Identification of a Reaction Intermediate and Mechanism of Action of Intermediary Enzymes in Plumbagin Biosynthetic Pathway Using Molecular Dynamics Simulation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Hypothetical Enzyme Reaction for Biosynthesis of Plumbagin
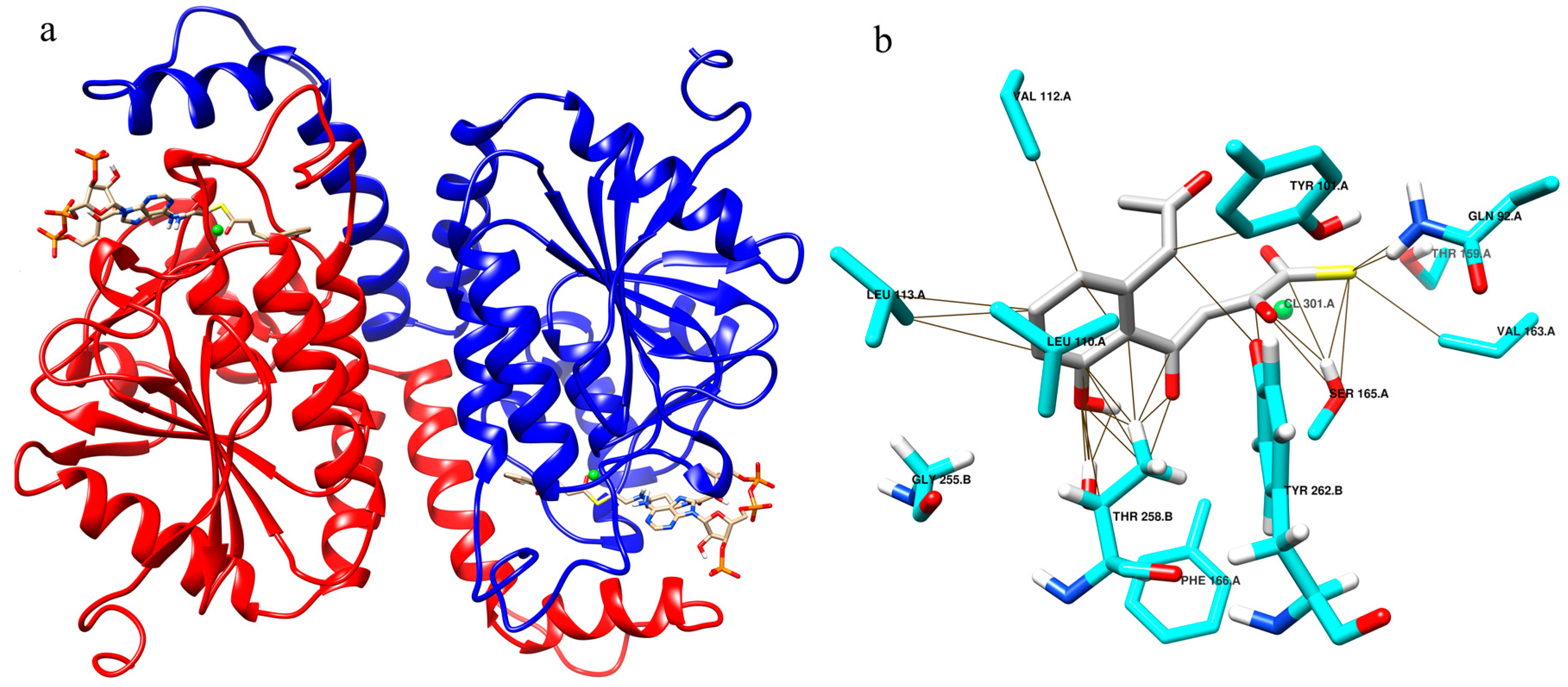
2.2. Enzyme Substrate Interaction of NS–OMB CoA
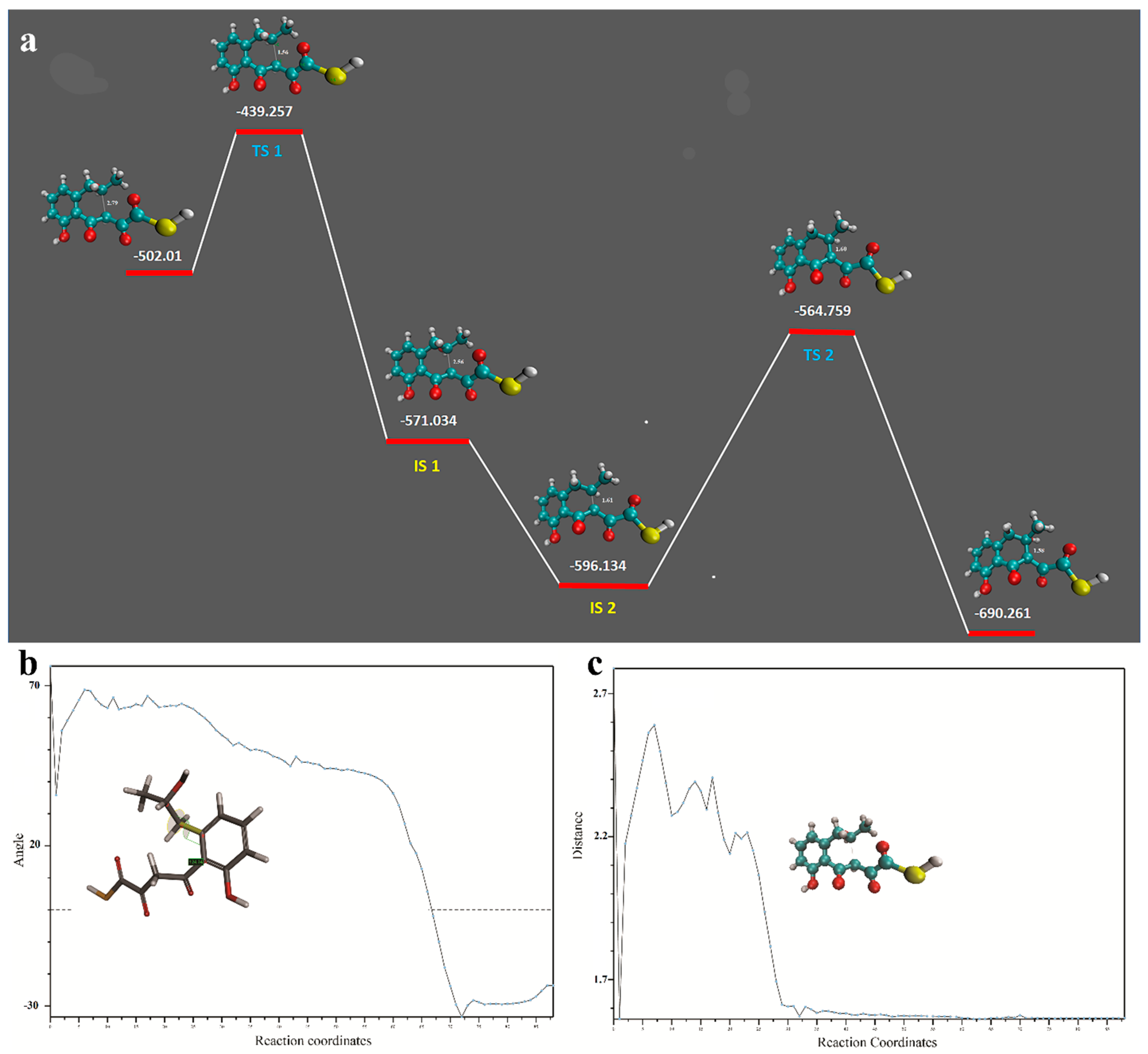
2.3. Transition and Intermediate Sates of the Intermediary Compound
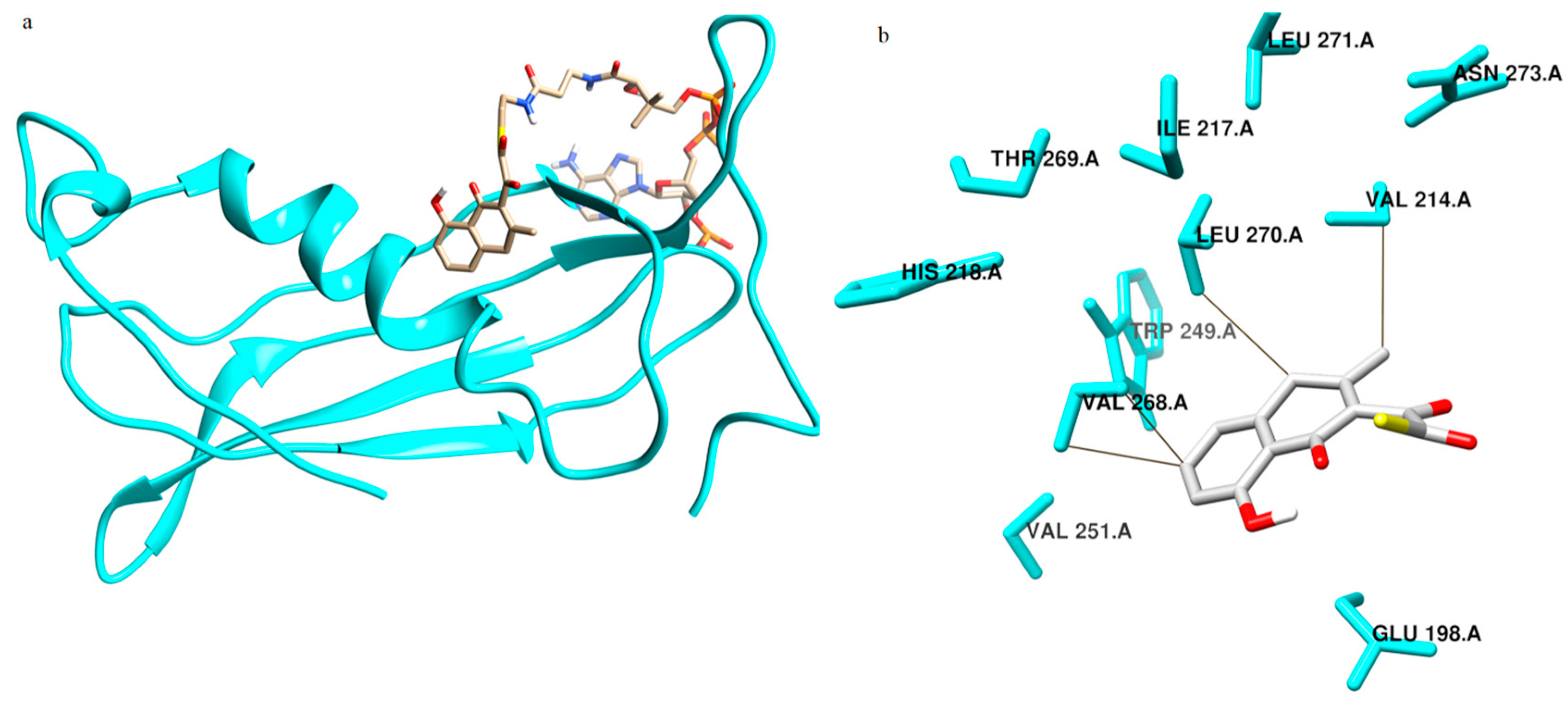
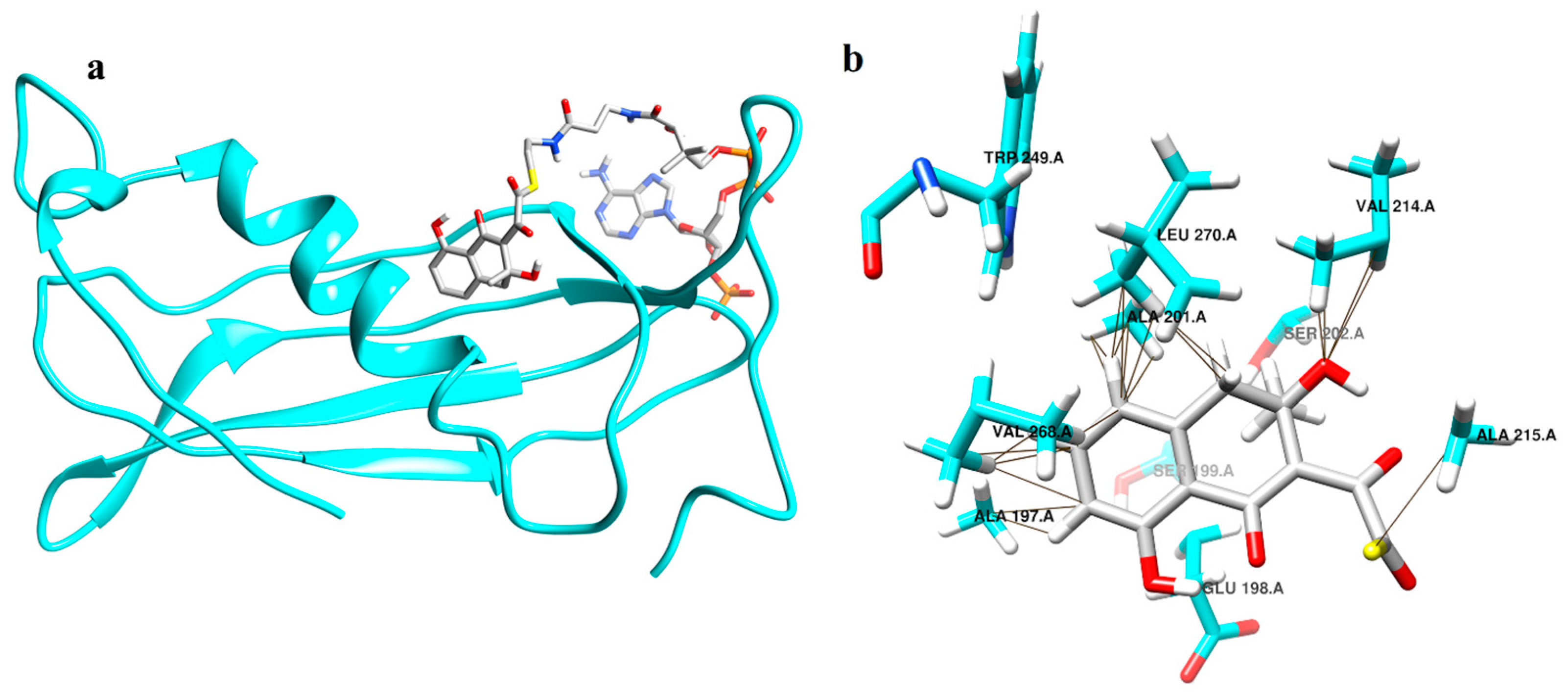
2.4. Enzyme Substrate Interaction Studies of Intermediate Structure 1 and 2 with Thioesterase
