
Kinetics of Strontium Carbonate Formation on a Ce-Doped SrFeO3 Perovskite


Center for Membrane Technology, Department of Chemistry and Bioscience, Aalborg University, DK-9220 Aalborg, Denmark
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(3), 265; https://doi.org/10.3390/catal12030265
Submission received: 4 February 2022
/
Revised: 19 February 2022
/
Accepted: 25 February 2022
/
Published: 26 February 2022
(This article belongs to the Section Catalytic Materials)
Abstract
:Some perovskites exhibit catalytic activity in the abatement of organic pollutants in water. However, their performance decreases over time, possibly due to forms of poisoning, such as carbonate formation. Here, we present the kinetics of carbonate formation on a Ce-doped SrFeO3 perovskite with formula Sr0.85Ce0.15FeO3−δ (SCF), which can act as a thermocatalyst for the degradation of organic pollutants. The carbonate formation was studied in air, in deionized water, and during degradation of bisphenol A. The formation of SrCO3 occurred for perovskites in aqueous environments, i.e., when dispersed in water or used as catalysts in the degradation of bisphenol A, while no SrCO3 was detected for samples stored in air for up to 195 days. SrCO3 formation was detected using both XRD and ATR-FT-IR, and from the XRD, the crystallite size was found to decrease when carbonates formed. The samples containing SrCO3 showed an increasing mass loss at >610 °C with increasing time used as catalysts or dispersed in water, showing that SCF reduces its own efficiency during catalytic use. The kinetics of carbonate formation based on the TGA measurements showed that SrCO3 forms approximately three times faster during the degradation of organic compounds in water compared to SCF dispersed in water. The formation of SrCO3 in SCF is thermally reversible; thus, the catalyst can resume its activity after heat treatment at 900 °C for 1 h.
1. Introduction
Perovskite-like materials have the formula ABX3 and are structurally similar to the original perovskite, CaTiO3 [1]. In the metal oxide area, X is oxygen, while A and B are 12-coordinated and 6-coordinated metals, respectively [2]. A-site cations are often alkaline or rare-earth metals, while B-site cations are transition metals. Perovskites exhibit a broad range of promising properties including piezoelectric, dielectric, superconductor, and ferromagnetic properties [3], which can be tuned through variations in their chemical composition. A more recently studied property is their catalytic activity in the abatement of aqueous pollutants [4].
Within the catalytic area, various oxide perovskites have shown thermocatalytic properties regarding the degradation of organic compounds by the formation of reactive oxygen species (oxygen-containing radicals) [5], which can be useful in the removal of micropollutants from wastewater in the future [6]. The catalytic site in perovskites is mainly located in the B-site (transition metal) due to their multiple oxidation states [7], covering, for example Ti [8,9], Fe [10,11], Co [12,13], and Mn [14]. However, doping and tuning the A-site can alter the band gap [15]. The specific perovskite that is the focus of the current study is Ce-doped SrFeO3, more precisely, Sr0.85Ce0.15FeO3−δ (SCF), which has been used to degrade bisphenol A (BPA) and dyes, such as Rhodamine B and Orange II [10,16].
A drawback of SCF is the potential for carbonate formation on its surface, which can block the active sites, a so-called poisoning of the catalyst caused by chemisorption on catalytic sites [17]. In the case of SCF, mainly SrCO3 is formed [16,18], and, previously, the amount of surface carbonates (SrCO3) reached up to 30% after one month due to interaction with CO2 from the atmosphere [18]. By contrast, Srilakshmi et al. [19] only observed carbonate formation when SrFeO3 was used as a catalyst for the degradation of nitrobenzene in water, and no carbonate formation was reported while SrFeO3 was stored in air. This might be attributed to the formation of carbonate species due to the dissolved CO2 in the water, which can form carbonates on the surface of SCF and related perovskites, such as SrO parts. Carbonate formation reduced the catalytic activity of a non-doped SrFeO3−δ [19] and might have been the reason for the slower kinetics of SCF in the degradation of BPA in aqueous samples when reused in four consecutive degradation cycles [10]. However, temperature-programmed desorption (TPD) was previously used to investigate the adsorption of CO2 on a BaxSr1−xCo0.8Fe0.2O3−δ perovskite, and the adsorption was shown to occur at temperatures between 400 °C and 700 °C [20], which is much higher temperature than room temperature. Yang et al. [21] showed minor carbonate formation when treating SrFe0.9Ti0.1O3−δ in a pure CO2 atmosphere at temperatures ≥ 600 °C and suggested the physisorption of CO2 to the surface rather than carbonate formation (chemisorption).
In this study, a systematic investigation of the formation of carbonates on Sr0.85Ce0.15FeO3−δ (SCF) was carried out to understand the decreasing efficiency of the catalytic activity over time. This was investigated by storing the SCF in air, dispersing the SCF in deionized water, and using the SCF in the thermocatalytic degradation of bisphenol A. The carbonate formation was studied using XRD and ATR-FT-IR, and the kinetics of carbonate formation were determined using TGA measurements.
2. Results and Discussion
The recorded XRD diffractograms of the SCF perovskites exposed to air or water or used as catalysts in the degradation of BPA for different durations are shown in Figure 1. The crystal structures of the fresh samples are denoted “Day 0” or “0 min”. All the perovskites dispersed in water or used for catalytic tests contained small amounts of segregated CeO2 (<1%), since the solubility of Ce in SrFeO3 is about 14 mol%, as previously reported [16,18]. However, the diffraction peaks assigned as segregated CeO2 did not change throughout the experiments.
During the 195 days of air storage, no changes in the XRD pattern of the SCF powder were observed (Figure 1a), indicating that no carbonate was chemically bound to any of the metal oxide sites in the SCF. This agrees with the results of carbonate formation studies on similar perovskites [19,20,21]. However, when SCF powder is dispersed in water, new diffraction peaks appear, proving the formation of new crystal phases. The new signals were all related to SrCO3, whereas no sign of carbonate formation on the iron oxide or cerium oxide sites was detected (Figure 1b). For comparison, an XRD pattern of pure SrCO3 is presented in the Supporting Information (Figure S1). The carbonate formation only on SrO sites can be attributed to weak metal oxygen bond strength [22]. The SrCO3 content increased with exposure time, as shown by the increase in the intensity ratio between the most intense diffraction peaks of SrCO3 (2θ = 25.2) and SCF (2θ = 32.6), respectively (Figure 1d). The same carbonate formation tendency was observed when SCF was used as the catalyst to degrade BPA (Figure 1c). Furthermore, no carbonate or hydroxide species other than SrCO3 were observed. Carbonate formation on SCF occurs faster when SCF is used as catalyst than when is simply dispersed in water (Figure 1d). This is shown by the increased intensity of SrCO3 peaks, while that of SCF decreases. As parts of the SCF converted to SrCO3, a change to a lower peak position by the most dominant peak of SCF (2θ = 32.6) was observed due to an expansion of the unit cell. The average crystallite size of all the samples was calculated from the full width half maxima (FWHM) using the most dominant diffraction peak of SCF (2θ = 32.6), using the Scherrer equation in Highscore Plus. The crystallite size was similar for all the samples stored in air, approximately 514 Å, while a significant reduction in crystallite size was found for the samples dispersed in water or used as catalysts (Table 1). Interestingly, the samples from the catalytic tests that showed the largest amount of SrCO3 did not have the largest change in crystallite size. Figure 1d indicates that the ratio of SrCO3 to SCF reached a plateau with sufficient time for carbonate formation. This suggests maximum carbonate formation, which relies on the amount of Sr available for forming SrCO3. With increasing SrCO3 formation, the perovskite’s structure possibly became unstable, which could have resulted in the formation of more favorable crystal structures of the remaining strontium, iron, and cerium oxides.
