
Triazolium Salt Organocatalysis: Mechanistic Evaluation of Unusual Ortho-Substituent Effects on Deprotonation



Abstract
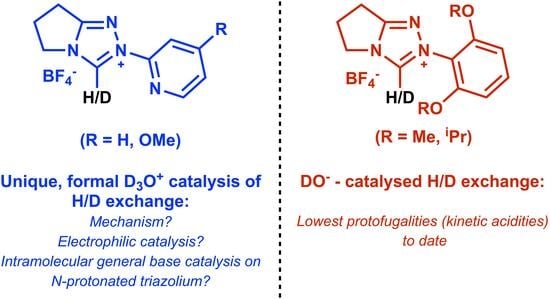
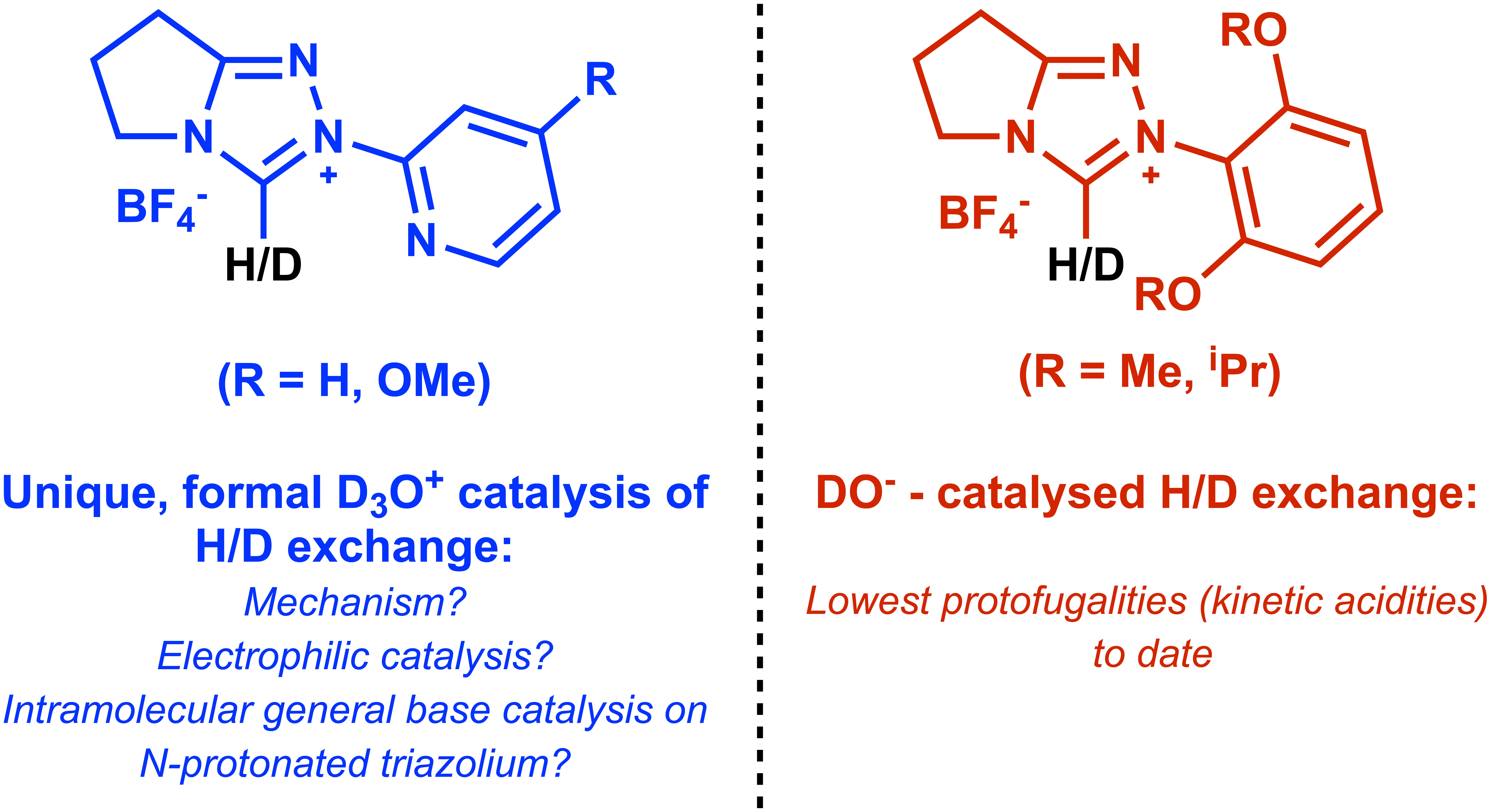
1. Introduction
2. Results and Discussion
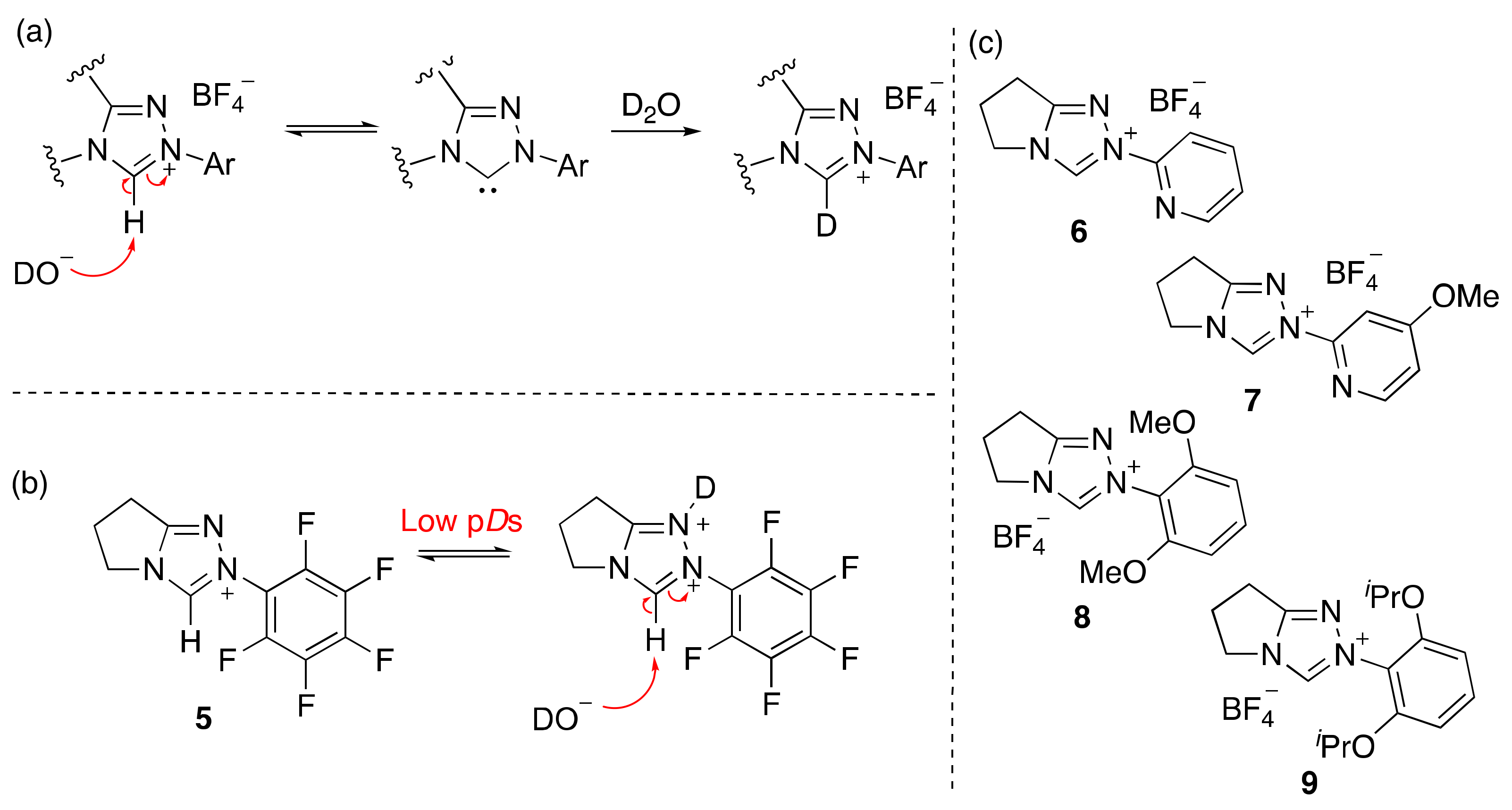
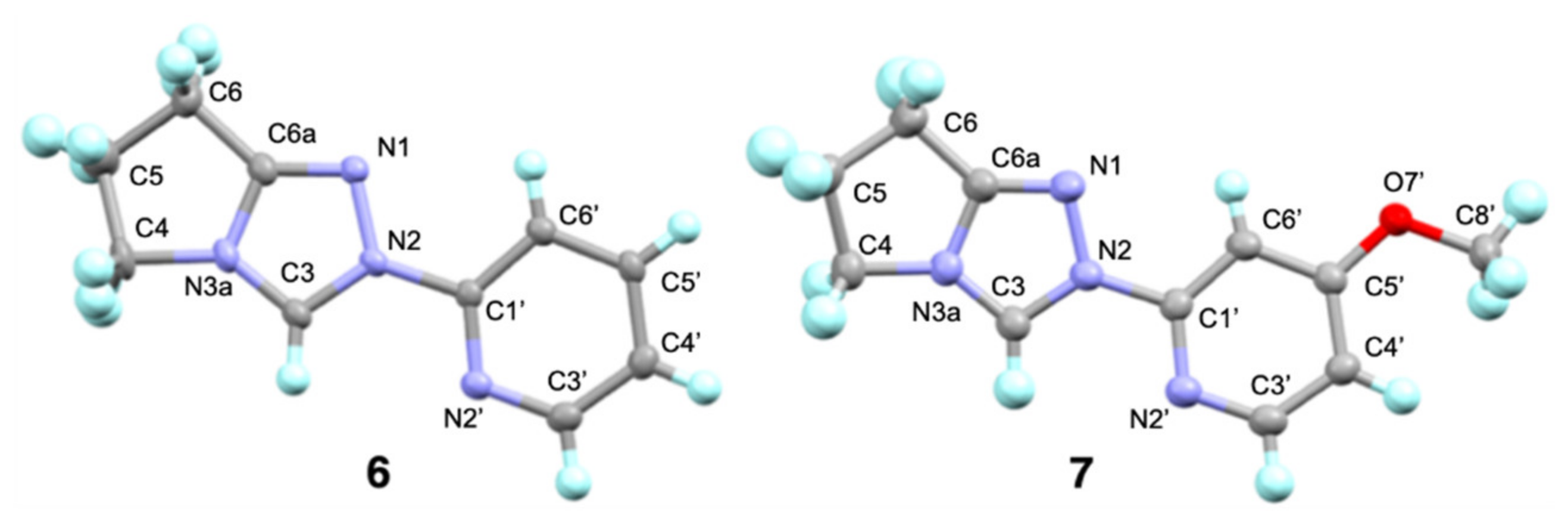
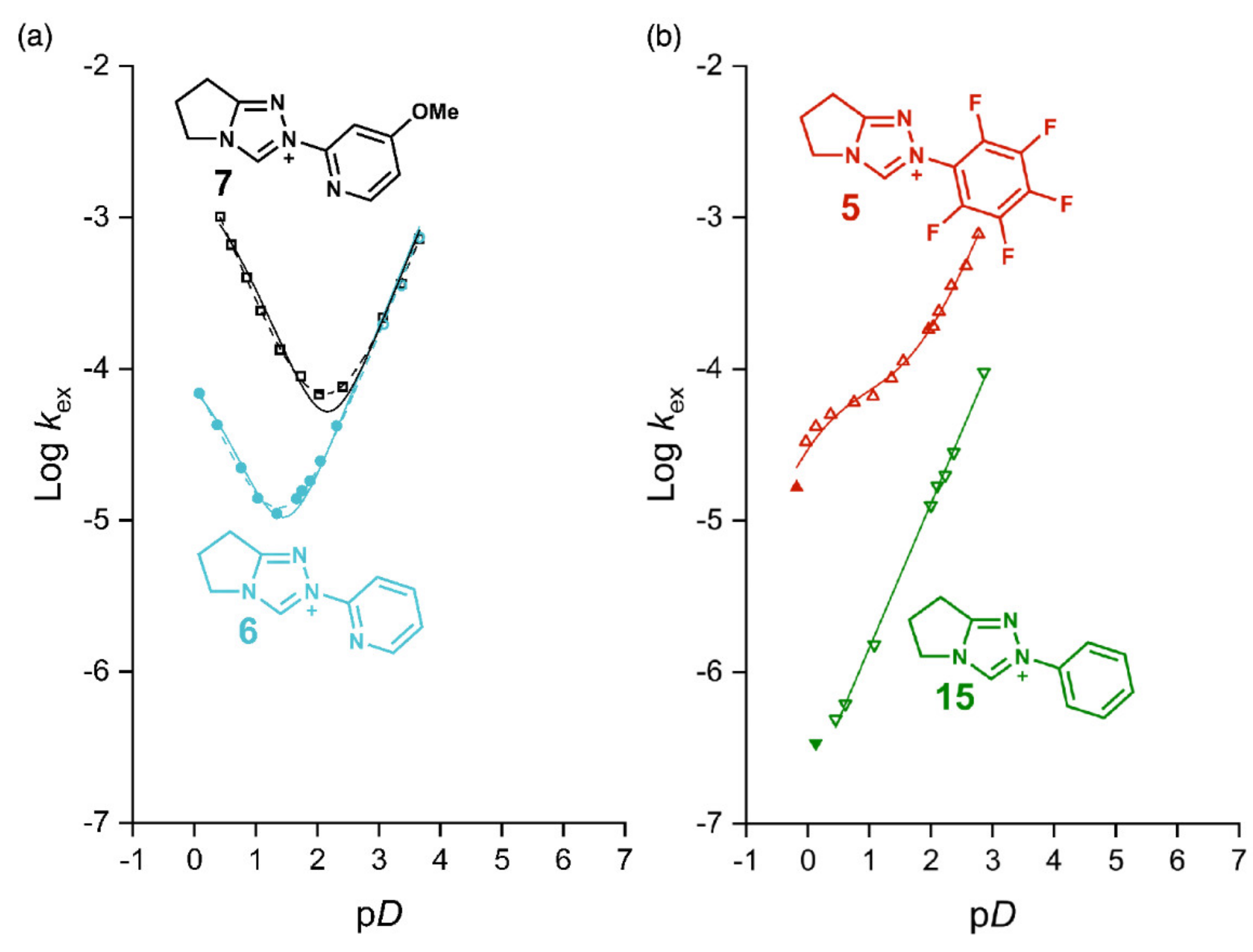
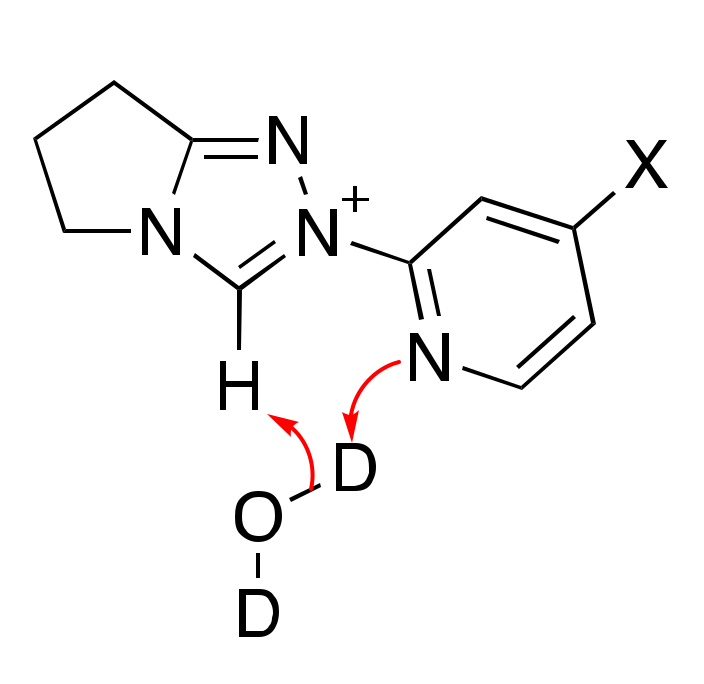
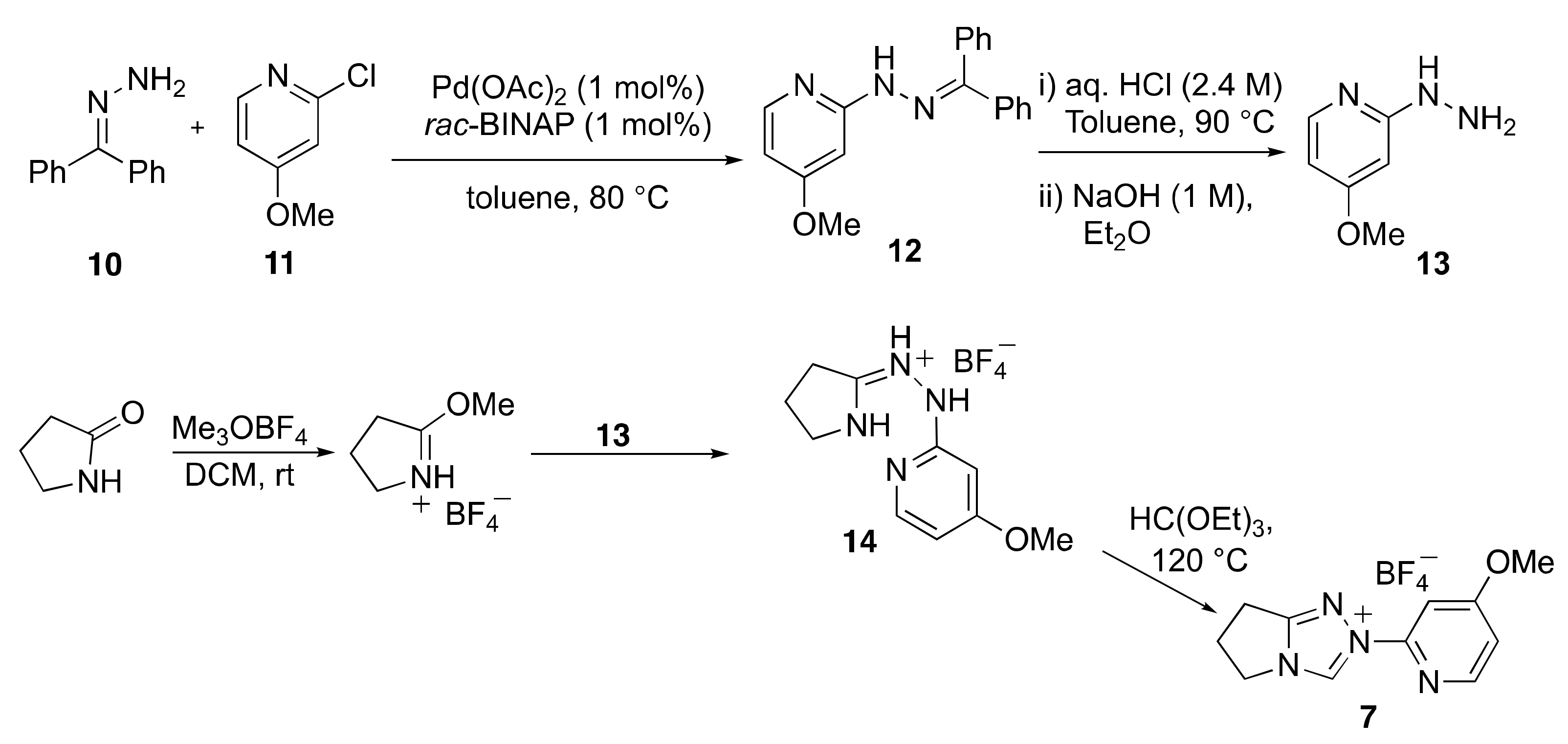
2.1. Kinetic Analysis of the C(3)-H/D Exchange Reactions of N-Pyridyl Triazolium Salts 6 and 7
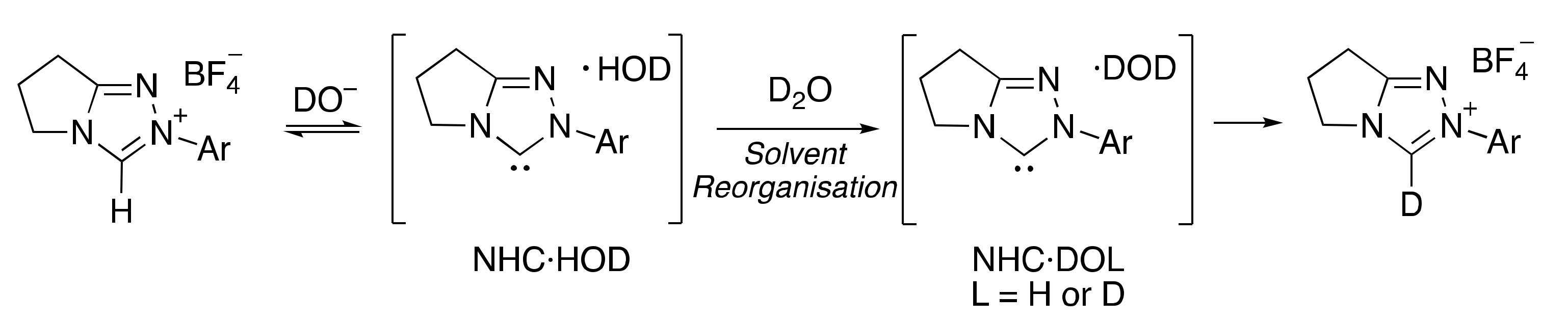
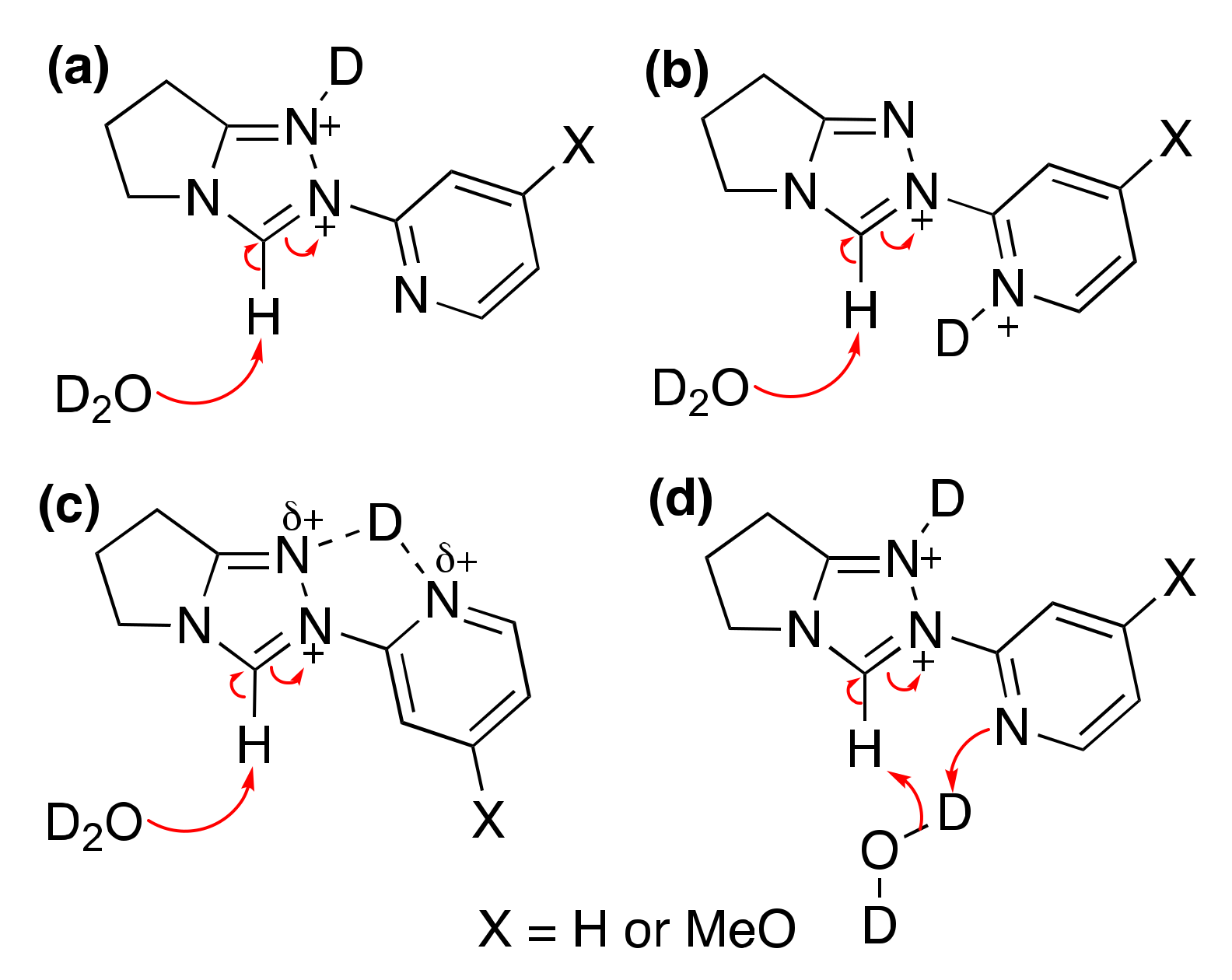
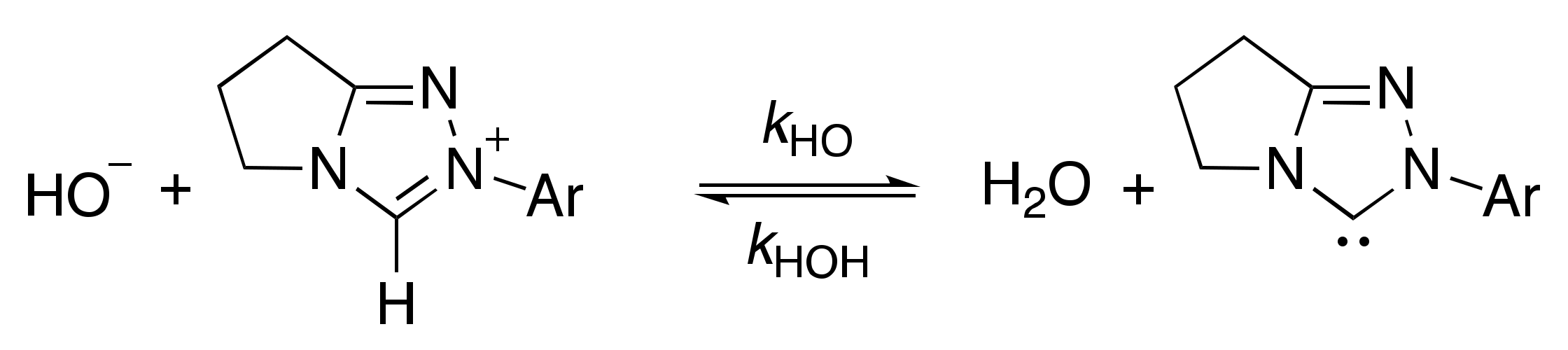
2.1.1. Mechanistic Options for the Region of + 1 Slope in log kex–pD Profiles at Higher pD Values (First-Order Dependence on DO−)
2.1.2. Mechanistic Options for the Region of –1 Slope in log kex–pD Profile (First-Order Dependence on D3O+)
2.1.3. Mechanistic Options for the (Potential) Region of Zero Slope in log kex–pD Profile (pD-Independent Region)
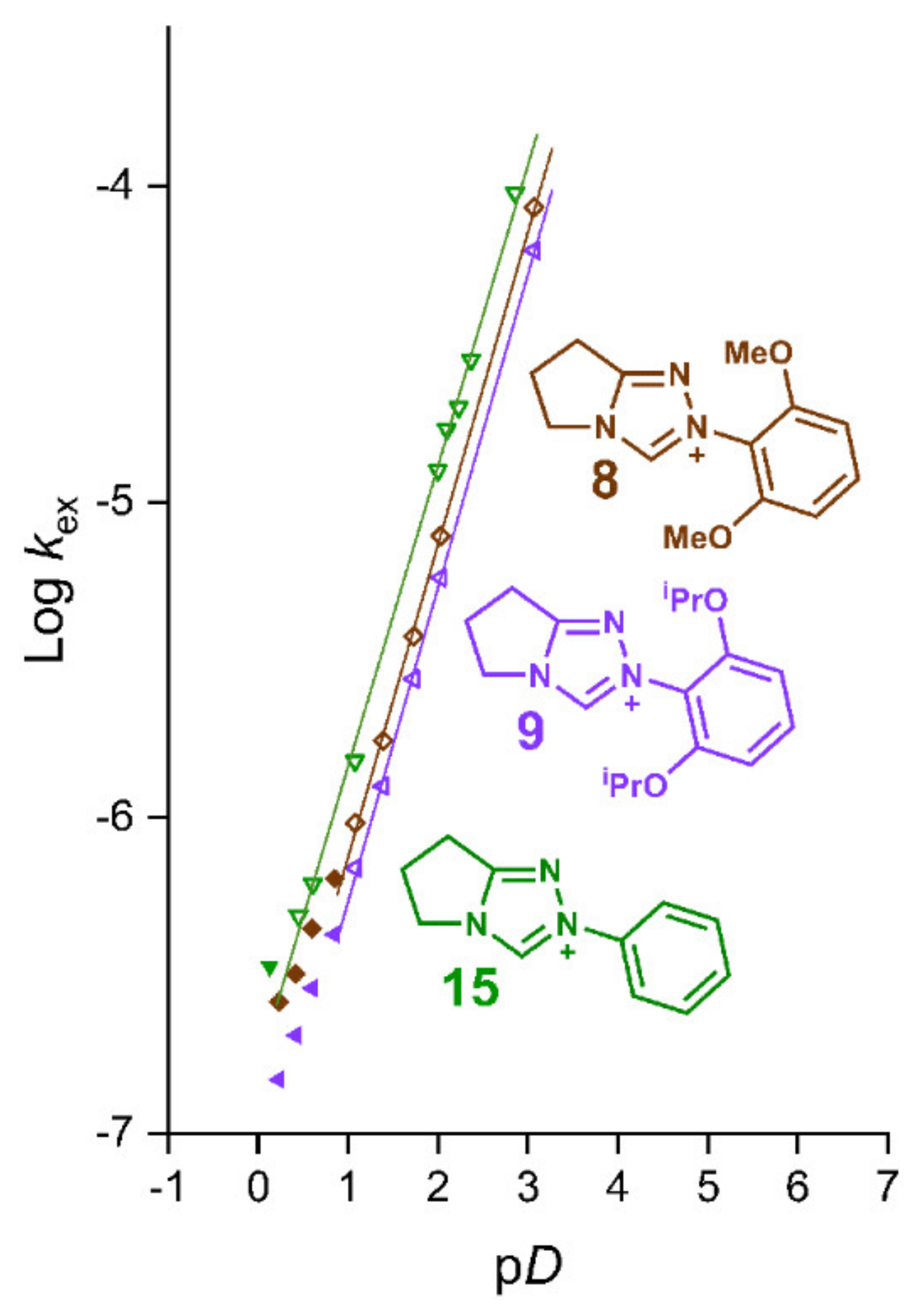
2.2. Kinetic Analysis of the C(3)-H/D Exchange Reactions of N-di-Ortho-Alkoxy Triazolium Salts 8 and 9
2.3. Estimation of Carbon Acid C(3)-H pKa Values
3. Conclusions
Supplementary Materials
 ) and the two dicationic forms afforded from N-protonation of the triazolyl N(1) (
) and the two dicationic forms afforded from N-protonation of the triazolyl N(1) ( ) or the pyridyl nitrogen (
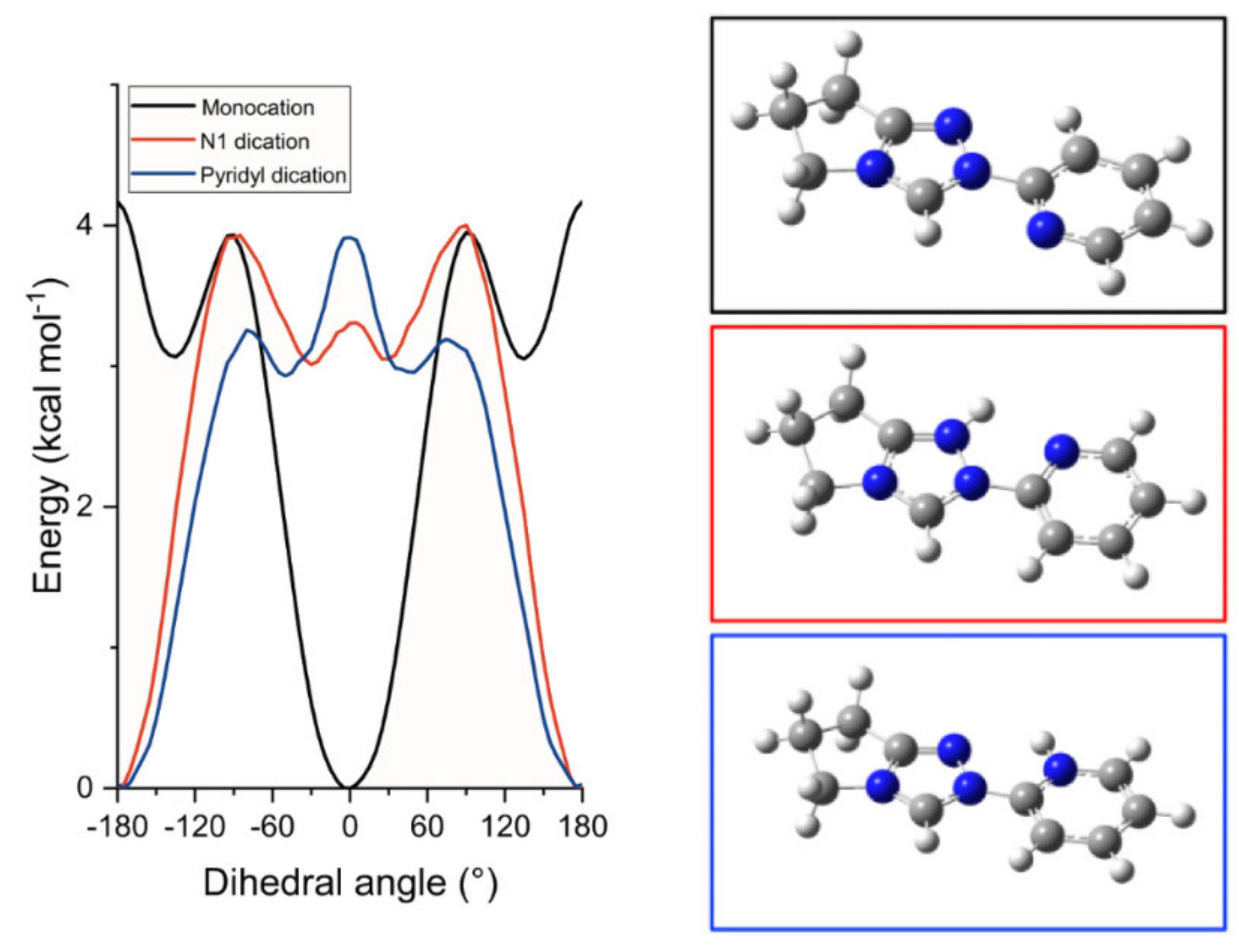
) or the pyridyl nitrogen ( ) obtained by DFT calculations using B3LYP (6-311G++(d,p) basis set, PCM methanol). Figure S16—The effect of dihedral angle between N-aryl and triazolium ring on the calculated energy of the dication of 6 resulting from pyrid-2-yl nitrogen protonation obtained using B3LYP/6-311g++ (d,p) or M062X/6-311g++ (d,p) with PCM (water). Points are calculated energies with the solid curve an interpolation between the data points.
) obtained by DFT calculations using B3LYP (6-311G++(d,p) basis set, PCM methanol). Figure S16—The effect of dihedral angle between N-aryl and triazolium ring on the calculated energy of the dication of 6 resulting from pyrid-2-yl nitrogen protonation obtained using B3LYP/6-311g++ (d,p) or M062X/6-311g++ (d,p) with PCM (water). Points are calculated energies with the solid curve an interpolation between the data points.Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- Breslow, R. On the mechanism of thiamine action. IV. 1 Evidence from studies on model systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Hachisu, Y.; Bode, J.W.; Suzuki, K. Catalytic intramolecular crossed aldehyde—Ketone benzoin reactions: A novel synthesis of functionalized preanthraquinones. J. Am. Chem. Soc. 2003, 125, 8432–8433. [Google Scholar] [CrossRef]
- Li, G.-Q.; Dai, L.-X.; You, S.-L. Thiazolium-derived N-heterocyclic carbene-catalyzed cross-coupling of aldehydes with unactivated imines. Chem. Commun. 2007, 852–854. [Google Scholar] [CrossRef]
