2. Enzyme Immobilization on QDs
QDs are luminescent semiconductor nanoscale crystals that have begun to be widely employed in both fundamental research and technical applications as a result of their unique optical and electronic properties, including size-dependent photoluminescence caused by the absorption of light generating excitons followed by electron–hole recombination. Colloidal QDs, which have a high surface area to volume ratio, can be synthesized from a range of semiconductor materials, but among the most common are CdSe, CdTe, and core/shell CdSe/ZnS and CdTe/ZnS. For the core/shell QDs, the core (e.g., CdSe) is surrounded by a shell (e.g., ZnS) that helps protect the core from oxidation and leaching, and improves the overall photoluminescence yield; thicker ZnS shells (e.g., 4–6 monolayers) give more protection whereas thinner ZnS shells (1–2 monolayers) produce higher photoluminescence. Hydrophilic ligands are then appended onto QDs to endow water solubility as well as additional stability and protection. In terms of their desirable photoluminescence properties, QDs can have a high quantum yield, high molar extinction coefficients, broad absorption windows, narrow photoluminescence windows over an overall broad range, large effective Stokes shifts, stability, resistance to photobleaching, and size-tunable photoluminescence, among other interesting properties [
86,
87,
88]. Several common applications utilizing QDs have emerged including theranostics, cellular imaging, in vitro and in vivo biosensing, and photodetection. Among the biological applications of QDs, one of the most promising is their bioconjugation to enzymes where their increase in study has been in part due to their recently-established relationship in the enhancement in activity for various enzymes, including beta-galactosidase [
40,
89], alkaline phosphatase [
41], trypsin [
90], phosphotriesterase [
39], and others.
In recent years, research groups have examined the effects that QDs have on enzymatic activity by immobilizing different enzymes on QDs—commonly CdSe/ZnS (core/shell)—with diameters generally smaller than other NP counterparts: ~3 nm to ~15 nm. The determination of diameters of QDs and NPs more generally is measured by transmission electron microscopy (TEM) and dynamic light scattering (DLS), where they are typically found to be uniform and devoid of large polydispersity due to their crystalline nature. In addition, since their spectral properties are tightly bound to their size, these properties can further enable quality control of the QDs used in experiments and in their various applications.
The most popular bioconjugation technique for immobilization involves the metal-affinity coordination of the QD with a polyhistidine tag engineered onto the enzyme, whose popularity may in part be due to the very common usage of polyhistidine tags in protein expression. This strategy also conveniently enables control over both the number and placement of enzymes on the QD surface in a site-specific manner allowing oriented enzyme immobilization, and although the enzyme movement has some degrees of freedom of movement around this site, this facilitates a largely tunable and controlled bioconjugation process [
35,
91,
92].
In order to increase the biocompatibility of the QD surface to enzymes, the QD surface is generally functionalized with a surface ligand, the most popular of which is where the QDs are solubilized with a dihydrolipoic acid-zwitterionic compact ligand (CL4) [
93,
94]. Although other coatings have been investigated in the context of QD-enzyme activity enhancement, including amine, acetyl, methoxy, hydroxy, carboxy, and polyethylene glycol-CL4 [
90], certain QD coatings such as CL4 demonstrate better solubility, high biocompatibility, maintain high quantum yields, and long-term stability across a broad pH range, each of which may be a relevant feature for consideration when investigating QD-associated impacts on enzymatic activity.
Accumulating work has demonstrated the enhancement of enzyme activity for both single-enzyme and multi-enzyme systems for cascade reactions following immobilization to QDs. In regards to single enzyme systems, recent work with phosphotriesterase (PTE) assembled to CdSe/ZnS core/shell QDs, which emitted at either 525 nm (4.3 ± 0.5 nm diameter) or 625 nm (9.2 ± 0.8 nm diameter) demonstrated that both QDs significantly enhanced immobilized enzymatic activity compared to enzyme in free solution; the
kcat increased ~4-fold while the enzymatic efficiency (
kcat/
KM) increased ~2-fold [
82]. Although the focus of this review is exclusively on the enhancement of enzymatic activity with enzymes immobilized to QDs, we note that several reports have also indicated striking improvements in enzymatic activity when the substrate is attached to QDs, particularly as proteolytic reporters [
87,
88,
90,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105,
106,
107].
In another detailed study on QD-associated enhancement of a single enzyme, Claussen et al. investigated the enhanced performance of the enzyme alkaline phosphatase (AP) immobilized on CdSe/ZnS core/shell QDs. They found that both the
Vmax and
kcat were improved with successful orientational control of AP placement relative to AP in free solution. Although the increase in these performance metrics varied in magnitude from ~5% to ~25%, both 525 nm and 625 nm QDs showed improvements at all QD:enzyme ratios examined. Interestingly, the enhancement on the smaller 525 nm QDs (4.2 ± 0.5 nm) ranged from 14% to 23%, while the larger 625 nm QDs (9.2 ± 0.8 nm) only saw improvements measuring slightly less than 10% [
41]. This study postulates that the higher surface curvature of the smaller QDs better promotes native enzyme configuration and lower enzyme-to-enzyme neighbor interactions than the larger QDs. The greatest enhancement in enzyme performance was noted when fewer enzymes were immobilized on the QDs and therefore further corroborates the aforementioned postulate. This report supports the idea that nanomaterial morphology, size, and orientation can significantly affect and improve enzymatic activity [
42].
