
Membrane-Less Ethanol Electrooxidation over Pd-M (M: Sn, Mo and Re) Bimetallic Catalysts

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
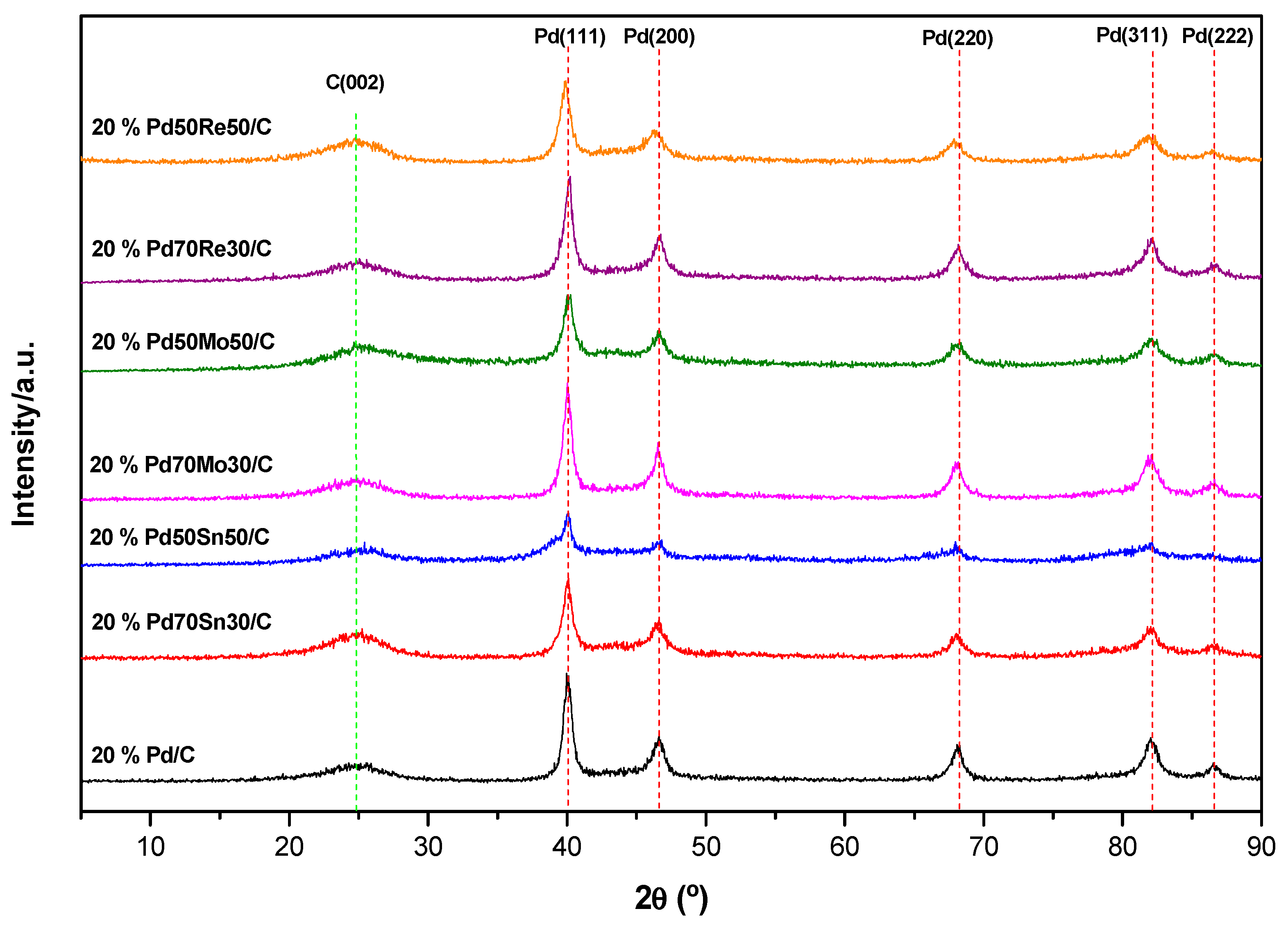
2.1. Structural Characterization
2.2. Electrochemical Measurements
2.2.1. CV in Aqueous KOH
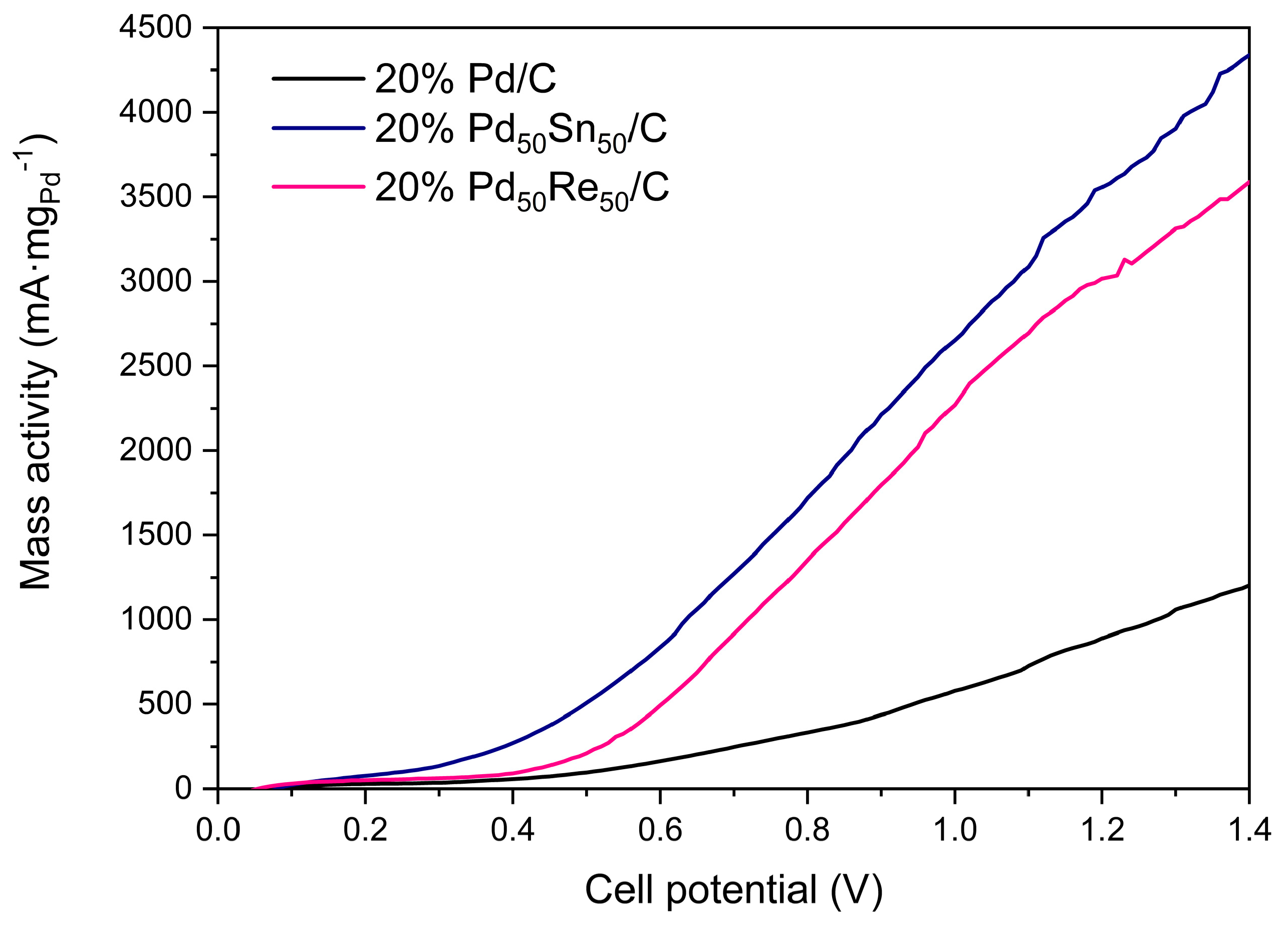
2.2.2. EOR Performance in KOH-EtOH Aqueous Solutions
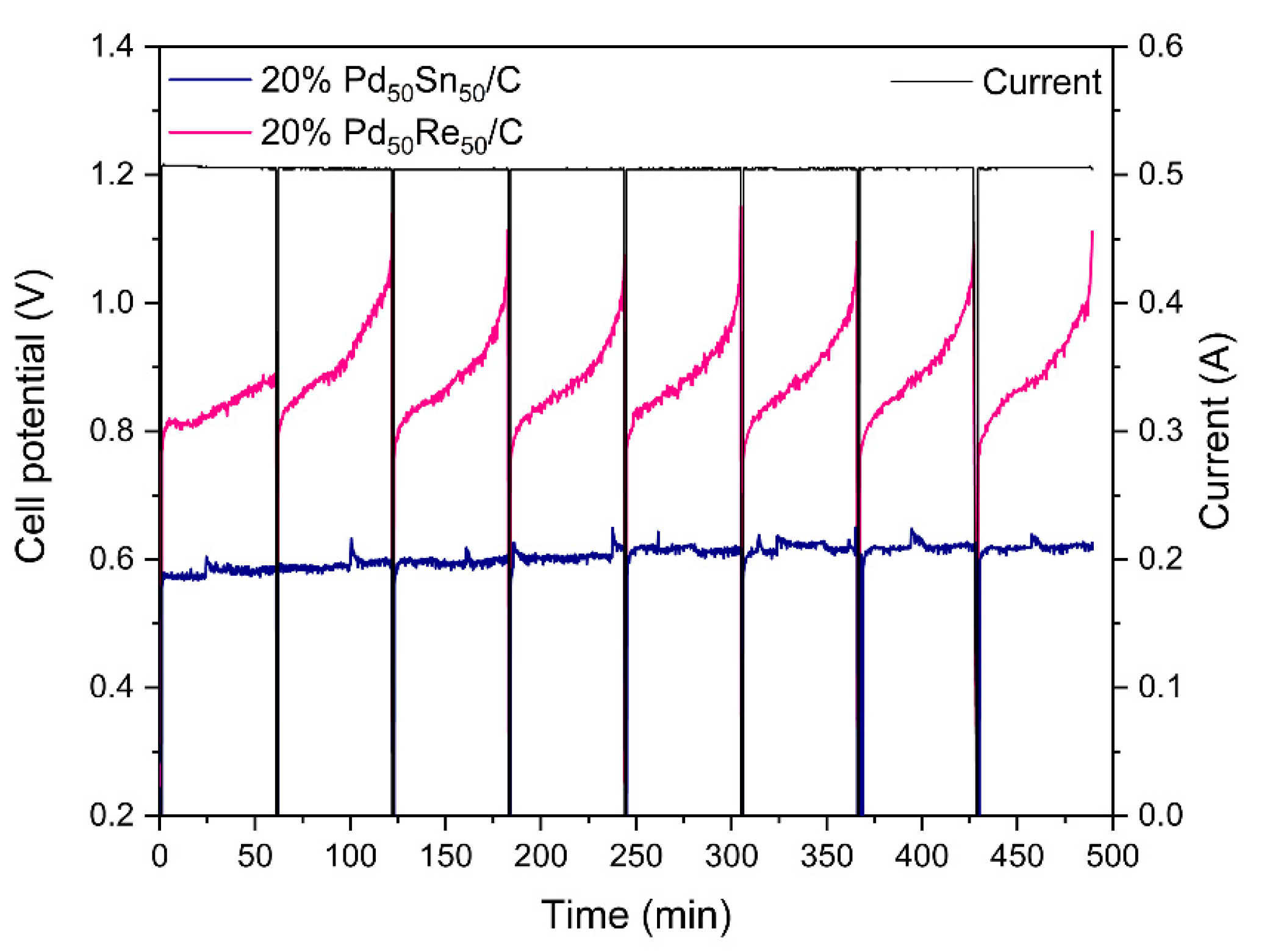
2.2.3. Membrane-Less Electrochemical Reforming Experiments.
3. Materials and Methods
3.1. Synthesis and Characterization of Pd-Based Catalyst Powders
3.2. Electrochemical Characterization in Half-Cell Configuration
3.3. Electrochemical Performance in a Membrane-Less Electrolyzer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dincer, I.; Acar, C. Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrogen Energy 2015, 40, 11094–11111. [Google Scholar] [CrossRef]
- Holladay, J.D.; Hu, J.; King, D.L.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Kumar, S.S.; Himabindu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Rashid, M.; Al Mesfer, M.; Naseem, H.; Danish, M. Hydrogen Production by Water Electrolysis: A Review of Alkaline Water Electrolysis, PEM Water Electrolysis and High Temperature Water Electrolysis. Int. J. Eng. Adv. Technol. 2015, 4, 2249–8958. [Google Scholar]
- Gutiérrez-Guerra, N.; Jiménez-Vázquez, M.; Serrano-Ruiz, J.; Valverde, J.; de Lucas-Consuegra, A. Electrochemical reforming vs. catalytic reforming of ethanol: A process energy analysis for hydrogen production. Chem. Eng. Process. Process. Intensif. 2015, 95, 9–16. [Google Scholar] [CrossRef]
- Ruiz-López, E.; Amores, E.; de la Osa, A.R.; Dorado, F.; de Lucas-Consuegra, A. Electrochemical reforming of ethanol in a membrane-less reactor configuration. Chem. Eng. J. 2020, 379, 122289. [Google Scholar] [CrossRef]
- Song, S.Q.; Zhou, W.J.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Leontidis, V.; Kontou, S.; Tsiakaras, P. Direct ethanol PEM fuel cells: The case of platinum based anodes. Int. J. Hydrogen Energy 2005, 30, 995–1001. [Google Scholar] [CrossRef]
