Quantum-Based Modeling of Dephosphorylation in the Catalytic Site of Serine/Threonine Protein Phosphatase-5 (PPP5C)

1
Department of Chemistry, University of South Alabama, Mobile, AL 36688, USA
2
Department of Biochemistry and Molecular Biology, College of Medicine, University of South Alabama, Mobile, AL 36688, USA
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(6), 674; https://doi.org/10.3390/catal10060674
Submission received: 30 May 2020
/
Revised: 12 June 2020
/
Accepted: 13 June 2020
/
Published: 16 June 2020
(This article belongs to the Special Issue Feature Papers to Celebrate "Computational Catalysis"—Trends and Outlook)
Abstract
:Serine/threonine protein phosphatase-5 (PP5; PPP5C) is a member of the phosphoprotein phosphatase (PPP) gene family. The PPP catalytic domains feature a bimetal system (M1/M2), an associated bridge hydroxide (W1(OH−)), an M1-bound water/hydroxide (W2), and a highly conserved core sequence. The PPPs are presumed to share a common mechanism: The seryl/threonyl phosphoryl group of the phosphoprotein coordinates the metal ions, W1(OH−) attacks the central phosphorous atom, rupturing the antipodal phosphoester bond and releasing the phosphate-free protein. Also, a histidine/aspartate tandem is responsible for protonating the exiting seryl/threonyl alkoxide. Here, we employed quantum-based computations on a large section of the PP5 catalytic site. A 33-residue, ONIOM(UB3LYP/6-31G(d):UPM7) model was built to perform computations using methylphosphate dianion as a stand-in substrate for phosphoserine/phosphothreonine. We present a concerted transition state (TS) in which W1(OH−) attacks the phosphate center at the same time that the exiting seryl/threonyl alkoxide is protonated directly by the His304/Asp274 tandem, with W2 assigned as a water molecule: W2(H2O). Arg275, proximal to M1, stabilizes the substrate and TS by binding both the ester oxygen (Oγ) and a phosphoryl oxygen (O1) in a bidentate fashion; in the product state, Tyr451 aids in decoupling Arg275 from O1 of the product phosphate ion. The reaction is exothermic (ΔH = −2.0 kcal/mol), occurs in a single step, and has a low activation barrier (ΔH‡ = +10.0 kcal/mol). Our work is an improvement over an earlier computational study that also found bond rupture and alkoxide protonation to be concerted, but concluded that Arg275 is deprotonated during the reactant and TS stages of the pathway. In that earlier study, the critical electron-withdrawal role that Arg275 plays during the hydroxide attack was not correctly accounted for.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Protein phosphorylation increases the functional diversity of the proteome and is critical to the regulation of numerous cellular processes. About 13,000 human proteins have one or more “p-sites”—amino acid residues where reversible phosphorylation occurs [1]. The presence or absence of a phosphoryl group at a p-site impacts protein structure, enzyme activity, interactions with other proteins, transport properties, and, ultimately, cell function and metabolism [2]. Countering the action of phosphoryl attachment by protein kinase enzymes (phosphorylation), the protein phosphatases catalyze the hydrolysis that liberates a phosphate ion and restores the p-site residue (dephosphorylation). The hydroxyl groups of serine, threonine, and tyrosine are amenable to phosphoryl attachment via a phosphoester bond, and most p-sites are one of these three amino acids, with serine dominant, threonine second, and tyrosine a distant third [3].
Our interest is the common mechanism by which the members of the phosphoprotein phosphatase (PPP) gene family (PPP1C, PPP2C, PPP3C/calcineurin, PPP4C, PPP5C, PPP6C, and PPPEF/PP7), a subcategory of the broader class of protein serine/threonine phosphatases, catalyze dephosphorylation at seryl and threonyl p-sites. The PPPs share a highly conserved catalytic core [4] featuring a bimetal system (M1/M2) as the site of substrate binding and hydrolysis. In this study, we focused on serine/threonine protein phosphatase-5 (PP5) as representative of the PPP family. PP5 expression is known to affect cell proliferation [5,6] and stress-induced responses [7,8,9], and its overexpression is implicated in human breast carcinoma [10], non-small cell lung cancer [11], and other cancers [12].
