
Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene



School of Chemical Engineering, Pusan National University, Busan 46241, Korea
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(9), 1074; https://doi.org/10.3390/catal11091074
Submission received: 6 August 2021
/
Revised: 2 September 2021
/
Accepted: 3 September 2021
/
Published: 7 September 2021
(This article belongs to the Special Issue Catalytic Pyrolysis for Environmental Applications)
Abstract
:In this work, sulfated and phosphated metal oxides were studied as catalysts for the Diels–Alder cycloaddition of biomass-derived 2,5-dimethylfuran (DMF) and ethylene to understand the effect of acid strength on the reaction. Four catalysts with varied acidity, namely sulfated SiO2, sulfated TiO2, phosphated SiO2, and phosphated TiO2, were prepared via wet impregnation using sulfuric acid and phosphoric acid as precursors, and their structural and acid properties were examined using X-ray diffraction, Brunauer–Emmett–Teller analysis, Fourier transform infrared spectroscopy, solid state 31P magic angle spinning nuclear magnetic resonance spectroscopy, and temperature programmed desorption of ammonia. The results revealed that the acidity of the catalysts was largely influenced by the type of the acid functional group and the support as well as the calcination temperature. The conversion of DMF and the selectivity toward p-Xylene (PX) were generally correlated with the total acid site density and the acid–metal oxide interaction strength, which in turn affected the acid strength. Overall, phosphated SiO2 and TiO2 calcined at 773 K were identified as the most active and selective catalysts, exhibiting a high PX selectivity of over 70% and DMF conversion of 80% at 523 K after 6 h. The origin of the stability of the highly active phosphated catalysts was also investigated in detail.
1. Introduction
The global production of chemicals largely relies on fossil fuels such as coal, natural gas, and petroleum, and the ever-increasing use of these chemicals accelerates the emission of greenhouse gases such as CO2 and air pollutants such as NOX and SOX. In this regard, lignocellulosic biomass has attracted significant attention as an alternative carbon source to fossil fuels owing to its carbon neutrality and low pollutant content [1]. Among the various petroleum-derived chemicals, aromatic chemicals such as benzene, toluene, and xylene (BTX) are the most widely used chemical feedstocks for the synthesis of a range of polymers including polystyrene, polyurethane, and polyethylene terephthalate [2]. Thus, the development of catalytic reactions for the conversion of biomass to BTX chemicals is of particular importance. The Diels–Alder cycloaddition of biomass-derived furans and ethylene is the most atom-efficient route for the production of BTX from biomass [3]. The chemistry of this reaction was first proposed by Timothy A. Brandvold (in UOP) using 2,5-dimethylfuran (DMF) as a reactant for the synthesis of p-Xylene (PX) [4]. Using various solid acid catalysts, such as zeolites and activated carbon, a decent yield of PX (>50%) was obtained. The DMF could be easily produced from solid biomass using an existing and well-established process: the solid biomass is first hydrolyzed into a C6 sugar, such as glucose, which is then converted into 2,5-hydroxymethylfurfural (HMF) through dehydration; the HMF is finally converted into DMF via hydrogenolysis in the presence of copper-based catalysts [5]. Therefore, this cycloaddition chemistry of DMF could be utilized for the production of renewable PX [6]. Later, cycloaddition chemistry was successfully extended to other C4–C5 furanic compounds such as 2-methylfuran and furan for the synthesis of toluene and benzene, demonstrating that the complete production of BTX chemicals from biomass is possible [7,8].
The Diels–Alder conversion of DMF and ethylene to PX proceeds via a two-step process and is catalyzed by BrØnsted and Lewis acid catalysts, as shown in Figure 1 [9]. The first step is the formation of a thermally stable oxanorbornene cycloadduct through Diels–Alder cycloaddition. Lewis acids can catalyze this step, whereas BrØnsted acids do not exhibit any catalytic effect on this step. The second step is the dehydrative aromatization of the cycloadduct intermediate to produce PX. This step cannot proceed in the absence of a catalyst. Both Lewis and BrØnsted acids are suitable for catalyzing the dehydrative aromatization. However, density functional theory calculations have shown that BrØnsted acids are significantly more effective for the dehydration than Lewis acids [10]. In addition to PX formation, diverse side reactions can occur. These side reactions include (1) hydrolysis of DMF to 2,5-hexanedione (HDO), (2) isomerization of the DMF/ethylene cycloadduct to dimethylcyclohexenone, and (3) alkylation of PX with ethylene [11,12]. Thus, achieving the selectivity toward PX over other side products is the key for obtaining high yields of PX.
To date, a wide range of solid acid catalysts have been investigated for the Diels–Alder conversion of DMF to PX, with the goal of obtaining high PX production rate and selectivity [2,6,9,13,14,15,16,17,18,19]. Williams et al. reported that a high PX selectivity (~75%) is achieved over H-Y zeolites when a hydrophobic solvent, such as n-heptane, is used [8]. Later, Chang et al. reported that the selectivity to PX could be increased to 90% using H-Beta zeolite in heptane [2]. In addition to zeolites, tungstated zirconia (WOx-ZrO2) exhibited a higher turnover rate for PX production, even higher than that in the presence of H-Y zeolites, with a high PX selectivity of ~80% [6]. Later, Wijaya et al. showed that heteropolytungstates, such as HSiW and HPW, supported on silica are highly active catalysts for PX production and exhibit turnover rates greater than six times those in the presence of conventional zeolites such as H-Beta (407.8 h−1 vs. 60.0 h−1) [13]. Overall, the general conclusion of these studies is that the presence of strong BrØnsted acid sites is the key for enhancing the formation rate of PX and that the second step, i.e., dehydration of the cycloadduct, is the rate-determining step of the overall reaction. Thus, the strength of the BrØnsted acid sites is correlated to the turnover rate for PX production.