- Almeida, T.D.S.; De Andrade, A.R. New Trends in Direct Ethanol Fuel Cells. New Future Dev. Catal. 2013, 429–452. [Google Scholar] [CrossRef]
- Yu, E.H.; Krewer, U.; Scott, K. Principles and Materials Aspects of Direct Alkaline Alcohol Fuel Cells. Energies 2010, 3, 1499–1528. [Google Scholar] [CrossRef]
- Xu, C.; Shen, P.K.; Liu, Y. Ethanol electrooxidation on Pt/C and Pd/C catalysts promoted with oxide. J. Power Sources 2007, 164, 527–531. [Google Scholar] [CrossRef]
- Liang, Z.; Zhao, T.; Xu, J.; Zhu, L. Mechanism study of the ethanol oxidation reaction on palladium in alkaline media. Electrochim. Acta 2009, 54, 2203–2208. [Google Scholar] [CrossRef]
- Takamura, T.; Minamiyama, K. Anodic Oxidation of Methanol at Palladium Electrode in Alkaline Solution. J. Electrochem. Soc. 1965, 112, 333. [Google Scholar] [CrossRef]
- Takamura, T.; Sato, Y. Specular reflectivity studies of the adsorption and the oxidation of methanol on palladium in an alkaline solution. Electrochim. Acta 1974, 19, 63–68. [Google Scholar] [CrossRef]
- Chen, L.; Lu, L.; Zhu, H.; Chen, Y.; Huang, Y.; Li, Y.; Wang, L. Improved ethanol electrooxidation performance by shortening Pd–Ni active site distance in Pd–Ni–P nanocatalysts. Nat. Commun. 2017, 8, 14136. [Google Scholar] [CrossRef] [Green Version]
- Monyoncho, E.A.; Ntais, S.; Soares, F.; Woo, T.K.; Baranova, E.A. Synergetic effect of palladium–ruthenium nanostructures for ethanol electrooxidation in alkaline media. J. Power Sources 2015, 287, 139–149. [Google Scholar] [CrossRef]
- Yi, Q.; Niu, F.; Song, L.; Liu, X.; Nie, H. Electrochemical Activity of Novel Titanium-Supported Porous Binary Pd-Ru Particles for Ethanol Oxidation in Alkaline Media. Electroanalysis 2011, 23, 2232–2240. [Google Scholar] [CrossRef]
- Maumau, T.; Modibedi, R.; Mathe, M. Electro-oxidation of alcohols using carbon supported gold, palladium catalysts in alkaline media. Mater. Today Proc. 2018, 5, 10542–10550. [Google Scholar] [CrossRef]