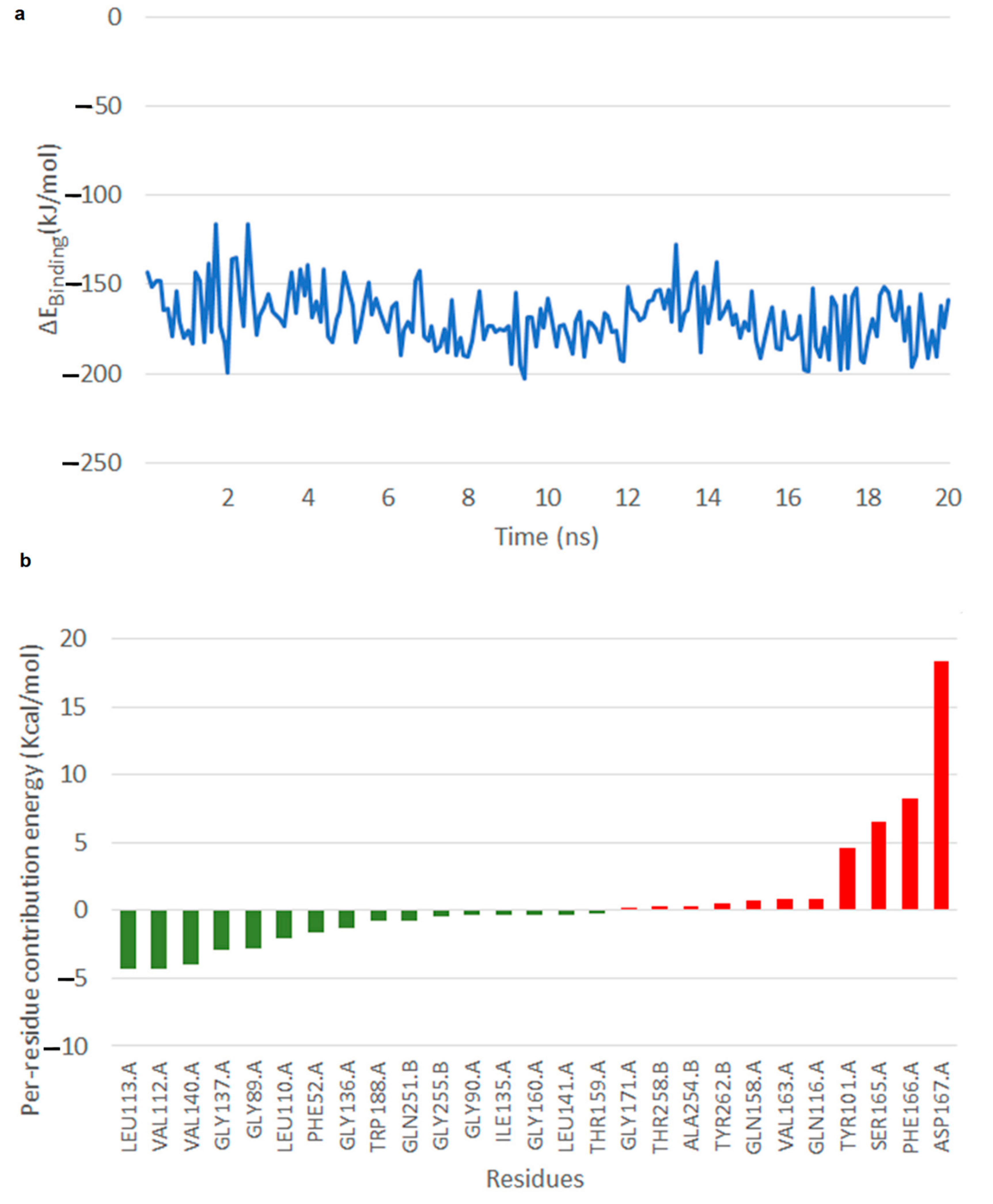
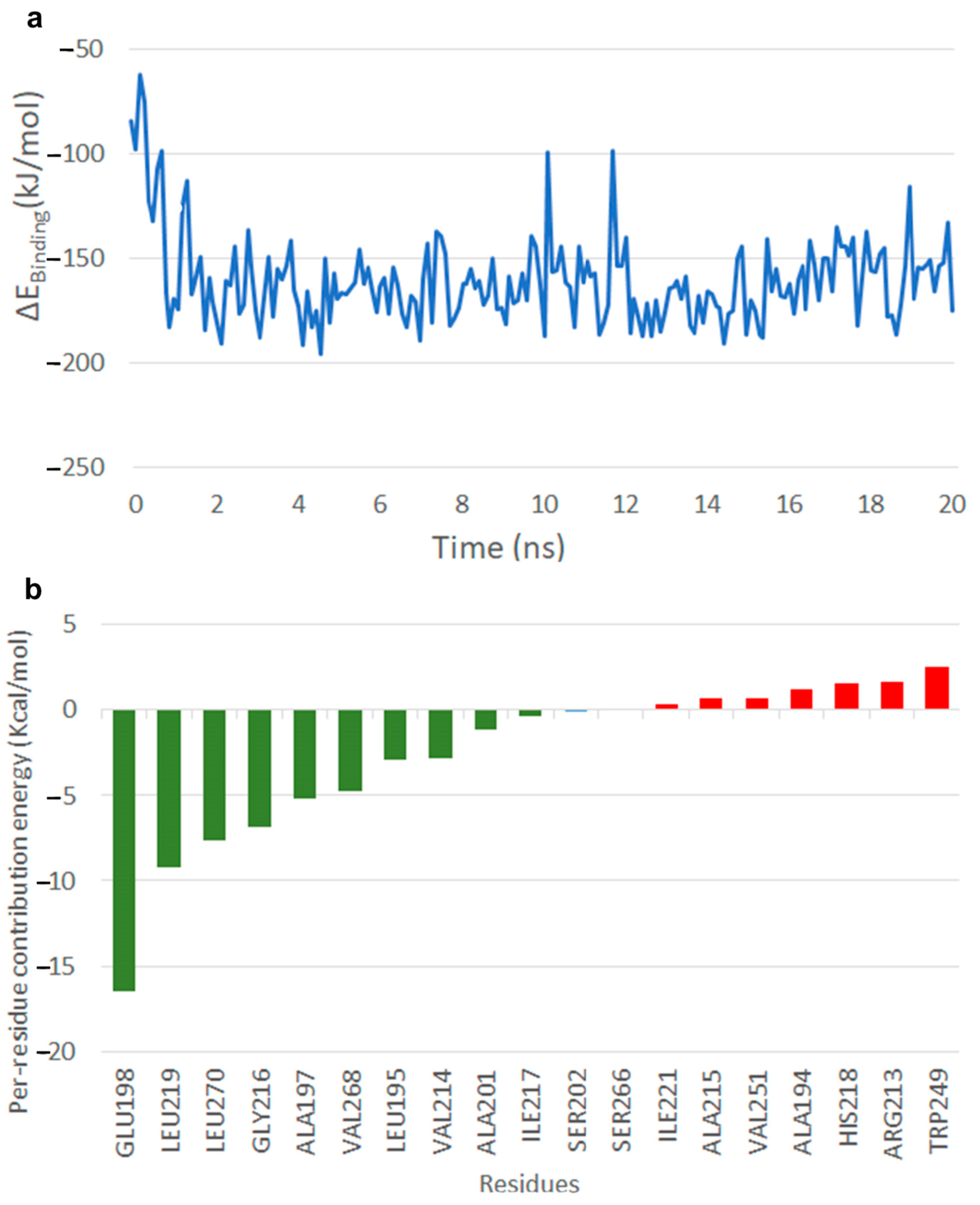
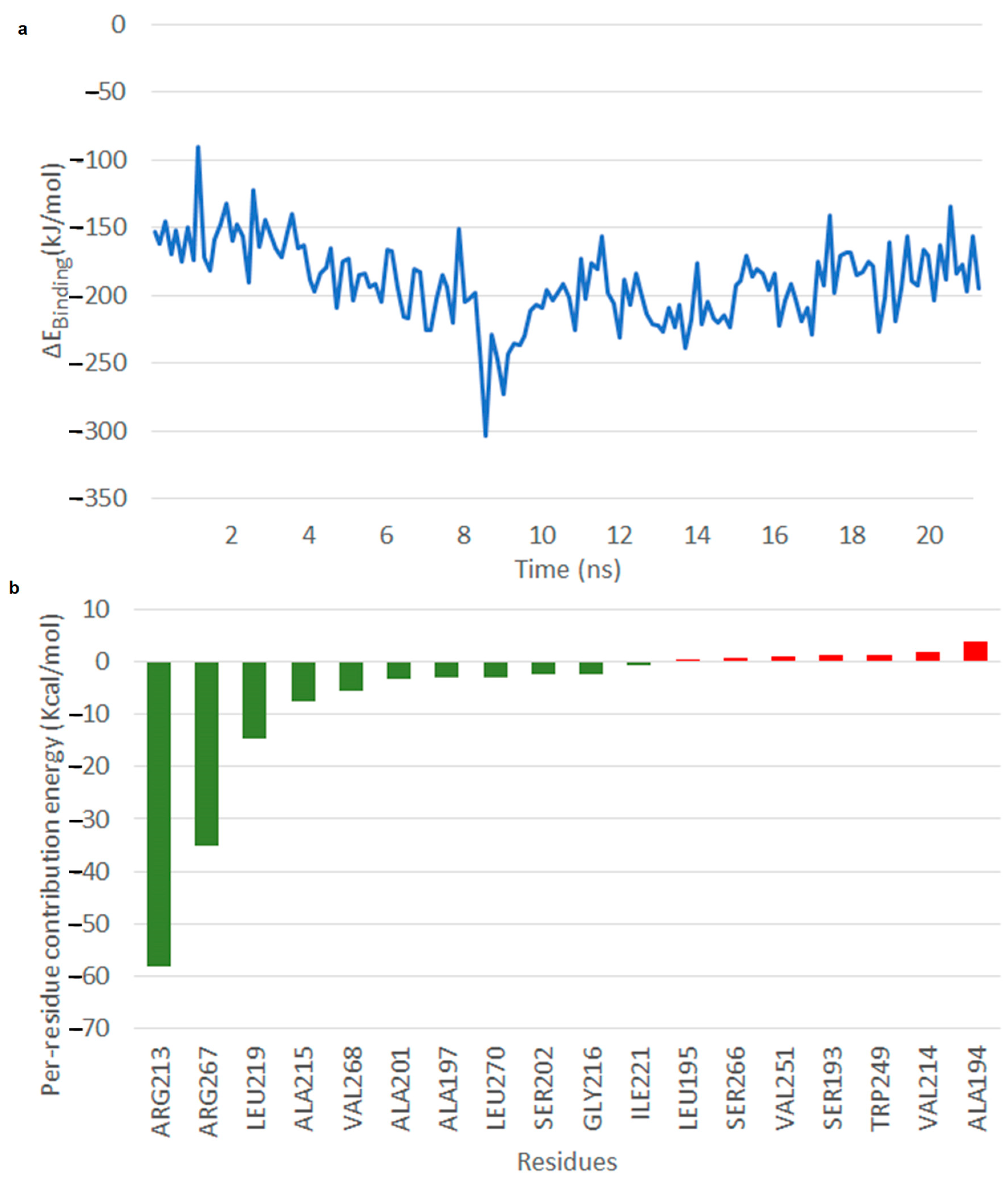
2.5. Results of Molecular Dynamics Simulation
2.5.1. Free Energy of Binding of OMB with Naphthoate Synthase after Simulations Reveals Stable Interactions
2.5.2. Simulations Reveal That Intermediate Structure 2 is More Stable in Its Interaction with Thioesterase
3. Materials and Methods
3.1. Molecular Modeling and Docking
3.2. Molecular Dynamics Simulations and Quantum Mechanics Optimization
3.3. Calculation of Free Energy of Binding Using the MM/PBSA Method