Co-crystal structures of PP5/phosphate [13], PP1/tungstate [14], and PP1/phosphate [15], reveal common PPP catalytic-site features, including: A bimetal system held in place by the same amino-acid scaffold of six residues, a bridge hydroxide/water (W1), a metal-bound phosphate (or tungstate) positioned by the same four residues, and a cooperative histidine/aspartate tandem. As shown for PP5/phosphate, Figure 1, the alignment of the phosphate with W1 strongly implies a nucleophilic attack on the P center by W1, with subsequent protonation of the exiting alkoxide at the O4 site by H+ transfer from Nϵ of His304. Furthermore, Swingle et al. argued that W1 is a hydroxide ion, and that the exiting alkoxide oxygen is probably not metal bound [13]. Of course, this inferred mechanistic scenario depends on a match between the phosphate ion’s binding mode seen in the PP5 co-crystal and the binding mode of the phosphoserine/phosphothreonine substrate, which is unknown. Notably, the co-crystals also reveal a second water/hydroxide (W2) coordinated to M1, a backbone carbonyl (PP5:His427) positioned to direct W1, and a backbone -NH group (PP5:Phe315) anchoring the His/Asp tandem at the backend, Figure 1. In PP5, Asp274 is also anchored by the nonconserved Asn310 (likewise in PP1 and PP7, threonine otherwise). Thus, these co-crystals, along with mutation studies [16,17] and analysis, established a plausible pathway for PPP dephosphorylation [13,14,15]. The substrate’s anticipated mode of binding to M1/M2 and the W1(OH−) attack on the P center were later corroborated by density functional theory (DFT) calculations by Ribeiro et al. on a cluster model of PP5/phosphoserine [18].
In their PP5 study [18], Ribeiro et al. presented pathways for two different systems, the first of which they endorsed: (I) W1(OH−)/W2(OH−) system (one step, exothermic): Arg275 loses a proton to the W2(OH−) pre-reaction. That is, W2 is actually a water and Arg275 is deprotonated during the reactant and TS stages of the pathway; the proton is returned to Arg275 in the product state. The seryl alkoxide is protonated by direct H+ transfer from His304 as W1 attacks. (II) W1(OH−)/W2(H2O) system (two steps, endothermic): The seryl alkoxide is protonated by Arg275 in step 1, in which the W1(OH−) attack also occurs. In step 2, H+ is transferred from His304 to Arg275 through the serine’s alcohol group.
Both pathways (I) and (II) contain modeling artifacts. The Ribeiro et al. model system comprised 10 amino acid moieties + M1/M2 + W1(OH−)+ W2 + phosphoserine. The amino acid moieties were terminated by frozen methyl groups. Asn310 and the Gly314-Phe315 peptide linkage, which together bind Asp274 (see Figure 1), Asp388, which binds Arg400 (see Figure 2), and Tyr451, which abuts and interacts with Arg275, were not represented. Consequently, the optimized positions of Asp274, Arg400, and Arg275 diverged significantly from their native PP5/phosphate co-crystal positions. It should be added that a molecular dynamics (MD) study of PP5/phosphoserine by Wang and Yan [19] concluded that W1(OH−)-based systems are stable, while W1(H2O)-based systems are not, in further support of the conclusion that W1 is a hydroxide. Additionally, they found that the W1(OH−)/W2(H2O) system has greater substrate affinity versus the W1(OH−)/W2(OH−) system. However, Arg275 and W2 were apparently not modeled in the latter system, as would be prescribed for the reactant stage of pathway (I), with Arg275 as a neutral species and W2 as a water molecule.
In the present work, we used a PP5 co-crystal (pdb entry 4ZX2 [20]) as foundation for a 33-residue model of the PP5 catalytic site (Figure 2). Our goal was to reexamine the W1(OH−)/W2(H2O) system, and our model was intended to include all relevant moieties that play a role in the reaction and/or serve as steric barriers to prevent nonnative reorientations during optimizations. Two types of calculations were performed: (1) Tests of the behavior of waters associated with the bimetal system and/or protonation states within the catalytic site and (2) tests of the W1(OH−)/W2(H2O) system for the reactant, transition state, and product stages of the reaction using methylphosphate dianion (CH3OPO32−) as a stand-in for phosphoserine. The dianionic species was used because kinetic isotope effect data indicated that PPPs act on the dianionic substrate [21] and because the protonated species would have enhanced acidity when metal bound.