- Adam, A.M.M.; Zhu, A.; Ning, L.; Deng, M.; Zhang, Q.; Liu, Q. Carbon supported PdSn nanocatalysts with enhanced performance for ethanol electrooxidation in alkaline medium. Int. J. Hydrog. Energy 2019, 44, 20368–20378. [Google Scholar] [CrossRef]
- Bambagioni, V.; Bianchini, C.; Marchionni, A.; Filippi, J.; Vizza, F.; Teddy, J.; Serp, P.; Zhiani, M. Pd and Pt–Ru anode electrocatalysts supported on multi-walled carbon nanotubes and their use in passive and active direct alcohol fuel cells with an anion-exchange membrane (alcohol=methanol, ethanol, glycerol). J. Power Sources 2009, 190, 241–251. [Google Scholar] [CrossRef]
- Na, H.; Zhang, L.; Qiu, H.; Wu, T.; Chen, M.; Yang, N.; Li, L.; Xing, F.; Gao, J. A two step method to synthesize palladium–copper nanoparticles on reduced graphene oxide and their extremely high electrocatalytic activity for the electrooxidation of methanol and ethanol. J. Power Sources 2015, 288, 160–167. [Google Scholar] [CrossRef]
- Douk, A.S.; Saravani, H.; Noroozifar, M. Novel fabrication of PdCu nanostructures decorated on graphene as excellent electrocatalyst toward ethanol oxidation. Int. J. Hydrog. Energy 2017, 42, 15149–15159. [Google Scholar] [CrossRef]
- Lv, H.; Sun, L.; Xu, D.; Suib, S.L.; Liu, B. One-pot aqueous synthesis of ultrathin trimetallic PdPtCu nanosheets for the electrooxidation of alcohols. Green Chem. 2019, 21, 2367–2374. [Google Scholar] [CrossRef]
- Mattarozzi, L.; Cattarin, S.; Comisso, N.; Gerbasi, R.; Guerriero, P.; Musiani, M.; Vázquez-Gómez, L. Preparation of compact and porous Pd-Ni alloys and study of their performances for ethanol oxidation in alkali. Electrochim. Acta 2019, 307, 503–511. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, T.S.; Liu, X.; Wang, X. Novel palladium–lead (Pd–Pb/C) bimetallic catalysts for electrooxidation of ethanol in alkaline media. J. Power Sources 2010, 195, 2619–2622. [Google Scholar] [CrossRef]