- Delany, E.G.; Connon, S.J. Enantioselective N-heterocyclic carbene-catalysed intermolecular crossed benzoin condensations: Improved catalyst design and the role of in situ racemisation. Org. Biomol. Chem. 2020, 19, 248–258. [Google Scholar] [CrossRef]
- Rose, C.A.; Gundala, S.; Fagan, C.L.; Franz, J.F.; Connon, S.J.; Zeitler, K. NHC-catalysed, chemoselective crossed-acyloin reactions. Chem. Sci. 2012, 3, 735–740. [Google Scholar] [CrossRef]
- O’Toole, S.E.; Rose, C.A.; Gundala, S.; Zeitler, K.; Connon, S.J. Highly chemoselective direct crossed aliphatic-aromatic acyloin condensations with triazolium-derived carbene catalysts. J. Org. Chem. 2011, 76, 347–357. [Google Scholar] [CrossRef]
- Ramanjaneyulu, B.T.; Mahesh, S.; Vijaya Anand, R. N-heterocyclic carbene catalyzed highly chemoselective intermolecular crossed acyloin condensation of aromatic aldehydes with trifluoroacetaldehyde ethyl hemiacetal. Org. Lett. 2015, 17, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Sunoj, R.B.; Pareek, M.; Reddi, Y. Tale of the Breslow Intermediate, a Central Player in N-Heterocyclic Carbene Organocatalysis: Then and Now. Chem. Sci. 2021, 12, 7973–7992. [Google Scholar]
- Berkessel, A.; Harnying, W.; Sudkaow, P.; Biswas, A. N-Heterocyclic Carbene/Carboxylic Acid Co-Catalysis Enables Oxidative Esterification of Demanding Aldehydes/Enals, at Low Catalyst Loading. Angew. Chem. Int. Ed. 2021, 60, 19631–19636. [Google Scholar]
- Grasa, G.A.; Kissling, R.M.; Nolan, S.P. N-heterocyclic carbenes as versatile nucleophilic catalysts for transesterification/acylation reactions. Org. Lett. 2002, 4, 3583–3586. [Google Scholar] [CrossRef]
- Nyce, G.W.; Lamboy, J.A.; Connor, E.F.; Waymouth, R.M.; Hedrick, J.L. Expanding the catalytic activity of nucleophilic N-heterocyclic carbenes for transesterification reactions. Org. Lett. 2002, 4, 3587–3590. [Google Scholar] [CrossRef]
- Zeitler, K. Stereoselective synthesis of (E)-alpha,beta-unsaturated esters via carbene-catalyzed redox esterification. Org. Lett. 2006, 8, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Grasa, G.A.; Güveli, T.; Singh, R.; Nolan, S.P. Efficient transesterification/acylation reactions mediated by N-heterocyclic carbene catalysts. J. Org. Chem. 2003, 68, 2812–2819. [Google Scholar] [CrossRef]
- Lai, C.L.; Lee, H.M.; Hu, C.H. Theoretical study on the mechanism of N-heterocyclic carbene catalyzed transesterification reactions. Tetrahedron Lett. 2005, 46, 6265–6270. [Google Scholar] [CrossRef]
- Singh, R.; Nolan, S.P. Synthesis of phosphorus esters by transesterification mediated by N-heterocyclic carbenes (NHCs). Chem. Commun. 2005, 5456–5458. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kissling, R.M.; Letellier, M.-A.; Nolan, S.P. Transesterification/acylation of secondary alcohols mediated by N-heterocyclic carbene catalysts. J. Org. Chem. 2004, 69, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Li, Y.; Hu, H.; Zeng, C.; Wang, S.; Xu, H.; Gao, Y. N-Heterocyclic Carbene Functionalized Covalent Organic Framework for Transesterification of Glycerol with Dialkyl Carbonates. Catalysts. 2021, 11, 423. [Google Scholar] [CrossRef]