The degradation of the BPA in water by the SCF was verified by HPLC analyses (Figure 2). Three separate experiments were stopped after different durations by removing the water by filtration, followed by drying and analyzing the SCF powder. The degradation of BPA was similar in all the experiments and all experiments showed a first-order degradation of bisphenol A with a reaction kinetic constant of k = 0.096 min−1 ± 0.005 within the first 10 min of each experiment. First-order reactions were also found in the literature for similar perovskites [10,16,23].
Figure 3 shows the ATR-FT-IR spectra of the samples exposed to air or water, or used as catalysts in the degradation of bisphenol A, for different durations. For all the samples, a significant ATR-FT-IR signal was visible around 575 cm−1, which was previously found for undoped SrFeO3 [19]. Other significant bands occurred around 1470 cm−1 and 860 cm−1 for the samples dispersed in water or used as catalysts. These bands were ascribed to SrCO3, in agreement with the literature [24], and a spectrum of pure SrCO3 found in the Supporting Information (Figure S2). As these signals are not visible in Figure 3a, it can be inferred that the SrCO3 does not form on SCF during storage in air for up to 195 days. Furthermore, the intensity of the SrCO3 signals increased with increasing time in water, supporting the hypothesis that carbonate formation is caused by reaction of Sr sites from perovskite with carbonate species in water formed by dissolved CO2.
The increased intensity centered around 3750 cm−1 is possibly attributable to the physisorption of water to the samples and partly to the formed Sr(OH)2 [25]. The small amounts of ceria segregated during the synthesis of the SCF are not visible in the ATR-FT-IR spectra as they should have been visible around 530 cm−1 [26], overlapping with general metal–oxygen bonds; however, the signal between 3500–4000 cm−1 might have been partly by different ceria hydroxyl groups with different oxidation states of cerium [27]. By contrast, the ATR-FT-IR spectrum does not indicate any formation of iron hydroxides, iron carbonates, or iron hydroxyl carbonates [28,29,30]. The signals occurring in the range 2900–3000 cm−1 in Figure 3c could have been related to BPA or the compounds formed during the BPA degradation, as they can be attributed to C–H bonds [31]. These compounds might have been adsorbed to the SCF in water. However, as there were indications of similar signals for the SCF stored in air, they were attributed to noise or the ethanol and acetone used during cleaning.
The mass loss during the heating of the perovskite samples to 900 °C in the TGA analysis is shown in Figure 4. The TGA analysis was carried out on perovskites that were calcined after combustion; thus, citric acid and nitrates were removed. The mass loss is divided into three regions: (i) 50–360 °C, (ii) 360–610 °C, and (iii) 610–900 °C and the mass loss for all the samples is reported in Table 2. The first region is explained by the evaporation of physically or chemically adsorbed water to SCF as adsorbed water is removed from the Sr(OH)2 around 210 °C [32] and from the iron sites around 275 °C [33], in accordance with the hydroxyl signals from the IR spectra. In the second region, only a small mass loss was observed, which was not as easily ascribed to a specific removal process. However, it could have been the removal of chemisorbed water, such as Sr(OH)2, that decomposed, as suggested by the ATR-FT-IR spectra. This decomposition initiates around 530 °C and completes at 700 °C [32], within the second region. The small mass loss might also be explained by residual carbon species that were not removed during calcination. The third region is explained by the decomposition of the formed SrCO3 that was previously shown to initiate its mass loss around 800 °C in pure SrCO3 [34] and a decomposition temperature around 930 °C in an argon atmosphere [35]. The lower temperature observed for the mass loss initiation and decomposition compared to pure SrCO3 from the literature was due to the different atmospheres used during measurement. The oxygen-containing atmosphere in this study reduced the decomposition temperature compared to inert atmospheres [36]. Another reason for the lower decomposition temperatures of the SrCO3 in the SCF compared to pure SrCO3 is related to the mixed structure, which was plausibly more open than that of the pure SrCO3, as seen for the transition metal-doped CaCO3 [37,38]. This open structure can be related to a destabilized crystal lattice. By contrast, mixed alkaline earth carbonates, such as dolomite, decompose at a temperature between those of pure compounds [38].
Based on the TGA measurements of each sample, the mass losses in the three temperature ranges are reported in Table 2, and the two main mass losses are shown in Figure 5. The first temperature range (50–360 °C) shows a rapid mass loss based on adsorbed water followed by a fully covered surface, as the mass loss was relatively constant after a few hours, as shown for the samples dispersed in water. Considering the final temperature range (610–900 °C), around 1.5% of the mass was carbonate after 10–30 min in water (either pure water or BPA solution) and around 2.5% after 360 min of use in water or 90 min of use as catalyst. A fast initial carbonate formation is observed from the fresh calcined powder to the first storage duration, as seen from the mass loss. After the initial carbonate formation, the mass loss increased linearly with time, with varying kinetics, depending on the environment. This shows a significant faster carbonate formation on SCF when organic compounds (in this case, bisphenol A) are present compared to SCF dispersed in pure water. The kinetic constant is three times higher for catalytic use than for when dispersed in water. This is caused by the thermocatalytic mineralization of the BPA into CO2 and the carbonate species that was dissolved in the water; thus, these compounds adsorbed to the surface of the perovskite, forming SrCO3, as suggested in Ref. [19]. This also agrees with the increase in inorganic carbon during and after catalytic reactions found for catalytic wet-air oxidation using TiO2 or Ru(3 wt%)/TiO2 [39] and catalytic oil degradation using SCF [40]. Only limited carbonate formation is observed in the TGA for samples stored in air, in accordance with the XRD and ATR-FT-IR results.
From the mass loss in Region III, the number of Sr-sites blocked by carbonate formation was calculated. To this end, the entire mass loss was considered to be CO2; based on this, the number of moles was calculated. The total mass of the SCF was determined after mass loss. Subsequently, the number of moles SCF was calculated; the number of moles of strontium sites is 85% of this value due to the chemical formula of the SCF. The fraction of strontium sites blocked by carbonate formation was calculated based on the moles of CO2 released and the strontium sites. The fraction was relatively constant for the samples stored in air around 2–3%. For the samples dispersed in water or used in the degradation of bisphenol A, up to 13% of the Sr-sites were blocked by carbonate formation (Table 2).
