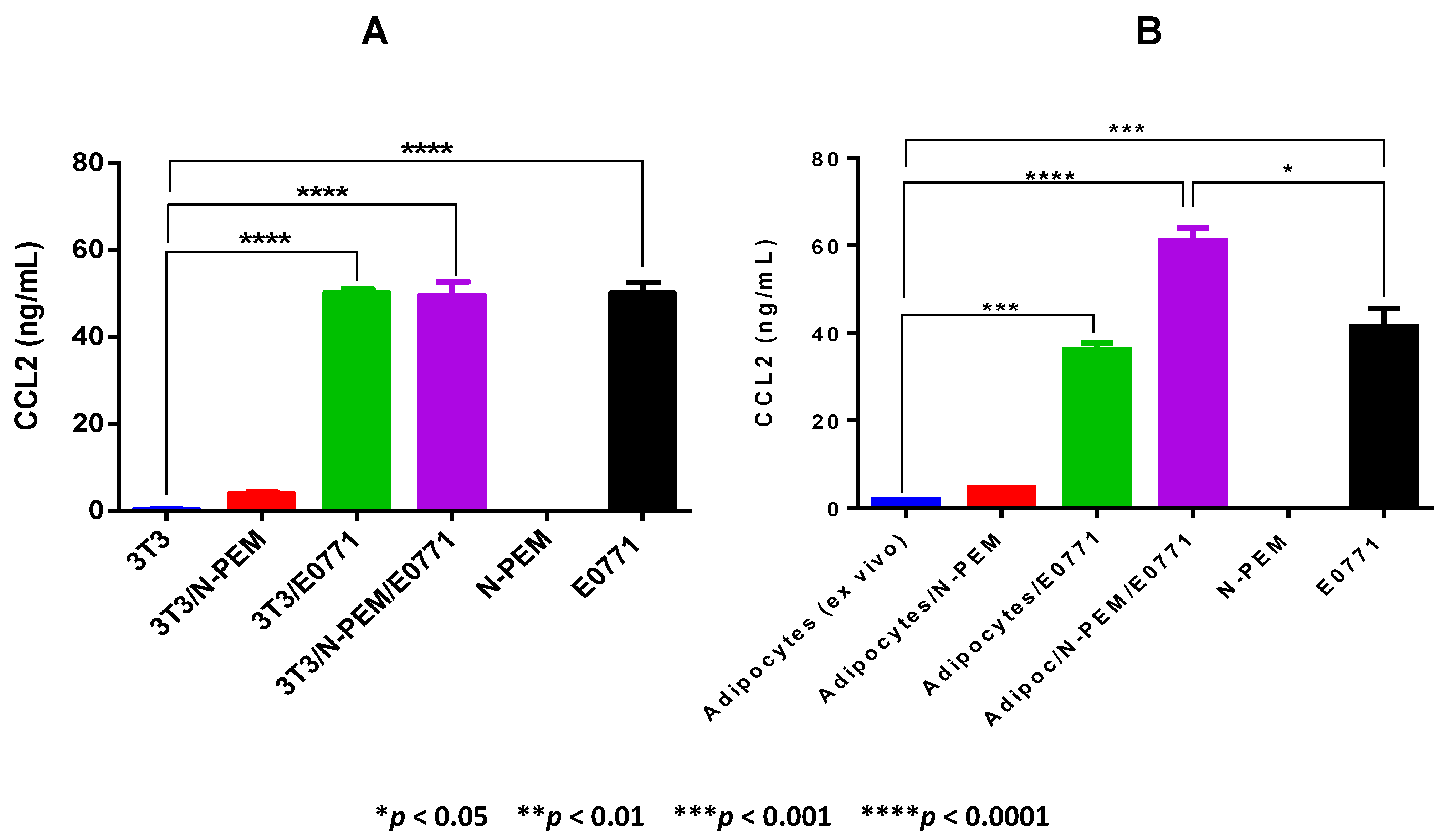
With respect to CCL2, our results demonstrate that the crosstalk between adipocytes from obese mice, macrophages and tumor cells—which is the setting that closely resembles the breast tumor microenvironment of obese females—is also the experimental condition that resulted in the highest amount of CCL2 detected among the cell co-cultures. Interestingly, as opposed to other tumor models where adipocytes are the main producers of CCL2 [
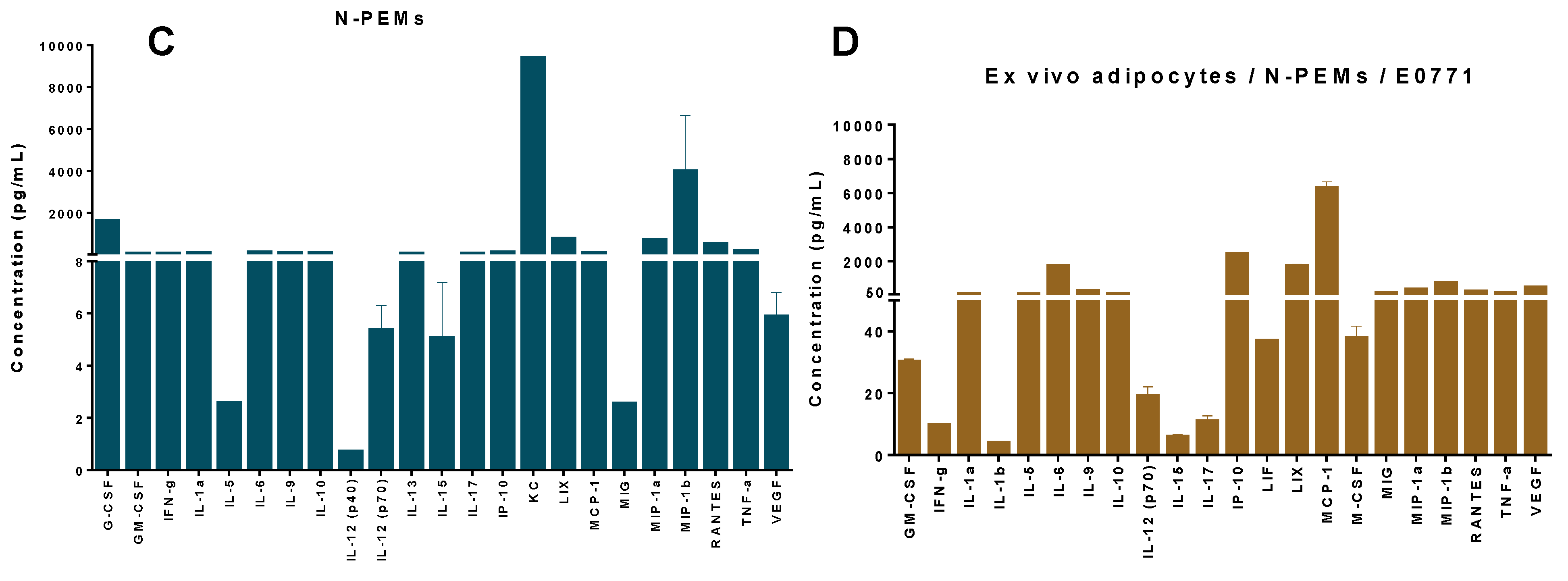
42], in our E0771 tumor model, even adipocytes from obese adipose tissues produced negligible amounts of CCL2, and it is the E0771 mammary tumor cell line that is the main producer of this chemokine. We thus confirmed the expression of these different paracrine factors in the E0771 tumor model in conditions of obesity, and concluded that the setting that mimics the mammary tumor microenvironment (co-culture of the three cell types) is the one in general that favors the production of the highest concentrations of these paracrine molecules.
