
Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
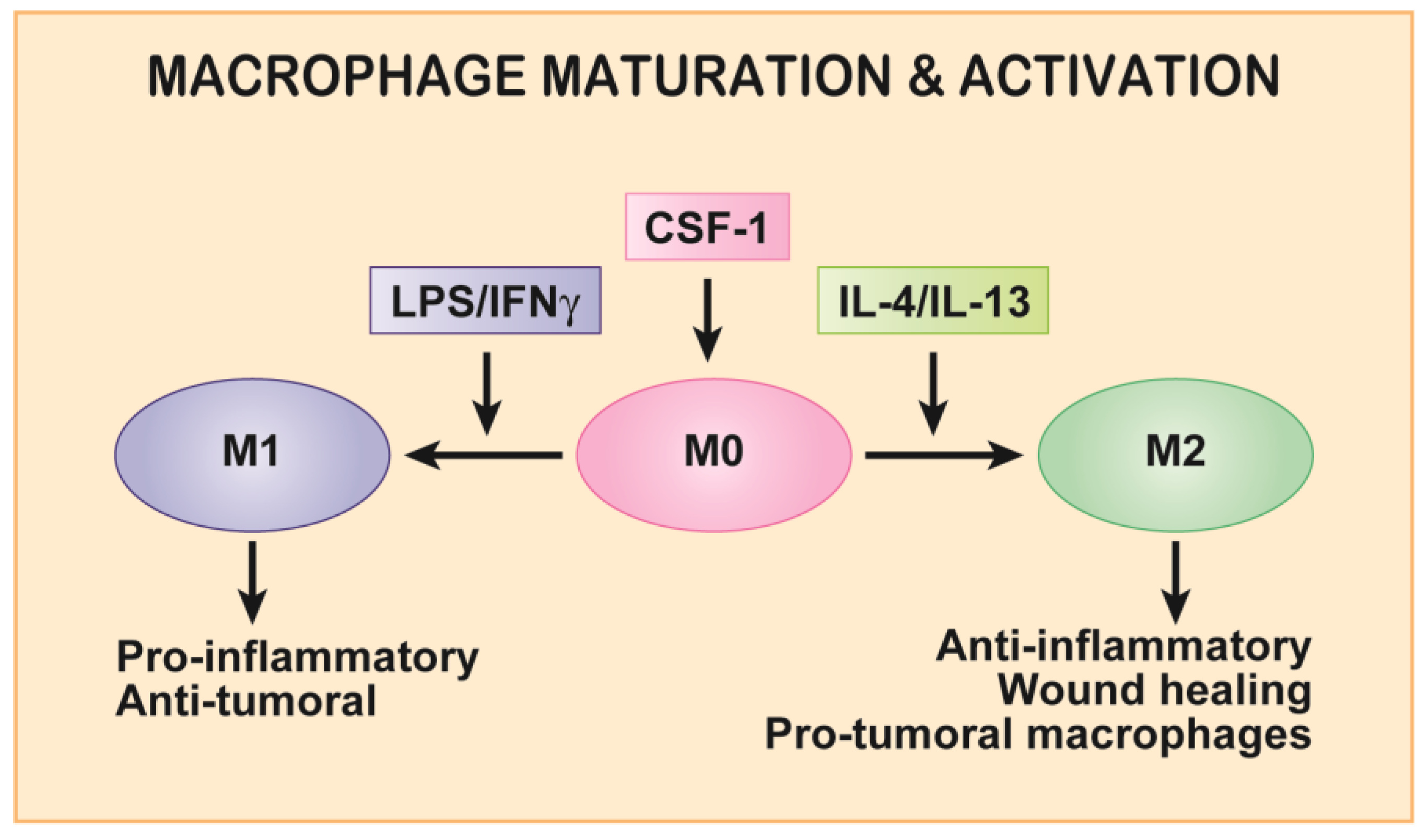
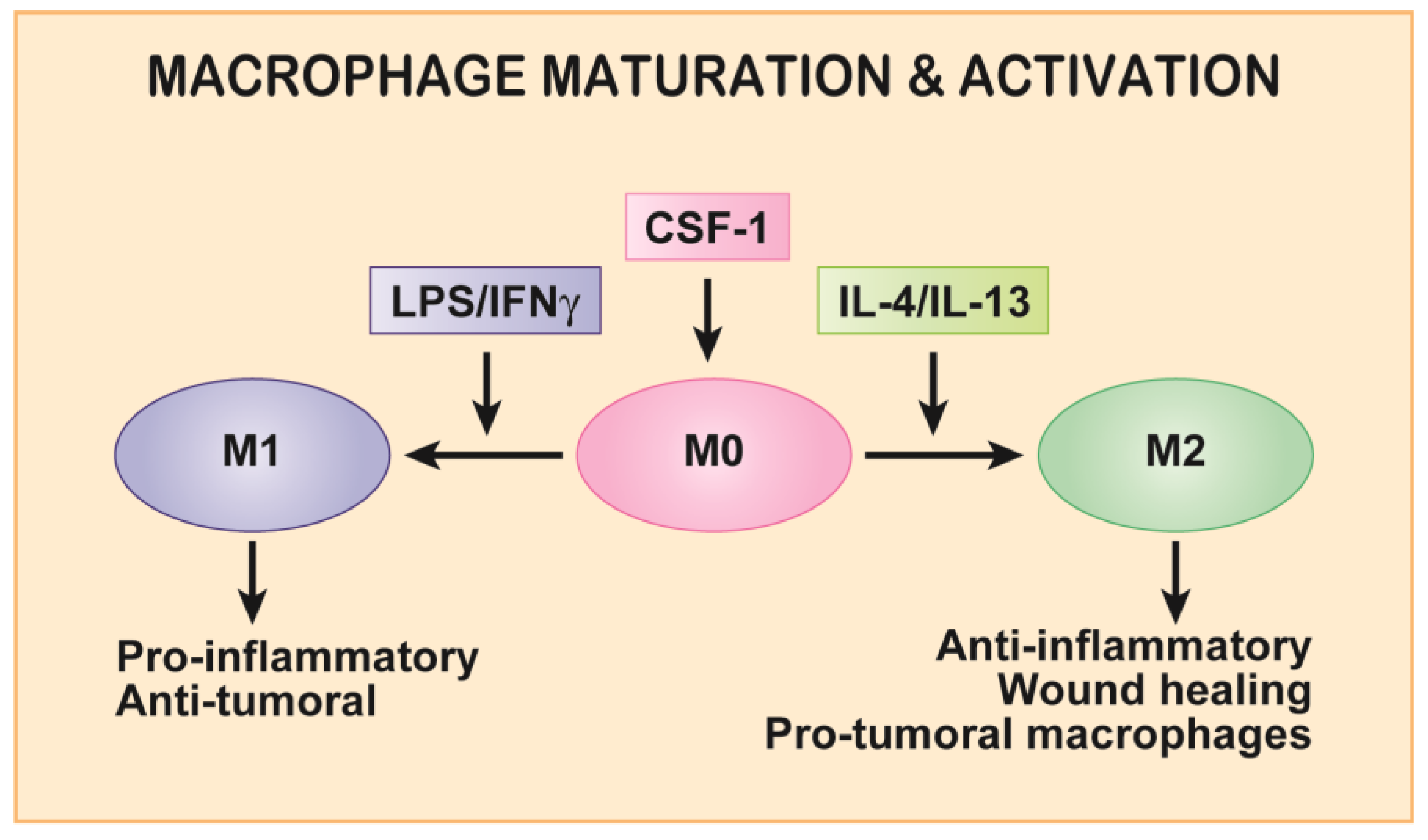
2. Macrophage Motility and CSF-1/CSF-1R Axis in Development and Homeostasis
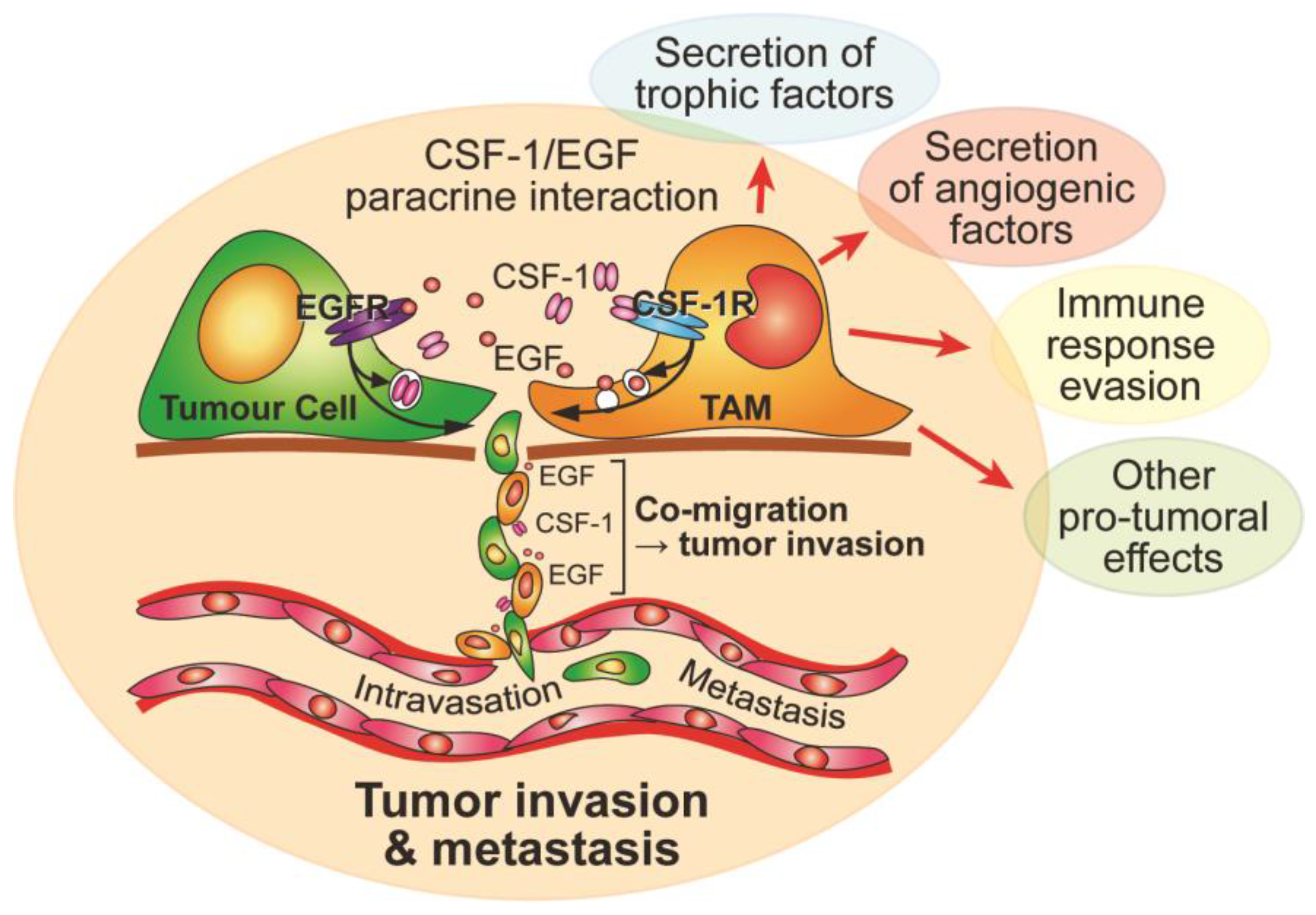
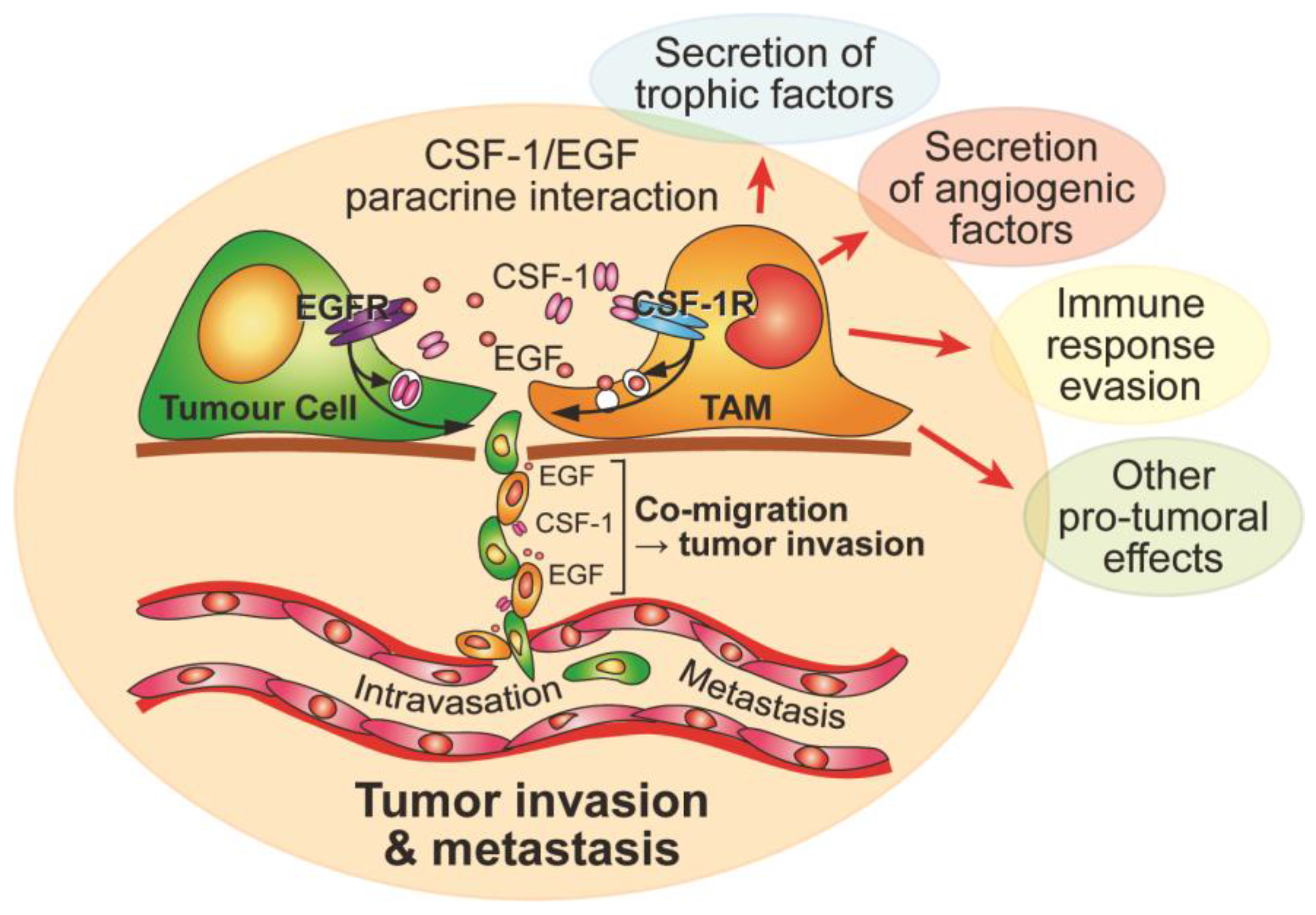
3. Macrophage Motility and CSF-1 in Cancer
4. Regulation of Macrophage Motility by CSF-1R Signaling
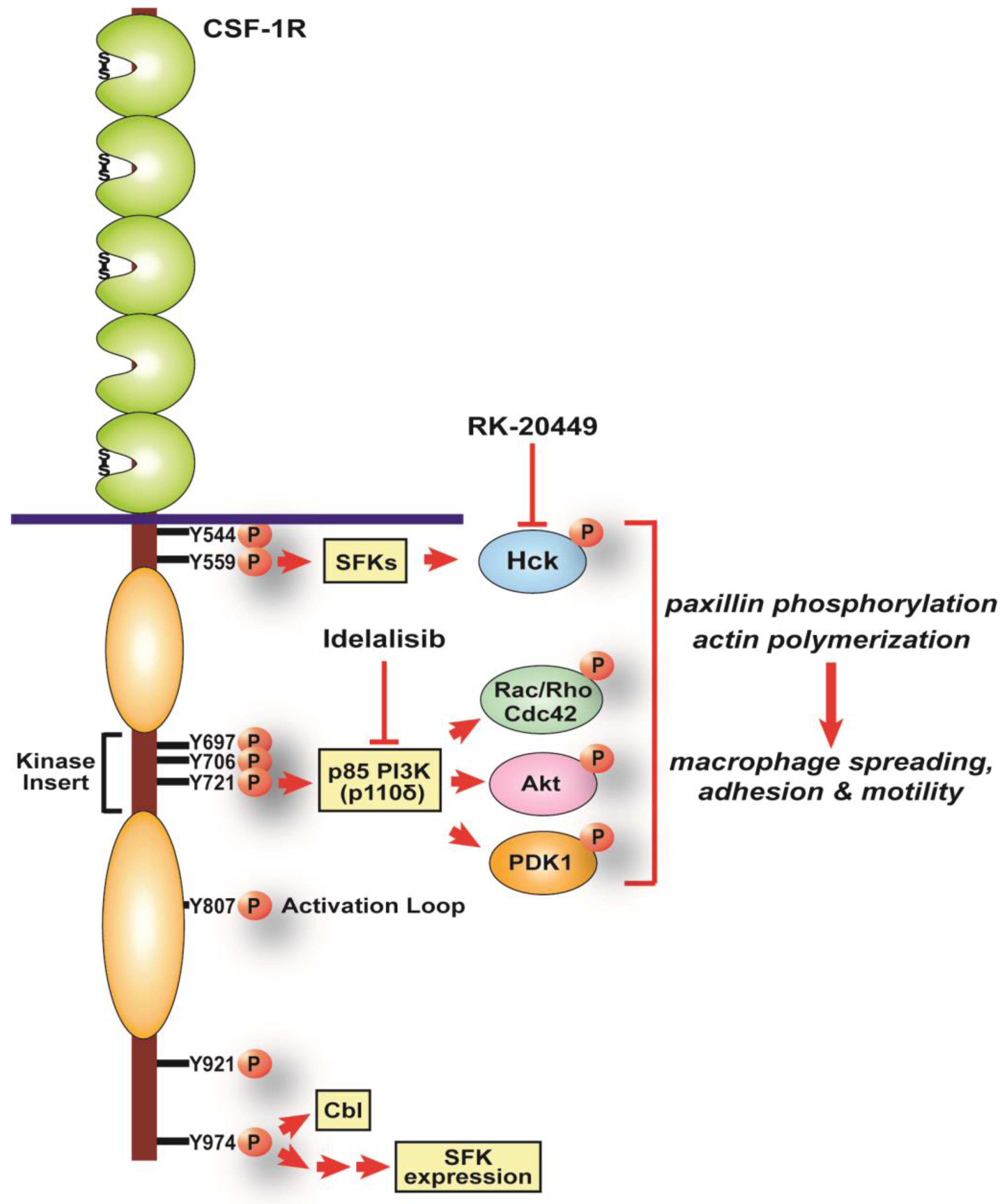
5. CSF-1R pY721, PI3K Signaling and Macrophage Motility
6. CSF-1R pY974, SFKs and Macrophage Motility
7. Inhibiting CSF-1-induced Macrophage Motility to Prevent Tumor Invasion
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2015, 17, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2015, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pixley, F.J. Macrophage migration and its regulation by CSF-1. Int. J. Cell. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ingman, W.V.; Wyckoff, J.; Gouon-Evans, V.; Condeelis, J.; Pollard, J.W. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev. Dyn. 2006, 235, 3222–3229. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.R.; Berg, K.L.; Einstein, D.B.; Lee, P.S.; Pixley, F.J.; Wang, Y.; Yeung, Y.G. Biology and action of colony-stimulating factor-1. Mol. Reprod. Dev. 1997, 46, 4–10. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Mouchemore, K.A.; Steer, J.H.; Sunderland, A.J.; Sampaio, N.G.; Greenland, E.L.; Joyce, D.A.; Pixley, F.J. Src family kinase expression and subcellular localization in macrophages: Implications for their role in CSF-1-induced macrophage migration. J. Leukoc. Biol. 2016, 100, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Griffin, J.D.; Rambaldi, A.; Chen, Z.G.; Mantovani, A. Induction of monocyte migration by recombinant macrophage colony-stimulating factor. J. Immunol. 1988, 141, 575–579. [Google Scholar] [PubMed]
- Webb, S.E.; Pollard, J.W.; Jones, G.E. Direct observation and quantification of macrophage chemoattraction to the growth factor CSF-1. J. Cell Sci. 1996, 109, 793–803. [Google Scholar] [PubMed]