2. Results
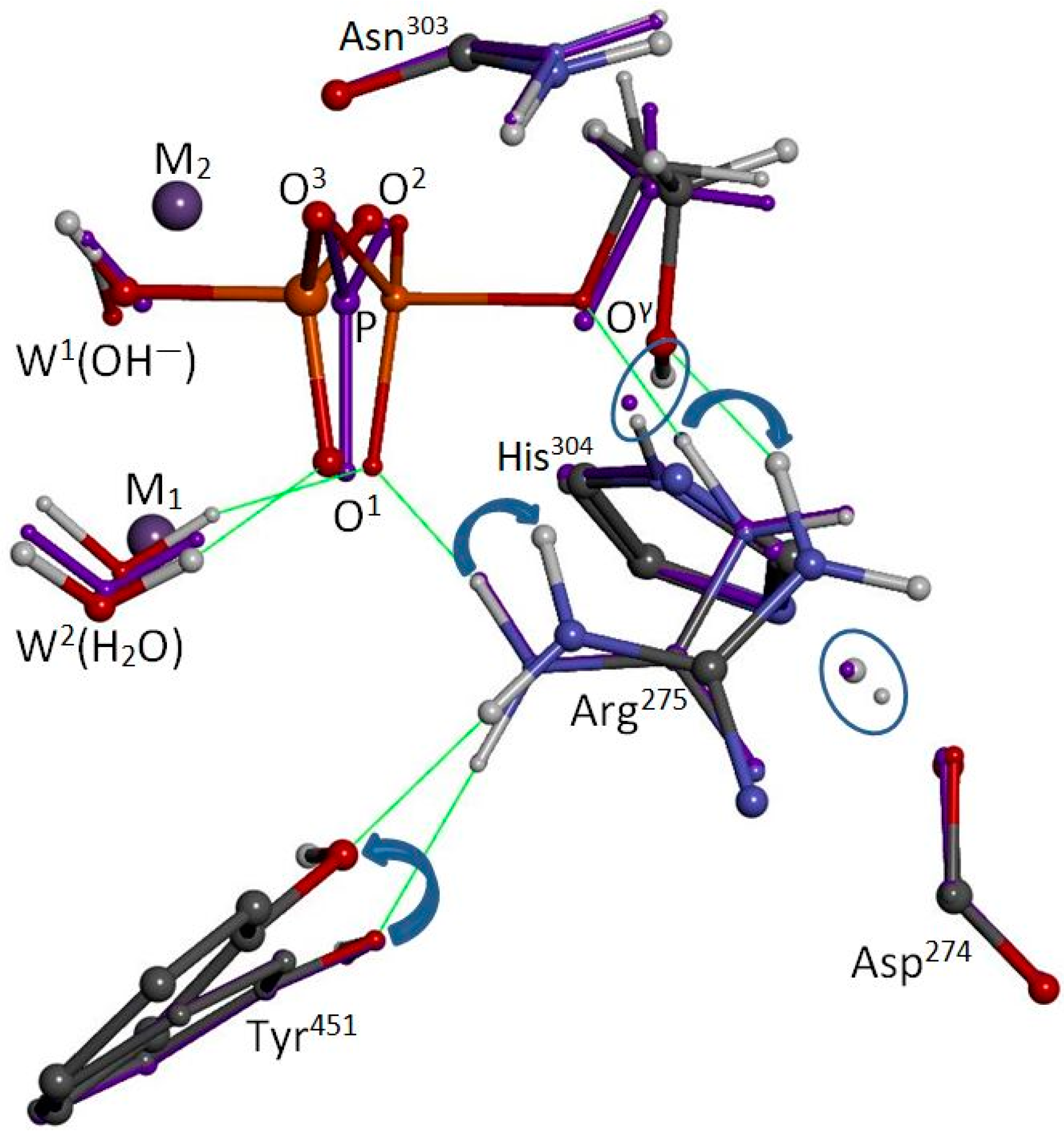
The reactant, TS, and product states for the W1(OH−)/W2(H2O) system are presented in a truncated form in Figure 3. According to both gas-phase and solvent-corrected energies, the reaction is exothermic (ΔH ≈ ΔE = −2.5 and −2.0 kcal/mol), with a low activation barrier (ΔH‡ ≈ ΔE‡ = +8.6 and +10.0 kcal/mol). Vibrational frequency analysis confirms the reactant and product structures as minima and assigns exactly one imaginary frequency (187i cm−1) to the TS. Free energy estimates under the rigid rotor/harmonic oscillator approximation afford ΔG = −7.2 and ΔG‡ = +6.0 kcal/mol. The coordinates of the optimized structures and an animation of the imaginary mode are available in the Supplementary Materials. W2(H2O) helps to stabilize the substrate via H-bonding with O1, and this interaction is present in the TS and product states as well. The other expected stabilizing interactions for the phosphate group are also present in all three states, specifically, O2 with Asn303 and O3 with Arg400. Importantly, Arg275 binds the substrate and TS in a bidentate fashion at O1 and Oγ, just as it binds the phosphate ion in the co-crystal. In short, the reactant state is very similar to the geometry of the PP5/phosphate co-crystal, with no unusual reorientations of the five critical residues that were allowed to move during the optimization. The most significant difference between the co-crystal and the optimized reactant state is that the face of Arg275 is turned about 49° to establish an H-bond with Tyr451. We add that the reactant state’s coordination distance for W2, r(W2 − M1) = 2.323 Å, is consistent with the X-ray crystal value of 2.295 Å and indicates that the assignment of W2 as a water molecule is correct.
As in the previous report [18], the TS is concerted; that is, W1(OH−) attacks the phosphorus center at the same time that the exiting seryl/threonyl alkoxide is protonated directly by the His304/Asp274 tandem. The primary atomic motion over the course of the reaction is the inversion of the P center along with the transfer of H+ from His304 to Oγ. As stated above, Arg275 stabilizes the TS in a bidentate fashion through interactions with O1 and Oγ. Post-TS, however, as the product alcohol recedes, Arg275 turns to maintain the Oγ interaction, and the O1 interaction is ended. Also post-TS, Tyr451 rotates inward to maintain its H-bond with Arg275. In the product state, Arg275 is isolated from the HPO42− product, which is ligated to M1/M2 through O1, O2, and now the bridge hydroxyl oxygen, formerly of the bridge hydroxide W1(OH−). As expected with the loss of formal negative charge, the bridge oxygen’s metal coordinations are longer in the product state than in the reactant state; meanwhile, the product alcohol lingers, engaged in an H-bonding trio along with Arg275 and His304.