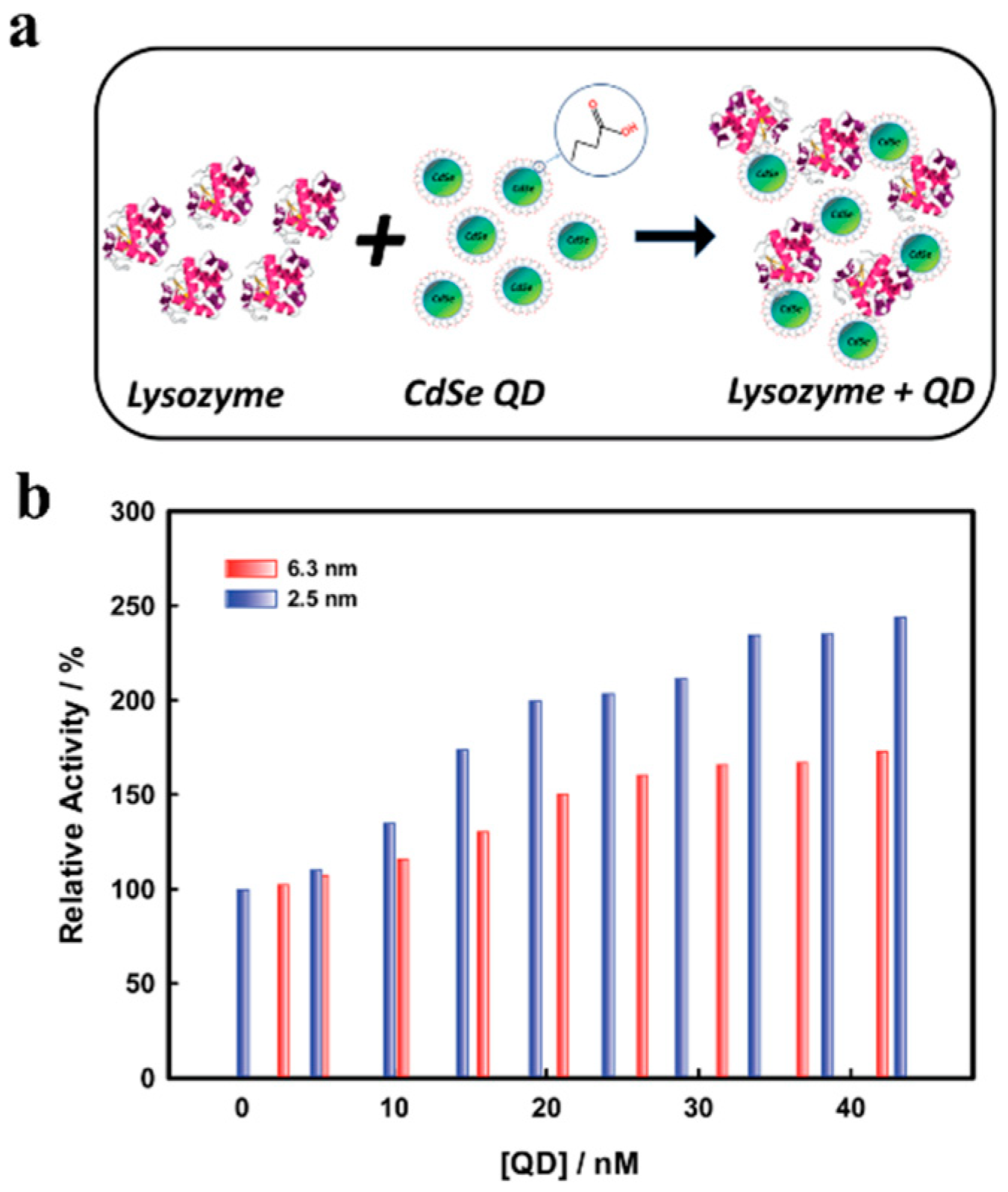
Further in-depth studies have been recently carried out by Das et al., where size-dependent CdSe QD-lysozyme interactions has been studied to establish the effect of adsorption-directed alterations to lysozyme secondary structure and the resulting impact to enzymatic activity, using a range of techniques including static and synchronous fluorescence spectroscopy to quantify QD–lysozyme binding isotherms, and circular dichroism. Lysozyme was assembled on 2.5 and 6.3 nm diameter CdSe QDs and investigated for resulting conformational changes depending on QD size and whether the QDs impacted lysozyme activity (
Figure 7a). Their results showed that conformational changes in lysozyme occurred for both QDs and that the smaller, higher curvature 2.5 nm QDs were found to promote more protein α-helical structure (
via circular dichroism) and greater enzymatic activity—here, lysis of Gram-positive bacterium
Micrococcus lysodeikticus—compared to larger QDs [
108] (
Figure 7b). Specifically, increasing the 2.5 nm QD concentration from 20 nM to 50 nM decreased the percent helix content of lysozyme from 32.89% to 24.09%. Interestingly, the researchers observed that despite the relatively poor binding of lysozyme by the smaller 2.5 nm QDs, they produced greater enhancement of enzymatic activity compared with the larger 6.3 nm QDs, which in turn exhibited stronger binding but which probably resulted in more protein denaturation across the larger NP surface. This observation is supported by previous work by Vertegel et al., showing that the activity of lysozyme on silica NPs is size-dependent, where there was a clear correlation between surface curvature of NPs and protein α-helix structure, and resulting enzymatic activity [
30]. Finally, in another publication in regards to QD:enzyme ratios, Tsai et al. showed that cellulase activity was enhanced only under certain parameters, namely smaller QDs were used at specific ratios, such as 1:5 (QD:enzyme) [
109].
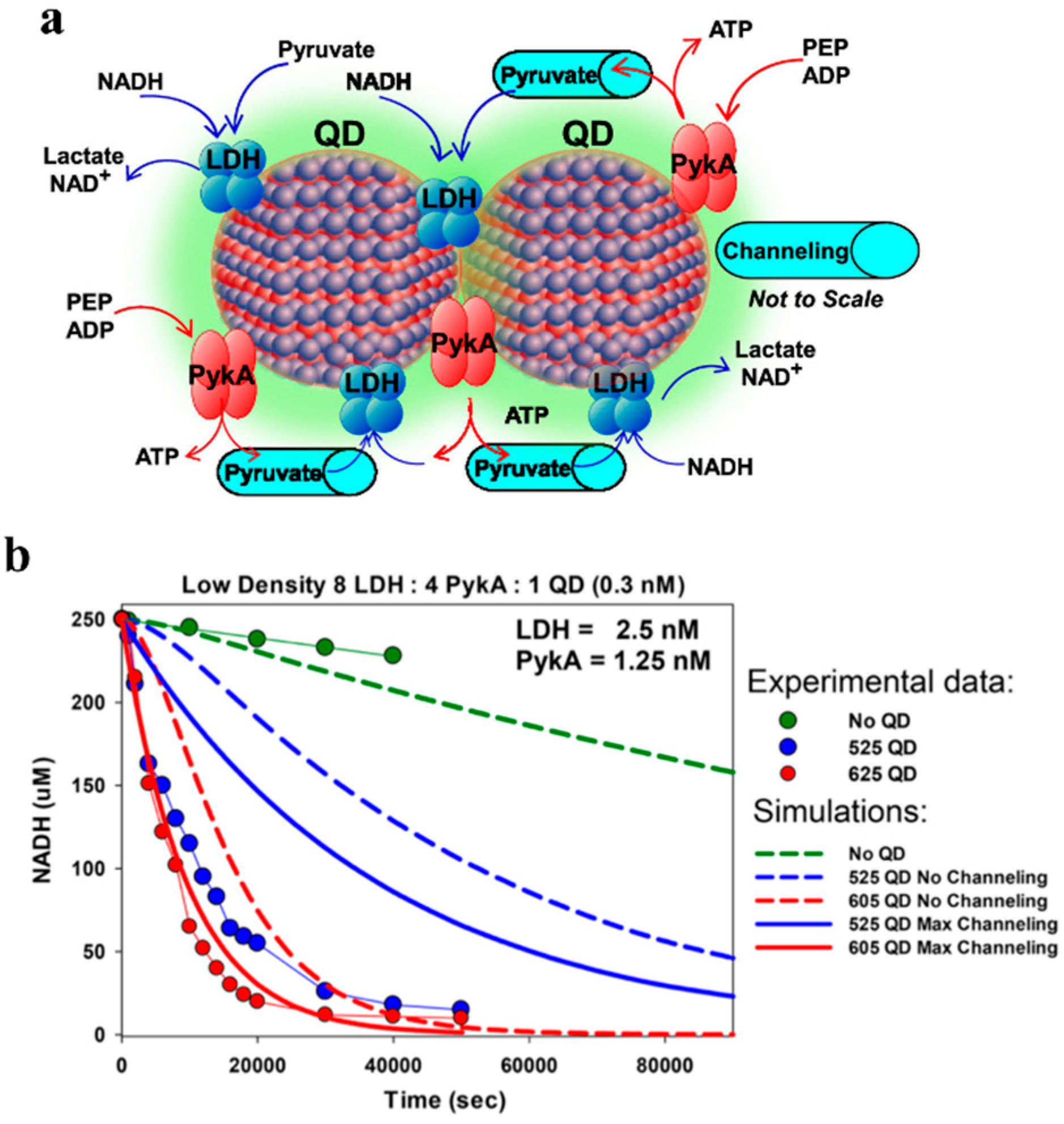
In regards to multistep enzymatic cascades, Vranish et al. recently demonstrated the value of QDs for pathway enhancement. The authors used a model glycolysis-derived dual enzyme system composed of pyruvate kinase (PykA) and lactate dehydrogenase (LDH), where the first enzyme catalyzes the reaction of adenosine diphosphate (ADP) and phosphoenol pyruvate (PEP) to pyruvate and adenosine triphosphate (ATP) and the second enzyme converts the pyruvate intermediate plus nicotinamide adenine dinucleotide (NADH) to lactate and NAD+. This coupled glycolysis system was co-localized to the surface of QDs emitting at either 525 (4.3 ± 0.6 nm diameter) or 605 nm (10.1 ± 0.1 nm × 4.5 ± 0.4, length × width) (
Figure 8). Assembled to either of the QDs, the group showed that attachment of tetrameric LDH to both the spherical 525 nm QDs and rod-like 605 QDs yielded a significant activity enhancement in turnover of as much as 50-fold. Critically, when this complex was paired with PykA on the QD surface, their coupled activity as measured by
kcat was enhanced, by over 100-fold greater than their free enzyme counterparts. The scale of this enhancement is significant compared to previously reported figures which were normally between ~2- and 5-fold (see
