
Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts



1
Foundation for Research & Technology-Hellas, Institute of Chemical Engineering Sciences (FORTH/ICE-HT), GR-26504 Patras, Greece
2
Department of Chemistry, University of Patras, GR-26504 Patras, Greece
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(4), 396; https://doi.org/10.3390/catal12040396
Submission received: 9 March 2022
/
Revised: 29 March 2022
/
Accepted: 30 March 2022
/
Published: 1 April 2022
(This article belongs to the Special Issue Exclusive Papers of the Editorial Board Members (EBMs) of Catalysts)
Abstract
:Dimethyl ether (DME) is considered an alternative hydrogen carrier with potential use in fuel cells and automotive and domestic applications. Dimethyl ether hydrolysis to methanol is a thermodynamically limited reaction catalyzed by solid-acid catalysts, mainly Al2O3 and zeolites. Moreover, it is the rate-limiting step of the DME steam reforming reaction, which is employed for the production of hydrogen fuel for fuel cell feeding. In the present study, the performance of WO3/Al2O3 catalysts (0–44% wt. WO3) was tested in DME hydrolysis reaction. The catalysts were characterized by means of N2-physisorption, XRD, Raman spectroscopy, XPS, NH3-TPD and 2,6-di-tert-butylpyridine adsorption experiments. The reaction rate of DME hydrolysis exhibited a volcanic trend as a function of tungsten surface density, while the best-performing catalyst was 37WO3/Al2O3, with a tungsten surface density of 7.4 W/nm2, noting that the theoretical monolayer coverage for the specific system is 4–5 W/nm2. Brønsted acidity was directly associated with the catalytic activity, following the same volcanic trend as a function of tungsten surface density. Blocking of Brønsted acid sites with 2,6-di-tert-butylpyridine led to a dramatic decrease in hydrolysis rates by 40 times, proving that Brønsted acid sites are primarily responsible for the catalytic activity. Thus, the type and strength rather than the concentration of acid sites are the key factors influencing the catalytic activity.

1. Introduction
In recent years, the utilization of dimethyl ether (DME) as an alternative vessel for energy storage and automotive and residential applications is gaining ground. DME has been considered as an ideal candidate fuel in fuel cells due to the fact its high hydrogen-to-carbon ratio, energy density and ease in transportation and handling [1,2]. Compared to methanol, DME is non-toxic and non-corrosive. The physical properties of DME are akin to those of liquified petroleum gas (LPG); thus, the existing infrastructure can be utilized with minimal alterations for its wide distribution [3,4,5]. Moreover, automotive DME-fueled processors demonstrate higher overall efficiencies compared to those with gasoline, methane or ethanol [6]. It is advantageous that DME does not contain a C–C bond; therefore, it can be fully oxidized without byproducts. Additionally, DME can be used as propellant in the cosmetics industry as well as in the production of important chemicals such as dimethoxymethane (DMM), methyl acetate (MA), formaldehyde and ethylene [7,8,9,10,11,12,13].
DME steam reforming (SR) is regarded as the most effective way to produce high-quality reformate gas as feed for fuel cells. Reformates can be also produced by employing partial oxidation (POx) and autothermal reforming (AR) reactions [4]. DME steam reforming (Equation (1)) occurs in the temperature range 250–400 °C and involves two sequential endothermic reactions: DME hydrolysis to methanol (Equation (2)) and methanol steam reforming to hydrogen and carbon dioxide (Equation (3)).
DME Steam Reforming
CH3OCH3 + 3H2O ⇌ 6H2 + 2CO2 ΔHr0 = +36.6 kJ mol−1, ΔGr0 = +15.9 kJ mol−1
DME Hydrolysis
CH3OCH3 + H2O ⇌ 2CH3OH ΔHr0 = +35 kJ mol−1, ΔGr0 = +15 kJ mol−1
Methanol Steam Reforming
CH3OH + H2O ⇌ 3H2 + CO2 ΔHr0 = +49.1 kJ mol−1, ΔGr0 = −0.4 kJ mol−1
Methanol steam reforming is a well-documented reaction that takes place over Cu-, Ni-, Pt- and Pd-based catalysts [14,15,16,17,18]. DME hydrolysis is an equilibrium-limited, acid-catalyzed reaction and the rate-determining step of the overall DME reforming process [1]. Hence, the development of more efficient acid catalysts for DME hydrolysis in conjunction with the rapid elimination of methanol via steam reforming will contribute to the improvement of DME reforming efficiency.
Usually, the DME hydrolysis reaction is investigated along with DME steam reforming, where most of the attention is focused on the reforming catalyst, whereas the acidic one is typically Al2O3, being active above 250 °C [19,20,21,22,23,24,25]. Due to the fact of their acidic properties, zeolites have also been employed in DME hydrolysis, where they proved to be particularly active in the temperature range 150–300 °C [5,26,27,28,29,30,31]. High reaction temperatures (>300 °C) are not recommended, especially for ZSM-5 zeolites, since a methanol-to-gasoline (MTG) reaction is favored due to the fact of its high acidity [29]. Thus, care must be taken in controlling the amount, type and strength of acid sites of the employed catalysts. Other catalysts proposed as the acidic component in hybrid catalytic systems for DME steam reforming have been ZrO2 [32], WO3/ZrO2 [31,33] and HPA/Al2O3 [34].