In addition to achieving high activity, ensuring the catalyst durability is a major concern in the Diels–Alder conversion of DMF to PX. It has been shown that catalysts with strong BrØnsted acid sites, such as zeolites and heteropolytungstates, are deactivated significantly after repeated reaction cycles in a batch reactor as well as in a continuous flow reactor [13]. The catalyst deactivation mainly occurs owing to carbon deposits on the catalyst surface, which result from the oligomerization of reaction intermediates such as HDO. Although highly crystalline catalysts, such as zeolites, can be reused after removing coke through high-temperature calcination, it is desirable to increase the catalyst durability to the maximum possible extent to reduce the processing cost. Because strong BrØnsted acids likely accelerate the formation of undesirable carbon deposits as well as the desired PX, the acid strength of the catalyst should be carefully tuned to achieve a balance between the catalyst activity and durability. In this regard, the effect of BrØnsted acid strength on the Diels–Alder reaction selectivity and stability using the catalysts with carefully tuned BrØnsted acid strength would be worth investigating for designing more robust catalysts. However, to the best of our knowledge, no such studies have been conducted yet.
Sulfuric acid and phosphoric acid, which are inorganic liquid acids, are representative strong and weak acid catalysts, respectively. Although these are homogeneous acid catalysts, these can be easily heterogenized after immobilization onto a metal oxide support. It was reported that liquid acids anchored to the metal oxide supports retain their acid strength and that the support type can subtly impact the acid strength because of the varied acid molecule–support interaction [11]. Therefore, these can be good model catalysts for understanding the influence of acid strength. In a few studies, liquid acid-anchored metal oxides have been used as catalysts for the Diels–Alder conversion of DMF to PX [11,18,19]. Cho et al. reported the conversion of DMF to PX, with a product yield exceeding 90%, using phosphoric acid-anchored siliceous beta zeolites [18]. Feng et al. reported a high PX selectivity of >80% using mesoporous silica anchored with sulfonic acid [11].
In this study, the activity and stability of sulfated and phosphated catalysts for the Diels–Alder conversion of DMF to PX were comprehensively investigated for the first time to elucidate the role of acid strength in the reaction. Four different catalysts with varying acidity, namely sulfated SiO2, sulfated TiO2, phosphated SiO2, and phosphated TiO2, were prepared via wet impregnation using sulfuric acid and phosphoric acid as precursors, and their structural and acid properties were examined using X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) analysis, Fourier transform infrared (FT-IR) spectroscopy, solid state 31P magic-angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy, and temperature programmed desorption of ammonia. The acidity–activity correlation and the stability of the liquid acid-anchored catalysts were then investigated in detail.
2. Results and Discussion
2.1. Catalyst Characterization
XRD patterns of the sulfated and phosphated catalysts are shown in Figure 2. The parent silica and titania support are also included in the figure. All the silica-based catalysts exhibited a broad diffraction peak at 2θ = 22.8°, corresponding to that for amorphous silica. For the titania-based catalysts, the diffraction peaks at 2θ = 25.2 and 38.0° could be assigned to the anatase structure (JCPDS card no. 21-1272), whereas that at 2θ = 28.0° could be assigned to the rutile structure (JCPDS card no. 21-1276). This indicated that the titania support has a mixed crystal structure. Both the silica and titania catalysts did not show any diffraction peaks corresponding to the sulfate and phosphate framework structure.
The N2 adsorption/desorption isotherms of the sulfated and phosphated catalysts are shown in Figure S1. All the catalysts exhibited a type IV isotherm with a hysteresis loop in a relative pressure (P/P0) range of 0.9–1.0. The BET surface area and the total pore volume and micropore volume of the catalysts were also calculated and are listed in Table 1. When the neat support was treated with sulfuric acid and phosphoric acid, the surface area and pore volume generally decreased, probably owing to the pore blockage induced by the acid functionalities attached to the oxide surface. The decreases in surface area and pore volume were more significant in the phosphated catalysts than in the sulfated catalysts. The calcination temperature also influenced the surface area of the catalysts. SiP 500 and TiP 500 had lower pore volumes and lower surface areas than those of SiP 300 and TiP 300, suggesting that a higher calcination temperature led to a greater reduction in the catalyst surface area.
The chemical states of the acid molecules anchored onto the support were determined via solid state NMR spectroscopy and FT-IR spectroscopy. Figure 3 shows the 31P MAS NMR spectra of the phosphated silica and titania catalysts. SiP 300 and SiP 500 exhibited distinct peaks at −0.44 and −0.2 ppm, along with very weak peaks at −11.19 and −11.13 ppm, respectively (Figure 3a). The peaks at −0.44 and −0.2 ppm correspond to neat H3PO4, which is not chemically bound to the surface [20,21], whereas that at −11 ppm can be assigned to pyrophosphate (H3P2O7), which is chemically bound to the silica matrix [21]. This result suggests that phosphoric acid is not chemically bound to the silica surface effectively and exists mainly as a separated active site in the catalyst. The peak intensity of SiP 500 at −0.2 ppm was considerably lower than that of SiP 300 at −0.44 ppm, indicating that a higher calcination temperature decreased the concentration of H3PO4 on the silica surface owing to its loss through decomposition. The NMR spectra of the TiP 300 and TiP 500 catalysts are also shown in Figure 3b. Various peaks were observed for these catalysts, unlike the phosphated silica catalysts. TiP 300 exhibited four peaks at 0.6, −4.6, −10.3, and −17.6 ppm. The peak at 0.6 ppm corresponds to unbound H3PO4 [21]. According to a previous study on titanium phosphate, the peak at −4.6 ppm can be assigned to the H2PO4 group bound to TiO2 as the layered structure, whereas the peaks at −10.3 and −17.6 ppm can be assigned to the H2PO4 groups bound to TiO2 as the framework structure, e.g., α-TiP and γ-TiP [22,23]. The appearance of intense peaks in a lower downfield indicated that H3PO4 is more strongly bound to the TiO2 support than to the SiO2 support. However, we cannot rule out the possibility that the peaks at −10.3 and −17.6 ppm originate from unbound pyrophosphate and polyphosphate. Upon increasing the calcination temperature of TiP to 500 °C, the NMR peak distribution slightly changed, with three peaks appearing at −0.02, −5.6, and −11.3 ppm. Compared to the peak intensity of TiP 300, the peak intensity of the unbound H3PO4 decreased, whereas that of the chemically bound H3PO4 increased, suggesting that a higher calcination temperature leads to enhanced coordination of H3PO4 with TiO2. Notably, the absolute peak intensity of the TiP catalysts is fairly low, indicating that the amount of bound H3PO4 is small.