- Pixley, F.J.; Lee, P.S.; Condeelis, J.S.; Stanley, E.R. Protein tyrosine phosphatase φ regulates paxillin tyrosine phosphorylation and mediates colony-stimulating factor 1-induced morphological changes in macrophages. Mol. Cell. Biol. 2001, 21, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Pixley, F.J.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trend. Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Lichanska, A.M.; Hume, D.A. Origins and functions of phagocytes in the embryo. Exp. Hematol. 2000, 46, 46–52. [Google Scholar] [CrossRef]
- Rae, F.; Woods, K.; Sasmono, T.; Campanale, N.; Taylor, D.; Ovchinnikov, D.A.; Grimmond, S.M.; Hume, D.A.; Ricardo, S.D.; Little, M.H. Characterisation and trophic functions of murine embryonic macrophages based upon the use of a Csf1r-EGFP transgene reporter. Dev. Biol. 2007, 308, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperoes for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.; Turmaine, M.; Weber, R.; Camp, V.; Maki, R.A.; McKercher, S.R.; Martin, P. Mesenchymal cells engulf and clear apoptotic footplate cells in macrophageless PU.1 null mouse embryos. Development 2000, 127, 5245–5252. [Google Scholar]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Stefater, J.A.; Ren, S.; Lang, R.A.; Duffield, J.S. Metchnikoff’s policemen: Macrophages in development, homeostasis and regeneration. Trends Mol. Med. 2011, 17, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Herbomel, P.; Thisse, B.; Thisse, C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina and epidermis through a M-CSF receptor-dependent invasive process. Dev. Biol. 2001, 238, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W.J.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2012, 44, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Stabile, C.; Taglia, I.; Battisti, C.; Bianchi, S.; Federico, A. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): Update on molecular genetics. Neurol. Sci. 2016, 37, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Tada, M.; Tada, M.; Koyama, A.; Nozaki, H.; Harigaya, Y.; Nishimiya, J.; Matsunaga, A.; Yoshikura, N.; Ishihara, K.; et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2014, 82, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Gokhan, S.; Gulinello, M.; Branch, C.A.; Patil, M.; Basu, R.; Stoddart, C.; Mehler, M.F.; Stanley, E.R. Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol. Dis. 2015, 74, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Konno, T.; Tada, M.; Tezuka, T.; Miura, T.; Mezaki, N.; Okazaki, K.; Arakawa, M.; Kyoko, I.; Toru, Y.; et al. Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids. Ann. Neurol. 2016, 80, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Parichy, D.M.; Turner, J.M. Temporal and cellular requirements for Fms signaling during zebrafish adult pigment pattern development. Development 2003, 130, 817–833. [Google Scholar] [CrossRef]
- Eom, D.S.; Parichy, D.M. A macrophage relay for long-distance signaling during post-embryonic tissue remodeling. Science 2017, 355, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Sasmono, R.T.; Oceandy, D.; Pollard, J.W.; Tong, W.; Pavli, P.; Wainwright, B.J.; Ostrowski, M.C.; Himes, S.R.; Hume, D.A. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 2003, 101, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Haldar, M.; Murphy, K.M. Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 2014, 262, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic growth factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Nandi, S.; Chitu, V.; Yeung, Y.G.; Yu, W.; Huang, M.; Williams, L.T.; Lin, H.; Stanley, E.R. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J. Leukoc. Biol. 2010, 88, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D. IL-34 is a tissue-restricted ligand for CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, Y.; Udagawa, N.; Takahashi, N. IL-34 and CSF-1: Similarities and differences. J. Bone Miner. Metab. 2013, 31, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, J.; Xiong, Y.; Pixley, F.J.; Dai, X.M.; Yeung, Y.G.; Stanley, E.R. CSF-1 receptor structure/function in MacCsf1r−/− macrophages: Regulation of proliferation, differentiation, and morphology. J. Leukoc. Biol. 2008, 84, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Mouchemore, K.A.; Pixley, F.J. CSF-1 signaling in macrophages: Pleiotrophy through phosphotyrosine-based signaling pathways. Crit. Rev. Clin. Lab. Sci. 2012, 49, 49–61. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Nat. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.; Heijnen, P.D.; Breur, M.; De Vries, H.E.; Toll, A.T.; Amor, S.; Dijkstra, C.D. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. J. Neuroinflamm. 2014, 11, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hsu, H.C.; Mountz, J.D. Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 2012, 14, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Li, M.O. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer 2016, 2, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.R.; Pixley, F.J. CSF-1R signaling in health and disease: A focus on the mammary gland. J. Mammary Gland Biol. Neoplasia 2014, 19, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [PubMed]
- Roussos, E.T.; Balsamo, M.; Alford, S.K.; Wyckoff, J.B.; Gligorijevic, B.; Wang, Y.; Pozzuto, M.; Stobezki, R.; Goswami, S.; Segall, J.E.; et al. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J. Cell Sci. 2011, 124, 2120–2131. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Harris, A.L. Tumor-associated macrophages in breast cancer. J. Mammary Gland Biol. Neoplasia 2002, 7, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-associated macrophages in breast cancer: Distinct subsets, distinct functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.M.; Pallud, C.; Beuvon, F.; Hacene, K.; Stanley, E.R.; Rohrschneider, L.; Tang, R.; Pouillart, P.; Lidereau, R. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J. Natl. Cancer Inst. 1994, 86, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Kübler, K.; Ayub, T.H.; Weber, S.K.; Zivanovic, O.; Abramian, A.; Keyver-Paik, M.D.; Mallmann, M.R.; Kaiser, C.; Serçe, N.B.; Kuhn, W.; et al. Prognostic significance of tumor-associated macrophages in endometrial adenocarcinoma. Gynecol. Oncol. 2014, 135, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Qian, Y.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Hao, W. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol. Lett. 2015, 10, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Skrzypski, M.; Dziadziuszko, R.; Jassem, E.; Szymanowska-Narloch, A.; Gulida, G.; Rzepko, R.; Biernat, W.; Taron, M.; Jelitto-Gorska, M.; Marjanski, T.; et al. Main histologic types of non-small-cell lung cancer differ in expression of prognosis-related genes. Clin. Lung Cancer 2013, 14, 666–673.e2. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Mitsunaga, S.; Kinoshita, T.; Konishi, M.; Takahashi, S.; Gotohda, N.; Kato, Y.; Aizawa, M.; Ochiai, A. Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012, 103, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Gadea, B.B.; Wang, H.W.; Gocheva, V.; Hunter, K.E.; Tang, L.H.; Joyce, J.A. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Hoves, S.; Cannarile, M.A.; Rüttinger, D. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr. Opin. Pharmacol. 2015, 23, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro. Oncol. 2016, 18, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Swierczak, A.; Cook, A.D.; Lenzo, J.C.; Restall, C.M.; Doherty, J.P.; Anderson, R.L.; Hamilton, J.A. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol. Res. 2014, 2, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C (high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, N.G.; Yu, W.; Cox, D.; Wyckoff, J.; Condeelis, J.; Stanley, E.R.; Pixley, F.J. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. J. Cell Sci. 2011, 124, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.G.; Stanley, E.R. Proteomic approaches to the analysis of early events in colony-stimulating factor-1 signal transduction. Mol. Cell. Proteom. 2003, 2, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, J.; Xiong, Y.; Pixley, F.J.; Yeung, Y.G.; Stanley, E.R. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J. Biol. Chem. 2012, 287, 13694–13704. [Google Scholar] [CrossRef] [PubMed]
- Boocock, C.A.; Jones, G.E.; Stanley, E.R.; Pollard, J.W. Colony-stimulating factor-1 induces rapid behavioural responses in the mouse macrophage cell line, BAC1.2F5. J. Cell Sci. 1989, 93, 447–456. [Google Scholar] [PubMed]