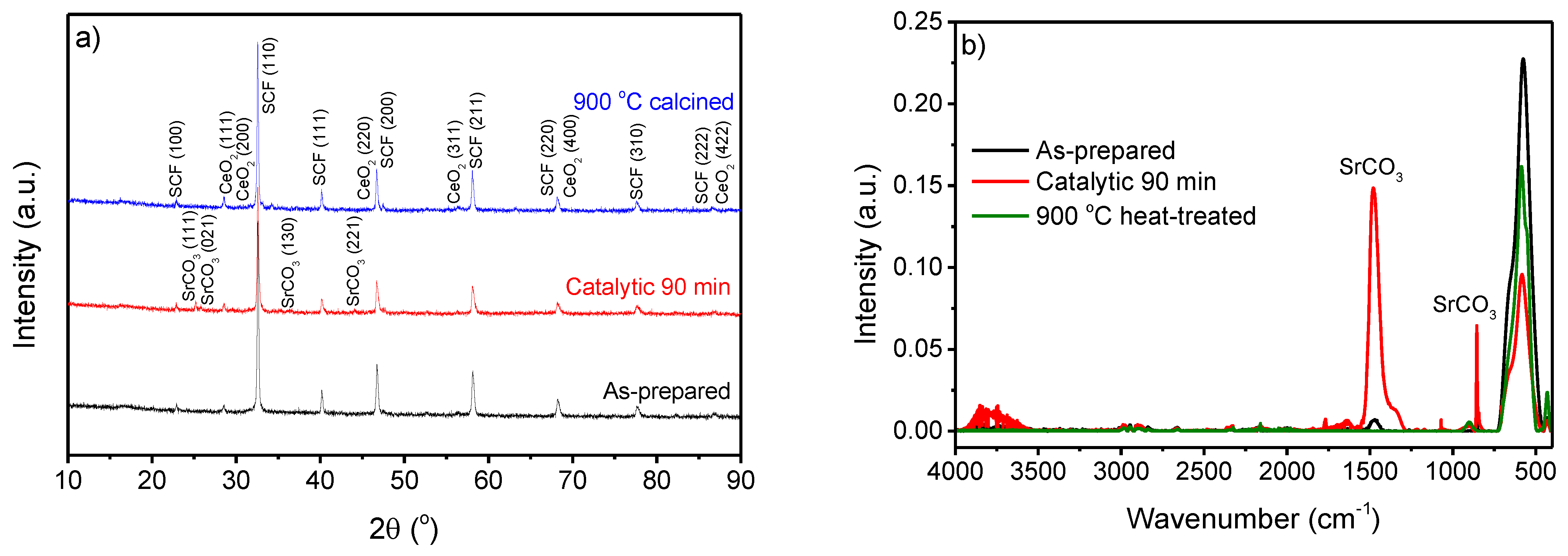
To elucidate whether the carbonate formation is reversible and confirm that the mass loss up to 900 °C was due to decarbonation, a SCF sample used as a catalyst for 90 min was heat-treated at 900 °C for 1 h in air and reanalyzed (Figure 6). The SrCO3 diffraction peaks and IR signals did not appear after thermal treatment; thus, the SCF can regain its original structure after carbonate formation. A previous study stated that a mild temperature treatment could remove the carbonate [18]; however, a specific temperature was not stated. In contrast to the current study, the previous study found carbonate formation by storage in air within one month; this was detected by XPS, which is more sensitive in the outermost layer. Here, SrCO3 was only found when the SCF was dispersed in water or used as a catalyst in water for the degradation of bisphenol A, showing a fast carbonate formation. Carbonate formation in SCF possibly results in morphological changes, as previously suggested by Tummino et al. [16]. The thermally induced reversibility of the carbonation is important if SCF needs to be reused as a catalyst to maintain the highest reaction rate possible, as the carbonate species block the active sites of the catalyst. The fast deactivation of the catalyst is problematic in terms of commercial use [17]. However, the carbonate formation and reactivation provide important knowledge for the future design of oxide perovskite catalysts for the degradation of organic species in aqueous environments. This research suggests the design of perovskite catalysts with A-sites that show less of a tendency to form carbonates to prevent poisoning, and, thus, the deactivation of the catalyst. This could maintain the catalytic activity. Alkaline earth metal oxides, in general, show a significant tendency to form carbonates. Therefore, a higher-stability perovskite catalyst might be found by substituting the A-site alkaline earth metal by a lanthanide metal, as is partly found in SCF.
3. Materials and Methods
3.1. Perovskite Oxide Synthesis
Ce-doped SrFeO3 with composition Sr0.85Ce0.15FeO3−δ (SCF) was synthesized by solution combustion synthesis. Stoichiometric amounts of Sr(NO3)2 (Acros Organics, purity 99+%, Karlsruhe, Germany), Ce(NO3)3·6H2O (Sigma Aldrich, purity 99%, Steinheim, Germany), and Fe(NO3)3·9H2O (Sigma Aldrich, purity ≥ 98%, Steinheim, Germany) were dissolved in deionized water. Citric acid (Carl Roth, purity ≥ 99.5%, Karlsruhe, Germany) was added as organic fuel with a citric acid-to-metal cations molar ratio of 2. NH4NO3 (Sigma Aldrich, purity ≥ 99.5%, Steinheim, Germany) was added as additional oxidant to achieve a reducer-to-oxidizer ratio of 1, and NH4OH solution (25 wt%) was added to adjust the pH to 6 to have a better citrate-metal cations complexation, according to the literature [41]. The solution was placed on a hot plate at 80 °C for evaporation of the water under magnetic stirring until a sticky gel was formed. The temperature of the hot plate was increased until the spontaneous self-ignition of the gel was initiated. The as-burned powder was crushed, followed by calcination at 1000 °C for 5 h heated at 2 °C min−1. The calcined powder was washed in deionized water under stirring for an hour followed by centrifugation for 15 min at 7000 rpm, and this was repeated twice. Washing removes residual carbon species and enhances the surface area [16]. Finally, the water was removed by filtration and the powder was left to dry overnight at 105 °C.
3.2. Formation of Carbonates
SCF was stored in air at room temperature (relative humidity 40% ± 5), dispersed in deionized water while stirring at 60 °C, or used as catalyst for degradation of bisphenol A at 60 °C in deionized water to investigate whether carbonation occurred. For samples dispersed in water or used as catalysts, SCF was added in a concentration of 1 mg mL−1. In the catalytic degradation study, bisphenol A (BPA) was added in a concentration of 10 mg L−1. Three catalytic experiments were performed to collect perovskite powder after different durations of use as catalyst. For all three types of storage, SCF was analyzed by X-ray diffraction (XRD), attenuated total reflectance FT-IR (ATR-FT-IR), and thermogravimetric analysis (TGA) after different durations of exposure to air, water, and degradation of BPA in water, in order to obtain kinetics of the carbonate formation, as described below. The samples subjected to water were filtrated to remove the water followed by drying for at least 48 h at room temperature to evaporate remaining water without removing potential hydroxyl or carbonate species by heat treatment. Additionally, the degradation of BPA was analyzed through HPLC with UV detection at 230 nm (Summit-Dionex, with a Luna 5u C18 100 Å column (250 × 6.60 mm)) and a mobile phase with a flow of 1 mL min−1 (water/acetonitrile = 60/40). A calibration curve with concentrations between 0 and 10 mg L−1 BPA was used to determine the concentrations of BPA during degradation.