The chemical state of sulfuric acid attached to the oxide support was analyzed using FT-IR spectroscopy, and the results are presented in Figures S3 and S4 for the silica- and titania-based catalysts, respectively. In Figure S3, the infrared (IR) bands in the region from 900 to 1200 cm−1 correspond to Si-OH and Si-O-Si stretching vibrations, whereas the IR band around 800 cm−1 corresponds to the Si-O bending vibration [24,25,26]. The IR band for SiO2-SO3H appears around 1170 cm−1, which is related to the Si-O-SO3H stretching vibration [26]. Thus, the IR band of the sulfonic acid group overlaps with the Si-O-Si stretching vibration band, and hence, the presence of the sulfonic acid groups grafted onto silica cannot be confirmed using IR spectroscopy. In the case of the titania-based catalysts, broad and intense IR bands in the region from 500 to 800 cm−1 were observed, as shown in Figure S4. This is a characteristic feature of TiO2 samples with mixed rutile and anatase structures [27]. Compared to the neat TiO2 support, sulfated TiO2 samples showed an additional small IR band at 1140 cm−1 related to the S=O stretching vibration, suggesting the presence of the sulfonic acid group on the TiO2 support [28]. The additional IR bands at 1620 and 3300 cm−1 for the sulfated TiO2 samples arise owing to the H-O-H bending and OH stretching vibrations of the water adsorbed onto TiO2 [27,28].
To further confirm the presence of the sulfonic acid groups on the SiO2 and TiO2 supports, the sulfated catalysts were subjected to elemental analysis. As shown in Table 2, the sulfur content in the sulfated silica samples, SiS 300 and SiS 500, was remarkably low (<0.02 wt. %), indicating almost no sulfation of silica. Compared to sulfated silica, the sulfur content in sulfated titania was nearly 20-fold higher (>0.34 wt. %), suggesting that sulfation of TiO2 was more effective than that of SiO2. The sulfur content of TiS 300 (0.53 wt. %) was higher than that of TiS 500 (0.34 wt. %). The lower sulfur content of the latter can be attributed to the decomposition of the sulfonic acid group at a higher calcination temperature. Overall, the elemental analysis clearly demonstrated the presence of varying amounts of sulfur species on the sulfated catalysts.
The acid properties of the sulfated and phosphated oxide catalysts were also examined via NH3-TPD. Figure 4 shows the TPD profiles of the silica- and titania-based catalysts, and the calculated acid site densities are listed in Table 1. The TPD profiles of the sulfated samples, i.e., SiS 500 and TiS 500, are almost similar to those of the neat support materials, suggesting that the acidity of the sulfated samples is very low. This result is in good agreement with the elemental analysis result, which suggested that the concentration of the sulfonic acid groups grafted on the oxide support is very low. In contrast, the phosphated catalysts exhibited various new peaks corresponding to ammonia desorption, depending on the type of support and the calcination temperature. In the case of silica, both SiP 300 and SiP 500 showed a broad and distinct desorption peak in the temperature range of 100–350 °C, with the peak maximum at 200 °C, indicating that the presence of H3PO4 on the silica surface induces the formation of new acid sites with medium strength. The total acid densities for SiP 300 and SiP 500 were 0.676 and 0.424 mmol/g, respectively. The acid site density of SiP 500 is lower probably owing to the decomposition of H3PO4 at a higher calcination temperature, and this observation is in good agreement with the 31P NMR analysis result. In the case of phosphated titania, TiP 300 shows an intense peak at 100 °C and a very broad peak in the temperature range of 150–350 °C. Except for the intense peak at 100 °C, the ammonia desorption profile was very similar to that of the neat TiO2 support, suggesting that the overall acidity of the TiO2 support did not change remarkably upon phosphation. In contrast, TiP 500 showed two intense peaks at 110 and 320 °C, with intensities considerably higher than that of the TiO2 support. This indicated that a higher calcination temperature resulted in more effective phosphation of TiO2, thereby forming new acid sites effectively. The total acid site density of TiP 500 was 0.934 mmol/g, which is considerably higher than that of TiP 300 (0.432 mmol/g).
Overall, the characterization studies indicated that phosphation of the metal oxide surface was more successful than its sulfation under the employed conditions, resulting in high density of the available acid sites. The concentration of the sulfonic acid groups was almost negligible in SiO2, while some sulfonic acid groups could be detected on the TiO2 surface. In the case of phosphation, the phosphoric acid was mostly physically bound to the silica surface and was trapped inside the micropores. This hypothesis is supported by the large reduction in the micropore volume of the silica and the unchanged chemical shift of H3PO4 after grafting onto the silica surface. In contrast, the phosphoric acid was grafted more effectively to the TiO2 surface, as evidenced by the various peaks appearing downfield in the 31P NMR spectrum. Especially, calcination of the phosphated titania at 500 °C significantly increased the overall acidity of the catalyst, presumably owing to the higher dispersion of phosphoric acid.
