
ErbB Family Signalling: A Paradigm for Oncogene Addiction and Personalized Oncology



{kind=link}
Abstract
:1. Signal Transduction of ErbB Receptor Tyrosine Kinases
2. ErbB Proteins and Oncogene Addiction
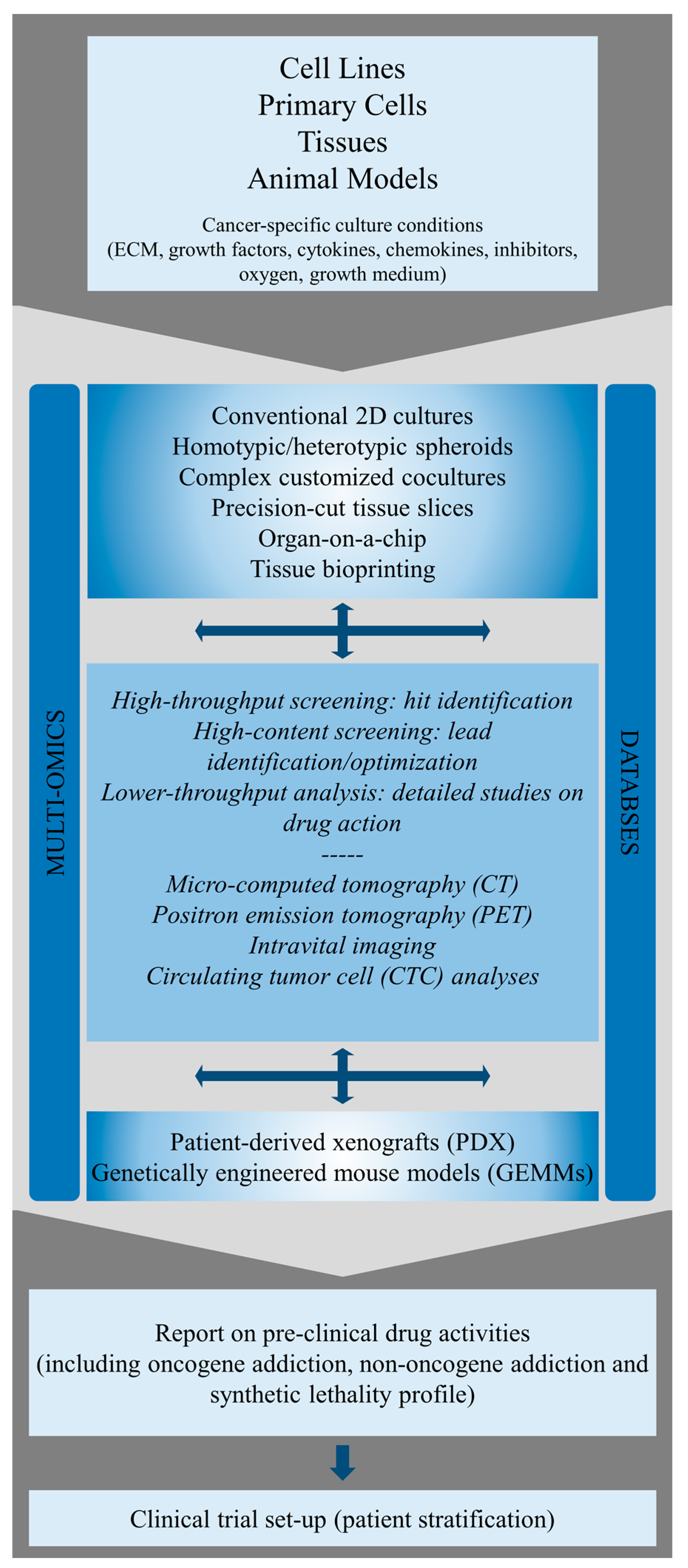
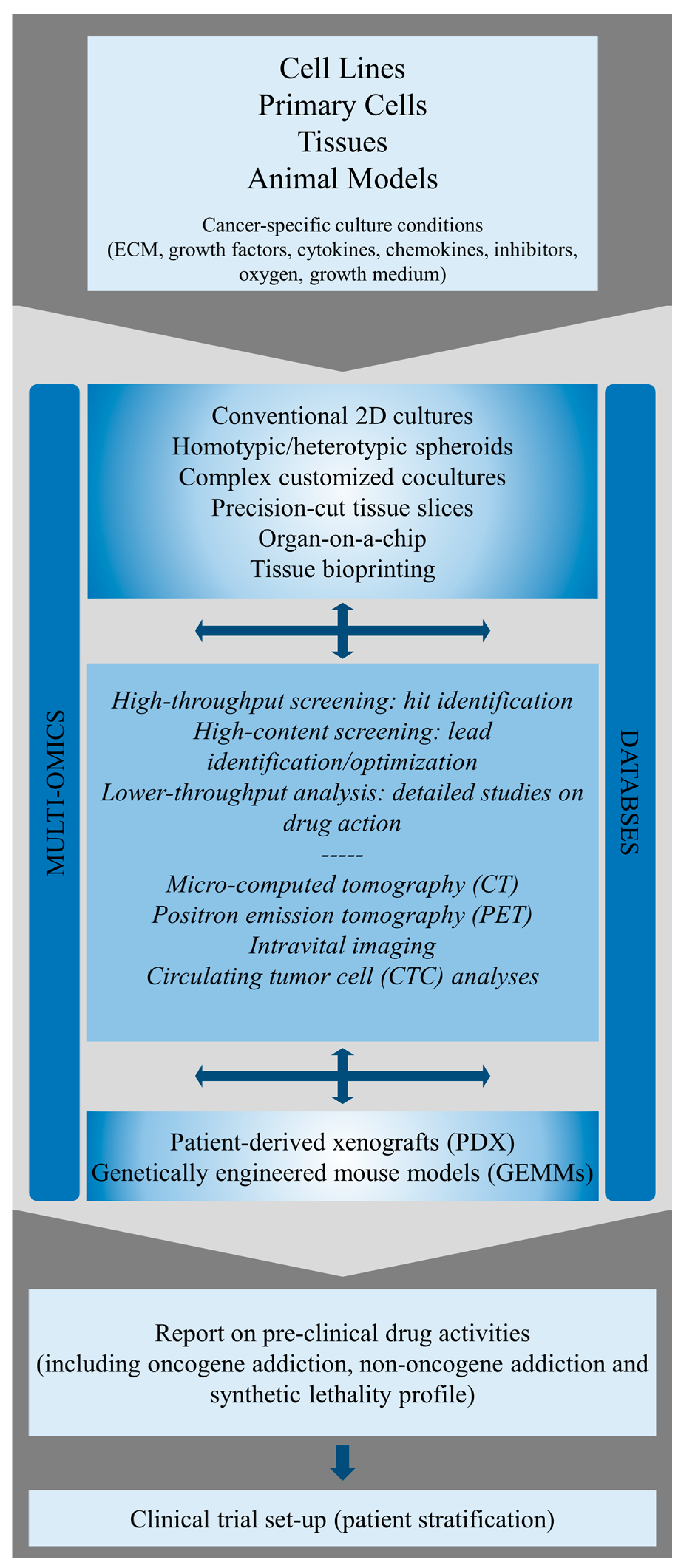
3. Identification and Molecular Characterization of ErbB Oncogene Addiction in Preclinical Cancer Models
4. Targeted ErbB Therapies and Evidence-Based Treatments in Precision Medicine
5. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Roskoski, R., Jr. The erbb/her family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Holbro, T.; Civenni, G.; Hynes, N.E. The erbb receptors and their role in cancer progression. Exp. Cell Res. 2003, 284, 99–110. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. Erbb receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. Erbb receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Reinholz, M.; Weidler, J.; Yolanda, L.; Paquet, A.; Whitcomb, J.; Lingle, W.; Jenkins, R.B.; Chen, B.; Larson, J.S.; et al. Comparison of central her2 testing with quantitative total her2 expression and her2 homodimer measurements using a novel proximity-based assay. Am. J. Clin. Pathol. 2010, 134, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Schrock, A.B.; Bocharov, E.V.; Klempner, S.J.; Haddad, C.K.; Steinecker, G.; Johnson, M.; Gitlitz, B.J.; Chung, J.; Campregher, P.V.; et al. Her2 transmembrane domain (tmd) mutations (v659/g660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J. Thorac. Oncol. 2017, 12, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Mujoo, K.; Choi, B.; Huang, Z.; Zhang, N.; An, Z. Regulation of erbb3/her3 signaling in cancer. Oncotarget 2014, 5, 10222–10236. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Ligand-induced erbb receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The erbb network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.J.; Telesco, S.E.; Radhakrishnan, R. Analysis of somatic mutations in cancer: Molecular mechanisms of activation in the erbb family of receptor tyrosine kinases. Cancers 2011, 3, 1195–1231. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the egfr signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Pala, A.; Karpel-Massler, G.; Kast, R.E.; Wirtz, C.R.; Halatsch, M.E. Epidermal to mesenchymal transition and failure of egfr-targeted therapy in glioblastoma. Cancers 2012, 4, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Ujue Latasa, M.; Urtasun, R.; Goni, S.; Elizalde, M.; Garcia-Irigoyen, O.; Azcona, M.; Prieto, J.; Avila, M.A. Epidermal growth factor receptor (egfr) crosstalks in liver cancer. Cancers 2011, 3, 2444–2461. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, V.; Stang, E. The mysterious ways of erbb2/her2 trafficking. Membranes 2014, 4, 424–446. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal growth factor receptor in pancreatic cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ma, J.; Lyu, H.; Huang, J.; Kim, A.; Liu, B. Role of erbb3 receptors in cancer therapeutic resistance. Acta Biochim. Biophys. Sin. 2014, 46, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.R.; Gupta, G.K.; Sharma, A.K.; Batra, N.; Sharma, D.K.; Joshi, A.; Sharma, A.K. Pi3k/akt/mtor intracellular pathway and breast cancer: Factors, mechanism and regulation. Curr. Pharm. Des. 2016. Epub ahead of print. [Google Scholar]
- Choo, A.Y.; Blenis, J. Torgeting oncogene addiction for cancer therapy. Cancer Cell 2006, 9, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. The pkb/akt pathway in cancer. Curr. Pharm. Des. 2010, 16, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, N.; Liu, P.; Xie, X. Ampk and cancer. EXS 2016, 107, 203–226. [Google Scholar] [PubMed]
- Hatakeyama, M.; Kimura, S.; Naka, T.; Kawasaki, T.; Yumoto, N.; Ichikawa, M.; Kim, J.H.; Saito, K.; Saeki, M.; Shirouzu, M.; et al. A computational model on the modulation of mitogen-activated protein kinase (mapk) and akt pathways in heregulin-induced erbb signalling. Biochem. J. 2003, 373, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Nakakuki, T.; Yumoto, N.; Naka, T.; Shirouzu, M.; Yokoyama, S.; Hatakeyama, M. Topological analysis of mapk cascade for kinetic erbb signaling. PLoS ONE 2008, 3, e1782. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.M.; Fry, D.W. Akt, mapk (erk1/2), and p38 act in concert to promote apoptosis in response to erbb receptor family inhibition. J. Biol. Chem. 2001, 276, 14842–14847. [Google Scholar] [CrossRef] [PubMed]
- Fang, B. Ras signaling and anti-ras therapy: Lessons learned from genetically engineered mouse models, human cancer cells, and patient-related studies. Acta Biochim. Biophys. Sin. 2016, 48, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. Ras isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Zhang, S. Past, present, and future of targeting ras for cancer therapies. Mini Rev. Med. Chem. 2016, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Rossner, F.; Gieseler, C.; Morkel, M.; Royer, H.D.; Rivera, M.; Blaker, H.; Dietel, M.; Schafer, R.; Sers, C. Uncoupling of egfr-ras signaling and nuclear localization of ybx1 in colorectal cancer. Oncogenesis 2016, 5, e187. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. Kras mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [PubMed]
- Campos-Parra, A.D.; Zuloaga, C.; Manriquez, M.E.; Aviles, A.; Borbolla-Escoboza, J.; Cardona, A.; Meneses, A.; Arrieta, O. Kras mutation as the biomarker of response to chemotherapy and egfr-tkis in patients with advanced non-small cell lung cancer: Clues for its potential use in second-line therapy decision making. Am. J. Clin. Oncol. 2015, 38, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Drilon, A. Kras-mutant lung cancers in the era of targeted therapy. Adv. Exp. Med. Biol. 2016, 893, 155–178. [Google Scholar] [PubMed]
- Umelo, I.A.; De Wever, O.; Kronenberger, P.; Van Deun, J.; Noor, A.; Singh, K.; Teugels, E.; Chen, G.; Bracke, M.; De Greve, J. Combined targeting of egfr/her promotes anti-tumor efficacy in subsets of kras mutant lung cancer resistant to single egfr blockade. Oncotarget 2015, 6, 20132–20144. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, Y.; Wang, Y.; Meng, Y.; Zhu, J.; Jin, H.; Li, J.; Zhang, D.; Yu, Y.; Wu, X.R.; et al. A new tumour suppression mechanism by p27kip1: Egfr down-regulation mediated by jnk/c-jun pathway inhibition. Biochem. J. 2014, 463, 383–392. [Google Scholar]