3. Discussion
Crystal structure superposition shows that, in the modeling of Ribeiro et al. [18], Arg275 took a position that is partially occupied by Tyr451 in the enzyme. This nonnative reorientation occurred because of the absence of Tyr451 in their model, and it permitted the transfer of H+ from Arg275 to W2(OH−). In the enzyme, Tyr451 blocks Arg275 from contact with W2 when a phosphate-bearing substrate is also ligated to the M1/M2 system. Also, the aforementioned reorientation of Asp274 did not occur in our model because we included Asn310 and the -NH peptide moiety needed to anchor Asp274 (see Figure 1); we froze Arg400, but the presence of Asp388 in our model (see Figure 2) would have prevented an unrealistic reorientation for Arg400 as well. Future improvements to our computational model would allow Arg400 and the M1/M2 system to move because of their direct contacts with the substrate, TS, and product phosphate ion. Only subtle changes would be expected, and the states, as shown in Figure 3, should remain qualitatively the same, but the reaction energetics might be impacted. Regarding movement of the M1/M2 system, an increase of about 0.12 Å in the M1…M2 separation distance did occur for pathways (I) and (II), upon going from reactant to the transition state(s) [18].
Arg275 is mobile and is known to take different positions depending upon the ligand present in the co-crystal [9]. Additionally, we now see that Tyr451 interacts with Arg275 during the course of the reaction and that their motions are coupled. Tyr451 moves to aid the disengagement of Arg275 from the product phosphate, which, of course, must be achieved as part of displacing the product phosphate for site regeneration. After the TS, Tyr451 follows Arg275 while keeping contact with the face of His244 and moves only a small distance. Generally, however, Tyr451 is part of the flexible β12-β13 loop and is evidently somewhat mobile. Depending upon the ligand present, Tyr451 is sometimes vertically farther away from His244 and is not always engaged in H-bonding with Arg275 in various crystal structures. Interestingly, PP2A:Arg89 (counterpart of PP5′s Arg275) can reorient to engage in an apparent salt-bridge interaction at the interface between the catalytic and regulatory domains of PP2A, at least in the presence of microcystin-LR (pdb entry 3FGA [22]). In this orientation, PP2A:Arg89 still contacts PP2A:Tyr265 (counterpart of PP5′s Tyr451). If a natural positioning of PP2A:Arg89 is truly represented in this crystal structure, it seems reasonable to assume that PP2A does not function efficiently when in such a state.
Quantum-based modeling of some small phosphopeptides as PP5 substrates may be possible in the model we have developed here. Of particular interest, Oberoi et al. [23] investigated the likely positions of the residues adjacent to the p-site serine of Cdc37 (pSer13) by appending a mimic sequence (15 residues) to the catalytic domain of PP5. The resulting crystal structure (pdb entry 5HPE) indicates that the backbone –C=O and –NH groups of Cdc37:Val12 interact with Arg400, while the carboxylate of Cdc37:Asp14 stacks parallel to the face of Arg400. Also, the backbone –C=O of Cdc37:Asp14 interacts with Arg275. A substrate longer than a Val-pSer-Asp tripeptide, however, will probably require a larger model of the catalytic site to incorporate the additional contacting residues.
Our results are an improvement over both pathways presented in the earlier computational report by Ribeiro et al. [18], as it is now practical computationally to enlarge the PP5 model system. In their report, they endorsed the W1(OH−)/W2(OH−) system and identified W2 as a hydroxide ion: W2(OH−). The mechanism they presented, pathway (I), follows an initial H+ extraction from Arg275 by W2(OH−). The charge-zero Arg275 binds the substrate and TS at O1 but not Oγ, and its position overlaps with Tyr451 (absent in the model) in order to interact with W2. In short, Arg275‘s mode of interaction with the substrate/TS in their model is not optimal for substrate/TS stabilization and does not match the expected bidentate binding seen in the PP5/phosphate complex. Moreover, we argue that an unprotonated arginine (pKb ≈ 1.5) that is H-bonded to an electron-rich phosphate moiety would have a very low pKb and a strong tendency to gain a proton from solvent. Of course, a protonated arginine would also be more effective in drawing the substrate’s electron density and, thus, promote hydroxide attack.