Table 6), possibly in part due to an increase in tetramer stability, in addition to other factors. Of note was the importance of the ratio of QD to enzyme, and the relative ratio of the PykA and LDH enzymes to one another. The best-observed specific ratio was found to be 8:4:1 (LDH/PykA/QD), as determined by NADH consumption. In their study, both the experimental data and kinetic simulations strongly indicated that the enhancement was the result of substrate channeling between the enzymes [
19] (
Figure 8b). Interestingly, unlike several other studies reporting the enhanced enzymatic activity at the interface of smaller rather than large NPs, Vranish et al. found the greatest increase in
kcat when the coupled PykA−LDH enzyme system colocalized on a QD surface of the larger, oblong 605 QDs with length × width of 10.1 ± 1.0 nm × 4.5 ± 0.4 nm rather than the spherical 525 QDs of diameter 4.3 ± 0.6 nm. However, they did observe that the lowest ratios of enzymes on QDs displayed the highest activities for the coupled system, a trend consistent with that seen by Breger et al., with the AuNP-PTE system [
76].
Another example of an in vitro cascaded reaction with QD-associated enhancement involved three enzymes found in the menaquinone biosynthetic pathway. In this study, Kang et al. reported the effect that localization of the three enzymes—MenF, MenD, and MenH—of the menaquinone biosynthetic pathway on CdSe/ZnS core/shell QDs with a diameter of ~3.5 nm had on activity. In the menaquinone biosynthetic pathway, MenF isomerizes chorismate to isochorismate, MenD (with thiamine diphosphate coenzyme) catalyzes a conjugate addition of α-ketoglutarate to isochorismate to produce 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexadiene-1-carboxylate (SEPHCHC), and MenH SEPHCHC then undergoes pyruvate elimination catalyzed by MenH to give 2-succinyl-6- hydroxy-2,4-cyclo-hexadiene-1-carboxylate (SHCHC). SHCHC can lead to menaquinone through several additional steps. They demonstrated that the efficiency of the cascade reaction was dependent on the both the total number of enzymes per particle and the relative ratio of the three enzymes per particle, similar to the results seen in the dual enzyme system reported by Vranish et al. [
19]. The reaction was observed to be more efficient when each particle contained a mixture of the three enzymes than when each particle contained only one type of enzyme and was most active when MenF was in excess over the other two enzymes—in a ratio of 20:5:5:2 (MenF/MenD/MenH/QD)—as determined by production of SHCHC from chorismate substrate (
Figure 9). An interesting point was that complexation with QDs did not affect the catalytic activity of MenF, MenD, or MenH alone. In addition, the enzymatic rate of the three-enzyme system on QDs, when immobilized on QDs at equal amounts, performed worse than the equivalent free enzyme mixture, clearly highlighting the need for enzyme:QD ratio optimization, and inter-enzyme ratio optimization if working in a multi-enzyme cascade. Altogether, this study demonstrates the enhancement via co-localization of the pathway’s enzymes and their relative inter-enzyme distances. They hypothesized that, because the enzymes were tightly packed on the QD surface, the surface itself was unlikely to have an effect on their activity—serving only as a scaffold for co-localization of the enzymes [
110]. Critically, since Kang et al. did not report enhancement of single enzymes when immobilized, their study supports a distinct mechanism of enhancement for cascades on QD scaffolds relating to favorable proximity.