- Li, J.Y.; Wang, H.; May, S.; Song, X.; Fueyo, J.; Fuller, G.N.; Wang, H. Constitutive activation of c-jun n-terminal kinase correlates with histologic grade and egfr expression in diffuse gliomas. J. Neurooncol. 2008, 88, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Z.; Li, W.; Xu, S.; Wang, T.; Wang, T.; Niu, M.; Zhang, S.; Jia, L.; Li, S. Topk promotes lung cancer resistance to egfr tyrosine kinase inhibitors by phosphorylating and activating c-jun. Oncotarget 2016, 7, 6748–6764. [Google Scholar] [PubMed]
- Nottingham, L.K.; Yan, C.H.; Yang, X.; Si, H.; Coupar, J.; Bian, Y.; Cheng, T.F.; Allen, C.; Arun, P.; Gius, D.; et al. Aberrant ikkalpha and ikkbeta cooperatively activate nf-kappab and induce egfr/ap1 signaling to promote survival and migration of head and neck cancer. Oncogene 2014, 33, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Antoun, G.; Keir, S.T.; Friedman, H.; Bigner, D.D.; Ali-Osman, F. Phosphorylation of glutathione s-transferase p1 (gstp1) by epidermal growth factor receptor (egfr) promotes formation of the gstp1-c-jun n-terminal kinase (jnk) complex and suppresses jnk downstream signaling and apoptosis in brain tumor cells. J. Biol. Chem. 2015, 290, 30866–30878. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Stockinger, A.; Schaffhauser, B.; Beug, H.; Foisner, R. Epithelial mesenchymal transition by c-fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J. Cell Biol. 2000, 148, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, E.; Schwarz, H.; Deiner, E.M.; Leitner, I.; Eilers, M.; Berger, J.; Busslinger, M.; Beug, H. Activation of an inducible c-foser fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell 1992, 71, 1103–1116. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Badache, A.; Daly, J.M.; Hynes, N.E. An essential role for src kinase in erbb receptor signaling through the mapk pathway. Exp. Cell Res. 2001, 267, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Balz, L.M.; Bartkowiak, K.; Andreas, A.; Pantel, K.; Niggemann, B.; Zanker, K.S.; Brandt, B.H.; Dittmar, T. The interplay of her2/her3/pi3k and egfr/her2/plc-gamma1 signalling in breast cancer cell migration and dissemination. J. Pathol. 2012, 227, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, G.; Deng, X.Y.; He, X.B.; Chen, L.J.; Wu, C.; Shi, Y.; Wu, K.P.; Mei, L.J.; Lu, J.X.; et al. Activated human hydroxy-carboxylic acid receptor-3 signals to map kinase cascades via the plc-dependent pkc and mmp-mediated egfr pathways. Br. J. Pharmacol. 2012, 166, 1756–1773. [Google Scholar] [CrossRef] [PubMed]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. Stat-mediated egfr signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Han, Y.; Wang, X.; Wang, W.; Wang, X.; Wen, M.; Xia, J.; Xing, H.; Li, X.; Zhang, Z. Correlation of egfr del 19 with fn14/jak/stat signaling molecules in non-small cell lung cancer. Oncol. Rep. 2016, 36, 1030–1040. [Google Scholar] [PubMed]
- Yang, S.; Park, K.; Turkson, J.; Arteaga, C.L. Ligand-independent phosphorylation of y869 (y845) links mutant egfr signaling to stat-mediated gene expression. Exp. Cell Res. 2008, 314, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Lee, M.H.; Lim, D.Y.; Lee, S.Y.; Jeong, C.H.; Kim, J.E.; Lim, T.G.; Chen, H.; Bode, A.M.; Lee, H.J.; et al. Butein, a novel dual inhibitor of met and egfr, overcomes gefitinib-resistant lung cancer growth. Mol. Carcinog. 2015, 54, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Okamoto, I.; Okamoto, W.; Tanizaki, J.; Nakagawa, K.; Nishio, K. Targeting met amplification as a new oncogenic driver. Cancers 2014, 6, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Presutti, D.; Santini, S.; Cardinali, B.; Papoff, G.; Lalli, C.; Samperna, S.; Fustaino, V.; Giannini, G.; Ruberti, G. Met gene amplification and met receptor activation are not sufficient to predict efficacy of combined met and egfr inhibitors in egfr tki-resistant nsclc cells. PLoS ONE 2015, 10, e0143333. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Rothstein, M.E.; Keohavong, P.; Lenzner, D.; Land, S.R.; Gaither-Davis, A.L.; Kim, K.J.; Kaminski, N.; Siegfried, J.M. Targeting of both the c-met and egfr pathways results in additive inhibition of lung tumorigenesis in transgenic mice. Cancers 2010, 2, 2153–2170. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of met amplification in egfr mutant nsclc. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Crombet, T.; de Leon, J.; Moreno, E. A view on egfr-targeted therapies from the oncogene-addiction perspective. Front. Pharmacol. 2013, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—the achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Torti, D.; Trusolino, L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med. 2011, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Semenova, E.A.; Berns, A. Drugging the addict: Non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016, 17, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Choi, P.S.; Felsher, D.W. Oncogene addiction: Resetting the safety switch? Oncotarget 2014, 5, 7986–7987. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Synthetic lethality: A framework for the development of wiser cancer therapeutics. Genome Med. 2009, 1, 99. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Wong, K.K.; Engelman, J.A. Differences underlying egfr and her2 oncogene addiction. Cell Cycle 2010, 9, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Settleman, J. Oncogene addiction. Curr. Biol. 2012, 22, R43–R44. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Gajowniczek, P.; Way, I.P.; Lee, D.Y.; Jiang, J.; Yuza, Y.; Classon, M.; Haber, D.A.; Settleman, J. A common signaling cascade may underlie "addiction" to the src, bcr-abl, and egf receptor oncogenes. Cancer Cell 2006, 10, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Albanell, J.; Gascon, P. Small molecules with egfr-tk inhibitor activity. Curr. Drug Targets 2005, 6, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Roy, V.; Perez, E.A. Beyond trastuzumab: Small molecule tyrosine kinase inhibitors in her-2-positive breast cancer. Oncologist 2009, 14, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Azuma, K.; Nagai, H.; Kim, Y.H.; Togashi, Y.; Sesumi, Y.; Chiba, M.; Shimoji, M.; Sato, K.; Tomizawa, K.; et al. Characterization of egfr t790m, l792f, and c797s mutations as mechanisms of acquired resistance to afatinib in lung cancer. Mol. Cancer Ther. 2016, 16, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; D'Argento, E.; Basso, M.; Strippoli, A.; Dadduzio, V.; Cerchiaro, E.; Martini, M.; Cassano, A.; Barone, C. Different egfr gene mutations in exon 18, 19 and 21 as prognostic and predictive markers in nsclc: A single institution analysis. Mol. Diagn. Ther. 2016, 20, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, W.; Zhi, Q.; Jiang, M. Lapatinib resistance in her2+ cancers: Latest findings and new concepts on molecular mechanisms. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vavala, T.; Follador, A.; Tiseo, M.; Galetta, D.; Morabito, A.; Di Maio, M.; Martelli, O.; Caffo, O.; Piovano, P.L.; Cortinovis, D.; et al. Be-positive: Beyond progression after tyrosine kinase inhibitor in egfr- positive non small cell lung cancer patients: Results from a multicenter italian observational study. Lung Cancer 2016, 95, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting egfr t790m mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Maiello, M.R.; Chicchinelli, N.; Iannaccone, A.; Esposito, C.; De Cecio, R.; D’Alessio, A.; De Luca, A. Targeting the egfr t790m mutation in non-small-cell lung cancer. Expert Opin. Ther. Targets 2017, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Settleman, J. Erbbs in lung cancer. Exp. Cell Res. 2009, 315, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Gefitinib (iressa) in oncogene-addictive cancers and therapy for common cancers. Cancer Biol. Ther. 2004, 3, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Kobayashi, K.; Usui, K.; Maemondo, M.; Okinaga, S.; Mikami, I.; Ando, M.; Yamazaki, K.; Saijo, Y.; Gemma, A.; et al. First-line gefitinib for patients with advanced non-small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J. Clin. Oncol. 2009, 27, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Mukohara, T.; Engelman, J.A.; Hanna, N.H.; Yeap, B.Y.; Kobayashi, S.; Lindeman, N.; Halmos, B.; Pearlberg, J.; Tsuchihashi, Z.; Cantley, L.C.; et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J. Natl. Cancer Inst. 2005, 97, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing egfr mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel d761y and common secondary t790m mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Osimertinib making a breakthrough in lung cancer targeted therapy. Onco Targets Ther. 2016, 9, 5489–5493. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in egfr t790m-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The egfrviii variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Parker, B.A.; Schwab, R.; Kurzrock, R. Her2 aberrations in cancer: Implications for therapy. Cancer Treat Rev. 2014, 40, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A. Breast cancer: Her2—A good addiction. Nat. Rev. Clin. Oncol. 2012, 9, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Mustacchi, G.; Biganzoli, L.; Pronzato, P.; Montemurro, F.; Dambrosio, M.; Minelli, M.; Molteni, L.; Scaltriti, L. Her2-positive metastatic breast cancer: A changing scenario. Crit. Rev. Oncol. Hematol. 2015, 95, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Shi, J.; Li, Z.; Feng, A.; Ye, Q. Comparison of the 2007 and 2013 asco/cap evaluation systems for her2 amplification in breast cancer. Pathol. Res. Pract. 2015, 211, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Noske, A. Impact of modified 2013 asco/cap guidelines on her2 testing in breast cancer. One year experience. PLoS ONE 2015, 10, e0140652. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J. The distinctive nature of her2-positive breast cancers. N. Engl. J. Med. 2005, 353, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for her2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Clark, E.; Baselga, J. Treatment of her2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 1964–1965. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Barry, W.T.; Dang, C.T.; Yardley, D.A.; Moy, B.; Marcom, P.K.; Albain, K.S.; Rugo, H.S.; Ellis, M.; Shapira, I.; et al. Adjuvant paclitaxel and trastuzumab for node-negative, her2-positive breast cancer. N. Engl. J. Med. 2015, 372, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Diver, E.J.; Foster, R.; Rueda, B.R.; Growdon, W.B. The therapeutic challenge of targeting her2 in endometrial cancer. Oncologist 2015, 20, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Von Hoff, D.D.; Neubauer, M.; Anderson, T.; Fleming, M.; Nagarwala, Y.; Mahoney, J.M.; Midwinter, D.; Vocila, L.; Zaks, T.Z. Target-specific, histology-independent, randomized discontinuation study of lapatinib in patients with her2-amplified solid tumors. Investig. New Drugs 2012, 30, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating her2 mutations in her2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Reis-Filho, J.S. Activating mutations in her2: Neu opportunities and neu challenges. Cancer Discov. 2013, 3, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Kourie, H.R.; Chaix, M.; Gombos, A.; Aftimos, P.; Awada, A. Pharmacodynamics, pharmacokinetics and clinical efficacy of neratinib in her2-positive breast cancer and breast cancer with her2 mutations. Expert Opin. Drug Metab. Toxicol. 2016, 12, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Ryan, E.D.; Cardiff, R.D.; Muller, W.J. Elevated expression of activated forms of neu/erbb-2 and erbb-3 are involved in the induction of mammary tumors in transgenic mice: Implications for human breast cancer. EMBO J. 1999, 18, 2149–2164. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, E.; Seeboeck, R.; Jacobi, N.; Obrist, P.; Huter, S.; Klein, C.; Oender, K.; Wiesner, C.; Hundsberger, H.; Eger, A. The combinatorial approach of laser-captured microdissection and reverse transcription quantitative polymerase chain reaction accurately determines her2 status in breast cancer. Biomark Res. 2016, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Travis, A.; Pinder, S.E.; Robertson, J.F.; Bell, J.A.; Wencyk, P.; Gullick, W.J.; Nicholson, R.I.; Poller, D.N.; Blamey, R.W.; Elston, C.W.; et al. C-erbb-3 in human breast carcinoma: Expression and relation to prognosis and established prognostic indicators. Br. J. Cancer 1996, 74, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wong, P.; Salvaggio, C.; Salhi, A.; Osman, I.; Bedogni, B. Synchronized targeting of notch and erbb signaling suppresses melanoma tumor growth through inhibition of notch1 and erbb3. J. Investig. Dermatol. 2016, 136, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chang, Y.; Rios, A.; An, Z. Her3/erbb3, an emerging cancer therapeutic target. Acta Biochim. Biophys. Sin. 2016, 48, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Rudloff, U.; Samuels, Y. A growing family: Adding mutated erbb4 as a novel cancer target. Cell Cycle 2010, 9, 1487–1503. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Killian, K.J.; Samuels, Y.; Rudloff, U. Erbb4 mutation analysis: Emerging molecular target for melanoma treatment. Methods Mol. Biol. 2014, 1102, 461–480. [Google Scholar] [PubMed]