- Zeng, T.; Song, G.; Li, C.-J. Separation, recovery and reuse of N-heterocyclic carbene catalysts in transesterification reactions. Chem. Commun. 2009, 41, 6249–6251. [Google Scholar] [CrossRef]
- Du, G.-F.; Guo, H.; Wang, Y.; Li, W.-J.; Shi, W.-J.; Dai, B. N-heterocyclic carbene catalyzed synthesis of dimethyl carbonate via transesterification of ethylene carbonate with methanol. J. Saudi Chem. Soc. 2015, 19, 112–115. [Google Scholar] [CrossRef]
- Sohn, S.S.; Rosen, E.L.; Bode, J.W. N-heterocyclic carbene-catalyzed generation of homoenolates: Gamma-butyrolactones by direct annulations of enals and aldehydes. J. Am. Chem. Soc. 2004, 126, 14370–14371. [Google Scholar] [CrossRef]
- He, M.; Bode, J.W. Catalytic Synthesis of gamma-Lactams via direct annulations of enals and N-sulfonylimines. Org. Lett. 2005, 7, 3131–3134. [Google Scholar] [CrossRef]
- Rommel, M.; Fukuzumi, T.; Bode, J.W. Cyclic ketimines as superior electrophiles for NHC-catalyzed homoenolate additions with broad scope and low catalyst loadings. J. Am. Chem. Soc. 2008, 130, 17266–17267. [Google Scholar] [CrossRef]
- Sun, L.-H.; Shen, L.-T.; Ye, S. Highly diastereo- and enantioselective NHC-catalyzed [3+2] annulation of enals and isatins. Chem. Commun. 2011, 47, 10136–10138. [Google Scholar] [CrossRef]
- Guo, C.; Fleige, M.; Janssen-Müller, D.; Daniliuc, C.G.; Glorius, F. Switchable selectivity in an NHC-catalysed dearomatizing annulation reaction. Nature Chem. 2015, 7, 842. [Google Scholar] [CrossRef]
- Yetra, S.R.; Mondal, S.; Mukherjee, S.; Gonnade, R.G.; Biju, A.T. Enantioselective Synthesis of Spirocyclohexadienones by NHC-Catalyzed Formal [3+3] Annulation Reaction of Enals. Angew. Chem. Int. Ed. 2016, 55, 268–272. [Google Scholar] [CrossRef]
- Xie, Y.; Li, L.; Sun, S.; Wu, Z.; Lang, M.; Jiang, D.; Wang, J. Enantioselective NHC-Catalyzed [3+3] Annulation of α-Bromoenals with 2-Aminobenzimidazoles. Org. Lett. 2020, 22, 391–394. [Google Scholar] [CrossRef]
- Liu, L.; Guo, D.; Wang, J. NHC-Catalyzed Asymmetric α-Regioselective [4+2] Annulation to Construct α-Alkylidene-δ-lactones. Org. Lett. 2020, 22, 7025–7029. [Google Scholar] [CrossRef]
- Lyngvi, E.; Bode, J.W.; Schoenebeck, F. A computational study of the origin of stereoinduction in NHC-catalyzed annulation reactions of α, β-unsaturated acyl azoliums. Chem. Sci. 2012, 3, 2346–2350. [Google Scholar] [CrossRef]
- Nguyen, X.B.; Nakano, Y.; Duggan, N.M.; Scott, L.; Breugst, M.; Lupton, D.W. N-Heterocyclic Carbene Catalyzed (5+1) Annulations Exploiting a Vinyl Dianion Synthon Strategy. Angew. Chem. Int. Ed. 2019, 58, 11483–11490. [Google Scholar] [CrossRef]
- Draskovits, M.; Kalaus, H.; Stanetty, C.; Mihovilovic, M.D. Intercepted dehomologation of aldoses by N-heterocyclic carbene catalysis–a novel transformation in carbohydrate chemistry. Chem. Commun. 2019, 55, 12144–12147. [Google Scholar] [CrossRef]