3. Enzyme Immobilization on AuNPs
Similarly to QDs, AuNPs have a high surface area to volume ratio. They have been extensively-studied (see
Table 7) in part due to their relatively direct and low-cost, size-tunable synthesis from gold (III) chloride reduction and stabilization (e.g., by citrate). Applications utilizing AuNPs have included chemistry, physics, medicine, and biology. AuNPs have many inherent benefits, including that they can be (1) highly disperse; (2) biocompatible; (3) stable at small scales; (4) synthesized at specific sizes; (5) have a surface plasmon resonance (SPR) band at ~520 nm useful for allowing characterization of sizes, concentration, and binding of molecules; (6) fairly easily (bio)functionalized and stabilized through Au-S bonds, citrate, and non-covalent ionic and hydrophobic interactions; and as NPs have a relatively (7) low cost; (8) are easy to produce; and (9) are environmentally-benign [
15,
29,
51,
52,
60,
61,
63] (and refs therein). As mentioned above, enzymes can be immobilized onto AuNPs in a variety of ways. Two of the most common methods for specific conjugation is the use of Ni
2+-NTA on the surface of AuNPs to bind to His
6-tagged enzymes, and the use of free cysteines (or free thiol tags) to bind to the AuNP surface. Below, we review a few representative, recent examples of enzyme immobilization onto AuNPs, in addition to the excellent work highlighted in the above section “Factors affecting immobilizing benefits”.
Proteases are enzymes that can degrade proteins and peptides, and have been a well-studied enzyme class for immobilization onto NPs. The Lämmerhofer laboratory has completed a series of manuscripts on protease immobilization onto AuNPs, including pepsin, papain, and trypsin [
15,
60,
111,
112,
118,
119]. These could be important for a variety of applications, and have been highlighted by Lämmerhofer and colleagues for sample preparation for liquid chromatography-mass spectrometry (LC-MS) in particular [
119]. An early report focused on immobilizing trypsin onto AuNPs [
60] (trypsin has been immobilized onto AuNPs in other reports as well, such as in [
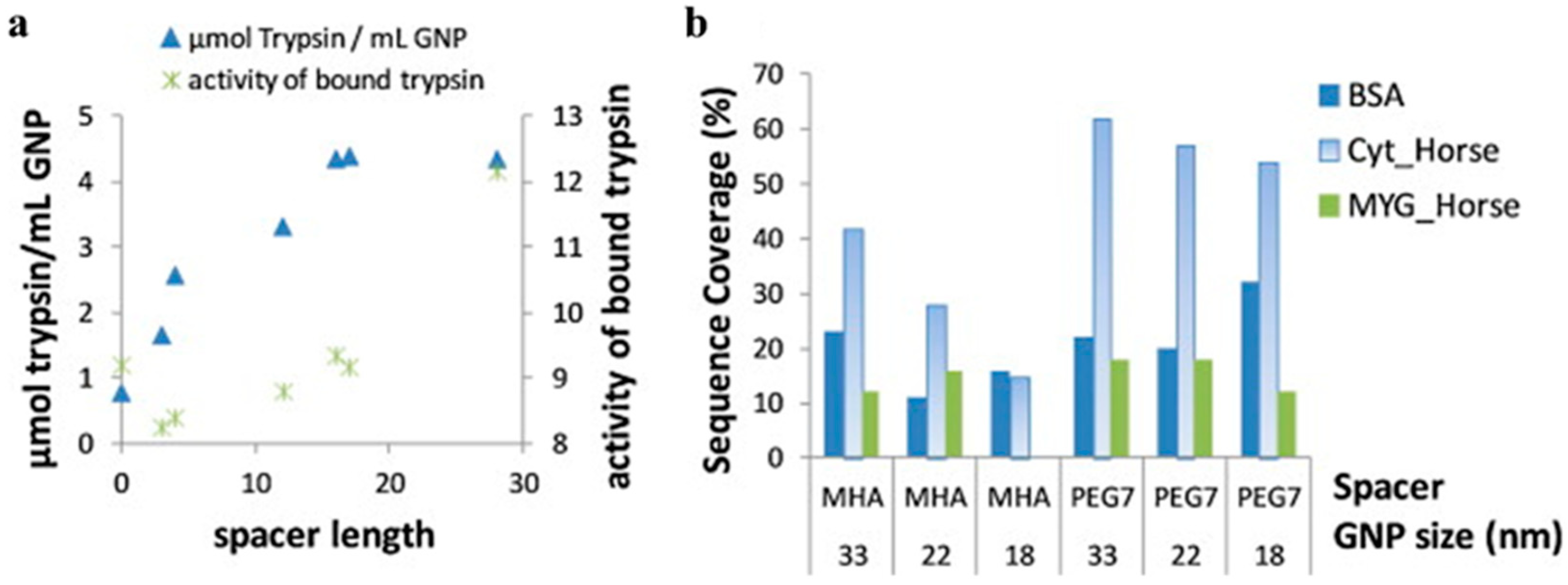
120]). Immobilization was accomplished using one of two modalities. First, trypsin was immobilized on differently-sized citrate-stabilized AuNPs (~15, 16.3, and 26.7 nm) using adsorption (ionic/hydrophobic interactions). Second, different length spacers of either mercapto-alkanoic acid or mercapto-(PEG)n-carboxylic acid were decorated onto 65 nm AuNPs, giving spacer lengths (number of atoms in final chain) of 3, 4, 12, 16, 17, or 28, in addition to “0” for the adsorption case. These spacers were chemically cross-linked to trypsin using EDC/NHS to couple trypsin amines to the carboxylic acids. The amount of trypsin immobilized onto AuNPs increased with spacer length, approximately plateauing at a spacer length of 16. Overall activity (against a peptide mimic, N
α-benzoyl-
DL-arginine-4-nitroanilide hydrochloride, BApNA) decreased from the adsorption case to the smallest-sized covalent spacer, then increased to about the same level as the adsorption case, but with much more trypsin immobilized on the AuNP, indicating lower activity per enzyme (
Figure 10a). The authors also investigated the use of trypsin-AuNP for digestion of three proteins (bovine serum albumin, BSA; cytochrome C, CYC; and myoglobin, MYG) for LC-MS, using differently sized AuNPs and different linkers (
Figure 10b). In general, increasing size increased sequence coverage, and the hydrophilic PEG linkers often increased surface coverage more, especially for CYC. Post-optimization, it was found that digestion could also be performed much faster using AuNP-trypsin, with digestion time decreasing from 19 h to ~1 h. It was also found that AuNP-trypsin was generally stable, had less auto-degradation, and could be removed by centrifugation and filtering. This study was significant in demonstrating how to optimize conditions to achieve these desirable benefits to the enzyme via immobilization.