- Williams, C.S.; Bernard, J.K.; Demory Beckler, M.; Almohazey, D.; Washington, M.K.; Smith, J.J.; Frey, M.R. Erbb4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis 2015, 36, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Kurppa, K.J.; Denessiouk, K.; Johnson, M.S.; Elenius, K. Activating erbb4 mutations in non-small cell lung cancer. Oncogene 2016, 35, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Pan, S.Y.; Huang, Q.L.; Lu, Y.N.; Wu, X.H.; Chang, J.H.; Liu, Z.B.; Cai, X.W.; Liu, Q.; Wang, J.L.; et al. Pa-msha in combination with egfr tyrosine kinase inhibitor: A new strategy to overcome the drug resistance of non-small cell lung cancer cells. Oncotarget 2016, 7, 49384–49396. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kambrick, S.; Fu, S.; Naing, A.; Subbiah, V.; Blumenschein, G.R.; Glisson, B.S.; Kies, M.S.; Tsimberidou, A.M.; Wheler, J.J.; et al. Advanced malignancies treated with a combination of the vegf inhibitor bevacizumab, anti-egfr antibody cetuximab, and the mtor inhibitor temsirolimus. Oncotarget 2016, 7, 23227–23238. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Madeddu, D.; Tiseo, M.; Vivo, V.; Galetti, M.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Falco, A.; et al. Combination of gefitinib and pemetrexed prevents the acquisition of tki resistance in nsclc cell lines carrying egfr-activating mutation. J. Thorac. Oncol. 2016, 11, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, I.; Wouters, A.; Van den Bossche, J.; Peeters, M.; Pauwels, P.; Deschoolmeester, V.; Vermorken, J.B.; Lardon, F. Preclinical and clinical studies on afatinib in monotherapy and in combination regimens: Potential impact in colorectal cancer. Pharmacol. Ther. 2016, 166, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Gomes, J.; Cruz, M.R. Combination of afatinib with cetuximab in patients with egfr-mutant non-small-cell lung cancer resistant to egfr inhibitors. Onco Targets Ther. 2015, 8, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, E.; Hotta, K.; Nogami, N.; Kuyama, S.; Kishino, D.; Fujii, M.; Kozuki, T.; Tabata, M.; Harada, D.; Chikamori, K.; et al. Phase ii trial of gefitinib in combination with bevacizumab as first-line therapy for advanced non-small cell lung cancer with activating egfr gene mutations: The okayama lung cancer study group trial 1001. J. Thorac. Oncol. 2015, 10, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Torka, R.; Penzes, K.; Gusenbauer, S.; Baumann, C.; Szabadkai, I.; Orfi, L.; Keri, G.; Ullrich, A. Activation of her3 interferes with antitumor effects of axl receptor tyrosine kinase inhibitors: Suggestion of combination therapy. Neoplasia 2014, 16, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Roscilli, G.; Fattore, L.; Malpicci, D.; Marra, E.; Luberto, L.; D'Andrilli, A.; Coluccia, P.; Giovagnoli, M.R.; et al. Combination therapy with anti-erbb3 monoclonal antibodies and egfr tkis potently inhibits non-small cell lung cancer. Oncotarget 2013, 4, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, N.; Seddon, A.M.; Dalgleish, A.; Mackintosh, D.; Modjtahedi, H. Treatment with a combination of the erbb (her) family blocker afatinib and the igf-ir inhibitor, nvp-aew541 induces synergistic growth inhibition of human pancreatic cancer cells. BMC Cancer 2013, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, S.; Lyu, H.; Cai, B.; Yang, X.; Wang, J.; Liu, B. The anti-erbb3 antibody mm-121/sar256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol. Cancer 2013, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Hochart, A.; Leblond, P.; Le Bourhis, X.; Meignan, S.; Tulasne, D. Met receptor inhibition: Hope against resistance to targeted therapies? Bull. Cancer 2017, 104, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating erbb3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Disorders in cell circuitry during multistage carcinogenesis: The role of homeostasis. Carcinogenesis 2000, 21, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Brysch, W.; Magal, E.; Louis, J.C.; Kunst, M.; Klinger, I.; Schlingensiepen, R.; Schlingensiepen, K.H. Inhibition of p185c-erbb-2 proto-oncogene expression by antisense oligodeoxynucleotides down-regulates p185-associated tyrosine-kinase activity and strongly inhibits mammary tumor-cell proliferation. Cancer Gene Ther. 1994, 1, 99–105. [Google Scholar] [PubMed]
- Colomer, R.; Lupu, R.; Bacus, S.S.; Gelmann, E.P. Erbb-2 antisense oligonucleotides inhibit the proliferation of breast carcinoma cells with erbb-2 oncogene amplification. Br. J. Cancer 1994, 70, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Schmidt, U.; Botefur, I.C.; Holland, J.F.; Ohnuma, T. Marked inhibition of glioblastoma target cell tumorigenicity in vitro by retrovirus-mediated transfer of a hairpin ribozyme against deletion-mutant epidermal growth factor receptor messenger rna. J. Neurosurg. 2000, 92, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Hudziak, R.M.; Lewis, G.D.; Winget, M.; Fendly, B.M.; Shepard, H.M.; Ullrich, A. P185her2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 1989, 9, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Luwor, R.B.; Johns, T.G.; Murone, C.; Huang, H.J.; Cavenee, W.K.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (egfr) but not wild-type egfr. Cancer Res. 2001, 61, 5355–5361. [Google Scholar] [PubMed]
- Yamazaki, H.; Kijima, H.; Abe, Y.; Oshika, Y.; Tsuchida, T.; Tokunaga, T.; Tamaoki, N.; Nakamura, M.; Tsugu, A.; Ohnishi, Y.; et al. Inhibition of tumor growth by ribozyme-mediated suppression of aberrant epidermal growth factor receptor gene expression. J. Natl. Cancer Inst. 1998, 90, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Mullin, R.J.; Keith, B.R.; Liu, L.H.; Ma, H.; Rusnak, D.W.; Owens, G.; Alligood, K.J.; Spector, N.L. Anti-tumor activity of gw572016: A dual tyrosine kinase inhibitor blocks egf activation of egfr/erbb2 and downstream erk1/2 and akt pathways. Oncogene 2002, 21, 6255–6263. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Chen, Y.J.; Yen, C.J.; Chen, W.S.; Huang, W.C. Hbx sensitizes hepatocellular carcinoma cells to lapatinib by up-regulating erbb3. Oncotarget 2016, 7, 473–489. [Google Scholar] [PubMed]
- Ohnishi, Y.; Nakamura, H.; Yoshimura, M.; Tokuda, Y.; Iwasawa, M.; Ueyama, Y.; Tamaoki, N.; Shimamura, K. Prolonged survival of mice with human gastric cancer treated with an anti-c-erbb-2 monoclonal antibody. Br. J. Cancer 1995, 71, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant egf receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of hki-272, an orally active, irreversible inhibitor of the her-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [PubMed]