- Pixley, F.J.; Xiong, Y.; Yu, R.Y.; Sahai, E.A.; Stanley, E.R.; Ye, B.H. BCL6 suppresses RhoA activity to alter macrophage morphology and motility. J. Cell Sci. 2005, 118, 1873–1883. [Google Scholar] [CrossRef] [PubMed]
- Mouchemore, K.A.; Sampaio, N.G.; Murrey, M.W.; Stanley, E.R.; Lannutti, B.J.; Pixley, F.J. Specific inhibition of PI3K p110δ inhibits CSF-1-induced macrophage spreading and invasive capacity. FEBS J. 2013, 280, 5228–5236. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.E.; Zicha, D.; Ridley, A.J.; Jones, G.E. A role for Cdc42 in macrophage chemotaxis. J. Cell Biol. 1998, 141, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, M.; Liu, X.; van der Geer, P.; Letwin, K.; Waterfield, M.D.; Hunter, T.; Pawson, T. Tyr721 regulates specific binding of the CSF-1 receptor kinase insert to PI 3′-kinase SH2 domains: A model for SH2-mediated receptor-target interactions. EMBO J. 1992, 11, 1365–1372. [Google Scholar] [PubMed]
- Faccio, R.; Takeshita, S.; Colaianni, G.; Chappel, J.; Zallone, A.; Teitelbaum, S.L.; Ross, F.P. M-CSF regulates the cytoskeleton via recruitment of a multimeric signaling complex to c-Fms Tyr-559/697/721. J. Biol. Chem. 2007, 282, 18991–18999. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Koch, A.; Wilms, R.; Tamura, T. c-Cbl associates directly with the C-terminal tail of the receptor for macrophage colony-stimulating factor, c-Fms, and down-modulates this receptor but not the viral onco-gene v-Fms. J. Biol. Chem. 2002, 277, 14635–14640. [Google Scholar] [CrossRef] [PubMed]
- Papakonstanti, E.A.; Zwaenepoel, O.; Bilancio, A.; Burns, E.; Nock, G.E.; Houseman, B.; Shokat, K.; Ridley, A.J.; Vanhaesebroeck, B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J. Cell Sci. 2008, 121, 4124–4133. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Whitehead, M.A.; Pineiro, R. Molecules in medicine minireview: Isoforms of PI3K in biology and disease. J. Mol. Med. 2016, 94, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Anderson, K.E.; Davidson, K.; Stephens, L.R. Signalling through Class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006, 34, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Kölsch, V.; Charest, P.G.; Firtel, R.A. The regulation of