- Zhao, L.-L.; Li, X.-S.; Cao, L.-L.; Zhang, R.; Shi, X.-Q.; Qi, J. Access to dihydropyridinones and spirooxindoles: Application of N-heterocyclic carbene-catalyzed [3+3] annulation of enals and oxindole-derived enals with 2-aminoacrylates. Chem. Commun. 2017, 53, 5985–5988. [Google Scholar] [CrossRef]
- Schedler, M.; Wurz, N.E.; Daniliuc, C.G.; Glorius, F. N-Heterocyclic carbene catalyzed umpolung of styrenes: Mechanistic elucidation and selective tail-to-tail dimerization. Org. Lett. 2014, 16, 3134–3137. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J.H.; Melder, J.P.; Ebel, K.; Brode, S. Preparation, Structure, and Reactivity of 1,3,4-Triphenyl-4,5-Dihydro-1h-1,2,4-Triazol-5-Ylidene, a New Stable Carbene. Angew. Chem. Int. Ed. 1995, 34, 1021–1023. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Raabe, G.; Simonet, J.; Ghanimi, A.; Stegmann, H.B.; Teles, J.H. A stable carbene as π-acceptor electrochemical reduction to the radical anion. Tetrahedron Lett. 1997, 38, 2833–2836. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Teles, J.; Ebel, K. 1, 3, 4-Triphenyl-4, 5-dihydro-1H-1, 2, 4-triazol-5-ylidene–applications of a stable carbene in synthesis and catalysis. J. Prakt. Chem. 1997, 339, 397–399. [Google Scholar] [CrossRef]
- Nair, V.; Vellalath, S.; Babu, B.P. Recent advances in carbon–carbon bond-forming reactions involving homoenolates generated by NHC catalysis. Chem. Soc. Rev. 2008, 37, 2691–2698. [Google Scholar] [CrossRef]
- Zhao, C.; Blaszczyk, S.A.; Wang, J. Asymmetric reactions of N-heterocyclic carbene (NHC)-based chiral acyl azoliums and azolium enolates. Green Synth. Catal. 2021, 2, 198–215. [Google Scholar] [CrossRef]
- Song, R.; Jin, Z.; Chi, Y.R. NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions. Chem. Sci. 2021, 12, 5037–5043. [Google Scholar] [CrossRef]
- Dai, L.; Ye, S. Recent advances in N-heterocyclic carbene-catalyzed radical reactions. Chin. Chem. Lett. 2020, 32, 660–667. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-heterocyclic carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef]
- Grossmann, A.; Enders, D. N-heterocyclic carbene catalyzed domino reactions. Angew. Chem. Int. Ed. 2012, 51, 314–325. [Google Scholar] [CrossRef]
- Chen, X.Y.; Liu, Q.; Chauhan, P.; Enders, D. N-Heterocyclic Carbene Catalysis via Azolium Dienolates: An Efficient Strategy for Remote Enantioselective Functionalizations. Angew. Chem. Int. Ed. 2018, 57, 3862–3873. [Google Scholar] [CrossRef]
- Knight, R.L.; Leeper, F.J. Comparison of chiral thiazolium and triazolium salts as asymmetric catalysts for the benzoin condensation. J. Chem. Soc.-Perkin Trans. 1 1998, 1891–1893. [Google Scholar] [CrossRef]