- Shepard, H.M.; Lewis, G.D.; Sarup, J.C.; Fendly, B.M.; Maneval, D.; Mordenti, J.; Figari, I.; Kotts, C.E.; Palladino, M.A.; Ullrich, A.; et al. Monoclonal antibody therapy of human cancer: Taking the her2 protooncogene to the clinic. J. Clin. Immunol. 1991, 11, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, Y.; Ohnishi, Y.; Shimamura, K.; Iwasawa, M.; Yoshimura, M.; Ueyama, Y.; Tamaoki, N.; Tajima, T.; Mitomi, T. In vitro and in vivo anti-tumour effects of a humanised monoclonal antibody against c-erbb-2 product. Br. J. Cancer 1996, 73, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.W.; Lee, F.Y.; Yu, C.; Luo, F.R.; Oppenheimer, S.; Zhang, H.; Smykla, R.A.; Mastalerz, H.; Fink, B.E.; Hunt, J.T.; et al. Preclinical antitumor activity of bms-599626, a pan-her kinase inhibitor that inhibits her1/her2 homodimer and heterodimer signaling. Clin. Cancer Res. 2006, 12, 6186–6193. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the egf receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wang, L.; Zhang, Y.; Yu, X.; Wang, C.; Wang, H.; Yang, Y.; Chong, X.; Xia, T.; Meng, Y.; et al. An anti-erbb2 fully human antibody circumvents trastuzumab resistance. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, II; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Lin, Z.; Wang, F.; Zhao, W.; Dong, X. Four-membered heterocycles-containing 4-anilino-quinazoline derivatives as epidermal growth factor receptor (egfr) kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5385–5388. [Google Scholar] [CrossRef] [PubMed]
- Kugel, C.H., 3rd; Hartsough, E.J.; Davies, M.A.; Setiady, Y.Y.; Aplin, A.E. Function-blocking erbb3 antibody inhibits the adaptive response to raf inhibitor. Cancer Res. 2014, 74, 4122–4132. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xu, Y.; Feng, S.; Luo, S.; Zheng, R.; Yang, J.; Wang, L.; Zhong, L.; Yang, H.Y.; Wang, B.L.; et al. Sklb1206, a novel orally available multikinase inhibitor targeting egfr activating and t790m mutants, erbb2, erbb4, and vegfr2, displays potent antitumor activity both in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to egfr inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.K.; Kleinheinz, T.L.; Peterson, D.; Moffat, J.G. A simple high-content cell cycle assay reveals frequent discrepancies between cell number and atp and mts proliferation assays. PLoS ONE 2013, 8, e63583. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, H.; Gage, J.; Leonard, F.; Srinivasan, S.; Souza, G.R.; Dave, B.; Godin, B. Three-dimensional in vitro co-culture model of breast tumor using magnetic levitation. Sci. Rep. 2014, 4, 6468. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Lioni, M.; Herlyn, M. Life isn't flat_taking cancer biology to the next dimension. In Vitro Cell. Dev. Biol. Anim. 2006, 42, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, Q.; Voss, T.C.; Quick, K.L.; LaBarbera, D.V. High-throughput imaging: Focusing in on drug discovery in 3d. Methods 2016, 96, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Thoma, C.R.; Stroebel, S.; Rosch, N.; Calpe, B.; Krek, W.; Kelm, J.M. A high-throughput-compatible 3d microtissue co-culture system for phenotypic rnai screening applications. J. Biomol. Screen. 2013, 18, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Howes, A.L.; Richardson, R.D.; Finlay, D.; Vuori, K. 3-dimensional culture systems for anti-cancer compound profiling and high-throughput screening reveal increases in egfr inhibitor-mediated cytotoxicity compared to monolayer culture systems. PLoS ONE 2014, 9, e108283. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Krausz, E.; de Hoogt, R.; Gustin, E.; Cornelissen, F.; Grand-Perret, T.; Janssen, L.; Vloemans, N.; Wuyts, D.; Frans, S.; Axel, A.; et al. Translation of a tumor microenvironment mimicking 3d tumor growth co-culture assay platform to high-content screening. J. Biomol. Screen. 2013, 18, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Pickl, M.; Ries, C.H. Comparison of 3d and 2d tumor models reveals enhanced her2 activation in 3d associated with an increased response to trastuzumab. Oncogene 2009, 28, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. Her2 signaling pathway activation and response of breast cancer cells to her2-targeting agents is dependent strongly on the 3d microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Eimer, S.; Dugay, F.; Airiau, K.; Avril, T.; Quillien, V.; Belaud-Rotureau, M.A.; Belloc, F. Cyclopamine cooperates with egfr inhibition to deplete stem-like cancer cells in glioblastoma-derived spheroid cultures. Neuro-Oncology 2012, 14, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Ekert, J.E.; Johnson, K.; Strake, B.; Pardinas, J.; Jarantow, S.; Perkinson, R.; Colter, D.C. Three-dimensional lung tumor microenvironment modulates therapeutic compound responsiveness in vitro--implication for drug development. PLoS ONE 2014, 9, e92248. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; Mersch, S.; Deenen, R.; Schmidt, S.; Messner, I.; Schafer, K.L.; Baldus, S.E.; Huckenbeck, W.; Piekorz, R.P.; Knoefel, W.T.; et al. Impact of the 3d microenvironment on phenotype, gene expression, and egfr inhibition of colorectal cancer cell lines. PLoS ONE 2013, 8, e59689. [Google Scholar] [CrossRef] [PubMed]
- Unger, C.; Kramer, N.; Walzl, A.; Scherzer, M.; Hengstschlager, M.; Dolznig, H. Modeling human carcinomas: Physiologically relevant 3d models to improve anti-cancer drug development. Adv. Drug Deliv. Rev. 2014, 79–80, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Dolznig, H.; Walzl, A.; Kramer, N.; Rosner, M.; Garin-Chesa, P.; Hengstschläger, M. Organotypic spheroid cultures to study tumor–stroma interaction during cancer development. Drug Discov. Today Dis. Model. 2011, 8, 113–119. [Google Scholar] [CrossRef]
- Kunz-Schughart, L.A.; Knuechel, R. Tumor-associated fibroblasts (part i): Active stromal participants in tumor development and progression? Histol. Histopathol. 2002, 17, 599–621. [Google Scholar] [PubMed]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.; Kleef, J. Tumor microenvironment and progression of pancreatic cancer. Expe. Oncol. 2010, 32, 128–131. [Google Scholar]