The pathway offered in [18] for the W1(OH−)/W2(H2O) system is endothermic with a high activation energy (>25 kcal/mol), and the assignment option of W2(H2O) was consequently discarded [18]. The mechanism presented for this system in [18], pathway (II), involves two steps, with an initial proton transfer to the alkoxide coming from Arg275, not from His304. Instead, in this study of the W1(OH−)/W2(H2O) system, we have presented a concerted transition state (TS) in which W1(OH−) attacks the phosphate center at the same time that the exiting seryl/threonyl alkoxide is protonated directly by the His304/Asp274 tandem. Arg275, proximal to M1, stabilizes the substrate and TS in a bidentate fashion, and post-TS movement of the adjacent Tyr451 decouples Arg275 from the product phosphate ion. The reaction is exothermic (ΔH = −2.0 kcal/mol), as expected, and occurs in one step with a low activation barrier (ΔH‡ = +10.0 kcal/mol).
4. Materials and Methods
First, our model was built from pdb entry 4ZX2 [20] (PP5 catalytic domain, 1.23 Å res.) by removal of the inhibitor (a modified norcantharidin) and by selection of proximal residues of the catalytic site. Second, we applied aldehyde (–CHO) and neutral amine (–NH2) terminations where we had broken the enzyme’s peptide backbone. Third, hydrogen atoms were added, including one for protonation of His304. After placement of the methylphosphate dianion, the bridge hydroxide (W1), and a water of hydration (W2), the added species and all hydrogens were coarsely optimized using the PM7 semi-empirical method as implemented in Gaussian16 [24]. Finally, we selected the atoms of the “high-level” region of our hybrid computational system, as indicated in Figure 2, and carried out ONIOM(UB3LYP/6-31G(d):UPM7) [25,26,27,28,29,30] partial optimizations. (Of course, depending upon the nature of each computational test, we inserted other waters and removed the hydroxide, the His304 proton, or substrate, as appropriate.) The system, as shown in Figure 2, has zero total charge. During optimizations, the following residues were allowed to move: His304, Asp274, Arg275, Tyr451, and Asn303, as were any added species. The first two residues are the His/Asp tandem. The second two, Arg275 and Tyr451, are known to freely move to accommodate ligand binding and were adjusted to approximate their PP5/phosphate co-crystal positions. The last, Asn303, coordinates M2 and the substrate’s phosphoryl group at O2. All calculations were carried out using the Gaussian16 suite of programs [24].
The Mn2+/Mn2+ system is an antiferromagnetic singlet state (5 “up”/5 “down”). The proper “high-level” B3LYP electronic state was attained by first constructing an appropriate spin-unrestricted guess wavefunction, as in earlier work [18,31]. The “low-level” PM7 wavefunctions could not be manipulated in the same manner to get antiferromagnetic states in the current implementation of Gaussian16; instead, we converged to stable spin-unrestricted open-shell singlets (1 “up”/1 “down”) generated from closed-shell wavefunctions (STABLE = opt). PM7 convergence was aided by using the quadratic convergence option (SCF = yqc). The transition state search was initiated by first carrying out a partial optimization with the inversion dihedral frozen in a nearly planar conformation (GEOM = addgic). Optimizations of the reactant, TS, and product systems were completed using INT = grid = superfine. Optimizations were judged complete when the RMS force value met the default target, as satisfying the full set of default convergence criteria was not practical for a system of this size. Analytic frequencies were computed to confirm the curvature of the energy surface at the two stable structures and the transition state. Single-point calculations using the default continuum solvation model in Gaussian16 (SCRF = solvent = water, sas) were used to estimate solvent-corrected energies.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/6/674/s1, File S1: Superposition_of_States.pdb, Video S1: PP5_TS_Animation.gif.
Author Contributions
Concept development, E.A.S. and A.W.; computations and draft preparation, E.A.S.; computational resources, A.W.; draft review and editing, A.W. and R.E.H.; project administration and funding acquisition, R.E.H. All authors have read and agree to the published version of the manuscript.
Funding
This research and the APC were funded by Grant NIH CA 60750 (R.E.H.).