The Lämmerhofer laboratory has published additional articles relating to proteases on AuNPs, summarized here. Pepsin was immobilized onto AuNPs coated with HS-PEG7-COOH using EDC coupling [
15]. The final AuNP-PEG7-pepsin had a greater diameter by DLS (105.3 nm) than the AuNP-PEG7 (52.1 nm) or the AuNP-citrate (31.4 nm); for comparison, AuNP-citrate-pepsin by adsorption (66.5 nm) and pepsin alone (3.9 nm) were also smaller. Pepsin on AuNP had a final concentration of 7.24 µM. For the cleavage of CYC, AuNP-pepsin compared to free pepsin had a smaller
KM (73%),
Vmax (78%), and
kcat (78%) to give an ultimately higher
Kcat/
KM (107%) as compared to unconjugated enzyme. The AuNP-pepsin could also be recycled multiple times, although score and sequence coverage decreased. Follow-up work developed an assay using amino acid analysis to quantitate the amount of pepsin adsorbed onto AuNPs [
118]. Further work investigated other methods to characterize AuNP-pepsin [
111]. Here, pepsin adsorbed onto 44.1 nm citrate-stabilized AuNP gave diameters of ~64 nm average by DLS (one outlier of 79.7 nm). Multiple methods were utilized to examine pepsin surface coverage of AuNPs. Lowry assay of the supernatant (i.e., calculating amount not bound to AuNP) gave 16,002–314,673 pepsins per AuNP depending on the amount of pepsin added. Resonance mass measurement using a microfluidic system to measure the buoyant mass of the pepsin-AuNP gave a direct measurement of surface coverage. From this measurement, it was found that: (1) a population of AuNPs were either unmodified or had low surface coverage; and (2) for a specific treatment of AuNP, there were 73,513 pepsins per AuNP on average (compared to 51,745 pepsins per AuNP by the Lowry assay for similar conditions). Taylor dispersion analysis (TDA) was used to more accurately determine size differences of the pepsin-AuNPs produced from different amounts of added pepsin. From this analysis, the diameter of the pepsin-AuNP ranged from 17.0 to 23.7 nm, and the calculated surface coverage was 47,321–128,219 pepsins per AuNP. A binding isotherm indicated that the effective dissociation constant was 30.7 ± 4.1 µM. Finally, the bioactivity was compared between free pepsin, adsorbed pepsin on AuNPs, and covalently-bound pepsin on AuNPs (amide coupling with PEG spacer as in [
15]). Relative to free pepsin,
KM and
kcat decreased for both immobilized samples, while
kcat/
KM increased (
Table 8).
Finally, the Lämmerhofer laboratory also investigated papain immobilized on AuNPs [
112]. In this case, the authors started with citrate-stabilized AuNPs and then used a layer-by-layer process to add on the polyelectrolytes poly (allylamine hydrochloride) (PAH; Mw. ~17,500; cationic) and poly (acrylic acid, sodium salt) (PAA; Mw. ~15,000; anionic) for (1) increased colloidal stability and (2) multiple anchor groups for covalently-attaching papain using EDC/sulfo-NHS amine coupling between polyelectrolyte carboxylic acids and papain amines (sulfo-NHS,
N-hydroxysulfosuccinimide sodium). The authors note that immobilization on the first-layered AuNP-PAH failed due to aggregation in phosphate buffer. DLS measurements indicated that the diameter of the citrate-AuNP (36.4 nm) increased with PAH (127.6 nm), stayed similar with addition of PAA (126.3 nm for AuNP-PAH-PAA), and then increased with increasing concentrations of papain added for immobilization (176.2–1314.7 nm). Resonant mass measurement was used to determine the surface coverage and concentration of AuNPs. For 0.2 mg mL
−1 added papain, ~23,900 bound to each AuNP; for 1 mg mL
−1 added papain (~5× increase), this jumped to ~2,060,000 (~100× increase). The authors speculated this was due to conformational arrangement and morphology. The authors also note that given the calculated surface area of the AuNP, it is likely that there is multilayer binding of the papain to the AuNPs. The peptide mimic BApNA was again used for activity measurements (vide supra). In this case, immobilization was striking in its enhancement of papain activity (
Table 9). This enhancement can save significant amounts of enzyme. As the authors note, in the preparation of papain-AuNP using 1 mg mL
−1 papain, only 21.3 µg mL
−1 is conjugated (the rest could be recycled); yet, this smaller amount of enzyme (1/0.0213 = ~47× less) still produces a higher
Vmax than free papain (143% increase,
Table 9, for a total “savings” of ~67× of enzyme. In terms of applications, the authors show that papain-AuNPs can cleave IgG (for LC-MS analysis) and that papain does not bleed off from the AuNP to interfere with analysis.
In regards to papain immobilization, Sahoo et al. and Homaei et al. examined immobilization using other gold-associated nanocomposites [
52,
113]. Sahoo et al. used magnetic Fe
3O
4 NP surrounded by linkers that ultimately chemisorbed to citrate-stabilized AuNPs (which they termed magnetic gold nanocomposites) which then had papain adsorbed [
52]. The magnetic gold nanocomposites had a diameter of 55–85 nm. Importantly, the immobilized papain could be separated with a magnet, washed, and reused up to 5 times while maintaining 70% of its initial catalytic activity. Homaei et al. used gold nanorods with adsorbed papain and measured casein hydrolysis [
113]. The immobilized papain tended to show slightly lower activity overall, but showed considerably higher activity at pH ≥ 9 and temperatures ≥70 °C. The greatest benefit was seen in stability, with the immobilized papain being considerably more stable over time (
Figure 11), and less sensitive to inhibition by selected metal ions.
In addition to proteases, other enzymes have been immobilized on AuNPs, including pyrophosphatase (PPase). PPase is an important recycling enzyme responsible for hydrolyzing pyrophosphate (PPi) into inorganic phosphate (Pi). A series of papers by the Chen and Yuan laboratories have explored immobilization of PPase on AuNPs with and without organic polymers [
114,
115,
116,
121]. In Liu et al., the authors investigated the effects of orientation and surface density on PPase activity when bound to an ~18 nm diameter AuNP (
Figure 12a) [
114]. Three types of PPase were used: wild-type (WT) which bound by adsorption; MT1, which was mutated to have a free cysteine near the active site to bind the AuNP; and MT2, which was mutated to have a free cysteine away from the active site. The authors also added different amounts of PPase to the AuNP to modulate surface density; upon protein binding the diameter increased by ~3–4 nm. In general, MT1 and MT2 has somewhat higher amounts bound to the AuNP than WT. All enzymes lost activity compared to free PPase (“relative activity”), but MT2 was the most active of the bound PPase enzymes, indicating orientation was important (
Figure 12b). Increased surface density also improved activity, but generally to a lesser degree (
Figure 12b).