- Lv, H.; Wang, Y.; Lopes, A.; Xu, D.; Liu, B. Ultrathin PdAg single-crystalline nanowires enhance ethanol oxidation electrocatalysis. Appl. Catal. B Environ. 2019, 249, 116–125. [Google Scholar] [CrossRef]
- Caglar, A.; Kivrak, H. Highly active carbon nanotube supported PdAu alloy catalysts for ethanol electrooxidation in alkaline environment. Int. J. Hydrog. Energy 2019, 44, 11734–11743. [Google Scholar] [CrossRef]
- Geraldes, A.N.; Furtunato da Silva, D.; Martins da Silva, J.C.; Antonio de Sá, O.; Spinacé, E.V.; Neto, A.O.; Coelho dos Santos, M. Palladium and palladium–tin supported on multi wall carbon nanotubes or carbon for alkaline direct ethanol fuel cell. J. Power Sources 2015, 275, 189–199. [Google Scholar] [CrossRef]
- da Silva, S.G.; Assumpção, M.H.M.T.; Silva, J.C.M.; de Souza, R.F.; Spinacé, E.V.; Neto, A.O.S.; Buzzo, G.S. PdSn/C Electrocatalysts with Different Atomic Ratios for Ethanol Electro-Oxidation in Alkaline Media. Int. J. Electrochem. Sci. 2014, 9, 5416–5424. [Google Scholar]
- Du, W.; MacKenzie, K.E.; Milano, D.F.; Deskins, N.A.; Su, D.; Teng, X. Palladium–Tin Alloyed Catalysts for the Ethanol Oxidation Reaction in an Alkaline Medium. ACS Catal. 2012, 2, 287–297. [Google Scholar] [CrossRef]
- Zhang, Z.; Ge, J.; Ma, L.; Liao, J.; Lu, T.; Xing, W. Highly Active Carbon-supported PdSn Catalysts for Formic Acid Electrooxidation. Fuel Cells 2009, 9, 114–120. [Google Scholar] [CrossRef]
- Pech-Rodríguez, W.; González-Quijano, D.; Vargas-Gutiérrez, G.; Morais, C.; Napporn, T.; Rodríguez-Varela, F. Electrochemical and in situ FTIR study of the ethanol oxidation reaction on PtMo/C nanomaterials in alkaline media. Appl. Catal. B Environ. 2017, 203, 654–662. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, P.K. Glycerol electrooxidation on highly active Pd supported carbide/C aerogel composites catalysts. Int. J. Hydrog. Energy 2013, 38, 2257–2262. [Google Scholar] [CrossRef]