Acknowledgments
This work was made possible in part by a grant of high performance computing resources and technical support from the Alabama Supercomputer Authority.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Vlastaridis, P.; Kyriakidou, P.; Chaliotis, A.; Van De Peer, Y.; Oliver, S.G.; Amoutzias, G. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. GigaScience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Johnson, L.N.; Barford, D. The effects of phosphorylation on the structure and function of proteins. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 199–232. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Honkanen, R.E. Molecular Cloning, Expression, and Characterization of a Novel Human Serine/Threonine Protein Phosphatase, PP7, That Is Homologous to Drosophila Retinal Degeneration C Gene Product (rdgC). J. Biol. Chem. 1998, 273, 1462–1468. [Google Scholar] [CrossRef] [Green Version]
- Von Kriegsheim, A.; Pitt, A.R.; Grindlay, G.J.; Kolch, W.; Dhillon, A.S. Regulation of the Raf–MEK–ERK pathway by protein phosphatase 5. Nat. Cell Biol. 2006, 8, 1011–1016. [Google Scholar] [CrossRef]
- Shah, B.H.; Catt, K.J. Protein phosphatase 5 as a negative key regulator of Raf-1 activation. Trends Endocrinol. Metab. 2006, 17, 382–384. [Google Scholar] [CrossRef]
- Morita, K.-I.; Saitoh, M.; Tobiume, K.; Matsuura, H.; Enomoto, S.; Nishitoh, H.; Ichijo, H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001, 20, 6028–6036. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, F.; Tsuchiya, M.; Shimamoto, S.; Fujimoto, T.; Tokumitsu, H.; Tokuda, M.; Kobayashi, R. Oxidative Stress Impairs the Stimulatory Effect of S100 Proteins on Protein Phosphatase 5 Activity. Tohoku J. Exp. Med. 2016, 240, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Golden, T.; Swingle, M.; Honkanen, R.E. The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev. 2008, 27, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Golden, T.; Aragon, I.V.; Rutland, B.; Tucker, J.A.; Shevde, L.A.; Samant, R.S.; Zhou, G.; Amable, L.; Skarra, D.; Honkanen, R.E. Elevated levels of Ser/Thr protein phosphatase 5 (PP5) in human breast cancer. Biochim. Biophys. Acta (BBA)—Bioenerg. 2008, 1782, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, F.-S.; Hung, M.-H.; Wang, C.-Y.; Chen, Y.-L.; Hsiao, Y.-J.; Tsai, M.-H.; Li, J.-R.; Chen, L.-J.; Shih, C.-T.; Chao, T.-I.; et al. Inhibition of protein phosphatase 5 suppresses non-small cell lung cancer through AMP-activated kinase activation. Lung Cancer 2017, 112, 81–89. [Google Scholar] [CrossRef]
- Sager, R.A.; Dushukyan, N.; Woodford, M.; Mollapour, M. Structure and function of the co-chaperone protein phosphatase 5 in cancer. Cell Stress Chaperones 2020, 25, 383–394. [Google Scholar] [CrossRef]
- Swingle, M.R.; Honkanen, R.E.; Ciszak, E.M. Structural Basis for the Catalytic Activity of Human Serine/Threonine Protein Phosphatase-5. J. Biol. Chem. 2004, 279, 33992–33999. [Google Scholar] [CrossRef] [Green Version]
- Egloff, M.-P.; Cohen, P.T.; Reinemer, P.; Barford, D. Crystal Structure of the Catalytic Subunit of Human Protein Phosphatase 1 and its Complex with Tungstate. J. Mol. Biol. 1995, 254, 942–959. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Huang, H.-B.; Kwon, Y.-G.; Greengard, P.; Nairn, A.; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Brew, K.; Lee, E.Y.C. Mutational Analysis of the Catalytic Subunit of Muscle Protein Phosphatase-1. Biochemistry 1996, 35, 6276–6282. [Google Scholar] [CrossRef] [PubMed]
- Mondragon, A.; Griffith, E.; Sun, L.; Xiong, F.; Armstrong, C.; Liu, J.O. Overexpression and Purification of Human Calcineurin α from Escherichia coli and Assessment of Catalytic Functions of Residues Surrounding the Binuclear Metal Center. Biochemistry 1997, 36, 4934–4942. [Google Scholar] [CrossRef]
- Ribeiro, A.J.M.; Alberto, M.E.; Ramos, M.J.; Fernandes, P.A.; Russo, N. The Catalytic Mechanism of Protein Phosphatase 5 Established by DFT Calculations. Chem. A Eur. J. 2013, 19, 14081–14089. [Google Scholar] [CrossRef]
- Wang, L.; Yan, F. Deprotonation states of the two active site water molecules regulate the binding of protein phosphatase 5 with its substrate: A molecular dynamics study. Protein Sci. 2017, 26, 2010–2020. [Google Scholar] [CrossRef]
- Chattopadhyay, D.; Swingle, M.R.; Salter, E.A.; Wood, E.; D’Arcy, B.; Zivanov, C.; Abney, K.; Musiyenko, A.; Rusin, S.F.; Kettenbach, A.; et al. Crystal structures and mutagenesis of PPP-family ser/thr protein phosphatases elucidate the selectivity of cantharidin and novel norcantharidin-based inhibitors of PP5C. Biochem. Pharmacol. 2016, 109, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Hengge, A.C.; Martin, B.L. Isotope Effect Studies on the Calcineurin Phosphoryl-Transfer Reaction: Transition State Structure and Effect of Calmodulin and Mn2+. Biochemistry 1997, 36, 10185–10191. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Cetin, B.; Anger, M.; Cho, U.-S.; Helmhart, W.; Nasmyth, K.; Xu, W. Structure and Function of the PP2A-Shugoshin Interaction. Mol. Cell 2009, 35, 426–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberoi, J.; Dunn, D.; Woodford, M.R.; Mariotti, L.; Schulman, J.; Bourboulia, D.; Mollapour, M.; Vaughan, C.K. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl. Acad. Sci. USA 2016, 113, 9009–9014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dapprich, S.; Komáromi, I.; Byun, K.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461, 1–21. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- Hehre, W.J. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Windus, T.L.; Pople, J.A.; Ratner, M.A. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Salter, E.A.; Honkanen, R.E.; Wierzbicki, A. Modeling the antiferromagnetic MnIIMnII system within the protein phosphatase-5 catalytic site. J. Mol. Model. 2015, 21, 14. [Google Scholar] [CrossRef]
Figure 1.