2.2. Catalytic Performance
The activities of the well-characterized sulfated and phosphated catalysts were evaluated for the Diels–Alder reaction of DMF and ethylene. The reaction conditions were as follows: temperature = 523 K, ethylene pressure = 30 bar, DMF = 11.1 wt. %, catalyst weight = 300 ± 5 mg, and reaction time = 6 h. Figure 5 shows the conversion of DMF and the product selectivity for the sulfated catalysts. The highest DMF conversion (~40%) was achieved with TiS 300, whereas similar DMF conversions (~28%) were achieved with the other three catalysts. This indicated that the sulfated catalysts exhibited very similar activities regardless of the support type. However, distinct differences were observed in the PX selectivity. The PX selectivity decreased in the following order: TiS 300 > TiS 500 > SiS 300 > SiS 500; the sulfated silica had a significantly lower selectivity to PX (<5%) than that of sulfated titania (10–18%). Overall, the PX selectivity trend correlated well with the concentration of the sulfonic acid groups on the catalyst surface, i.e., the sulfur content measured using elemental analysis. Thus, the higher the sulfur content of the catalyst, the higher the PX selectivity. These results indicated that the strong acid sites were essential in producing PX from DMF via the Diels–Alder reaction with ethylene, as reported previously [13]. This is because the cycloadduct formed after the Diels–Alder reaction of DMF and ethylene undergoes dehydrative aromatization for conversion to PX, and the dehydration of the cycloadduct requires strong acid sites such as BrØnsted acid sites.
Figure 6 shows the conversion of DMF and the product selectivity over phosphated catalysts under identical reaction conditions. Typically, the activities of the phosphated catalysts were significantly higher than those of the sulfate catalysts. All the phosphated catalysts, except TiP 300, exhibited very high DMF conversions (80–85%) and high PX selectivity (60–70%). This high activity of the phosphated catalysts is perhaps linked to the more effective grafting of phosphoric acid onto the metal oxide surface than sulfuric acid under the employed conditions. The phosphated catalysts also showed enhanced selectivity to HDO. HDO is produced via the hydrolysis of DMF over acid sites and can be dehydrated back to DMF. The facile formation of HDO over phosphated catalysts supports their enhanced acidity more than that in the case of sulfate catalysts. Among the phosphate catalysts, the PX selectivity decreased in the following order: TiP 500 (71%) > SiP 500 (69%) > SiP 300 (61%) ≫ TiP 300 (15%). The very low activity and PX selectivity of TiP 300, compared to those of the other phosphated catalysts, can be attributed to its low acid site density, as measured using NH3-TPD. The overall acidity of TiP 300 was similar to that of the neat TiO2 sample, indicating that its acid character was almost unaltered by phosphation. In contrast, the other phosphated catalysts exhibited considerably enhanced acidity after phosphation, and this could be attributed to their high acidity. Importantly, when the reactions for SiP 500 and TiP 500 were conducted during 12 h reaction time, the conversion was reached 100% and the PX selectivity increased to ~80%, as shown in Figure S5.
It should be noted that most of the phosphoric acid was physically bound to the silica surface and thus could be easily leached into the solution during the reaction. Previous studies suggest that the leached H3PO4 could act as a homogeneous acid catalyst in the conversion of DMF to PX [18]. To check this possibility, we evaluated the catalytic effect of H3PO4 on the reaction under identical reaction conditions. Indeed, H3PO4 was fairly active for the conversion of DMF to PX, and a DMF conversion of ~72% and PX selectivity of ~39% were achieved.
Because H3PO4 is leached from the phosphated catalysts during the reaction, the stability and reusability of some selected catalysts, i.e., SiP 500 and TiP 500, were investigated (Figure 7). For this, the spent catalyst after the first reaction during 6 h reaction time was collected using filtration and reused for the second reaction. For the reusability test, the spent catalyst after the first reaction was calcined at 773 K for 4 h in flowing air to remove coke and then reused for the second reaction. The activities of the fresh, spent, and regenerated catalysts are shown in Figure 7. For both SiP 500 and TiP 500, the catalytic activity decreased significantly after the first reaction, with DMF conversions of ~20 and ~32% and PX selectivity of ~5 and ~28% for SiP 500 and TiP 500, respectively. The extent of decrease in the catalytic activity was higher for SiP 500, possibly owing to high phosphorus leaching induced by the significantly weaker binding of phosphate onto the silica than onto titania. However, the decrease in the catalytic activity could also result from the formation of heavy byproducts or owing to the coke deposited on the catalyst surface, which blocks the catalytically active sites. Thus, an examination of the catalytic activity of the spent-calcined catalyst could provide better information on the degree of leaching. The results showed that the activity of the spent SiP 500 catalyst was only partially recovered after calcination, with a DMF conversion of ~40% and PX selectivity of ~10%. Thus, the degree of phosphorous leaching in SiP 500 was significant, resulting in the irreversible deactivation of the catalyst. In contrast, the spent TiP 500 catalysts regained 80% of their original activity after calcination, with a DMF conversion of ~58% and PX selectivity of ~58%. Thus, TiP 500 is more resistant to leaching, and this is consistent with the 31P NMR analysis result, which suggests that phosphorous is more effectively grafted onto the TiO2 support than onto the SiO2 support.
Overall, it can be concluded that the conversion of DMF and the selectivity to PX are generally correlated to the total acid site density of the catalyst, which in turn depends on the acid molecule–support interaction strength. Sulfation of metal oxides was less successful than phosphation, and only a small amount of the sulfonic acid groups were anchored on the metal oxide surface. The low acidity of the sulfonated catalysts generally resulted in poor activity for PX production. In contrast, the phosphated catalysts had a higher acid site density owing to the more effective coordination of the phosphoric acid, and this was responsible for the considerably higher activity of these catalysts for PX production than the sulfate catalysts. Importantly, the leaching of phosphoric acid from the catalysts occurred during the reaction, and the leached species could act as an acid catalyst for PX production. Compared to the phosphated silica, the phosphated titania was found to be far more resistant to leaching, as evident from its reusability after recalcination. It is likely that the formation of denser Ti-P framework structures through advanced synthesis techniques such as the sol-gel method could lead to enhanced activity and stability of the TiP catalysts.