3.4. Prediction of Intermediates by Quantum Mechanics Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aziz, M.H.; Dreckschmidt, N.E.; Verma, A.K. Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res. 2008, 68, 9024–9032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichihara, A.; Ubukata, M.; Sakamura, S. Synthesis of Plumbagin by the Retro-Diels-Alder Reaction. Agric. Biol. Chem. 1980, 44, 211–213. [Google Scholar] [CrossRef]
- Mbaveng, A.T.; Kuete, V. Review of the Chemistry and Pharmacology of 7-Methyljugulone. Afr. Health Sci. 2014, 14, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, R.M.; Utturkar, S.M.; Crook, J.W.; Thimmapuram, J.; Widhalm, J.R. The origin and biosynthesis of the naphthalenoid moiety of juglone in black walnut. Hortic. Res. 2008, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.J.; Li, X.K.; Liu, N.N.; Zhang, H.N.; Truglio, J.J.; Mishra, S.; Kisker, C.; Garcia-Diaz, M.; Tonge, P.J. Mechanism of the Intramolecular Claisen Condensation Reaction Catalyzed by MenB, a Crotonase Superfamily Member. Biochemistry 2011, 50, 9532–9544. [Google Scholar] [CrossRef] [Green Version]
- Tzin, V.; Galili, G. The Biosynthetic Pathways for Shikimate and Aromatic Amino Acids in Arabidopsis thaliana. Arabidopsis Book/Am. Soc. Plant Biol. 2010, 8, e0132. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Truglio, J.J.; Theis, K.; Feng, Y.; Gajda, R.; Machutta, C.; Tonge, P.J.; Kisker, C. Crystal structure of Mycobacterium tuberculosis MenB, a key enzyme in vitamin K2 biosynthesis. J. Biol. Chem. 2003, 278, 42352–42360. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindah, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.; Couch, G.; Greenblatt, D.M.; Meng, E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Alexeev, Y.; Mazanetz, M.P.; Ichihara, O.; Fedorov, D.G. GAMESS as a Free Quantum-Mechanical Platform for Drug Research. Curr. Top. Med. Chem. 2012, 12, 2013–2033. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for highthroughput MM-PBSA calculations open source drug discovery consortium. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.N.; Su, S.J.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Sun, Y.; Song, H.; Li, J.; Li, Y.; Jiang, M.; Zhou, J.; Guo, Z. Structural basis of the induced-fit mechanism of 1,4-dihydroxy-2-naphthoyl coenzyme A synthase from the crotonase fold superfamily. PLoS ONE 2013, 8, e63095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 2nd ed.; John Wiley and Sons, Ltd.: West Sussex, UK, 2002. [Google Scholar] [CrossRef]
- Aphacha, J.; Karin, S.; Jürgen, S.; Wanchai, D.-E.; Toni, M.K. Pyrone polyketides synthesized by a type III polyketide synthase from Drosophyllum lusitanicum. Phytochemistry 2008, 69, 3043–3053. [Google Scholar] [CrossRef]