- Campbell, C.D.; Collett, C.J.; Thomson, J.E.; Slawin, A.M.; Smith, A.D. Organic base effects in NHC promoted O-to C-carboxyl transfer; chemoselectivity profiles, mechanistic studies and domino catalysis. Org. Biomol. Chem. 2011, 9, 4205–4218. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Concellón, C.; Smith, A.D. Catalytic enantioselective Steglich rearrangements using chiral N-heterocyclic carbenes. Tetrahedron Asymmetry 2011, 22, 797–811. [Google Scholar] [CrossRef]
- Baragwanath, L.; Rose, C.A.; Zeitler, K.; Connon, S.J. Highly enantioselective benzoin condensation reactions involving a bifunctional protic pentafluorophenyl-substituted triazolium precatalyst. J. Org. Chem. 2009, 74, 9214–9217. [Google Scholar] [CrossRef]
- DiRocco, D.A.; Oberg, K.M.; Dalton, D.M.; Rovis, T. Catalytic asymmetric intermolecular stetter reaction of heterocyclic aldehydes with nitroalkenes: Backbone fluorination improves selectivity. J. Am. Chem. Soc. 2009, 131, 10872–10874. [Google Scholar] [CrossRef]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Collett, C.J.; Massey, R.S.; Maguire, O.R.; Batsanov, A.S.; O’Donoghue, A.C.; Smith, A.D. Mechanistic insights into the triazolylidene-catalysed Stetter and benzoin reactions: Role of the N-aryl substituent. Chem. Sci. 2013, 4, 1514–1522. [Google Scholar] [CrossRef]
- Collett, C.J.; Massey, R.S.; Taylor, J.E.; Maguire, O.R.; O’Donoghue, A.C.; Smith, A.D. Rate and equilibrium constants for the addition of N-heterocyclic carbenes into benzaldehydes: A remarkable 2-substituent effect. Angew. Chem. Int. Ed. 2015, 54, 6887–6892. [Google Scholar] [CrossRef]
- Massey, R.S.; Murray, J.; Collett, C.J.; Zhu, J.; Smith, A.D.; O’Donoghue, A.C. Kinetic and structure–activity studies of the triazolium ion-catalysed benzoin condensation. Org. Biomol. Chem. 2021, 19, 387–393. [Google Scholar] [CrossRef]
- Collett, C.J.; Young, C.M.; Massey, R.S.; O’Donoghue, A.C.; Smith, A.D. Kinetic and Structure-Activity Studies of the Triazolium Ion-catalyzed Intramolecular Stetter Reaction. Eur. J. Org. Chem. 2021, 26, 3670–3675. [Google Scholar] [CrossRef]
- Wu, S.; Liu, C.; Luo, G.; Jin, Z.; Zheng, P.; Chi, Y.R. NHC-Catalyzed Chemoselective Reactions of Enals and Aminobenzaldehydes for Access to Chiral Dihydroquinolines. Angew. Chem. Int. Ed. 2019, 58, 18410–18413. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.S.; Biju, A.T.; Nair, V. Recent advances in N-heterocyclic carbene (NHC)-catalysed benzoin reactions. Beilstein J. Org. Chem. 2016, 12, 444–461. [Google Scholar] [CrossRef]
- Mahatthananchai, J.; Bode, J.W. The effect of the N-mesityl group in NHC-catalyzed reactions. Chem. Sci. 2012, 3, 192–197. [Google Scholar] [CrossRef]
- Delany, E.G.; Connon, S.J. Highly chemoselective intermolecular cross-benzoin reactions using an ad hoc designed novel N-heterocyclic carbene catalyst. Org. Biomol. Chem. 2018, 16, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Raed, A.A.; Dhayalan, V.; Barkai, S.; Milo, A. N-Heterocyclic Carbene Triazolium Salts Containing Brominated Aromatic Motifs: Features and Synthetic Protocol. CHIMIA 2020, 74, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Mayr, H.; Ofial, A.R. Philicities, fugalities, and equilibrium constants. Acc. Chem. Res. 2016, 49, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Higgins, E.M.; Sherwood, J.A.; Lindsay, A.G.; Armstrong, J.; Massey, R.S.; Alder, R.W.; O’Donoghue, A.C. pKas of the conjugate acids of N-heterocyclic carbenes in water. Chem. Commun. 2011, 47, 1559–1561. [Google Scholar] [CrossRef]