Following up on this work, in another publication Liu et al. added a temperature-responsive polymer with a free thiol, poly(
N-isopropylacrylamide) or pNIPAM, to an AuNP with PPase [
115]. The goal was to modulate PPase activity dependent on temperature. When the temperature increases above a reversible lower critical solution temperature or LCST (~32 °C), pNIPAM goes from an extended hydrophilic state to a collapsed hydrophobic state (
Figure 12c). In theory, when extended, the polymer would sterically block the bound PPase, but when the temperature increased and the polymer shrunk the PPase would be exposed for activity. The size of the construct increased from 13.1 nm for the AuNP, to 15.5 with PPase (~58 PPase/AuNP, leaving free space for pNIPAM), to 24.1 nm with pNIPAM additionally added, and then decreased to 18.1 nm when the temperature was raised from 25 °C to 45 °C (AuNP-pNIPAM alone was 27.1 nm). Since the Au-S bound is unstable at >60 °C, only temperatures ≤60 °C were investigated. It was found that at 25 °C, the activity of AuNP-PPase-pNIPAM was only 46.4% of free PPase at 25 °C, but as temperature increased to 50 °C and the polymer collapsed exposing the enzyme, the activity jumped to 110% of free PPase at 50 °C, an increase of 280% for the AuNP-PPase-pNIPAM (
Figure 12d). The authors optimized the system and found that for the conditions they tried, a 10 kDa pNIPAM worked best for off/on switching, a ratio of 1.4:1 PPase: pNIPAM was best on the AuNP, and orientation using a mutated PPase with a free cysteine away from the active site worked best. Finally, the authors also showed that a benefit of the polymer was protection from proteolysis: upon treatment with trypsin for 90 min, the relative specific activity for free PPase dropped by ~50% and 70% at 25 °C and 45 °C, respectively, while AuNP-PPase-pNIPAM activity dropped by only ~25 and 30% respectively.
Building on this work, two other polymers were used instead to give AuNP-PPase pH responsiveness [
116] (
Figure 12e). To a 14.2 nm diameter AuNP was added PPase to give the 18.4 nm diameter AuNP-PPase. To this was added the polymer poly (methacrylic acid) or PMAA with a free thiol to bind to the AuNP for a diameter of 35.1 nm. This polymer has carboxylic acid groups and therefore at pH 7–9 is mostly negatively charged. To this was added the polymer poly(2-(dimethylamino)ethyl methacrylate or PDMAEMA. This polymer has a p
Ka between 7.5 and 8, so that at pH 7 it is positively charged, binds to the negatively-charged AuNP-PPase-PMAA, decreasing surface charge of the complex leading to instability and aggregates with a diameter of 560 nM. Conversely, at pH 9 PDMAEMA is near neutral and can dissociate, allowing AuNP-PPase-PMAA to disperse (diameter of 41.7 nm). This leads to a drop of relative activity at pH 7 of 16.9% (“off”) and an increase to 97.9% at pH 9 (“on”) (
Figure 12f). The authors found that from their conditions tested, a PMAA of 19.9 kDa and a feed ratio of 150:1 for PPase:AuNP with a 14 nm diameter was best for modulating activity. Importantly, this system could be cycled multiple times,
albeit with a slight decrease in overall activity with each cycle perhaps due to irreversible aggregation of a small amount of AuNPs. The system was also able to protect PPase from trypsin digestion: after 90 min, the relative activity was ~40% for free PPase, ~50% for AuNP-PPase, ~60% for AuNP-PPase-PMAA, and 86.4% for AuNP-PPase-PMAA/PDMAEMA, or ~2× that of free PPase (pH 7 for 90 min of trypsin treatment then pH 9 for activity reading). Finally, Li et al. extended this system to a surface for pH responsive capture and release [
121]. PDMAEMA was bound to AuNP layers, and was used to bind AuNP-PPase-PMAA at pH 7 and release it at pH 10, and this could be cycled on and off. Horseradish peroxidase (HRP) bound to AuNP was also used, to demonstrate that (1) AuNP-PPase-PMAA could be bound then released from the surface, followed by binding of AuNP-HRP-PMAA and release, and (2) AuNP-PPase-PMAA and AuNP-HRP-PMAA could be bound to the surface simultaneously.
In addition to the above referenced papers, a few more examples are worth mentioning. In Mohammadi et al., the enzyme inulinase, which hydrolyzes fructose-containing polymers and inulin, was investigated [
51]. Magnetic iron NPs were made, and mixed with gold to produce Fe
3O
4-Au magnetic NPs, which were capped with glutathione (18.71 nm diameter). Glutaraldehyde was then used to immobilize inulinase onto the NPs (22.95 nm diameter). Enzyme activity after immobilization was 83%. The immobilized inulinase had a higher
KM (6.8 vs. 5.4 mg mL
−1), a lower
Vmax (3.03 vs. 3.55 µmol min
−1 mL
−1), a lower
kcat (841.67 vs. 986.11 min
−1), and a lower
kcat/
KM (123.77 vs. 182.61 min
−1 mg
−1 mL). However, the immobilized inulinase had higher temperature stability, keeping >80% of the relative activity vs. <40% for free inulinase at 80 °C. Importantly, the authors state that the food industry prefers reactions at higher temperature because of increased inulin solubility, lower microbial contamination, and better rates. Both free and immobilized inulinase kept ca. the same relative activity over 8 h at 50 °C and 60 °C. Importantly, the immobilized inulinase could be recycled after separation from the reaction mixture by a magnetic field up to 10 times while maintaining >70% relative activity. Finally, the stability of immobilized inulinase at 28 °C after 45 days (73% remaining relative activity) was higher than free inulinase (31% remaining relative activity).
Shikha et al. also investigated the heat stability of an immobilized enzyme, in this case lipase [
63]. AuNP was capped with cysteamine (25 nm diameter) and linked to lipase using EDC/NHS. The immobilized lipase had a lower apparent
KM (2.76 vs. 6.70 mM), a lower
Vmax (3.71 vs. 4.98 U µg
−1 protein), a lower
kcat (2059.05 vs. 2763.30 s
−1), and a greater
kcat/
KM, app (744.95 vs. 412.38 s
−1 mM
−1). While enzyme activity of free lipase over the range of 20–80 °C was generally higher than or similar to immobilized lipase, at 60 °C the activity of the immobilized lipase (65% of maximum) was significantly higher than free lipase (~20% maximum activity). This was also shown through a temperature sensitivity assay over 10 h, where the immobilized lipase retained more activity than the free lipase at 40, 50, and 60 °C; after 4h, the immobilized lipase still had >50% relative activity at 40 and 50 °C whereas free lipase relative activity was ~30%.