- Widhalm, J.R.; Rhodes, D. Biosynthesis and molecular actions of specialized 1,4-naphthoquinone natural products produced by horticultural plants. Hortic. Res. 2016, 3, 16046. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
K S, M.; Lalitha, R.; Girija, S.; Kumar R, P.; P S, A.; N Swamy, M.; M, N.; Jayanthi, M. Identification of a Reaction Intermediate and Mechanism of Action of Intermediary Enzymes in Plumbagin Biosynthetic Pathway Using Molecular Dynamics Simulation. Catalysts 2020, 10, 280. https://doi.org/10.3390/catal10030280
K S M, Lalitha R, Girija S, Kumar R P, P S A, N Swamy M, M N, Jayanthi M. Identification of a Reaction Intermediate and Mechanism of Action of Intermediary Enzymes in Plumbagin Biosynthetic Pathway Using Molecular Dynamics Simulation. Catalysts. 2020; 10(3):280. https://doi.org/10.3390/catal10030280
Chicago/Turabian StyleK S, Muralidharan, Roopa Lalitha, Shanmugam Girija, Pravin Kumar R, Akshai P S, Meghana N Swamy, Nayana M, and Malaiyandi Jayanthi. 2020. "Identification of a Reaction Intermediate and Mechanism of Action of Intermediary Enzymes in Plumbagin Biosynthetic Pathway Using Molecular Dynamics Simulation" Catalysts 10, no. 3: 280. https://doi.org/10.3390/catal10030280