- Massey, R.S.; Collett, C.J.; Lindsay, A.G.; Smith, A.D.; O’Donoghue, A.C. Proton transfer reactions of triazol-3-ylidenes: Kinetic acidities and carbon acid pKa values for twenty triazolium salts in aqueous solution. J. Am. Chem. Soc. 2012, 134, 20421–20432. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.C.; Massey, R.S. Contemporary carbene chemistry. In Contemporary Carbene Chemistry; Moss, R.A., Doyle, M.P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Tucker, D.E.; Quinn, P.; Massey, R.S.; Collett, C.J.; Jasiewicz, D.J.; Bramley, C.R.; Smith, A.D.; O’Donoghue, A.C. Proton transfer reactions of N-aryl triazolium salts: Unusual ortho-substituent effects. J. Phys. Org. Chem. 2015, 28, 108–115. [Google Scholar] [CrossRef]
- Massey, R.S.; Quinn, P.; Zhou, S.; Murphy, J.A.; O’Donoghue, A.C. Proton transfer reactions of a bridged bis-propyl bis-imidazolium salt. J. Phys. Org. Chem. 2016, 29, 735–740. [Google Scholar] [CrossRef][Green Version]
- Amyes, T.L.; Diver, S.T.; Richard, J.P.; Rivas, F.M.; Toth, K. Formation and stability of N-heterocyclic carbenes in water: The carbon acid pKa of imidazolium cations in aqueous solution. J. Am. Chem. Soc. 2004, 126, 4366–4374. [Google Scholar] [CrossRef]
- Magill, A.M.; Cavell, K.J.; Yates, B.F. Basicity of nucleophilic carbenes in aqueous and nonaqueous solvents theoretical predictions. J. Am. Chem. Soc. 2004, 126, 8717–8724. [Google Scholar] [CrossRef]
- Konstandaras, N.; Dunn, M.H.; Guerry, M.S.; Barnett, C.D.; Cole, M.L.; Harper, J.B. The impact of cation structure upon the acidity of triazolium salts in dimethyl sulfoxide. Org. Biomol. Chem. 2020, 18, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, J.; Lee, J.K. The importance of N-heterocyclic carbene basicity in organocatalysis. Org. Biomol. Chem. 2018, 16, 8230–8244. [Google Scholar] [CrossRef]
- Paul, M.; Detmar, E.; Schlangen, M.; Breugst, M.; Neudörfl, J.M.; Schwarz, H.; Berkessel, A.; Schäfer, M. Intermediates of N-Heterocyclic Carbene (NHC) Dimerization Probed in the Gas Phase by Ion Mobility Mass Spectrometry: C− H⋅⋅⋅:C Hydrogen Bonding Versus Covalent Dimer Formation. Chem. Eur. J. 2019, 25, 2511–2518. [Google Scholar] [CrossRef]
- Dunn, M.H.; Konstandaras, N.; Cole, M.L.; Harper, J.B. Targeted and Systematic Approach to the Study of pKa Values of Imidazolium Salts in Dimethyl Sulfoxide. J. Org. Chem. 2017, 82, 7324–7331. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Cheng, J.-P. An Acidity Scale of Triazolium-Based NHC Precursors in DMSO. J. Org. Chem. 2017, 82, 9675–9681. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef]