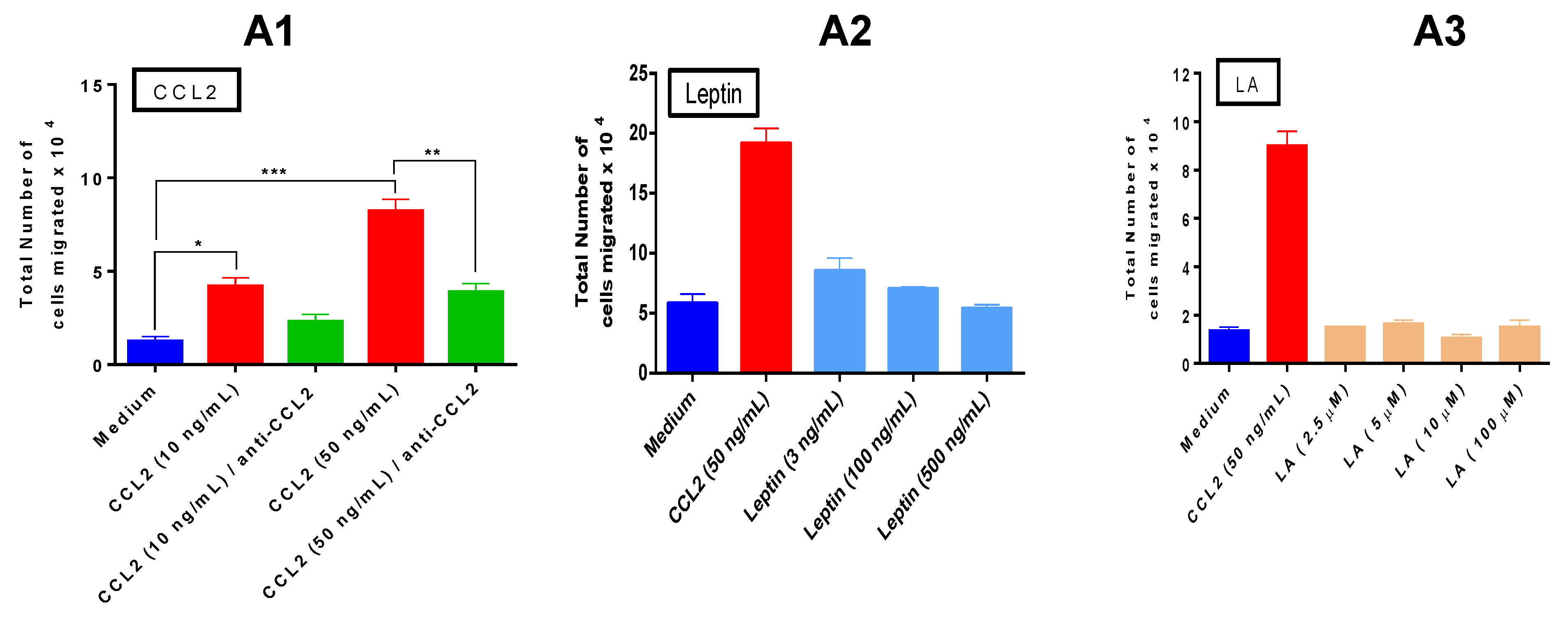
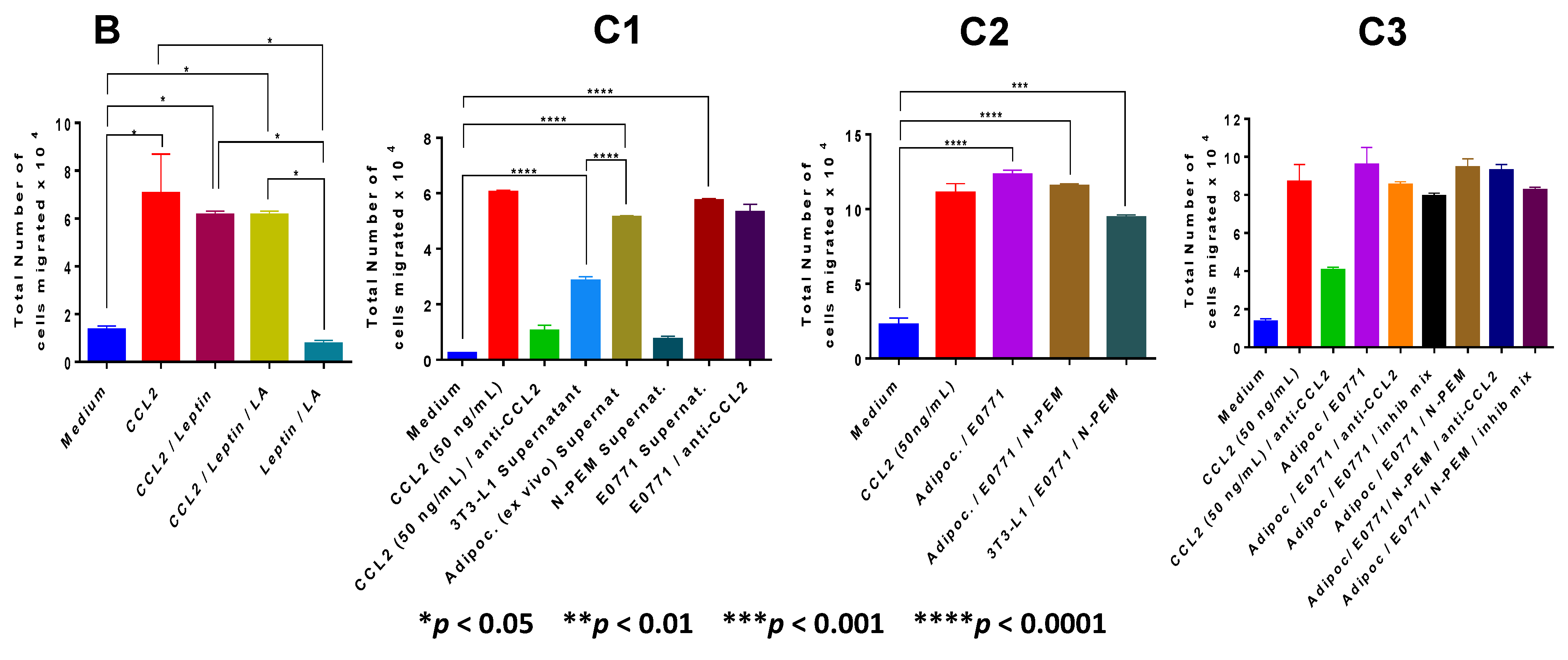
Recruitment of macrophages to the tumor microenvironment is a critical step in breast tumor inflammation. Analysis of monocyte chemotaxis by our three paracrine factors revealed that CCL2 is the major chemotactic factor, with lauric acid lacking such activity. With leptin, we detected a mild and non significant chemotaxis on THP1 monocytes at the lowest concentration used (3 ng/mL). Our results are in agreement with others who have reported that leptin is a chemoattractant at concentrations as low as 1 pg/mL with maximal effects at 1 ng/mL (which is why we did not see a significant chemotactic effect), and that when concentrations of 10–100 ng/mL are reached, leptin chemotaxis declined [
43]. Also in agreement with Gruen
et al. we found no evidence of additive chemotactic effects with the combination of leptin and CCL2 [
43]. The crucial role of CCL2 in macrophage recruitment to the fat tissue in postmenopause has been reported in ovarectomized mice, where subcutaneous fat depots demonstrated elevated macrophage recruitment and increased expression of CCL2 compared to control mice, suggesting CCL2 may enhance inflammation in postmenopausal women [
44]. CCL2 expression in tumors is correlated with higher histological grade breast cancer and is a significant indicator of early relapse [
45]. Elevated CCL2 is also associated with TAMs infiltration in tumors [
46]. In agreement with our results, it has been shown that obesity promotes breast cancer by CCL2-mediated macrophage recruitment [
42]. We conclude that CCL2 is a major molecular player in our system. In terms of chemotaxis exerted by the cell supernatants, our results compellingly demonstrate that it is the co-culture of the three cell lines, resembling conditions in the breast tumor microenvironment, the setting that induces the highest monocyte recruitment.