3. Experimental Section
3.1. Catalyst Preparation
Silica (SiO2), titania (TiO2), H2SO4, H3PO4 were purchased from Sigma-Aldrich. 2,5-Dimethylfuran (DMF), 2,5-hexanedione (HDO), p-Xylene (PX), n-tridecane, n-dodecane were obtained from Alfa Aesar (Seoul, Korea).
All catalysts were prepared via incipient wetness impregnation. For a typical synthesis, 1 g of SiO2 (or TiO2) was added to 6 mL of acetone, and mixed vigorously for 15 min. 0.2 mL of H2SO4 (or H3PO4) solution mixed with 5 mL of acetone was then added to the support material solution and stirred for 4 h at 303 K. The paste was dried in the oven at 373 K for 24 h. After drying, the obtained solid powder was washed several times with a deionized water to remove the excess acid molecules, which do not adsorb on the surface, and was dried at 373 K overnight [24]. Finally, the powder was calcined in flowing air at 573 K (or 773 K) for 6 h. The catalysts’ names were differentiated by the type of support and calcination temperature (SiP 300, SiP 500, SiS 300, SiS 500, TiP 300, TiP 500, TiS 300, TiP 500).
3.2. Catalyst Characterization
The powder X-ray diffraction was carried out with X’Pert-MPD System (PHILIPS, PANalytical Company, Almelo, The Netherlands) using Cu-Kα (λ = 0.15406 nm) operated at 40 kV and 30 mA in the 2θ range 5–50° with the scanning rate of 0.013° s−1 to determine the crystallinity of catalysts.
The surface area and porosity of the catalysts were measured by nitrogen gas adsorption/desorption. The catalysts were heated under vacuum at 573 K for 2 h. The specific surface area (SBET) and the total pore volume (Vtotal) of catalysts were calculated by the BET equation. Moreover, the micropore volume (Vmicro) of catalysts was calculated by the t-plot method.
Temperature-programmed desorption of ammonia (NH3-TPD) was carried out using Micromeritics AutoChem II 2920 equipped with a thermal conductivity detector (Micromeritics Instrument Corporation, Norcross, GA, USA). The samples were pretreated in air at 573 K for 2 h followed by the exposure to ammonia gas diluted with He at 323 K for 1 h. The samples were then heated to 973 K at a rate of 10 K min−1.
Solid state 31P magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were obtained with a 500 MHz Bruker Avance III HD using 4 mm probes and 10 kHz spinning speed (Bruker Corporation, Billerica, MA, USA). The 85% phosphoric acid was used as the reference for the standard calibration.
The infrared spectra were obtained from Fourier transform–infrared spectroscopy (FT-IR) using a Spectrum GX (PIKE Technologies, Madison, WI, USA). The detecting range was from 500 to 4000 cm−1. Before measurements, the samples were mixed with KBr, and then pressure was applied to prepare a form of pellet.
The elemental analysis was carried out using Elementar Vario macro/micro Elemental analyzer (Elementar, Langenselbold, Hesse, Germany) to identify the amounts of sulfur contained in the sulfated catalysts.
3.3. Catalytic Test Reaction
The Diels–Alder reactions were performed in a 160 mL stainless-steel Parr reactor. For a typical reaction, the reaction solution was initially prepared with the mixing ratio of DMF, n-dodecane, and n-tridecane to 11.1 wt. %: 86.7 wt. %: 2.2 wt. %. Next, 0.3 g of a catalyst sample was mixed with the solution and then added to the reactor. After sealing the reactor, a leak test was conducted by pressurizing the reactor with nitrogen gas to 60 bar at room temperature. The pressure change was monitored for 1 h whether there was a leak or not. Once the leak test was passed, the reactor was purged with nitrogen gas a couple of times followed by pressurizing with ethylene to 30 bar for the reaction. The reactor temperature was then rapidly raised to 523 K using the combined use of a thermocartridge heater and an electric furnace while stirring the reaction solution at 300 rpm by an impeller. All of the reactions were performed for 6 h. After the reactions were completed, the product liquid was collected by syringe, filtered with a syringe filter, and analyzed by Agilent 7890 GC equipped with a flame ionization detector and HP-5 column (Agilent Technologies, Santa Clara, CA, USA).
For the stability and reusability test, the spent catalysts were collected and calcined in air at 773 K for 4 h at a rate of 3 K min−1. The spent catalysts without calcination and with calcination were tested for the reactions under the identical reaction condition to the fresh catalysts.
4. Conclusions
In conclusion, the activities of sulfated and phosphated catalysts for the Diels–Alder cycloaddition of biomass-derived 2,5-dimethylfuran (DMF) and ethylene to p-Xylene (PX) were comprehensively investigated for the first time. The results revealed that the sulfation of metal oxides was less successful than phosphation, resulting in a very low amount of sulfonic acid groups on the catalyst surface. The sulfated catalysts had a very low acid site density and low activity for PX production. In contrast, the phosphoric acid grafted onto the metal oxides could be clearly detected in the 31P NMR spectra of the phosphated catalysts. The overall acidities of the phosphated catalysts calcined at 500 °C (i.e., SiP 500 and TiP 500) were also significantly higher than those of the neat supports. Both the catalysts showed a high DMF conversion exceeding 80% and a remarkable PX selectivity of ~70%. However, SiP 500 experienced severe leaching of H3PO4 during the reaction due to the weak interaction of phosphoric acid with silica. The reusability test confirmed that the SiP 500 catalysts underwent irreversible deactivation due to leaching. In contrast, TiP 500 was more resistant to leaching and recovered 80% of its original activity after the reaction–regeneration cycle. This enhanced resistance to leaching is attributed to the more effective coordination between phosphate and TiO2, as evidenced from the 31P NMR analysis. Overall, our results suggest that titanium phosphate is a potential catalyst for the Diels–Alder reaction of DMF and ethylene to form PX if the formation of denser Ti-P framework structures is achieved using advanced synthetic techniques such as the sol-gel method. In addition, our results reinforce the findings of previous studies, which suggest that weak acids, such as phosphoric acid, are sufficient to catalyze the Diels–Alder cycloaddition of DMF to form PX.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11091074/s1, Figure S1: N2 adsorption/desorption isotherms silica-based catalysts., Figure S2: N2 adsorption/desorption isotherms titania-based catalysts., Figure S3: FT-IR spectra of sulfate silica catalysts., Figure S4: FT-IR spectra of sulfate titania catalysts, Figure S5: Conversion of DMF and selectivity of PX and HDO for the Diels-Alder reaction of DMF and ethylene over phosphated catalysts at calcined 773 K for 12 h.