In an interesting application, Ball et al. used a nitroreductase (NfnB) with an engineered histidine and cysteine tag (NfnB-cys) immobilized on an AuNP to activate the prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) for potential application in Directed Enzyme Prodrug Therapy (DEPT) for cancer treatment [
117]. The prodrug has two nitro groups at the 2 and 4 positions that can be reduced to NHOH. The authors found different 2-NHOH:4-NHOH product ratios for free NfnB-his tag (44:56), free NfnB-cys (32:68), and the immobilized NfnB-cys (13:87). At least for the free NfnB-his tag and free NfnB-cys, this was due to differences in rates of further non-enzymatic reduction to 2-NH2:4-NH2 and the rate of the enzymes before HPLC analysis. In terms of activity, the immobilized NfnB-cys vs. the free NfnB-cys had a lower
KM (1108.67 vs. 5078.37 µM), a lower
Vmax (10.89 vs. 19.37 µM s
−1), a higher
kcat (61.85 vs. 55.34 s
−1) and a higher
kcat/
KM (0.0558 vs. 0.0109 µM
−1 s
−1).
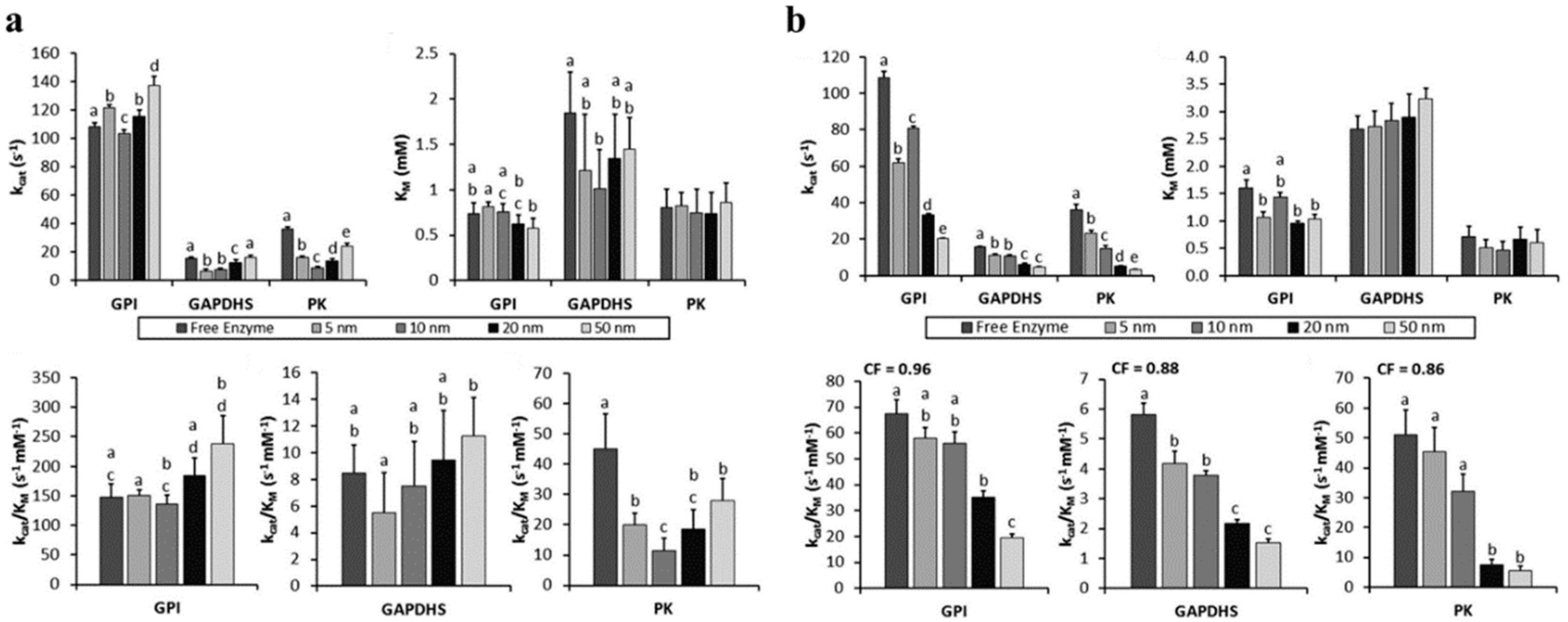
Finally, in another example of an enzyme conjugated onto AuNPs, Hondred et al. examined a trimer of PTE conjugated to 5, 10, and 20 nm AuNPs [
61]. The PTE trimer was constructed by each monomer containing PTE, a collagen-like triple helix domain, a trimerization domain, and a His
6 tag, and being allowed to trimerize [
82]. The His
6-tags could then bind a DHLA-Ni
2+-NTA coated AuNP. Conjugation onto the 20 nm AuNP resulted in the highest average turnover increase over free PTE trimer (~ 1.7× over 10 nm AuNPs; ~1.1× over 5 nm AuNPs) when holding the concentration of AuNPs constant and varying the amount of PTE trimer displayed. Relative enzyme efficiency (
kcat/
KM) over free PTE trimer was highest with the 10 nm AuNP in the fixed AuNP concentration method (~ 3.0×), but was highest with the 5 nm AuNP for a fixed PTE trimer concentration method (5.0×). Overall, the
Vmax was increased by ~17× using 20 nm AuNP at low PTE trimer concentration (5 pM) and at a low enzyme:scaffold ratio (1:1).
4. Discussion—Conclusions, Outlook, and Perspective
Enzymes are active accelerators of many biochemical processes and are widely used to catalyze reactions in a wide array of biological and industrial applications. NPs have been shown to enhance the effectiveness of immobilized enzymes, which has driven research interest in NP–enzyme systems. Although not all NP–enzyme conjugates have demonstrated an increase in enzymatic activity, many have, as described above. Among NPs, QDs and AuNPs are of particular popularity, potentially due to many of their shared properties owing to their being nanocrystalline, predominantly monodispersed, with certain favorable properties, including size-tunable photoluminescence and the capacity to act as excellent FRET donors and acceptors, in the case of QDs, and surface plasmon resonance for AuNPs [
90].
As can be seen with several representative examples presented in this review, while NP-associated enhancement is still not entirely predictable and additional research is required, the existing literature can provide a solid basis for a grasp of how enzymatic activity is enhanced in NP systems and for NP–enzyme system design. Here, in every case we covered, the NP acts as the central nano-scale scaffold for displaying the enzyme, which in turn reflects that NP quality, surface functionalization approaches for aqueous dispersal, and the chemistries to attach the enzymes and/or substrates to the NP are critically important for enzymatic activity. Especially since QDs and AuNPs are metallic in nature, they display no inherent solubility in water and must thus be stabilized as colloids through the addition of surface ligands to their surface, which can greatly impact enzymatic activity.