cell motility and chemotaxis by phospholipid signaling. J. Cell Sci. 2008, 121, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Cammer, M.; Gevrey, J.C.; Lorenz, M.; Dovas, A.; Condeelis, J.; Cox, D. The mechanism of CSF-1-induced Wiskott-Aldrich syndrome protein activation in vivo: A role for phosphatidylinositol 3-kinase and Cdc42. J. Biol. Chem. 2009, 284, 23302–23311. [Google Scholar] [CrossRef] [PubMed]
- Hind, L.E.; Mackay, J.L.; Cox, D.; Hammer, D.A. Two-dimensional motility of a macrophage cell line on microcontact-printed fibronectin. Cytoskeleton 2014, 71, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.E.; Jones, G.E.; Pollard, J.W.; Ridley, A.J. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J. Cell Sci. 1997, 110, 707–720. [Google Scholar] [PubMed]
- Wheeler, A.P.; Wells, C.M.; Smith, S.D.; Vega, F.M.; Henderson, R.B.; Tybulewicz, V.L.; Ridley, A.J. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 2006, 119, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Cantley, L.C. Idelalisib—A PI3Kδ inhibitor for B-cell cancers. N. Eng. J. Med. 2014, 370, 1061–1062. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell. Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Lowell, C.A. The diverse functions of Src family kinases in macrophages. Front. Biosci. 2007, 13, 4426–4450. [Google Scholar] [CrossRef]
- Boyce, B.F.; Yoneda, T.; Lowe, C.; Soriano, P.; Mundy, G. Requirement of pp60c-Src expression for osteoclasts to form ruffled borders and resorb bone in mice. J. Clin. Investig. 1992, 90, 1622. [Google Scholar] [CrossRef] [PubMed]
- Klinghoffer, R.A.; Sachsenmaier, C.; Cooper, J.A.; Soriano, P. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 1999, 18, 2459–2471. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Ellies, L.G.; Holme, A.L.; Pixley, F.J. A three-dimensional co-culture system to investigate macrophage-dependent tumor cell invasion. J. Biol. Meth. 2016, 3. [Google Scholar] [CrossRef]
- Meng, F.; Lowell, C.A. A beta 1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO J. 1998, 17, 4391–4403. [Google Scholar] [CrossRef] [PubMed]