- Apte, M.V. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol. Cancer Res. 2008, 6, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Sotgia, F. Oncogenes induce the cancer-associated fibroblast phenotype: Metabolic symbiosis and "fibroblast addiction" are new therapeutic targets for drug discovery. Cell Cycle 2013, 12, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Rinott, G.; Dotan, S.; Elkabets, M.; Rider, P.; Voronov, E.; Apte, R.N. Microenvironment-derived il-1 and il-17 interact in the control of lung metastasis. J. Immunol. 2011, 186, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Rupp, C.; Scherzer, M.; Rudisch, A.; Unger, C.; Haslinger, C.; Schweifer, N.; Artaker, M.; Nivarthi, H.; Moriggl, R.; Hengstschlager, M.; et al. Igfbp7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor-stroma interaction. Oncogene 2015, 34, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. Stellatum: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O'Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug discovery today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Bhadriraju, K.; Chen, S. Engineering cellular microenvironments to improve cell-based drug testing. Drug Discov. Today 2002, 7, 612–620. [Google Scholar] [CrossRef]
- Rudisch, A.; Dewhurst, M.R.; Horga, L.G.; Kramer, N.; Harrer, N.; Dong, M.; van der Kuip, H.; Wernitznig, A.; Bernthaler, A.; Dolznig, H.; et al. High emt signature score of invasive non-small cell lung cancer (nsclc) cells correlates with nfkappab driven colony-stimulating factor 2 (csf2/gm-csf) secretion by neighboring stromal fibroblasts. PLoS ONE 2015, 10, e0124283. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition (emt) gene signature predicts resistance to egfr and pi3k inhibitors and identifies axl as a therapeutic target for overcoming egfr inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Delmore, J.; Weisberg, E.; Negri, J.M.; Geer, D.C.; Klippel, S.; Mitsiades, N.; Schlossman, R.L.; Munshi, N.C.; Kung, A.L.; et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat. Med. 2010, 16, 483–489. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, I.A.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J.A.; Graeser, R.; de Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; van der Kuip, H.; Consortium, I.P. Three-dimensional models of cancer for pharmacology and cancer cell biology: Capturing tumor complexity in vitro/ex vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.J.; Dong, M.; Gutekunst, M.; Narhi, K.; van Zoggel, H.J.; Blom, S.; Nagaraj, A.; Metsalu, T.; Oswald, E.; Erkens-Schulze, S.; et al. Capturing complex tumour biology in vitro: Histological and molecular characterisation of precision cut slices. Sci. Rep. 2015, 5, 17187. [Google Scholar] [CrossRef] [PubMed]
- Koster, H. Comparison of in vitro preparations for prediction of in vivo drug metabolism. Drug Metab. Dispos. 2002, 30, 1129–1136. [Google Scholar]
- van der Kuip, H.; Murdter, T.E.; Sonnenberg, M.; McClellan, M.; Gutzeit, S.; Gerteis, A.; Simon, W.; Fritz, P.; Aulitzky, W.E. Short term culture of breast cancer tissues to study the activity of the anticancer drug taxol in an intact tumor environment. BMC Cancer 2006, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Perez-Vizcaino, F.; Harrington, L.; Faro, R.; Sturton, G.; Barnes, P.J.; Mitchell, J.A. Pharmacology of airways and vessels in lung slices in situ: Role of endogenous dilator hormones. Respir. Res. 2006, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lampe, J.; Lange, S.; Smirnow, I.; Konigsrainer, A.; Hann-von-Weyhern, C.; Fend, F.; Gregor, M.; Bitzer, M.; Lauer, U.M. Improved reproducibility in preparing precision-cut liver tissue slices. Cytotechnology 2009, 61, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Majumder, B.; Baraneedharan, U.; Thiyagarajan, S.; Radhakrishnan, P.; Narasimhan, H.; Dhandapani, M.; Brijwani, N.; Pinto, D.D.; Prasath, A.; Shanthappa, B.U.; et al. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat. Commun. 2015, 6, 6169. [Google Scholar] [CrossRef] [PubMed]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Moss, M.A.; Pollock, S.; Lane, S.; Shaaban, A.M.; Millican-Slater, R.; Nash, C.; Hanby, A.M.; Speirs, V. The practicalities of using tissue slices as preclinical organotypic breast cancer models. J. Clin. Pathol. 2013, 66, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Merz, F.; Gaunitz, F.; Dehghani, F.; Renner, C.; Meixensberger, J.; Gutenberg, A.; Giese, A.; Schopow, K.; Hellwig, C.; Schafer, M.; et al. Organotypic slice cultures of human glioblastoma reveal different susceptibilities to treatments. Neuro-Oncology 2013, 15, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, N.; Smolinska, V.; Stierschneider, A.; Klein, C.; Önder, K.; Lechner, P.; Kaiser, H.; Hundsberger, H.; Eger, A. Development of organotypic cancer models for the identification of individualized cancer therapies. In Forschungsforum der österreichischen Fachhochschulen; University of Applied Sciences BFI Vienna: Vienna, Austria, 2016. [Google Scholar]
- Malaney, P.; Nicosia, S.V.; Davé, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014, 344, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Amendt, C.; Staub, E.; Friese-Hamim, M.; Storkel, S.; Stroh, C. Association of egfr expression level and cetuximab activity in patient-derived xenograft models of human non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, J.; Zhen, R.; Lv, J.; Zheng, L.; Su, X.; Zhu, G.; Gavine, P.R.; Xu, S.; Lu, S.; et al. Trastuzumab anti-tumor efficacy in patient-derived esophageal squamous cell carcinoma xenograft (pdecx) mouse models. J. Transl. Med. 2012, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Qian, G.; Zhang, H.; Magliocca, K.R.; Nannapaneni, S.; Amin, A.R.; Rossi, M.; Patel, M.; El-Deiry, M.; Wadsworth, J.T.; et al. Her3 targeting sensitizes hnscc to cetuximab by reducing her3 activity and her2/her3 dimerization: Evidence from cell line and patient-derived xenograft models. Clin. Cancer Res. 2017, 23, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ye, Q.; Lv, J.; Ye, P.; Sun, Y.; Fan, S.; Su, X.; Gavine, P.; Yin, X. Evaluation of trastuzumab anti-tumor efficacy and its correlation with her-2 status in patient-derived gastric adenocarcinoma xenograft models. Pathol. Oncol. Res. 2015, 21, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, D.; Chen, L.; Shimamura, T.; Kobayashi, S.; McNamara, K.; Mahmood, U.; Mitchell, A.; Sun, Y.; Al-Hashem, R.; et al. The impact of human egfr kinase domain mutations on lung tumorigenesis and in vivo sensitivity to egfr-targeted therapies. Cancer Cell 2006, 9, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant iii mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822. [Google Scholar] [CrossRef] [PubMed]