3.3. Powder Characterization
The crystal structure of both washed and stored powders was analyzed by XRD on a PANalytical Empyrean diffractometer. The diffractometer used a Cu Kα radiation (λ = 1.5418 Å) and operated at 45 kV and 40 mA. Crystal phases were identified by comparing the diffraction patterns to patterns in the International Centre for Diffraction Data (ICDD) using Highscore Plus software (PANalytical). Highscore Plus was used to determine the crystallite size using the Scherrer equation.
ATR-FT-IR analysis was carried out by scanning from 400 to 4000 cm−1 using ATR mode on a Tensor II spectrometer (Bruker) to identify carbonate species. The resolution of the scans was 1 cm−1, and 128 spectra were collected and averaged. All spectra were baseline corrected using OPUS software.
TGA analyses were performed using a STA 449C Jupiter instrument (Netzsch, Selb, Germany). The powder samples (approx. 18 mg) were gently pressed by hand to enhance contact in a Pt crucible and heated at 10 °C min−1 to 900 °C, followed by cooling at 20 °C min−1 with a 40 mL min−1 air (N2/O2 = 80/20) gas flow. For TGA analyses, all samples exposed to water or used as catalysts in BPA degradation were analyzed, while samples stored in air were analyzed approximately every three weeks.
4. Conclusions
Carbonate formation on a Ce-doped SrFeO3 perovskite (SCF) was investigated after storage in air, dispersal in water, and use as a catalyst for the degradation of bisphenol A. SrCO3 formation was detected by XRD and ATR-FT-IR for the SCF dispersed in water or used as a catalyst in water, but not when the SCF was stored in air for up to 195 days. The crystallite size of the SCF decreased with carbonate formation. The TGA analyses showed mass losses related to the evaporation of adsorbed water and decarbonation. The kinetics of the carbonate formation were determined based on the decomposition of the SrCO3 in the temperature range of 610–900 °C, showing a three-times-faster carbonate formation when SCF was used as catalyst compared to when it was only dispersed in water, due to the formation of carbonate species during the degradation of bisphenol A. From the TGA results, it was calculated that up to 13% of all the Sr sites in the SCF were blocked by carbonate formation. Finally, by treating the carbonated SCF at 900 °C in air for 1 h, a thermally induced, reversible process of carbonate formation was shown, and the original SCF was regained. This information is important for catalytic use, since carbonate species can be removed, making the active sites available for reactions. These findings are expected to be relatable to other oxide perovskites containing Sr or other alkaline earth metals in A-sites.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12030265/s1, Figure S1: XRD pattern of SrCO3.; Figure S2: ATR-FT-IR spectrum of SrCO3.
Author Contributions
Conceptualization, M.B.Ø.; methodology, M.B.Ø. and M.K.J.; validation, M.B.Ø. and A.B.S.; formal analysis, M.B.Ø.; investigation, M.B.Ø. and A.B.S.; resources, M.K.J.; writing—original draft preparation, M.B.Ø.; writing—review and editing, A.B.S., V.B. and M.K.J.; visualization, M.B.Ø.; supervision, V.B. and M.K.J.; project administration, M.K.J.; funding acquisition, M.K.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Villum Foundation, grant number 00028236.
Data Availability Statement
The data are contained within the article.
Acknowledgments
The authors thank Francesca Deganello for discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tilley, R.J.D. Perovskites: Structure-Property Relationships, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Li, C.; Soh, K.C.K.; Wu, P. Formability of ABO3 Perovskites. J. Alloys Compd. 2004, 372, 40–48. [Google Scholar] [CrossRef]
- Twu, J.; Gallagher, P.K. Preparation of Bulk and Supported Perovskites. In Properties and Applications of Perovskite-Type Oxides; Tejuca, L.G., Fierro, J.L.G., Eds.; CRC Press: New York, NY, USA, 1993; pp. 1–23. [Google Scholar]
- Grabowska, E. Selected Perovskite Oxides: Characterization, Preparation and Photocatalytic Properties—A Review. Appl. Catal. B Environ. 2016, 186, 97–126. [Google Scholar] [CrossRef]
- Chen, H.; Ku, J.; Wang, L. Thermal Catalysis under Dark Ambient Conditions in Environmental Remediation: Fundamental Principles, Development, and Challenges. Chin. J. Catal. 2019, 40, 1117–1134. [Google Scholar] [CrossRef]
- Oturan, M.A.; Aaron, J. Advanced Oxidation Processes in Water/Wastewater Treatment: Principles and Applications. A Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in Catalysis and Electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharma, M.; Powar, S.; Kabachkov, E.N.; Vaish, R. Impact of Remnant Surface Polarization on Photocatalytic and Antibacterial Performance of BaTiO3. J. Eur. Ceram. Soc. 2019, 39, 2915–2922. [Google Scholar] [CrossRef]
- Srilakshmi, C.; Rao, G.M.; Saraf, R. Effect of the Nature of a Transition Metal Dopant in BaTiO3 Perovskite on the Catalytic Reduction of Nitrobenzene. RSC Adv. 2015, 5, 45965–45973. [Google Scholar] [CrossRef]
- Janowska, K.; Boffa, V.; Jørgensen, M.K.; Quist-Jensen, C.; Hubac, F.; Deganello, F.; Coelho, F.E.B.; Magnacca, G. Thermocatalytic Membrane Distillation for Clean Water Production. NPJ Clean Water 2020, 3, 1–7. [Google Scholar] [CrossRef]
- Markov, A.A.; Nikitin, S.S.; Leonidov, I.A.; Patrakeev, M.V. Oxygen and Electron Transport in Ce0.1Sr0.9FeO3−δ. Solid State Ion. 2020, 344, 115131. [Google Scholar] [CrossRef]
- Lu, Q.; Huberman, S.; Zhang, H.; Song, Q.; Wang, J.; Vardar, G.; Hunt, A.; Waluyo, I.; Chen, G.; Yildiz, B. Bi-Directional Tuning of Thermal Transport in SrCoOx with Electrochemically Induced Phase Transitions. Nat. Mater. 2020, 19, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Matras, D.; Vamvakeros, A.; Jacques, S.D.M.; Middelkoop, V.; Vaughan, G.; Agote Aran, M.; Cernik, R.J.; Beale, A.M. In Situ X-ray Diffraction Computed Tomography Studies Examining the Thermal and Chemical Stabilities of Working Ba0.5Sr0.5Co0.8Fe0.2O3−δ Membranes during Oxidative Coupling of Methane. Phys. Chem. Chem. Phys. 2020, 22, 18964–18975. [Google Scholar] [CrossRef]