- Suen, P.W.; Ilic, D.; Caveggion, E.; Berton, G.; Damsky, C.H.; Lowell, C.A. Impaired integrin-mediated signal transduction, altered cytoskeletal structure and reduced motility in Hck/Fgr deficient macrophages. J. Cell Sci. 1999, 112, 4067–4078. [Google Scholar] [PubMed]
- Karginov, A.V.; Tsygankov, D.; Berginski, M.; Chu, P.H.; Trudeau, E.D.; Yi, J.J.; Gomez, S.; Elston, T.C.; Hahn, K.M. Dissecting motility signaling through activation of specific Src-effector complexes. Nat. Chem. Biol. 2014, 10, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Inglese, M.; Scholz, G.M.; Harder, K.W.; Clay, F.J.; Bozinovski, S.; Waring, P.; Darwiche, R.; Kay, T.; Sly, P.; et al. Constitutive activation of the Src family kinase Hck results in spontaneous pulmonary inflammation and an enhanced innate immune response. J. Exp. Med. 2002, 196, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.R.; Pixley, F.J. Unpublished observations. 2015. [Google Scholar]
- Saito, Y.; Yuki, H.; Kuratani, M.; Hashizume, Y.; Takagi, S.; Honma, T.; Tanaka, A.; Shirouzu, M.; Mikuni, J.; Handa, N.; et al. A pyrrolo-pyrimidine derivative targets human primary AML stem cells in vivo. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Love, C.G.; Masson, F.; Preaudet, A.; Tsui, C.; Whitehead, L.; Monard, S.; Khakham, Y.; Burstroem, L.; Lessene, G.; et al. Inhibition of hematopoietic cell kinase activity suppresses myeloid cell-mediated colon cancer progression. Cancer Cell 2017, 31, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Guiet, R.; Van Goethem, E.; Cougoule, C.; Balor, S.; Valette, A.; Al Saati, T.; Lowell, C.A.; Le Cabec, V.; Maridonneau-Parini, I. The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumor cells. J. Immunol. 2011, 187, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers 2017, 9, 68. https://doi.org/10.3390/cancers9060068
Dwyer AR, Greenland EL, Pixley FJ. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers. 2017; 9(6):68. https://doi.org/10.3390/cancers9060068
Chicago/Turabian StyleDwyer, Amy R., Eloise L. Greenland, and Fiona J. Pixley. 2017. "Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling" Cancers 9, no. 6: 68. https://doi.org/10.3390/cancers9060068