The PP5/phosphate co-crystal (pdb entry 1S95 [13]) and its implications for phosphoester hydrolysis. The phosphate ion’s O1 and O2 are coordinated to two Mn2+ ions, M1 and M2, respectively. W1 is the oxygen of what is now accepted to be a bridge hydroxide. The hydroxide is oriented by the peptide carbonyl at left and is positioned along the P-O4 axis (solid line) for a backside attack, theoretically, on the central P atom of a similarly bound phosphoester. Nϵ of His304 is almost certainly protonated, and O4 marks what would be a seryl/threonyl Oγ requiring a proton as the phosphoester bond breaks. The His304/Asp274 tandem is coupled by an H-bond, and Asp274 is additionally H-bonded to Asn310 and the -NH moiety of the peptide link at right. W2 is the oxygen of a water/hydroxide ligated to M1. Dashed lines indicate H-bonds.
Figure 1.
The PP5/phosphate co-crystal (pdb entry 1S95 [13]) and its implications for phosphoester hydrolysis. The phosphate ion’s O1 and O2 are coordinated to two Mn2+ ions, M1 and M2, respectively. W1 is the oxygen of what is now accepted to be a bridge hydroxide. The hydroxide is oriented by the peptide carbonyl at left and is positioned along the P-O4 axis (solid line) for a backside attack, theoretically, on the central P atom of a similarly bound phosphoester. Nϵ of His304 is almost certainly protonated, and O4 marks what would be a seryl/threonyl Oγ requiring a proton as the phosphoester bond breaks. The His304/Asp274 tandem is coupled by an H-bond, and Asp274 is additionally H-bonded to Asn310 and the -NH moiety of the peptide link at right. W2 is the oxygen of a water/hydroxide ligated to M1. Dashed lines indicate H-bonds.
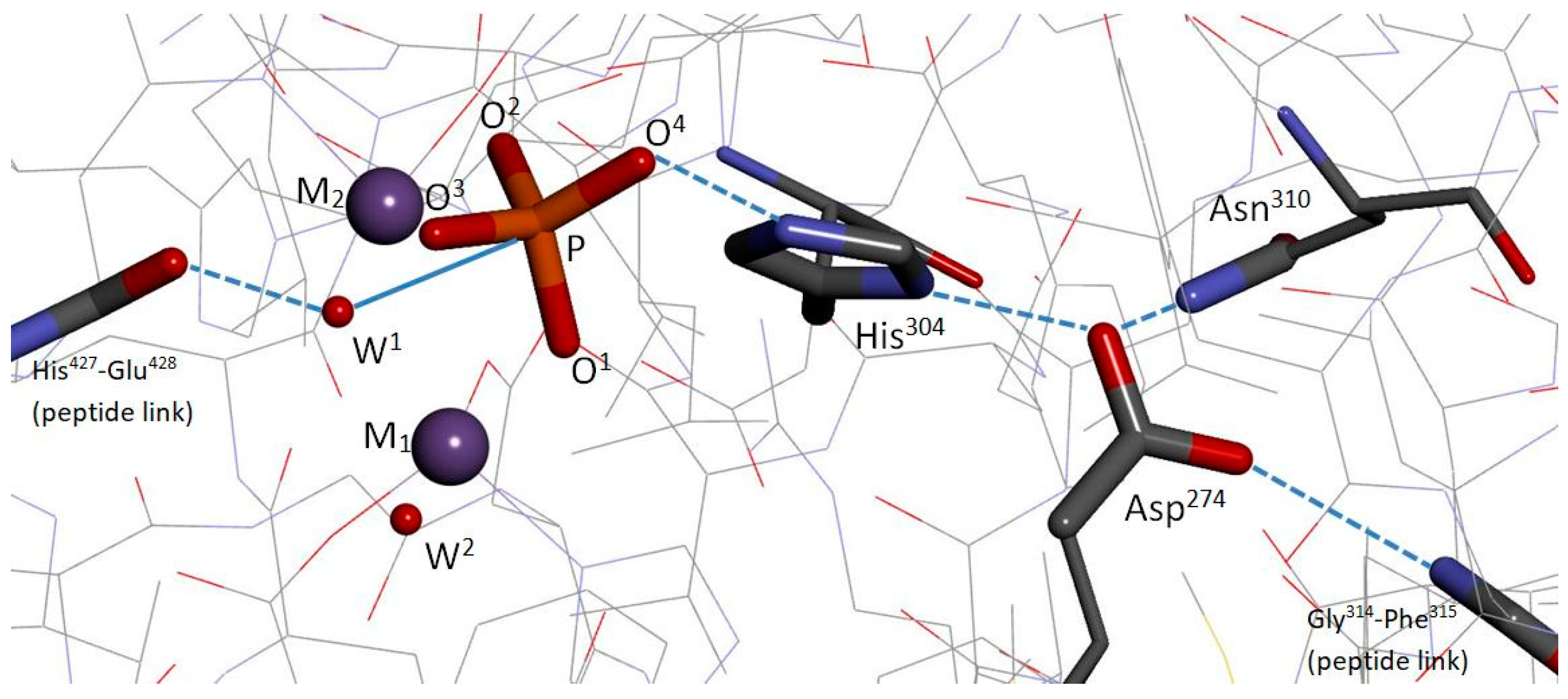
Figure 2.