- Colomer, M.T.; Ortiz, A.L.; López-Domínguez, V.; Alonso, J.M.; García, M.A. Preparation, Thermal and Phase Evolution and Functional Properties of Non-Stoichiometric Strontium-Doped Lanthanum Manganite Perovskite Ceramics. J. Eur. Ceram. Soc. 2017, 37, 3527–3533. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Liang, J.; Luo, Y.; Chen, G.; Shi, X.; Lu, S.; Gao, S.; Hu, J.; Liu, Q.; et al. A-Site Perovskite Oxides: An Emerging Functional Material for Electrocatalysis and Photocatalysis. J. Mater. Chem. A 2021, 9, 6650–6670. [Google Scholar] [CrossRef]
- Tummino, M.L.; Laurenti, E.; Deganello, F.; Bianco Prevot, A.; Magnacca, G. Revisiting the Catalytic Activity of a Doped SrFeO3 for Water Pollutants Removal: Effect of Light and Temperature. Appl. Catal. B Environ. 2017, 207, 174–181. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Deganello, F.; Liotta, L.F.; Longo, A.; Casaletto, M.P.; Scopelliti, M. Cerium Effect on the Phase Structure, Phase Stability and Redox Properties of Ce-Doped Strontium Ferrates. J. Solid State Chem. 2006, 179, 3406–3419. [Google Scholar] [CrossRef]
- Srilakshmi, C.; Saraf, R.; Shivakumara, C. Effective Degradation of Aqueous Nitrobenzene Using the SrFeO3−δ Photocatalyst under UV Illumination and Its Kinetics and Mechanistic Studies. Ind. Eng. Chem. Res. 2015, 54, 7800–7810. [Google Scholar] [CrossRef]
- Yan, A.; Liu, B.; Dong, Y.; Tian, Z.; Wang, D.; Cheng, M. A Temperature Programmed Desorption Investigation on the Interaction of Ba0.5Sr0.5Co0.8Fe0.2O3−δ Perovskite Oxides with CO2 in the Absence and Presence of H2O and O2. Appl. Catal. B Environ. 2008, 80, 24–31. [Google Scholar] [CrossRef]
- Yang, G.; Su, C.; Chen, Y.; Dong, F.; Tade, M.O.; Shao, Z. Cobalt-Free SrFe0.9Ti0.1O3−δ as a High-Performance Electrode Material for Oxygen Reduction Reaction on Doped Ceria Electrolyte with Favorable CO2 Tolerance. J. Eur. Ceram. Soc. 2015, 35, 2531–2539. [Google Scholar] [CrossRef]
- Rethwisch, D.G.; Dumesic, J.A. Effect of Metal-Oxygen Bond Strength on Properties of Oxides. 1. Infrared Spectroscopy of Adsorbed CO and CO2. Langmuir 1986, 2, 73–79. [Google Scholar] [CrossRef]
- Singh, K.P.; Singh, G.; Vaish, R. Utilizing the Localized Surface Piezoelectricity of Centrosymmetric Sr1-XFexTiO3 (X≤0.2) Ceramics for Piezocatalytic Dye Degradation. J. Eur. Ceram. Soc. 2021, 41, 326–334. [Google Scholar] [CrossRef]
- Wu, S.; Yin, S.; Cao, H.; Lu, Y.; Yin, J.; Li, B. Glucosan Controlled Biomineralization of SrCO3 Complex Nanostructures with Superhydrophobicity and Adsorption Properties. J. Mater. Chem. 2011, 21, 8734–8741. [Google Scholar] [CrossRef]
- Lutz, H.D.; Eckers, W.; Schneider, G.; Haeuseler, H. Raman and Infrared Spectra of Barium and Strontium Hydroxides and Hydroxide Hydrates. Spectrochim. Acta. Part A Mol. Spectrosc. 1981, 37, 561–567. [Google Scholar] [CrossRef]
- Grinter, D.C.; Allan, M.; Yang, H.J.; Salcedo, A.; Murgida, G.E.; Shaw, B.; Pang, C.L.; Idriss, H.; Ganduglia-Pirovano, M.V.; Thornton, G. Ce=O Terminated CeO2. Angew. Chem. 2021, 133, 13954–13958. [Google Scholar] [CrossRef]
- Mihaylov, M.Y.; Ivanova, E.Z.; Vayssilov, G.N.; Hadjiivanov, K.I. Revisiting Ceria-NOx Interaction: FTIR Studies. Catal. Today 2020, 357, 613–620. [Google Scholar] [CrossRef]
- Durães, L.; Costa, B.F.O.; Vasques, J.; Campos, J.; Portugal, A. Phase Investigation of As-Prepared Iron Oxide/Hydroxide Produced by Sol–Gel Synthesis. Mater. Lett. 2005, 59, 859–863. [Google Scholar] [CrossRef]
- Markov, L.; Blaskov, V.; Klissurski, D.; Nikolov, S. The Thermal Decomposition Mechanism of Iron(III) Hydroxide Carbonate to α-Fe2O3. J. Mater. Sci. 1990, 25, 3096–3100. [Google Scholar] [CrossRef]
- Savoye, S.; Legrand, L.; Sagon, G.; Lecomte, S.; Chausse, A.; Messina, R.; Toulhoat, P. Experimental Investigations on Iron Corrosion Products Formed in Bicarbonate/Carbonate-Containing Solutions at 90 °C. Corros. Sci. 2001, 43, 2049–2064. [Google Scholar] [CrossRef]
- Ullah, R.; Li, H.; Zhu, Y. Terahertz and FTIR Spectroscopy of ‘Bisphenol A’. J. Mol. Struct. 2014, 1059, 255–259. [Google Scholar] [CrossRef]
- Dinescu, R.; Preda, M. Thermal Decomposition of Strontium Hydroxide. J. Therm. Anal. 1973, 5, 465–473. [Google Scholar] [CrossRef]
- Balek, V.; Šubrt, J. Thermal Behaviour of Iron(III) Oxide Hydroxides. Pure Appl. Chem. 1995, 67, 1839–1842. [Google Scholar] [CrossRef]
- Ptáček, P.; Bartoníčková, E.; Švec, J.; Opravil, T.; Šoukal, F.; Frajkorová, F. The Kinetics and Mechanism of Thermal Decomposition of SrCO3 Polymorphs. Ceram. Int. 2015, 41, 115–126. [Google Scholar] [CrossRef]
- Robbins, S.A.; Rupard, R.G.; Weddle, B.J.; Maull, T.R.; Gallagher, P.K. Some Observations on the Use of Strontium Carbonate as a Temperature Standard for DTA. Thermochim. Acta 1995, 269, 43–49. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Zhu, M.; Cheng, F.; Zhang, D. Decomposition of Key Minerals in Coal Gangues during Combustion in O2/N2 and O2/CO2 Atmospheres. Appl. Therm. Eng. 2019, 148, 977–983. [Google Scholar] [CrossRef]
- Belete, T.T.; van de Sanden, M.C.M.; Gleeson, M.A. Effects of Transition Metal Dopants on the Calcination of CaCO3 under Ar, H2O and H2. J. CO2 Util. 2019, 31, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Reller, A.; Padeste, C.; Hug, P. Formation of Organic Carbon Compounds from Metal Carbonates. Nature 1987, 329, 527–529. [Google Scholar] [CrossRef]
- Pintar, A.; Berčič, G.; Besson, M.; Gallezot, P. Catalytic Wet-Air Oxidation of Industrial Effluents: Total Mineralization of Organics and Lumped Kinetic Modelling. Appl. Catal. B Environ. 2004, 47, 143–152. [Google Scholar] [CrossRef]
- Østergaard, M.B.; Strunck, A.B.; Jørgensen, M.K.; Boffa, V. Abatement of Oil Residues from Produced Water Using a Thermocatalytic Packed Bed Reactor. J. Environ. Chem. Eng. 2021, 9, 106749. [Google Scholar] [CrossRef]
- Deganello, F.; Marcí, G.; Deganello, G. Citrate-Nitrate Auto-Combustion Synthesis of Perovskite-Type Nanopowders: A Systematic Approach. J. Eur. Ceram. Soc. 2009, 29, 439–450. [Google Scholar] [CrossRef]
Figure 1.