In the
in vitro experiments which recreated the tumor microenvironment we used specific inhibitors for these paracrine factors, to reverse the specific functions identified: a novel antagonistic peptide—PEG-LPrA2—as a leptin signaling inhibitor which significantly delayed onset and decreased progression of mammary cancer in mice [
29], or specific inhibitors for CCL2 and TLR4. Our experiments with the mixture of those specific inhibitors of the paracrine factors suggested that additional unidentified paracrine factors could be promoting monocyte chemotaxis to the obese mammary tumor microenvironment. To identify some of these new factors we used Luminex analysis. Our Luminex data revealed that adipocytes from obese adipose tissue, macrophages and mammary tumor cells, by themselves or interacting altogether, express numerous cytokines, chemokines and growth factors with known chemotactic, pro-inflammatory, pro-angiogenic and tumor-promoting activities, all of which may contribute to a mammary tumor microenvironment reflecting those different properties. Importantly, prevailing among all the different molecules that were identified in the co-culture of the three cell types was CCL2, followed by IP-10 (CXCL10) and IL-6, confirming the central role of these molecules in this tumor model. Cytokines secreted by the adipocytes and the tumor-associated immune cells and vasculature may also promote epithelial-to-mesenchymal transition (EMT). Because EMT plays a critical role in metastasis, obese adipocytes in the breast tissue can play a critical role in tumor progression which may explain why their presence is likely an underlying mediator of poor outcome in obese breast cancer patients [
3,
36].
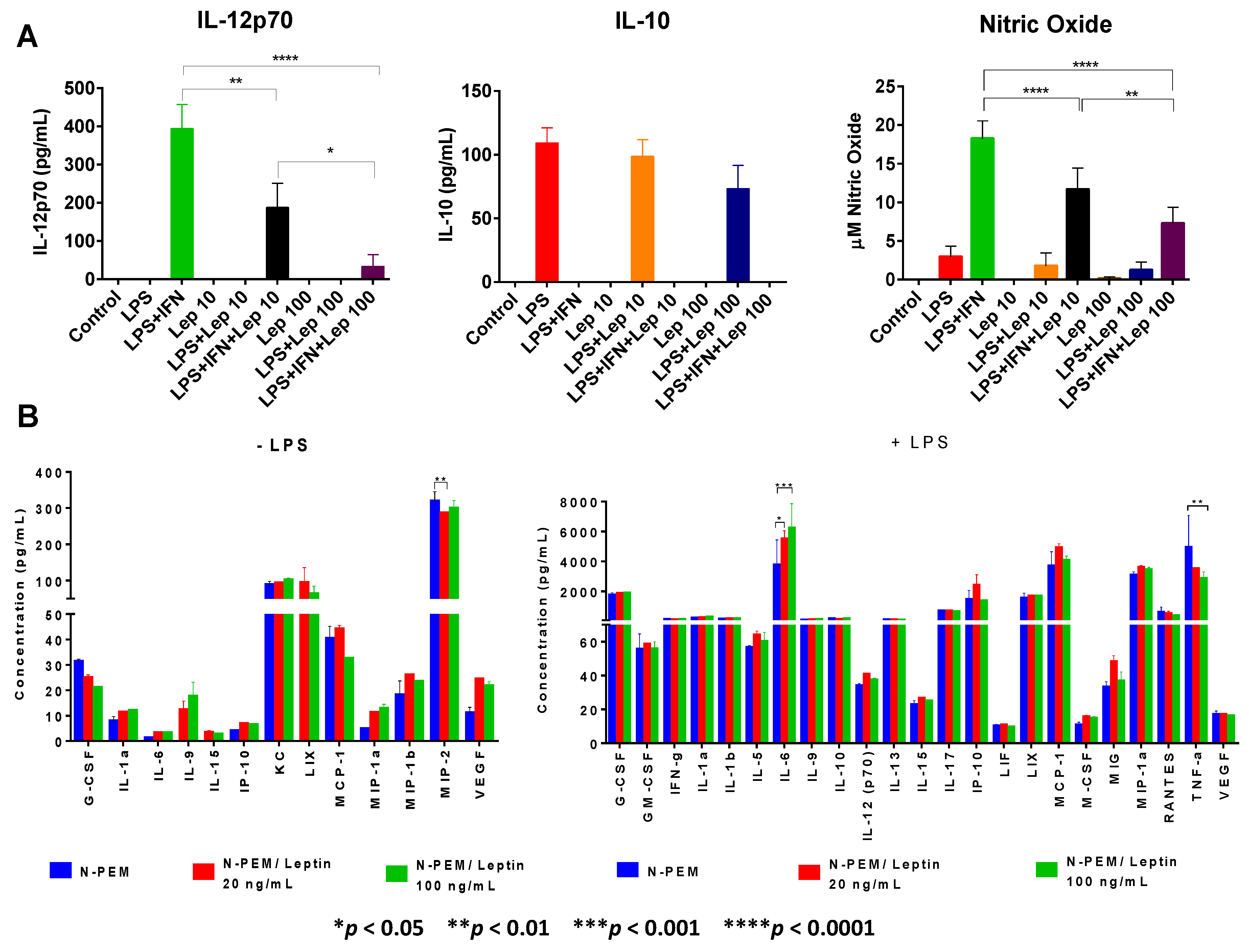
To further investigate other plausible roles of leptin on macrophages, we examined whether acting in the tumor microenvironment or systemically, this adipokine might modulate macrophages M1/M2 activation. Activation with LPS (a TLR4 ligand and a prototypical activator of macrophages) was included to mimic the TLR4 activation that occurs in the tumor microenvironment through the action of endogenous ligands that are generated in tumors, such as heat shock proteins (HSP), high mobility group box 1 (HMGB1) and proteoglycans (Versican, Heparin sulfate, Hyaluronic Acid fragments). In contrast to what has been reported in the literature, where leptin is described as a pro-inflammatory adipokine, our results in peritoneal elicited macrophages clearly revealed that leptin decreases the constitutive expression of pro-inflammatory chemokine MIP-2 (CXCL2) and in a dose-response fashion downregulates the LPS or LPS+IFNγ-induced expression of the critical pro-inflammatory cytokines IL-12 and TNFα, as well as of the pro-inflammatory free radical nitric oxide (NO). In contrast, other investigators have found that leptin indirectly increases production of TNFα and other cytokines in monocytes [
47]. Importantly, leptin induces the expression of inflammatory cytokines like IL-1 in tumor cells and non-tumor cells of the tumor stroma [
29]; however we were not able to demonstrate IL-1 upregulation by leptin in macrophages, although IL-1 is a major inducer of IL-6 in a variety of cells. Interestingly, our data also demonstrate that leptin upregulates expression of the pro-inflammatory and tumor-promoting cytokine IL-6 upon LPS stimulation of these macrophages, although in similar conditions it does not modify expression of the main anti-inflammatory molecule IL-10. Surprisingly, IL-6 upregulation has been reported in macrophages from leptin-deficient ob/ob obese mice [