- Chu, Y.; Deng, H.; Cheng, J.-P. An acidity scale of 1, 3-dialkylimidazolium salts in dimethyl sulfoxide solution. J. Org. Chem. 2007, 72, 7790–7793. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Streitwieser, A. Basicity of a Stable Carbene, 1, 3-Di-tert-butylimidazol-2-ylidene, in THF. J. Am. Chem. Soc. 2002, 124, 5757–5761. [Google Scholar] [CrossRef] [PubMed]
- Alder, R.W.; Allen, P.R.; Williams, S.J. Stable carbenes as strong bases. J. Chem. Soc. Chem. Commun. 1995, 12, 1267–1268. [Google Scholar] [CrossRef]
- Dhayalan, V.; Gadekar, S.C.; Alassad, Z.; Milo, A. Unravelling mechanistic features of organocatalysis with in situ modifications at the secondary sphere. Nat. Chem. 2019, 11, 543–551. [Google Scholar] [CrossRef]
- Zak, I.L.; Gadekar, S.C.; Milo, A. Designing the Secondary Coordination Sphere in Small-Molecule Catalysis. Synlett 2021, 32, 329–336. [Google Scholar] [CrossRef]
- Hartwig, J.F. Synthesis, Structure, and Reactivity of a Palladium Hydrazonato Complex: A New Type of Reductive Elimination Reaction to Form C−N Bonds and Catalytic Arylation of Benzophenone Hydrazone. Angew. Chem. Int. Ed. 1998, 37, 2090–2093. [Google Scholar] [CrossRef]
- Wagaw, S.; Yang, B.H.; Buchwald, S.L. A palladium-catalyzed strategy for the preparation of indoles: A novel entry into the Fischer indole synthesis. J. Am. Chem. Soc. 1998, 120, 6621–6622. [Google Scholar] [CrossRef]
- Wagaw, S.; Yang, B.H.; Buchwald, S.L. A palladium-catalyzed method for the preparation of indoles via the Fischer indole synthesis. J. Am. Chem. Soc. 1999, 121, 10251–10263. [Google Scholar] [CrossRef]
- Kerr, M.S.; Read de Alaniz, J.; Rovis, T. An efficient synthesis of achiral and chiral 1,2,4-triazolium salts: Bench stable precursors for N-heterocyclic carbenes. J. Org. Chem. 2005, 70, 5725–5728. [Google Scholar] [CrossRef] [PubMed]
- As pD = − log aD+ therefore 10−pD = aD+ = γD+[D+].
- In our previous publication in relation to 6, we used a rate constant k’ (s−1) rather than kH (s−1) to describe the H/D-exchange reaction of substrate in the region of close to −1 slope. Owing to the consideration of an additional pD-independent region in the present manuscript, and the need for a third rate constant to describe this region, we have changed k’ (s−1) to kH (s−1) for the −1 region. The latter more clearly denotes a region involving formal acid catalysis.
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Phys. Chem. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+ G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]






 ) and the two dicationic forms afforded from N-protonation of the triazolyl N(1) () or the pyridyl nitrogen () obtained by DFT calculations using a B3LYP (6–311G++(d,p) basis set [84,85,86], PCM water [87]). The inset ball and stick diagrams show the structures corresponding to the lowest energy conformations of N-pyrid-2-yl triazolium monocation 6 () and the two dicationic forms () or ().