- Ambriz-Peláez, O.; Durón, S.; Olivas, A.; Valdez, R.; Arriaga, L.G.; Álvarez-Contreras, L.; Guerra-Balcázar, M.; Arjona, N. Effect of molybdenum content on the morphology and electronic characteristics of Pd–MoO nanomaterials and activity evaluation for ethylene glycol electro–oxidation. Appl. Surf. Sci. 2019, 498, 143842. [Google Scholar] [CrossRef]
- Lim, E.J.; Kim, H.J.; Kim, W.B. Efficient electrooxidation of methanol and ethanol using MoOx-decorated Pd catalysts in alkaline media. Catal. Commun. 2012, 25, 74–77. [Google Scholar] [CrossRef]
- Xiao, J.; Puddephatt, R.J. Pt-Re clusters and bimetallic catalysts. Coord. Chem. Rev. 1995, 143, 457–500. [Google Scholar] [CrossRef]
- Tayal, J.; Rawat, B.; Basu, S. Effect of addition of rhenium to Pt-based anode catalysts in electro-oxidation of ethanol in direct ethanol PEM fuel cell. Int. J. Hydrog. Energy 2012, 37, 4597–4605. [Google Scholar] [CrossRef]
- Gopi, K.H.; Bhat, S.D.; Sahu, A.K.; Sridhar, P. Quaternized poly(phenylene oxide) anion exchange membrane for alkaline direct methanol fuel cells in KOH-free media. J. Appl. Polym. Sci. 2016, 133, 133. [Google Scholar] [CrossRef]
- LaConti, A.B.; Hamdan, M.; McDonald, R.C. Mechanisms of membrane degradation. In Handbook of Fuel Cells; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Collier, A.; Wang, H.; Yuan, X.Z.; Zhang, J.; Wilkinson, D.P. Degradation of polymer electrolyte membranes. Int. J. Hydrog. Energy 2006, 31, 1838–1854. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, M.; He, Y.; Ma, G.; Zhang, Z.; Wang, X. A comparative study of elemental additives (Ni, Co and Ag) on electrocatalytic activity improvement of PdSn-based catalysts for ethanol and formic acid electro-oxidation. Electrochim. Acta 2014, 148, 291–301. [Google Scholar] [CrossRef]
- Grdeń, M.; Czerwiński, A. EQCM studies on Pd–Ni alloy oxidation in basic solution. J. Solid State Electrochem. 2007, 12, 375–385. [Google Scholar] [CrossRef]