XRD pattern of freshly calcined perovskite (0 min/Day 0) and after storage for various durations in (a) air at room temperature, (b) dispersed in water at 60 °C, and (c) used as catalyst for degradation of bisphenol A at 60 °C. Diffraction peaks are marked with compound and Miller Indices of SCF (Sr0.85Ce0.15FeO3) (ID: 01–076–4076), SrCO3 (ID: 00–005–0418), and CeO2 (ID: 00–057–0401). (d) The ratio of the most significant peaks assigned to SrCO3 and SCF for samples dispersed in water or used as catalyst in degradation of bisphenol A. Inset in (d) shows a zoom-in on the XRD spectra of the samples used for catalytic degradation of bisphenol A to show the decreasing intensity of the SCF peak and increase in SrCO3 peak.
Figure 1.
XRD pattern of freshly calcined perovskite (0 min/Day 0) and after storage for various durations in (a) air at room temperature, (b) dispersed in water at 60 °C, and (c) used as catalyst for degradation of bisphenol A at 60 °C. Diffraction peaks are marked with compound and Miller Indices of SCF (Sr0.85Ce0.15FeO3) (ID: 01–076–4076), SrCO3 (ID: 00–005–0418), and CeO2 (ID: 00–057–0401). (d) The ratio of the most significant peaks assigned to SrCO3 and SCF for samples dispersed in water or used as catalyst in degradation of bisphenol A. Inset in (d) shows a zoom-in on the XRD spectra of the samples used for catalytic degradation of bisphenol A to show the decreasing intensity of the SCF peak and increase in SrCO3 peak.
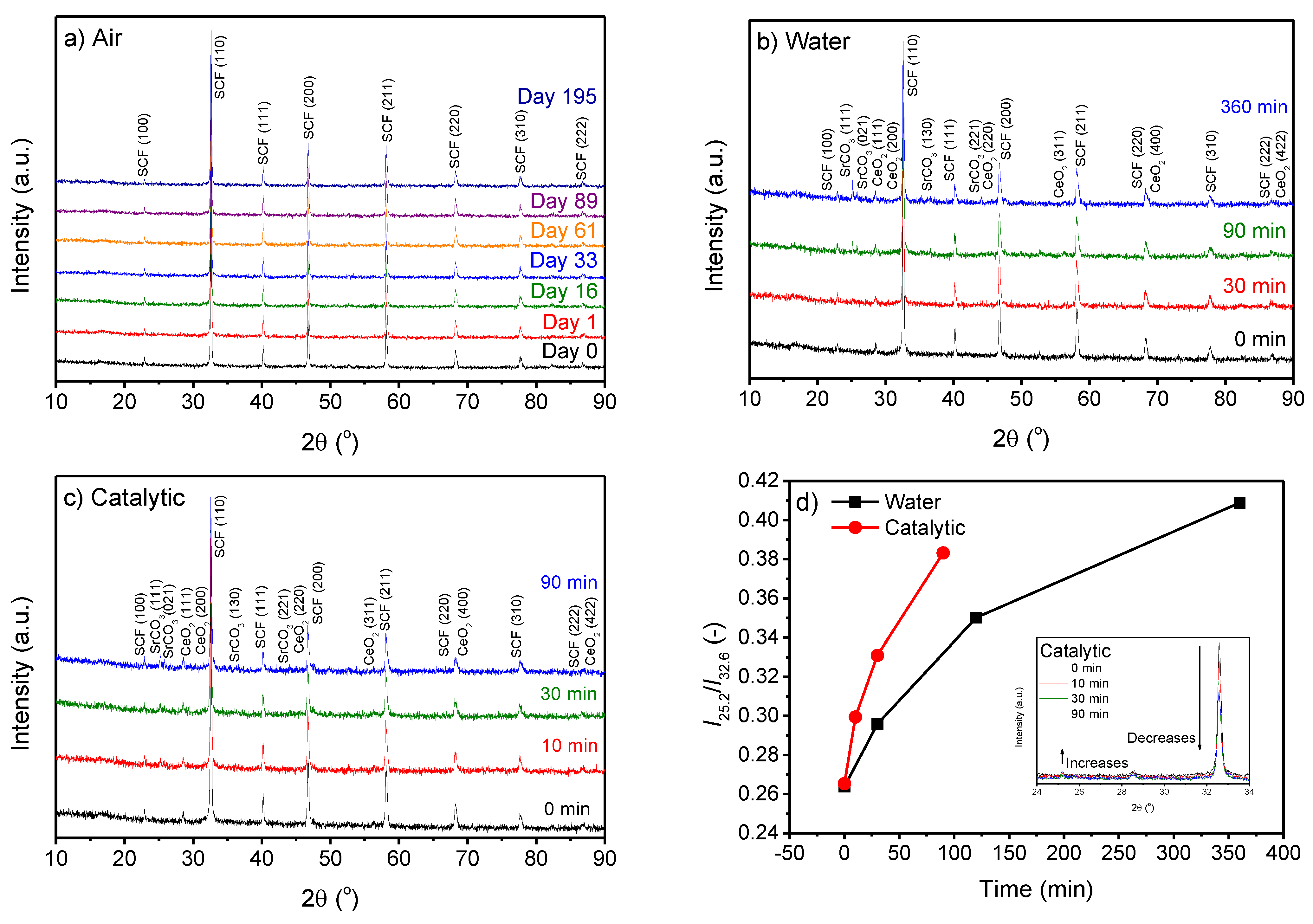
Figure 2.
Relative concentration of bisphenol A plotted against time during catalytic degradation using SCF. Three different experiments stopped after different times to collect SCF after different times of use as catalysts. Concentrations are based on HPLC analyses.
Figure 2.
Relative concentration of bisphenol A plotted against time during catalytic degradation using SCF. Three different experiments stopped after different times to collect SCF after different times of use as catalysts. Concentrations are based on HPLC analyses.

Figure 3.