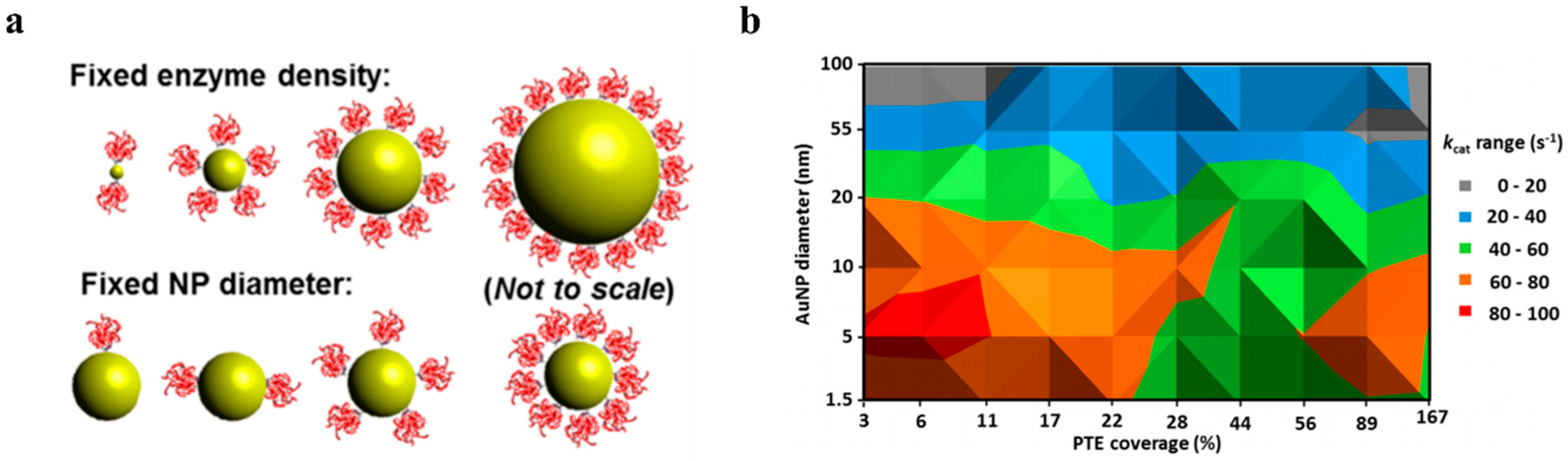
Other NP-related aspects such as NP size and enzyme:NP ratio were closely examined and discussed in detail. Investigation of each NP’s size, curvature, and morphology offer a better understanding of the enhancement of activity of enzymes immobilized on NPs and can be further investigated to optimize the design of new NP–enzyme systems for various biological and industrial applications. Furthermore, enzyme:NP ratio has a significant impact on enzymatic activity—either positive or negative. Recent work and an initial meta-analysis of available work with reported kinetic parameters suggests the hypothesis of a “sweet-spot” consisting of both a small diameter NP with fairly low enzyme coverage, consistent with low enzyme: NP ratios [
19,
22,
39,
81,
82]. Possible explanations are that this could be due to a relief of ligand crowding with an increase in curvature (and decrease in diameter) as a result of released steric hindrance or an increase in product solubility in the NP hydration layer which could also help with product partitioning or release. Nevertheless, with greater accumulation of work reporting the impact of NP attributes on kinetic parameters of enzymes they are immobilized to will significantly improve the confidence in the “sweet spot” hypothesis and other related trends which may allow for the better utilization of modeling or generation of design rules for NP–enzyme systems for optimal performance. In addition, future work will likely assist in determining whether NP immobilization of enzymes is cost-effective.
One can expand this into a cost–benefit analysis by determining the relative complete turnover rates for each sample immobilized vs. free in solution (
Table 4 and
Table 5, last two columns, as well as
Appendix A Table A3). Does it make sense to use AuNP immobilization? One can determine this from the last column ((Extra) PTE per AuNP); as long as the cost ratio of the NP to PTE exceeds this value, it is more economical to use immobilization. For illustration, using the 32:1 PTE:AuNP conjugate data from
Table 4, if a mole of PTE costs
$1, as long as a mole of NP costs <
$111.8, it is more economical to immobilize. From
Table 5, one can see that the cost ratio is even more beneficial for larger AuNPs at low coverage (i.e., the 100 nm AuNP can cost up to 2103.5× the cost of PTE, mol/mol). Which size AuNP and what percentage coverage makes the most sense to use will depend on the relative costs. For example, using the data from
Table 4, 5.9 of the most efficient 4:1 PTE:AuNP conjugates (=23.6 PTE total) gives the same approximate overall turnovers (5.9 × 4 × 79.2 = 1869 turnovers) as 1 of the 32:1 PTE:AuNP conjugates (=32 PTE total; 32 × 58.4 = 1869 turnovers), gaining ~8.4 “extra” PTEs at the cost of 4.9 more AuNPs. Dividing this out, if a mole of AuNPs costs <1.72× the cost of a mole of PTE, then low coverage (4:1) is better; otherwise, higher coverage (32:1) is better. As another example, using the data from
Table 5, 45.5 of the most efficient 10 nm PTE:AuNP conjugates (=273.1 PTE total) gives the same approximate overall turnovers (45.5 × 6 × 81.2 = 22,168 turnovers) as 1 of the 100 nm PTE:AuNP conjugates (=601 PTE total; 601 × 36.9 = 22,177), gaining ~327.9 “extra” PTEs at the cost of 44.5 more AuNPs. Dividing this out, if a mole of 10 nm AuNPs costs <7.37× the cost of a mole of PTE, then the smaller conjugate (10 nm) is better; otherwise, the larger conjugate (100 nm) is better (this is before considering that the 100 nm conjugate likely costs more than the 10 nm conjugate). Of course, the degree of benefit to enzymes will change depending on enzyme; this example illustrates the importance of trying conjugates at different enzyme:scaffold ratios (this becomes even more important with multienzyme cascades and investigating the ratios between enzymes as well as between each enzyme and the scaffold). Beyond the interest from the perspective of fundamental science, the reviewed literature suggests significantly less enzyme could be used to accomplish the same levels of activity and/or product production, when displayed on the surface of a NP, which may greatly inspire industrial applications and for usage in other applications, including cell-free synthetic biology systems, where a modular scaffold for any cascaded enzymatic system could greatly improve the state of cell-free synthesis.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