- Hannesdottir, L.; Tymoszuk, P.; Parajuli, N.; Wasmer, M.H.; Philipp, S.; Daschil, N.; Datta, S.; Koller, J.B.; Tripp, C.H.; Stoitzner, P.; et al. Lapatinib and doxorubicin enhance the stat1-dependent antitumor immune response. Eur. J. Immunol. 2013, 43, 2718–2729. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase ii trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (the ideal 1 trial) [corrected]. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (iressa survival evaluation in lung cancer). Lancet 2005, 366, 1527–1537. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N.Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. Egf receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Janne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic egfr mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Su, K.Y.; Chen, H.Y.; Li, K.C.; Kuo, M.L.; Yang, J.C.; Chan, W.K.; Ho, B.C.; Chang, G.C.; Shih, J.Y.; Yu, S.L.; et al. Pretreatment epidermal growth factor receptor (egfr) t790m mutation predicts shorter egfr tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Janne, P.A. Egfr mutations and resistance to irreversible pyrimidine-based egfr inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed]
- Villadolid, J.; Ersek, J.L.; Fong, M.K.; Sirianno, L.; Story, E.S. Management of hyperglycemia from epidermal growth factor receptor (egfr) tyrosine kinase inhibitors (tkis) targeting t790m-mediated resistance. Transl. Lung Cancer Res. 2015, 4, 576–583. [Google Scholar] [PubMed]
- Yu, H.A.; Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Cancer Netw. 2013, 11, 161–169. [Google Scholar]
- Ramalingam, S.S.; Blackhall, F.; Krzakowski, M.; Barrios, C.H.; Park, K.; Bover, I.; Seog Heo, D.; Rosell, R.; Talbot, D.C.; Frank, R.; et al. Randomized phase ii study of dacomitinib (pf-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 3337–3344. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. Azd9291, an irreversible egfr tki, overcomes t790m-mediated resistance to egfr inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Pirker, R. Third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr. Opin. Oncol. 2016, 28, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding mode of the breakthrough inhibitor azd9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Ohhara, Y.; Fukuda, N.; Takeuchi, S.; Honma, R.; Shimizu, Y.; Kinoshita, I.; Dosaka-Akita, H. Role of targeted therapy in metastatic colorectal cancer. World J. Gastrointest. Oncol. 2016, 8, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Khuri, F.R.; Herbst, R.S. Epidermal growth factor receptor biology (imc-c225). Curr. Opin. Oncol. 2001, 13, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Bianco, R.; Damiano, V.; Fontanini, G.; Caputo, R.; Pomatico, G.; De Placido, S.; Bianco, A.R.; Mendelsohn, J.; Tortora, G. Antiangiogenic and antitumor activity of anti-epidermal growth factor receptor c225 monoclonal antibody in combination with vascular endothelial growth factor antisense oligonucleotide in human geo colon cancer cells. Clin. Cancer Res. 2000, 6, 3739–3747. [Google Scholar] [PubMed]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. Kras mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type kras is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Nakagawa, K. Role of egfr monoclonal antibodies in the management of non-small cell lung cancer. Curr. Cancer Drug Targets 2015, 15, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martinez, A.; Canadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; Gonzalez, I.; et al. Emergence of multiple egfr extracellular mutations during cetuximab treatment in colorectal cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Braig, F.; März, M.; Schieferdecker, A.; Schulte, A.; Voigt, M.; Stein, A.; Grob, T.; Alawi, M.; Indenbirken, D.; Kriegs, M.; et al. Epidermal growth factor receptor mutation mediates crossresistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget 2015, 6, 12035–12047. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.; Rachiglio, A.M.; La Porta, M.L.; Sacco, A.; Roma, C.; Iannaccone, A.; Tatangelo, F.; Forgione, L.; Pasquale, R.; Barbaro, A.; et al. The s492r egfr ectodomain mutation is never detected in kras wild-type colorectal carcinoma before exposure to egfr monoclonal antibodies. Cancer Biol. Ther. 2013, 14, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Sickmier, E.A.; Kurzeja, R.J.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The panitumumab egfr complex reveals a binding mechanism that overcomes cetuximab induced resistance. PLoS ONE 2016, 11, e0163366. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, F.J.; Bellosillo, B.; Gelabert-Baldrich, M.; Dalmases, A.; Canadas, I.; Vidal, J.; Martinez, A.; Argiles, G.; Siravegna, G.; Arena, S.; et al. The first-in-class anti-egfr antibody mixture sym004 overcomes cetuximab resistance mediated by egfr extracellular domain mutations in colorectal cancer. Clin. Cancer Res. 2016, 22, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Siravegna, G.; Mussolin, B.; Kearns, J.D.; Wolf, B.B.; Misale, S.; Lazzari, L.; Bertotti, A.; Trusolino, L.; Adjei, A.A.; et al. Mm-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring egfr extracellular domain mutations. Sci. Transl. Med. 2016, 8, 324ra314. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Liu, E.T. Her2 as a predictor of therapeutic response in breast cancer. Breast Dis. 2000, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Owens, M.A.; Horten, B.C.; Da Silva, M.M. Her2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin. Breast Cancer 2004, 5, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.; Hollmen, M.; Junttila, T.T.; Kapanen, A.I.; Tommola, S.; Soini, Y.; Helin, H.; Salo, J.; Joensuu, H.; Sihvo, E.; et al. Amplification of her-2 in gastric carcinoma: Association with topoisomerase iialpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann. Oncol. 2005, 16, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Cuello, M.; Ettenberg, S.A.; Clark, A.S.; Keane, M.M.; Posner, R.H.; Nau, M.M.; Dennis, P.A.; Lipkowitz, S. Down-regulation of the erbb-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbb-2. Cancer Res. 2001, 61, 4892–4900. [Google Scholar] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. Pten activation contributes to tumor inhibition by trastuzumab, and loss of pten predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Xu, L.; di Tomaso, E.; Fukumura, D.; Jain, R.K. Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature 2002, 416, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [PubMed]