- Sankar, S.; Watanabe, N.; Anilkumar, G.M.; Nair, B.N.; Sivakamiammal, S.G.; Tamaki, T.; Yamaguchi, T.; Sasidharan, S.; Sailaja, G. Electro-oxidation competency of palladium nanocatalysts over ceria–carbon composite supports during alkaline ethylene glycol oxidation. Catal. Sci. Technol. 2018, 9, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Hamo, E.; Raviv, A.; Rosen, B.A. Influence of Nanocrystalline Palladium Morphology on Alkaline Oxygen Reduction Kinetics. Catalysts 2019, 9, 566. [Google Scholar] [CrossRef] [Green Version]
- Kepp, K.P. A Quantitative Scale of Oxophilicity and Thiophilicity. Inorg. Chem. 2016, 55, 9461–9470. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A. The Role of Oxophilic Metal Promoters in Bimetallic Hydrodeoxygenation Catalysts. Ph.D. Thesis, University of Colorado at Boulder, Boulder, CO, USA, 2016. [Google Scholar]
- Robinson, A.M.; Montemore, M.M.; Tenney, S.A.; Sutter, P.; Medlin, J.W. Interactions of Hydrogen, CO, Oxygen, and Water with Molybdenum-Modified Pt(111). J. Phys. Chem. C 2013, 117, 26716–26724. [Google Scholar] [CrossRef]
- Ordóñez, L.; Roquero, P.; Sebastian, P.; Ramírez, J. Carbon-supported platinum-molybdenum electro-catalysts for methanol oxidation. Catal. Today 2005, 107–108, 46–52. [Google Scholar] [CrossRef]
- Bonarowska, M.; Malinowski, A.; Karpiński, Z. Hydrogenolysis of C–C and C–Cl bonds by Pd–Re/Al2O3 catalysts. Appl. Catal. A Gen. 1999, 188, 145–154. [Google Scholar] [CrossRef]
- Calcerrada, A.B.; de la Osa, A.R.; Dole, H.A.E.; Dorado, F.; Baranova, E.A.; de Lucas-Consuegra, A. Stability Testing of Pt x Sn1 − x /C Anodic Catalyst for Renewable Hydrogen Production Via Electrochemical Reforming of Ethanol. Electrocatalysis 2018, 9, 293–301. [Google Scholar] [CrossRef]
- Xu, C.; Shen, P.K. Electrochamical oxidation of ethanol on Pt-CeO2/C catalysts. J. Power Sources 2005, 142, 27–29. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrog. Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Baranova, E.; Bock, C.; Ilin, D.; Wang, D.; MacDougall, B. Infrared spectroscopy on size-controlled synthesized Pt-based nano-catalysts. Surf. Sci. 2006, 600, 3502–3511. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst. | FWHM (°) | Pd mean Crystallite Size (nm) | ECSA (m2 g−1) |