ATR–FT–IR spectra of perovskites stored for various durations (a) in air at room temperature, (b) dispersed in water at 60 °C, and (c) used as catalysts for degradation of bisphenol A at 60 °C. SrCO3 signals are marked.
Figure 3.
ATR–FT–IR spectra of perovskites stored for various durations (a) in air at room temperature, (b) dispersed in water at 60 °C, and (c) used as catalysts for degradation of bisphenol A at 60 °C. SrCO3 signals are marked.

Figure 4.
TGA curves showing the mass loss vs. temperature of SCF stored under different conditions. The mass loss is divided into three temperature ranges: 50–360 °C (Region I), 360–610 °C (Region II), and 610–900 °C (Region III).
Figure 4.
TGA curves showing the mass loss vs. temperature of SCF stored under different conditions. The mass loss is divided into three temperature ranges: 50–360 °C (Region I), 360–610 °C (Region II), and 610–900 °C (Region III).
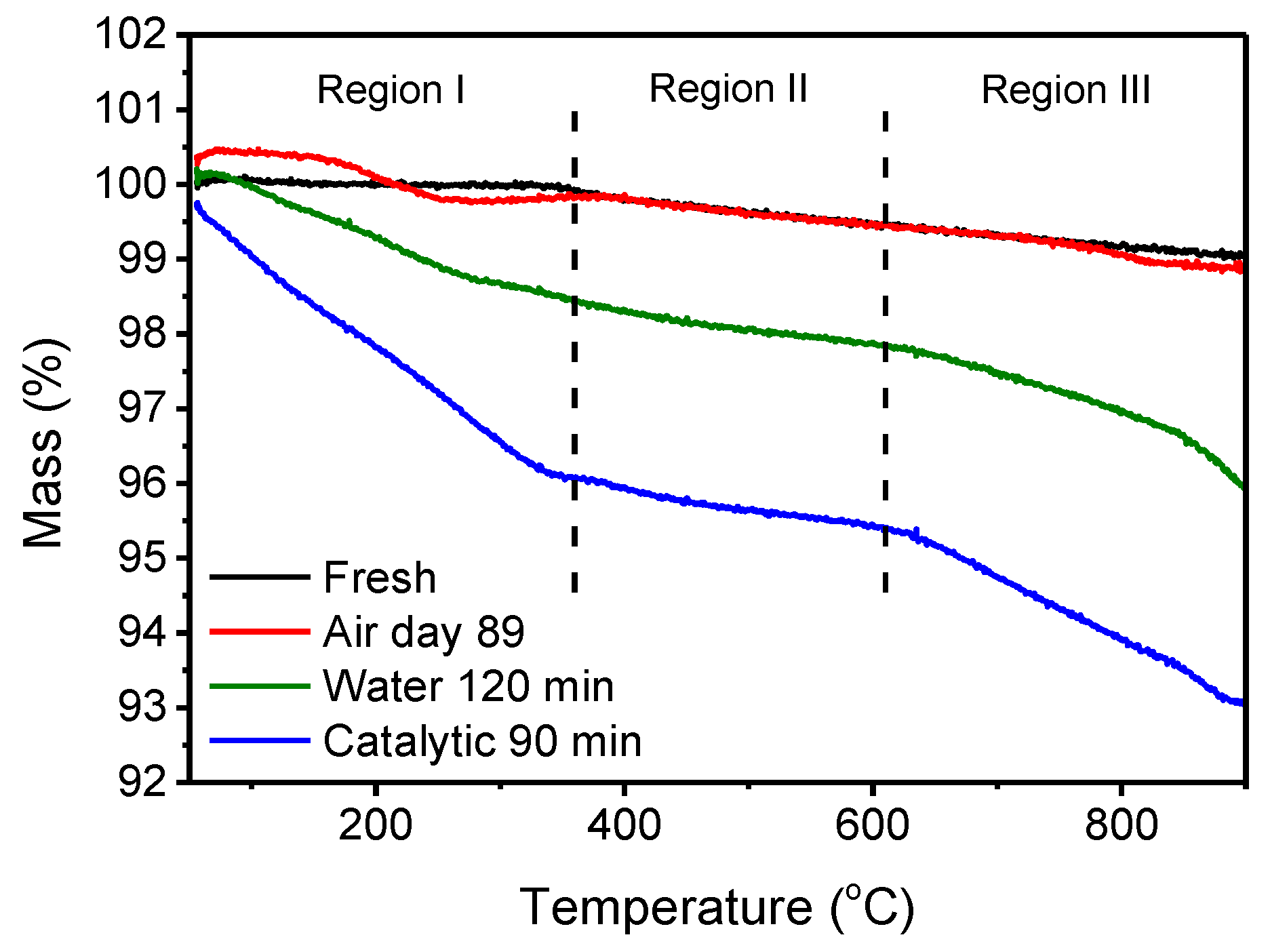
Figure 5.
Mass loss of SCF samples stored in in (a) air and (b) water, and (c) used as catalysts for the temperature regions 50–360 °C (symbol ■) and 610–900 °C (symbol ●) determined by TGA measurements. Lines are linear fits to determine the kinetic constant k after initial carbonate formation. The two kinetic constants k in (a) are calculated for same data with different units. Note the difference in scale between the x–axis and y–axis.
Figure 5.
Mass loss of SCF samples stored in in (a) air and (b) water, and (c) used as catalysts for the temperature regions 50–360 °C (symbol ■) and 610–900 °C (symbol ●) determined by TGA measurements. Lines are linear fits to determine the kinetic constant k after initial carbonate formation. The two kinetic constants k in (a) are calculated for same data with different units. Note the difference in scale between the x–axis and y–axis.