Author Contributions
H.K.: investigation, formal analysis, writing—original draft preparation; J.J.: conceptualization, methodology, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF) funded by the Korean Government (MSIT) (2020M1A2A2079801).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-C.; Green, S.K.; Williams, C.L.; Dauenhauer, P.J.; Fan, W. Ultra-selective cycloaddition of dimethylfuran for renewable p-Xylene with H-BEA. Green Chem. 2014, 16, 585–588. [Google Scholar] [CrossRef]
- Settle, A.E.; Berstis, L.; Rorrer, N.A.; Roman-Leshkóv, Y.; Beckham, G.T.; Richards, R.M.; Vardon, D.R. Heterogeneous Diels–Alder catalysis for biomass-derived aromatic compounds. Green Chem. 2017, 19, 3468–3492. [Google Scholar] [CrossRef]
- Brandvold, T.A. Carbohydrate Route to Para-Xylene and Terephthalic Acid. U.S. Patent 8314267B2, 20 November 2012. [Google Scholar]
- Román-Leshkov, Y.; Barrett, C.J.; Liu, Z.Y.; Dumesic, J.A. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates. Nature 2007, 447, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Osmundsen, C.M.; Taarning, E.; Dumesic, J.A. Selective production of aromatics from alkylfurans over solid acid catalysts. ChemCatChem 2013, 5, 2044–2050. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Kristianto, I.; Lee, H.; Jae, J. Production of renewable toluene from biomass-derived furans via Diels-Alder and dehydration reactions: A comparative study of Lewis acid catalysts. Fuel 2016, 182, 588–596. [Google Scholar] [CrossRef]
- Green, S.K.; Patet, R.E.; Nikbin, N.; Williams, C.L.; Chang, C.-C.; Yu, J.; Gorte, R.J.; Caratzoulas, S.; Fan, W.; Vlachos, D.G. Diels–Alder cycloaddition of 2-methylfuran and ethylene for renewable toluene. Appl. Catal. B Environ. 2016, 180, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.L.; Chang, C.-C.; Do, P.; Nikbin, N.; Caratzoulas, S.; Vlachos, D.G.; Lobo, R.F.; Fan, W.; Dauenhauer, P.J. Cycloaddition of biomass-derived furans for catalytic production of renewable p-Xylene. ACS Catal. 2012, 2, 935–939. [Google Scholar] [CrossRef]
- Nikbin, N.; Do, P.T.; Caratzoulas, S.; Lobo, R.F.; Dauenhauer, P.J.; Vlachos, D.G. A DFT study of the acid-catalyzed conversion of 2,5-dimethylfuran and ethylene to p-Xylene. J. Catal. 2013, 297, 35–43. [Google Scholar] [CrossRef]
- Feng, X.; Shen, C.; Tian, C.; Tan, T. Highly selective production of biobased p-Xylene from 2,5-dimethylfuran over SiO2–SO3H catalysts. Ind. Eng. Chem. Res. 2017, 56, 5852–5859. [Google Scholar] [CrossRef]
- Feng, X.; Cui, Z.; Ji, K.; Shen, C.; Tan, T. Ultra-selective p-Xylene production through cycloaddition and dehydration of 2,5-dimethylfuran and ethylene over tin phosphate. Appl. Catal. B Environ. 2019, 259, 118108. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Winoto, H.P.; Park, Y.-K.; Suh, D.J.; Lee, H.; Ha, J.-M.; Jae, J. Heteropolyacid catalysts for Diels-Alder cycloaddition of 2,5-dimethylfuran and ethylene to renewable p-Xylene. Catal. Today 2017, 293, 167–175. [Google Scholar] [CrossRef]
- Yin, J.; Shen, C.; Feng, X.; Ji, K.; Du, L. Highly selective production of p-Xylene from 2,5-dimethylfuran over hierarchical NbO x-based catalyst. ACS Sustain. Chem. Eng. 2018, 6, 1891–1899. [Google Scholar] [CrossRef]