48]. Together with CCL2 chemokine, IL-6 is the pro-inflammatory and tumor-promoting cytokine that we have seen consistently upregulated in our tumor model by the different cell types examined or their crosstalk. The pleiotropic cytokine IL-6 is known to play central roles in various types of cancers as a tumor growth factor, and is one of the main factors also responsible for cancer-induced cachexia. In breast cancer, it has recently been shown that IL-6 contributes to tumor progression [
49,
50].
Our work proves that leptin controls macrophage differentiation, as it downregulates the percentage of macrophages that express myeloid markers F4/80 and CD11b. We also reveal that leptin impairs protein expression of the central pro-inflammatory and tumor-promoting transcription factor NFκB in macrophages, yet strongly upregulates the expression of STAT3 and its active form (pSTAT3); both transcription factors operate in important tumor-promoting signaling pathways in macrophages and tumor cells. We showed that leptin does not regulate expression of pro-inflammatory STAT-1/pSTAT-1 yet increases IRAK-1 expression in macrophages, in agreement with others who have also reported leptin-induced increased expression of IRAK-1 in peritoneal macrophages [
51]. In contrast, others have reported increased NFκB signaling in the stromal vascular fraction (SVF) of the mammary gland of high fat diet-induced obese mice, a fraction that these authors use as representative of the macrophages in bAT [
22]. The SVF is actually comprised not only of macrophages but includes all the nonadipocyte cells in the adipose tissue, among them T and B cells, which also secrete cytokines, so this fraction may not identify exclusively macrophages. Leptin is reported to be anti-apoptotic in tumor cells [
52], consistent with an upregulation of NFκB and STAT3 anti-apoptotic transcription factors. However, our results in leptin-treated macrophages reveal the opposite for NFκB although confirm the upregulation of pSTAT3 observed by these authors in tumor cells exposed to leptin. We have previously reported a similar contrasting effect between macrophages from tumor hosts and tumor cells [
20] showing opposite expression patterns, e.g., decreased NFκB expression in TAMs and peritoneal macrophages as opposed to the constitutive NFκB upregulation that has been consistently reported in tumor cells [
53]. Importantly, we provide the first report of a Notch protein modification induced by leptin in macrophages. Using the same E0771 mammary tumor cells and several human breast cancer cell lines we recently reported that when exposed to leptin, breast cancer cells express higher protein levels of Notch 3 [
54], as we also found in the present study in macrophages exposed to low leptin concentrations. Interestingly, our results suggest that macrophages are more responsive than E0771 cells to the action of leptin on Notch 3 expression at lower levels of leptin, since 0.62 nM (10 ng/mL) leptin already upregulate Notch 3 in macrophages whereas a concentration of 6.2 nM (100 ng/mL), 10-fold higher, is required in E0771 cells to see a consistent increase in Notch 3 expression in these tumor cells [
54]. Alterations of the Notch signaling pathway can lead to a variety of disorders, including cancer. We have reported the existence of a crosstalk between oncogenic signaling pathways in breast tumor cells in humans and mice triggered by leptin, IL-1 and Notch (NILCO) critical to leptin-induced breast cancer promotion [
31].