) and the two dicationic forms afforded from N-protonation of the triazolyl N(1) () or the pyridyl nitrogen () obtained by DFT calculations using a B3LYP (6–311G++(d,p) basis set [84,85,86], PCM water [87]). The inset ball and stick diagrams show the structures corresponding to the lowest energy conformations of N-pyrid-2-yl triazolium monocation 6 () and the two dicationic forms () or ().
) and the two dicationic forms afforded from N-protonation of the triazolyl N(1) () or the pyridyl nitrogen () obtained by DFT calculations using a B3LYP (6–311G++(d,p) basis set [84,85,86], PCM water [87]). The inset ball and stick diagrams show the structures corresponding to the lowest energy conformations of N-pyrid-2-yl triazolium monocation 6 () and the two dicationic forms () or ().
) and the two dicationic forms afforded from N-protonation of the triazolyl N(1) () or the pyridyl nitrogen () obtained by DFT calculations using a B3LYP (6–311G++(d,p) basis set [84,85,86], PCM water [87]). The inset ball and stick diagrams show the structures corresponding to the lowest energy conformations of N-pyrid-2-yl triazolium monocation 6 () and the two dicationic forms () or ().




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triazolium Salt | kDO (M−1s−1) | kH (s−1) | kin (s−1) | Fit | pKa [C(3)-H] 3 |
|---|---|---|---|---|---|
 | 1.01 (±0.05) × 108 | 1.35 (±0.44) × 10−4 | - | Equation (3) (R2 = 0.991) | - |
| 8.79 (±0.30) × 107 | 4.36 (±3.52) × 10−4 | 1.97 × 10−5 1 | Equation (4) (R2 = 0.998) | 17.4 | |
 | 9.57 (±0.84) × 107 | 2.22 (±0.93) × 10−3 | - | Equation (3) (R2 = 0.976) | - |
| 8.02 (±0.22) × 107 | 2.30 (±35.9) × 10−1 | 2.59 × 10−3 1 | Equation (4) (R2 = 0.999) | 17.5 | |
 | 3.87 (±0.07) × 107 | - | - | Equation (5) 2 (R2 = 0.999) | 17.8 |
 | 2.87 (±0.13) × 107 | - | - | Equation (5) 2 (R2 = 0.999) | 17.9 |
 | 6.82 × 107 4 | - | - | Equation (5) | 17.5 4 |
 | 4.20 × 107 4 | - | - | Equation (5) | 17.8 4 |
 | 5.29 × 107 4 | - | - | Equation (5) | 17.7 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, P.; Smith, M.S.; Zhu, J.; Hodgson, D.R.W.; O’Donoghue, A.C. Triazolium Salt Organocatalysis: Mechanistic Evaluation of Unusual Ortho-Substituent Effects on Deprotonation. Catalysts 2021, 11, 1055. https://doi.org/10.3390/catal11091055
Quinn P, Smith MS, Zhu J, Hodgson DRW, O’Donoghue AC. Triazolium Salt Organocatalysis: Mechanistic Evaluation of Unusual Ortho-Substituent Effects on Deprotonation. Catalysts. 2021; 11(9):1055. https://doi.org/10.3390/catal11091055
Chicago/Turabian StyleQuinn, Peter, Matthew S. Smith, Jiayun Zhu, David R. W. Hodgson, and AnnMarie C. O’Donoghue. 2021. "Triazolium Salt Organocatalysis: Mechanistic Evaluation of Unusual Ortho-Substituent Effects on Deprotonation" Catalysts 11, no. 9: 1055. https://doi.org/10.3390/catal11091055
APA StyleQuinn, P., Smith, M. S., Zhu, J., Hodgson, D. R. W., & O’Donoghue, A. C. (2021). Triazolium Salt Organocatalysis: Mechanistic Evaluation of Unusual Ortho-Substituent Effects on Deprotonation. Catalysts, 11(9), 1055. https://doi.org/10.3390/catal11091055