|---|---|---|---|
| 20% Pd/C | 0.8826 | 9.580 | 6.78 |
| 20% Pd70Sn30/C | 0.9999 | 8.456 | 9.17 |
| 20% Pd50Sn50/C | 0.7537 | 11.218 | 67.61 |
| 20% Pd70Mo30/C | 0.8314 | 10.172 | 4.80 |
| 20% Pd50Mo50/C | 1.0331 | 8.186 | 5.52 |
| 20% Pd70Re30/C | 0.8633 | 9.798 | 2.44 |
| 20% Pd50Re50/C | 0.9738 | 8.680 | 21.22 |
| Catalyst | Eonset (mV) a | im (mA μgPd−1) b |
|---|---|---|
| 20% Pd/C | −700 | 0.063 |
| 20% Pd70Sn30/C | −725 | 0.216 |
| 20% Pd50Sn50/C | −750 | 0.292 |
| 20% Pd70Mo30/C | −670 | 0.063 |
| 20% Pd50Mo50/C | −660 | 0.068 |
| 20% Pd70Re30/C | −680 | 0.060 |
| 20% Pd50Re50/C | −740 | 0.237 |
| Catalyst Nomenclature | Pd Content (%*) | Sn Content (%*) | Mo Content (%*) | Re Content (%*) |
|---|---|---|---|---|
| 20% Pd/C | 100 | - | - | - |
| 20% Sn/C | - | 100 | - | - |
| 20% Mo/C | - | - | 100 | - |
| 20% Re/C | - | - | - | 100 |
| 20% Pd70Sn30/C | 70 | 30 | - | - |
| 20% Pd50Sn50/C | 50 | 50 | - | - |
| 20% Pd70Mo30/C | 70 | - | 30 | - |
| 20% Pd50Mo50/C | 50 | - | 50 | - |
| 20% Pd70Re30/C | 70 | - | - | 30 |
| 20% Pd50Re50/C | 50 | - | - | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-López, E.; Diaz-Perez, M.A.; de Lucas-Consuegra, A.; Dorado, F.; Serrano-Ruiz, J.C. Membrane-Less Ethanol Electrooxidation over Pd-M (M: Sn, Mo and Re) Bimetallic Catalysts. Catalysts 2021, 11, 541. https://doi.org/10.3390/catal11050541
Ruiz-López E, Diaz-Perez MA, de Lucas-Consuegra A, Dorado F, Serrano-Ruiz JC. Membrane-Less Ethanol Electrooxidation over Pd-M (M: Sn, Mo and Re) Bimetallic Catalysts. Catalysts. 2021; 11(5):541. https://doi.org/10.3390/catal11050541
Chicago/Turabian StyleRuiz-López, Estela, Manuel Antonio Diaz-Perez, Antonio de Lucas-Consuegra, Fernando Dorado, and Juan Carlos Serrano-Ruiz. 2021. "Membrane-Less Ethanol Electrooxidation over Pd-M (M: Sn, Mo and Re) Bimetallic Catalysts" Catalysts 11, no. 5: 541. https://doi.org/10.3390/catal11050541