We examined whether the presence of leptin might modulate the expression of its own receptor in macrophages. Macrophages, as other cells susceptible to leptin’s action, express two main forms of the leptin receptor,
i.e., the long, full-length form (Ob-Rb) and the short form (Ob-Ra), both involved in leptin signaling [
55]. We report for the first time that leptin profoundly downregulates in a clear dose-response fashion both forms of its own receptor in peritoneal macrophages as well as in TAMs. As we previously discussed, leptin’s concentration in the
in vitro-simulated murine obese mammary tumor microenvironment is decreased after adipocytes crosstalk with E0771 tumor cells (which do not produce leptin themselves) and with macrophages. Thus, leptin’s actual concentrations in the tumor microenvironment of the E0771 obese mammary tumor model may not be as high as the ones which experimentally downregulated its receptor in macrophages. In any case, it is interesting to note that the effects that we have described are induced by leptin in macrophages occur despite the downregulation of its receptor after exposure to leptin. Importantly, the chemotactic activity of leptin in THP1 monoctytic cells requires the expression of the long and the short forms of the leptin receptor [
43,
56], so this leptin receptor downregulation is not seen in monocytes. The protein downregulation of leptin receptor that we report in both types of macrophages was observed after 20 h stimulation with leptin. Signaling events occur in minutes, after which the receptor may be downregulated by mechanisms that remain to be elucidated and that are beyond the aims of this study.
Our animal experiments with DIO and lean C57BL6 mice bearing E0771 mammary tumors were specifically designed to examine
in vivo the role of leptin in TAMs recruitment to the mammary tumors and in tumor progression, as well as to assess the association of body weight with bAT inflammation in mice fed a HFD that results in obese and overweight mice. Mice were fed with a 33% Kcal/fat HFD. This diet was selected because it reproduces the current human Western diet. Mice fed this diet, as with humans, have a varied response in that some become obese, others overweight, and others that remain resistant to obesity. We also had animals fed a 10%Kcal/fat which remained as lean controls. Our results confirm that obesity significantly contributes to tumor size, with the obese mice bearing the largest tumors and the lean animals bearing the smaller ones. Interestingly, tumor sizes were comparable between overweight and obese mice, suggesting that being overweight already involves a level of tumor aggressiveness and progression similar to the one observed in obese mice. Using a different HFD (60%Kcal/fat) in the same E0771 tumor model we recently reported that obesity tends to positively increase the detection rate of mammary cancer in DIO mice [
54]. Injecting our mice with a designated treatment scheme (a single dose of 50 µL/100 µM i.v. in tail vein per week) of the same leptin receptor antagonist peptide (PEG-LPrA2) that was used in our
in vitro chemotaxis experiments at other appropriate concentrations, our
in vivo results demonstrate a non significant tendency of this peptide to reduce tumor sizes in obese and overweight mice. A significant increase in serum leptin levels was observed in obese but not in overweight mice when compared with obese-resistant mice. Interestingly, treatment with the leptin inhibitor significantly reduced those elevated serum leptin levels in obese mice only. Corroborating our
in vitro experiments mimicking the E0771 tumor microenvironment of obese mice, where we demonstrated that leptin is not produced by the tumor cells or macrophages but by obese adipocytes, overall our
in vivo results indicate that in this DIO tumor model, when mice are fed 33% HFD, leptin inhibition at the doses used was not enough to significantly reduce tumor growth, particularly when the tumor-promoting factors CCL2 and IL-6 are present. Interestingly, recent
in vivo studies from our group using the same mammary tumor model but a different HFD (60% instead of 33%, as we used here), and a more aggressive treatment scheme with the peptide inhibitor (two injections per week instead of one), showed significant results in tumor regression, suggesting a more relevant role for leptin in the same tumor model in mice fed a different diet and treated more aggressively with the leptin inhibitor [
54]. Taken together, these results illustrate how a different diet can regulate the biological characteristics of a tumor, making our conclusions in the present paper relevant to the diet and experimental conditions we used here.