Two-level ONIOM(UB3LYP/6-31G(d):UPM7) model of the PP5 catalytic site. The functional groups of the “high-level” region (tubes) are conserved across the PPP gene family; the “low-level” region (wireframe) spans 33 residues. (Only the residues Gly314 and Glu428 located at the peptide linkages are not conserved.) M1 and M2 are Mn2+ ions. The transition state for the backside attack by W1(OH−) on methylphosphate dianion (the stand-in substrate) is shown; the substrate’s P-Oγ bond is elongated and the phosphoryl group is flattened as part of a trigonal bipyramidal complex.
Figure 2.
Two-level ONIOM(UB3LYP/6-31G(d):UPM7) model of the PP5 catalytic site. The functional groups of the “high-level” region (tubes) are conserved across the PPP gene family; the “low-level” region (wireframe) spans 33 residues. (Only the residues Gly314 and Glu428 located at the peptide linkages are not conserved.) M1 and M2 are Mn2+ ions. The transition state for the backside attack by W1(OH−) on methylphosphate dianion (the stand-in substrate) is shown; the substrate’s P-Oγ bond is elongated and the phosphoryl group is flattened as part of a trigonal bipyramidal complex.

Figure 3.
Superposition of reactant, transition, and product states of methylphosphate hydrolysis. Representations are small ball-and-stick, violet ball-and-stick, and large ball-and-stick, respectively. Most atoms of the model are hidden from view. W1(OH−) at upper left attacks the M1/M2-bound methylphosphate dianion (CH3OPO32−), causing rupture of the P-Oγ bond. Reaction motion primarily involves inversion of the P center and transfer of H+ from His304 to Oγ. Post-TS withdrawal of the product alcohol (CH3OH) to the right induces Arg275 to swing away from O1, now an atom belonging to the hydrolysis product (HPO42−). At the same time, Tyr451 rotates inward to maintain an H-bond with Arg275. W2(H2O) is H-bonded to O1 throughout the reaction. Proton movements to and from His304 are circled; selected H-bonds are indicated by green lines.
Figure 3.
Superposition of reactant, transition, and product states of methylphosphate hydrolysis. Representations are small ball-and-stick, violet ball-and-stick, and large ball-and-stick, respectively. Most atoms of the model are hidden from view. W1(OH−) at upper left attacks the M1/M2-bound methylphosphate dianion (CH3OPO32−), causing rupture of the P-Oγ bond. Reaction motion primarily involves inversion of the P center and transfer of H+ from His304 to Oγ. Post-TS withdrawal of the product alcohol (CH3OH) to the right induces Arg275 to swing away from O1, now an atom belonging to the hydrolysis product (HPO42−). At the same time, Tyr451 rotates inward to maintain an H-bond with Arg275. W2(H2O) is H-bonded to O1 throughout the reaction. Proton movements to and from His304 are circled; selected H-bonds are indicated by green lines.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Salter, E.A.; Wierzbicki, A.; Honkanen, R.E. Quantum-Based Modeling of Dephosphorylation in the Catalytic Site of Serine/Threonine Protein Phosphatase-5 (PPP5C). Catalysts 2020, 10, 674. https://doi.org/10.3390/catal10060674
AMA Style
Salter EA, Wierzbicki A, Honkanen RE. Quantum-Based Modeling of Dephosphorylation in the Catalytic Site of Serine/Threonine Protein Phosphatase-5 (PPP5C). Catalysts. 2020; 10(6):674. https://doi.org/10.3390/catal10060674
Chicago/Turabian StyleSalter, E. Alan, Andrzej Wierzbicki, and Richard E. Honkanen. 2020. "Quantum-Based Modeling of Dephosphorylation in the Catalytic Site of Serine/Threonine Protein Phosphatase-5 (PPP5C)" Catalysts 10, no. 6: 674. https://doi.org/10.3390/catal10060674
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.