- Zhao, R.; Zhao, Z.; Li, S.; Parvulescu, A.N.; Müller, U.; Zhang, W. Excellent Performances of Dealuminated H-Beta Zeolites from Organotemplate-Free Synthesis in Conversion of Biomass-derived 2,5-Dimethylfuran to Renewable p-Xylene. ChemSusChem 2018, 11, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- McGlone, J.; Priecel, P.; Da Vià, L.; Majdal, L.; Lopez-Sanchez, J.A. Desilicated ZSM-5 zeolites for the production of renewable p-Xylene via Diels–Alder cycloaddition of dimethylfuran and ethylene. Catalysts 2018, 8, 253. [Google Scholar] [CrossRef] [Green Version]
- Wijaya, Y.P.; Suh, D.J.; Jae, J. Production of renewable p-Xylene from 2,5-dimethylfuran via Diels–Alder cycloaddition and dehydrative aromatization reactions over silica−alumina aerogel catalysts. Catal. Commun. 2015, 70, 12–16. [Google Scholar] [CrossRef]
- Cho, H.J.; Ren, L.; Vattipalli, V.; Yeh, Y.-H.; Gould, N.; Xu, B.; Gorte, R.J.; Lobo, R.; Dauenhauer, P.J.; Tsapatsis, M. Renewable p-Xylene from 2,5-dimethylfuran and ethylene using phosphorus-containing zeolite catalysts. ChemCatChem 2017, 9, 398–402. [Google Scholar] [CrossRef]
- Gulbinski, J.; Ren, L.; Vattipalli, V.; Chen, H.; Delaney, J.; Bai, P.; Dauenhauer, P.; Tsapatsis, M.; Abdelrahman, O.A.; Fan, W. Role of silica support in phosphoric acid catalyzed production of p-Xylene from 2,5-dimethylfuran and ethylene. Ind. Eng. Chem. Res. 2020, 59, 22049–22056. [Google Scholar] [CrossRef]
- Zhao, P.; Boekfa, B.; Nishitoba, T.; Tsunoji, N.; Sano, T.; Yokoi, T.; Ogura, M.; Ehara, M. Theoretical study on 31P NMR chemical shifts of phosphorus-modified CHA zeolites. Microporous Mesoporous Mater. 2020, 294, 109908. [Google Scholar] [CrossRef]
- Jain, S.K.; Tabassum, T.; Li, L.; Ren, L.; Fan, W.; Tsapatsis, M.; Caratzoulas, S.; Han, S.; Scott, S.L. P-Site Structural Diversity and Evolution in a Zeosil Catalyst. J. Am. Chem. Soc. 2021, 143, 1968–1983. [Google Scholar] [CrossRef]
- Bortun, A.I.; Bortun, L.; Clearfield, A.; Villa-García, M.A.; García, J.R.; Rodríguez, J. Synthesis and characterization of a novel layered titanium phosphate. J. Mater. Res. 1996, 11, 2490–2498. [Google Scholar] [CrossRef]
- Djafer, L.; Ayral, A.; Boury, B.; Laine, R.M. Surface modification of titania powder P25 with phosphate and phosphonic acids–Effect on thermal stability and photocatalytic activity. J. Colloid Interface Sci. 2013, 393, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Radwan, N.R.; Hagar, M.; Afifi, T.H.; Al-wadaani, F.; Okasha, R.M. Catalytic Activity of Sulfated and Phosphated Catalysts towards the Synthesis of Substituted Coumarin. Catalysts 2018, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Ghoreishi, K.; Asim, N.; Yarmo, M.; Samsudin, M. Mesoporous phosphated and sulphated silica as solid acid catalysts for glycerol acetylation. Chem. Pap. 2014, 68, 1194–1204. [Google Scholar] [CrossRef]
- Haihong, X.; Daishi, G.; Qizhong, J.; Zifeng, M.; Wanjun, L.; Zheng, W. Catalytic performance of sulfated silica MCM-41 for the cyclization of pseudoionone to ionones. Chin. J. Catal. 2006, 27, 1080–1086. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Sodagar, E.; Doroodmand, M.M. Nano sulfated titania as solid acid catalyst in direct synthesis of fatty acid amides. J. Org. Chem. 2011, 76, 2853–2859. [Google Scholar] [CrossRef]
- Bai, X.; Pan, L.; Zhao, P.; Fan, D.; Li, W. A new solid acid SO42−/TiO2 catalyst modified with tin to synthesize 1, 6-hexanediol diacrylate. Chin. J. Catal. 2016, 37, 1469–1476. [Google Scholar] [CrossRef]
Figure 1.
Overall schematic diagram of Diels–Alder reaction of DMF and ethylene.
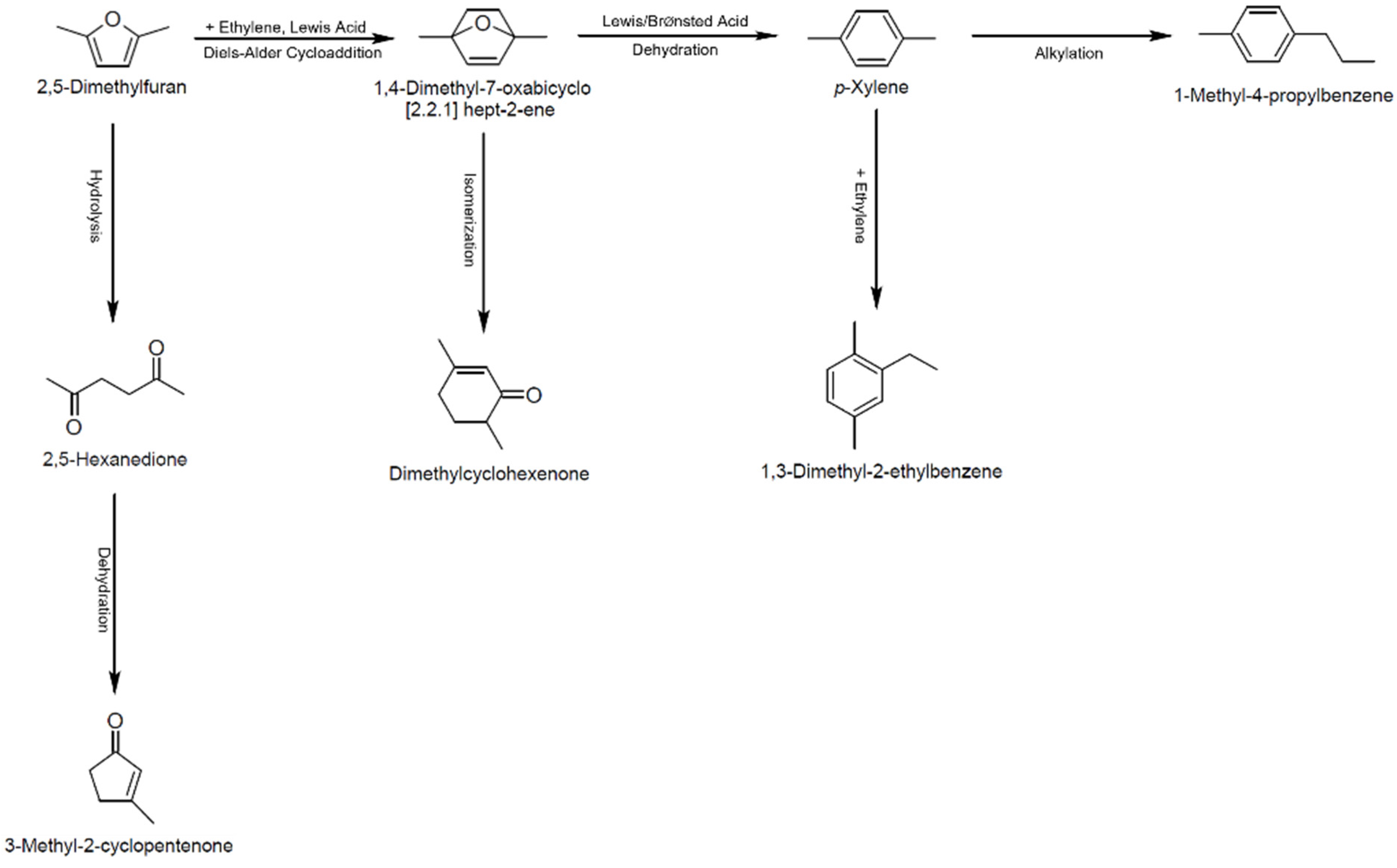
Figure 2.