We capitalized on the opportunity offered by the 33% HFD which generates obese and overweight mice to examine the association between body weight and bAT inflammation. We analyzed bAT within the mammary tumor microenvironment as well as in areas distal from the tumor in the mammary gland, to investigate whether body weight is related to bAT inflammation in the tumor microenvironment and also distant from it. Our results compellingly demonstrate that obese mice have higher numbers of macrophages and CLS than overweight mice in their bAT, and that lean animals have the least, strongly indicating that body weight is associated with bAT inflammation in the tumor microenvironment. Cinti
et al. [
35] reported a high occurrence of “crown-like structures” (CLS) in obese white fat of humans and mice. These CLS are histological configurations where dying adipocytes are surrounded by macrophages; these structures are considered a hallmark of chronic inflammation. More recently Subbaramaiah
et al. [
22] have demonstrated an association between CLS with NFκB activation, increased levels of pro-inflammatory mediators (TNF-α, IL-1β, Cox-2) and elevated levels of aromatase expression/activity in the breast and visceral adipose tissues of obese mice and women. We additionally showed that bAT from obese mice is accompanied by high numbers of isolated macrophages and hypertrophic adipocytes, as additional parameters of adipose tissue inflammation that should be considered. Importantly, we also report here that inflammation in the bAT distal from the tumor microenvironment is also associated with body weight. Our results reveal that bAT located far from the tumor contains higher numbers of macrophages/CLS and adipocyte hypertrophy in obese mice than in overweight mice, with the lean mice showing the lowest numbers of macrophages/adipocytes hypertrophy, an
in vivo observation that we report here for the first time. Interestingly, we verified that within each weight group there is higher inflammation in the bAT from the tumor microenvironment than in the tumor-distal bAT, strongly indicating that the contact with tumor cells increases bAT inflammation. We could not see any differences in the expression of IL-12 (M1)
vs. IL-10 (M2) macrophages in the mammary tumor microenvironments of obese, overweight or lean mice, suggesting that obesity is not particularly related to M1 or M2 macrophages in the mammary tumor microenvironment. This result was anticipated, since although obese visceral adipose tissues are colonized mainly by M1 macrophages, TAMs in tumor microenvironments have been reported to be principally M2 or mixtures of M1 and M2 [
19,
20]. Thus, bAT within a tumor microenvironment from an obese female should be expected to show mixtures of M1 and M2 macrophages. We were able to consistently confirm
in vivo in the tumor microenvironment our
ex vivo results where we found Ob-R downregulation in peritoneal macrophages and TAMs upon their pre-treatment with leptin. Our IHC results show that leptin-receptor expression decreases in the tumor microenvironment of overweight and obese mice. In summary, our work confirms our hypothesis that bAT within the tumor microenvironment of obese mammary tumor bearing females contains higher numbers of macrophages/CLS and hypertrophic adipocytes than bAT from lean tumor-bearing mice, indicating that the bAT is more inflamed in breast cancer in obesity. Also, our data demonstrate that the bAT by itself (distant from the tumor microenvironment) is more inflamed in obese than in lean mice, strongly suggesting that the contact with tumor cells increases the amount of macrophages/CLS in the breast fat.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