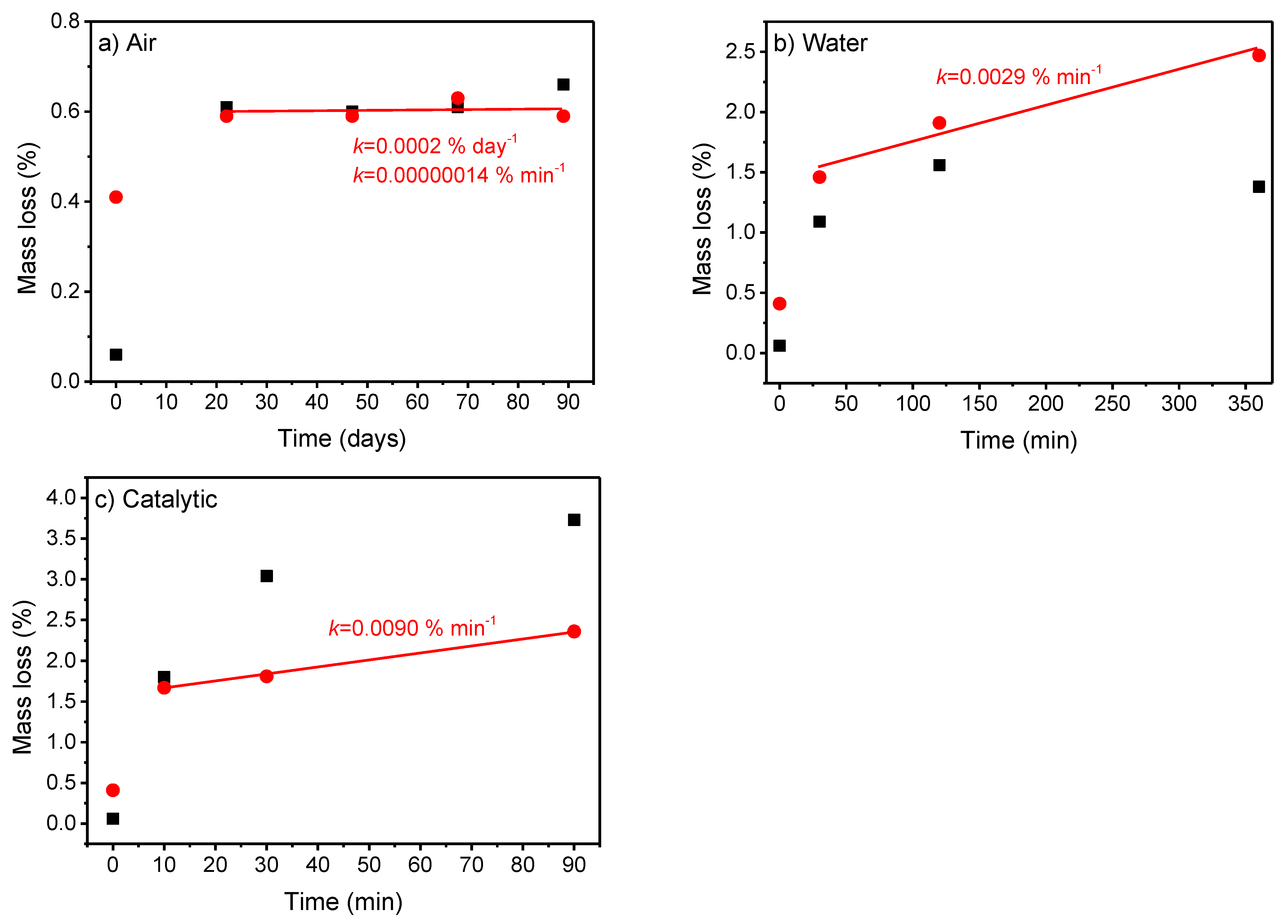
Figure 6.
Structural characterization of as-prepared SCF, used as catalyst for BPA degradation at 60 °C for 90 min, and heat-treated at 900 °C for 1 h in air using (a) XRD and (b) ATR-FT-IR. The diffraction peaks are assigned based on the compound and the Miller index of SCF (Sr0.85Ce0.15FeO3) (ID: 01–076–4076), SrCO3 (ID: 00–005–0418), and CeO2 (ID: 00–057–0401).
Figure 6.
Structural characterization of as-prepared SCF, used as catalyst for BPA degradation at 60 °C for 90 min, and heat-treated at 900 °C for 1 h in air using (a) XRD and (b) ATR-FT-IR. The diffraction peaks are assigned based on the compound and the Miller index of SCF (Sr0.85Ce0.15FeO3) (ID: 01–076–4076), SrCO3 (ID: 00–005–0418), and CeO2 (ID: 00–057–0401).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Peak position and full width half maxima (FWHM) of most dominant SCF peak at 2θ = 32.6 for calculation of crystallite size.
Table 1.
Peak position and full width half maxima (FWHM) of most dominant SCF peak at 2θ = 32.6 for calculation of crystallite size.
| Storage | Time | Peak Position | FWHM | Crystallite Size (Å) |
|---|---|---|---|---|
| Fresh perovskite | - | 32.599 | 0.166 | 514 |
| Air | 1 days | 32.603 | 0.170 | 502 |
| Air | 16 days | 32.606 | 0.167 | 511 |
| Air | 33 days | 32.607 | 0.166 | 514 |
| Air | 61 days | 32.607 | 0.167 | 511 |
| Air | 89 days | 32.602 | 0.163 | 524 |
| Water | 30 min | 32.583 | 0.207 | 410 |
| Water | 120 min | 32.576 | 0.230 | 368 |
| Water | 360 min | 32.584 | 0.229 | 369 |
| Catalytic | 10 min | 32.586 | 0.201 | 422 |
| Catalytic | 30 min | 32.560 | 0.205 | 414 |
| Catalytic | 90 min | 32.582 | 0.208 | 408 |
Table 2.
Calculated mass losses in different temperature ranges after storage of SCF in air, dispersed in water, and used as catalyst for degradation of bisphenol A. The fraction of blocked Sr-sites nSrCO3 is calculated based on the mass losses.
Table 2.
Calculated mass losses in different temperature ranges after storage of SCF in air, dispersed in water, and used as catalyst for degradation of bisphenol A. The fraction of blocked Sr-sites nSrCO3 is calculated based on the mass losses.
| Storage | Time | Mass Loss (%) | nSrCO3 (%) | |||
|---|---|---|---|---|---|---|
| 50–360 °C | 360–610 °C | 610–900 °C | Total | |||
| Fresh perovskite | - | 0.06 | 0.48 | 0.41 | 0.95 | 2.19 |
| Air | 26 days | 0.61 | 0.40 | 0.59 | 1.6 | 3.16 |
| Air | 47 days | 0.60 | 0.41 | 0.59 | 1.6 | 3.16 |
| Air | 68 days | 0.61 | 0.44 | 0.63 | 1.68 | 3.38 |
| Air | 89 days | 0.66 | 0.37 | 0.59 | 1.62 | 3.16 |
| Water | 30 min | 1.09 | 0.60 | 1.46 | 3.15 | 7.89 |
| Water | 120 min | 1.56 | 0.74 | 1.91 | 4.21 | 10.38 |
| Water | 360 min | 1.38 | 1.24 | 2.47 | 5.09 | 13.49 |
| Catalytic | 10 min | 1.80 | 0.60 | 1.67 | 4.07 | 9.05 |
| Catalytic | 30 min | 3.04 | 0.63 | 1.81 | 5.48 | 9.82 |
| Catalytic | 90 min | 3.73 | 0.68 | 2.38 | 6.79 | 12.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Østergaard, M.B.; Strunck, A.B.; Boffa, V.; Jørgensen, M.K. Kinetics of Strontium Carbonate Formation on a Ce-Doped SrFeO3 Perovskite. Catalysts 2022, 12, 265. https://doi.org/10.3390/catal12030265
AMA Style
Østergaard MB, Strunck AB, Boffa V, Jørgensen MK. Kinetics of Strontium Carbonate Formation on a Ce-Doped SrFeO3 Perovskite. Catalysts. 2022; 12(3):265. https://doi.org/10.3390/catal12030265
Chicago/Turabian StyleØstergaard, Martin B., Azeem B. Strunck, Vittorio Boffa, and Mads K. Jørgensen. 2022. "Kinetics of Strontium Carbonate Formation on a Ce-Doped SrFeO3 Perovskite" Catalysts 12, no. 3: 265. https://doi.org/10.3390/catal12030265
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.