XRD patterns of (a) silica-based catalysts and (b) titania-based catalysts.

Figure 3.
31P MAS NMR spectra of the phosphated (a) silica and (b) titania catalysts.

Figure 4.
NH3-TPD data of (a) silica-based catalysts and (b) titania-based catalysts.

Figure 5.
Conversion of DMF and selectivity of PX and HDO for the Diels–Alder reaction of DMF and ethylene over sulfated catalysts. Reaction condition; temperature = 523 K, reaction time = 6 h, C2H4 pressure = 30 bar, DMF = 11.1 wt. %, catalyst mass = 300 ± 5 mg.
Figure 5.
Conversion of DMF and selectivity of PX and HDO for the Diels–Alder reaction of DMF and ethylene over sulfated catalysts. Reaction condition; temperature = 523 K, reaction time = 6 h, C2H4 pressure = 30 bar, DMF = 11.1 wt. %, catalyst mass = 300 ± 5 mg.

Figure 6.
Conversion of DMF and selectivity of PX and HDO for the Diels–Alder reaction of DMF and ethylene over phosphated catalysts. Reaction condition; temperature = 523 K, reaction time = 6 h, C2H4 pressure = 30 bar, DMF = 11.1 wt. %, catalyst mass = 300 ± 5 mg.
Figure 6.
Conversion of DMF and selectivity of PX and HDO for the Diels–Alder reaction of DMF and ethylene over phosphated catalysts. Reaction condition; temperature = 523 K, reaction time = 6 h, C2H4 pressure = 30 bar, DMF = 11.1 wt. %, catalyst mass = 300 ± 5 mg.

Figure 7.
Conversion of DMF and selectivity of PX and HDO for the Diels–Alder reaction of DMF and ethylene for stability test over phosphated catalysts at 773 K calcined. F: Fresh, S: Spent, SC: Spent Calcined. Reaction condition; temperature = 523 K, reaction time = 6 h, C2H4 pressure = 30 bar, DMF = 11.1 wt. %, catalyst mass = 300 ± 5 mg.
Figure 7.
Conversion of DMF and selectivity of PX and HDO for the Diels–Alder reaction of DMF and ethylene for stability test over phosphated catalysts at 773 K calcined. F: Fresh, S: Spent, SC: Spent Calcined. Reaction condition; temperature = 523 K, reaction time = 6 h, C2H4 pressure = 30 bar, DMF = 11.1 wt. %, catalyst mass = 300 ± 5 mg.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Physical properties of sulfated and phosphated catalysts.
| Catalyst | SBET (m2/g) | Vtotal (cm3/g) | Vmicro (cm3/g) | Total NH3 Desorbed (mmol/g) |
|---|---|---|---|---|
| SiO2 | 175 | 1.45 | 0.27 | - |
| SiP 300 | 128 | 0.70 | 0.13 | 0.676 |
| SiP 500 | 75 | 0.53 | 0.16 | 0.424 |
| SiS 500 | 44 | 0.46 | 0.19 | - |
| TiO2 | 36 | 0.58 | 0.38 | - |
| TiP 300 | 31 | 0.56 | 0.18 | 0.432 |
| TiP 500 | 27 | 0.45 | 0.11 | 0.934 |
| TiS 500 | 47 | 0.54 | 0.21 | - |
Table 2.
Elemental analysis of sulfated catalysts.
| Catalyst | Mass Fraction (%) | |||
|---|---|---|---|---|
| C | H | N | S | |
| SiS 300 | 0.17 | 0.23 | 0.03 | 0.02 |
| SiS 500 | 0.14 | 0 | 0.07 | 0 |
| TiS 300 | 0.18 | 0.19 | 0 | 0.53 |
| TiS 500 | 0.19 | 0.19 | 0 | 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, H.; Jae, J. Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene. Catalysts 2021, 11, 1074. https://doi.org/10.3390/catal11091074
AMA Style
Kim H, Jae J. Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene. Catalysts. 2021; 11(9):1074. https://doi.org/10.3390/catal11091074
Chicago/Turabian StyleKim, Hanbyeol, and Jungho Jae. 2021. "Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene" Catalysts 11, no. 9: 1074. https://doi.org/10.3390/catal11091074
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.