- Barok, M.; Isola, J.; Palyi-Krekk, Z.; Nagy, P.; Juhasz, I.; Vereb, G.; Kauraniemi, P.; Kapanen, A.; Tanner, M.; Vereb, G.; et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic jimt-1 breast cancer xenografts despite intrinsic drug resistance. Mol. Cancer Ther. 2007, 6, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Valero, V.; Booser, D.; Guerra, L.T.; Murray, J.L.; Pusztai, L.; Cristofanilli, M.; Arun, B.; Esmaeli, B.; Fritsche, H.A.; et al. Phase ii study of weekly docetaxel and trastuzumab for patients with her-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against her2 for metastatic breast cancer that overexpresses her2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- de Melo Gagliato, D.; Jardim, D.L.; Marchesi, M.S.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-her2 therapies in her2+ breast cancer. Oncotarget 2016, 7, 64431–64446. [Google Scholar] [PubMed]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; de Vos, A.M.; Sliwkowski, M.X. Insights into erbb signaling from the structure of the erbb2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd. Dual kinase inhibition in the treatment of breast cancer: Initial experience with the egfr/erbb-2 inhibitor lapatinib. Oncologist 2004, 9, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Szoor, A.; Simon, L.; Yarden, Y.; Szollosi, J.; Vereb, G. The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MAbs 2016, 8, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Campbell, M.R.; Moasser, M.M. The role of her3, the unpretentious member of the her family, in cancer biology and cancer therapeutics. Semin. Cell Dev. Biol. 2010, 21, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Bezler, M.; Hengstler, J.G.; Ullrich, A. Inhibition of doxorubicin-induced her3-pi3k-akt signalling enhances apoptosis of ovarian cancer cells. Mol. Oncol. 2012, 6, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Desbois-Mouthon, C.; Baron, A.; Blivet-Van Eggelpoel, M.J.; Fartoux, L.; Venot, C.; Bladt, F.; Housset, C.; Rosmorduc, O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/her3/akt signaling pathway: Rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 5445–5456. [Google Scholar] [PubMed]
- Gijsen, M.; King, P.; Perera, T.; Parker, P.J.; Harris, A.L.; Larijani, B.; Kong, A. Her2 phosphorylation is maintained by a pkb negative feedback loop in response to anti-her2 herceptin in breast cancer. PLoS Biol. 2010, 8, e1000563. [Google Scholar] [CrossRef] [PubMed]
- Jathal, M.K.; Chen, L.; Mudryj, M.; Ghosh, P.M. Targeting erbb3: The new rtk(id) on the prostate cancer block. Immunol. Endocr. Metab. Agents Med. Chem. 2011, 11, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ordonez-Ercan, D.; Fan, Z.; Edgerton, S.M.; Yang, X.; Thor, A.D. Downregulation of erbb3 abrogates erbb2-mediated tamoxifen resistance in breast cancer cells. Int. J. Cancer 2007, 120, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Dugger, T.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and erbb ligands and remain dependent on the erbb receptor network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, X.; Lee, C.K.; Liu, B. Elevated expression of erbb3 confers paclitaxel resistance in erbb2-overexpressing breast cancer cells via upregulation of survivin. Oncogene 2010, 29, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Malm, M.; Frejd, F.Y.; Stahl, S.; Lofblom, J. Targeting her3 using mono- and bispecific antibodies or alternative scaffolds. MAbs 2016, 8, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Arnett, S.O.; Teillaud, J.L.; Wurch, T.; Reichert, J.M.; Dunlop, C.; Huber, M. Ibc’s 21st annual antibody engineering and 8th annual antibody therapeutics international conferences and 2010 annual meeting of the antibody society. December 5–9, 2010, San Diego, CA USA. MAbs 2011, 3, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Rossin, R.; Kohno, T.; Hagooly, A.; Sharp, T.; Gliniak, B.; Arroll, T.; Chen, Q.; Hewig, A.; Kaplan-Lefko, P.; Friberg, G.; et al. Characterization of 64cu-dota-conatumumab: A pet tracer for in vivo imaging of death receptor 5. J. Nucl. Med. 2011, 52, 942–949. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.; Janne, P.A.; Oliveira, M.; Rizvi, N.; Malburg, L.; Keedy, V.; Yee, L.; Copigneaux, C.; Hettmann, T.; Wu, C.Y.; et al. Phase i study of u3-1287, a fully human anti-her3 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 3078–3087. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.S.; Harclerode, D.; Gondo, M.; Stephenson, M.; Brown, R.W.; Younes, M.; Cagle, P.T. High c-erbb-3 protein expression is associated with shorter survival in advanced non-small cell lung carcinomas. Mod. Pathol. 1997, 10, 142–148. [Google Scholar] [PubMed]
- Bieche, I.; Onody, P.; Tozlu, S.; Driouch, K.; Vidaud, M.; Lidereau, R. Prognostic value of erbb family mrna expression in breast carcinomas. Int. J. Cancer 2003, 106, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Tanner, B.; Hasenclever, D.; Stern, K.; Schormann, W.; Bezler, M.; Hermes, M.; Brulport, M.; Bauer, A.; Schiffer, I.B.; Gebhard, S.; et al. Erbb-3 predicts survival in ovarian cancer. J. Clin. Oncol. 2006, 24, 4317–4323. [Google Scholar] [CrossRef] [PubMed]
- Beji, A.; Horst, D.; Engel, J.; Kirchner, T.; Ullrich, A. Toward the prognostic significance and therapeutic potential of her3 receptor tyrosine kinase in human colon cancer. Clin. Cancer Res. 2012, 18, 956–968. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Liu, X.; Fleming, E.; Yuan, K.; Piao, H.; Chen, J.; Moustafa, Z.; Thomas, R.K.; Greulich, H.; Schinzel, A.; et al. An activated erbb3/nrg1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell 2010, 17, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Iljin, K.; Kilpinen, S.; Sara, H.; Kallioniemi, O.P.; Elenius, K. Role of erbb4 in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 259–268. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobi, N.; Seeboeck, R.; Hofmann, E.; Eger, A. ErbB Family Signalling: A Paradigm for Oncogene Addiction and Personalized Oncology. Cancers 2017, 9, 33. https://doi.org/10.3390/cancers9040033
Jacobi N, Seeboeck R, Hofmann E, Eger A. ErbB Family Signalling: A Paradigm for Oncogene Addiction and Personalized Oncology. Cancers. 2017; 9(4):33. https://doi.org/10.3390/cancers9040033
Chicago/Turabian StyleJacobi, Nico, Rita Seeboeck, Elisabeth Hofmann, and Andreas Eger. 2017. "ErbB Family Signalling: A Paradigm for Oncogene Addiction and Personalized Oncology" Cancers 9, no. 4: 33. https://doi.org/10.3390/cancers9040033