WO3 is known to possess acidic properties; hence, it can be utilized in an effort to enhance the acidity of the conventional catalysts used in acid-catalyzed reactions [35,36]. Indeed, many research groups have reported on the catalytic performance of WOx-supported catalysts over alcohol dehydration reactions [37,38,39,40,41]. There is a consensus that the addition of WOx species leads to the formation of new or additional Brønsted acid sites, with suitable strength, that enhance the dehydration rate. Additionally, although they are less active than zeolites, they present very good stability, since they do not possess strong acidic sites that favor the MTG reaction and coke formation, resulting in their degradation. It has been reported that the activity of WOx-supported catalysts over dehydration and isomerization reactions is dependent on tungsten surface density and the nature of the formed WOx species [42]. Baertsch et al. studied the dehydration of 2-butanol over WOx/ZrO2 catalysts and concluded that the maximum dehydration rates were attained at intermediate tungsten surface densities, above the theoretical monolayer coverage. The improved catalytic activity was ascribed to the presence of Brønsted acid sites related to more easily reduced polytungstate species by 2-butanol. The highest reaction rates coincided with the highest density of Brønsted acid sites [43].
Due to the principle of microscopic reversibility, the catalysts employed in methanol dehydration are potential catalysts for the reverse reaction of DME hydrolysis. To the best of our knowledge, the hydrolysis of DME over WO3/Al2O3-supported catalysts has not previously been reported.
In this work, WO3/Al2O3-supported catalysts were synthesized via the incipient wetness impregnation method with various tungsten loadings (0–44% wt. WO3), and they were evaluated in the DME hydrolysis reaction. The effect of tungsten surface density on the catalytic activity of the samples was investigated, and the best-performing catalyst was identified. The physicochemical characteristics of the catalysts was determined by means of N2 physisorption, XRD, Raman spectroscopy, XPS, NH3-TPD and 2,6-di-tert-butylpyridine adsorption experiments. Thus, the correlation between the catalytic activity and the formation of Brønsted acid sites is discussed.
2. Results
2.1. Catalyst Characterization
2.1.1. Textural and Structural Properties
The specific surface area (SBET) and pore size distribution of the catalysts were determined by means of N2 sorption measurements. N2 adsorption–desorption isotherms and the pore size distribution of WO3/Al2O3 catalysts are illustrated in Figure 1a,b, respectively. Blank γ-Al2O3 was also included for comparison. All isotherms could be classified as type IV with H3 hysteresis loops (IUPAC classification) closing at P/P0 = 0.50, indicative of aggregated plate-like particles forming slit-like pores (Figure 1a). Regarding the pore size distribution estimated by the BJH method (Figure 1b), all samples were mesoporous with a mean pore diameter of 3.5–4 nm, whereas the addition of tungsten resulted in narrower pore size distribution. Table 1 summarizes the textural characteristics as well as the tungsten surface density of the supported catalysts expressed as the number of W atoms per nm2 of catalyst. It is apparent that the specific surface area as well as the total pore volume of the catalysts systematically diminished upon augmentation of the tungsten percentage. The decrease in specific surface area was mainly caused by the diminution of the pore volume as tungsten oxo-species were formed and deposited inside the pores of the samples leading to their gradual blockage. According to the literature, the decrease in specific surface area was expected upon an increase in tungsten content [41,42,44]. Nevertheless, Said et al. reported that doping with tungsten (below 10% w/w) resulted in higher specific surface areas than the support which was attributed to the diffusion of tungsten species in Al2O3, leading to alterations in the textural properties of the samples [45].
The XRD diffractograms of WO3/γ-Al2O3 catalysts are shown in Figure 2. For the supported samples up to 19% wt. WO3/Al2O3, the reflections observed at 2θ = 37.3, 42.8, 45.8 and 67.1° can be ascribed exclusively to γ-Al2O3 support. WO3 phases could not be detected leading to the conclusion that the tungsten species formed were either amorphous or highly dispersed on the support. Further increases in tungsten content resulted in the detection of additional reflections at 2θ = 23.5, 26.6, 28.6, 33.6, 41.4, 54.9, 60.4 and 62.4°, which could be attributed to the monoclinic WO3 structure (JCPDS 01-083-0950, space group P21/n). WO3 reflections became more prominent upon increment in WO3 loading, suggesting higher crystallinity of the samples and formation of bulk WO3. The monolayer coverage was exceeded in samples with 28% wt. WO3 loading or higher was manifested by the detection of a crystalline WO3 phase. According to the literature, the monolayer coverage was attained at a tungsten surface density of 4–5 W atoms/nm2 of catalyst, which is in close agreement with our results, since the tungsten surface density of 28WO3/Al2O3 was 5.1 W/nm2 [41,46,47,48].
Raman spectra of the supported catalysts recorded under ambient conditions are presented in Figure 3. The pure WO3 Raman spectrum w included for comparison. The only bands detected are associated with tungsten species, whereas there are no additional Raman vibrations attributed to γ-Al2O3. All tungsten-containing samples exhibit a characteristic band in the region 975–986 cm−1, which is assigned to the symmetrical stretching mode (W = O) of tetrahedral monotungstate species. An increase in the intensity of this band was observed upon an increase in tungsten surface coverage, along with a systematic shift of its position from 975 (10% wt. WO3) to 986 cm−1 (37 and 44% wt. WO3) due to the polymerization of the amorphous monotungstates to distorted octahedral polytungstates, leading to less hydrated tungsten species on the catalyst surface [39,48]. Apart from the mono- and polytungstate species, crystalline WO3 was also detected in the catalysts with higher tungsten loading (≥28% wt.), demonstrated by the appearance of additional bands at 275, 716 and 810 cm−1 attributed to the W–O–W deformation mode and W–O bending and stretching modes, respectively [49,50,51]. The higher the tungsten content, the sharper the bands, indicating the increase in the crystallinity of the samples. Many research groups have reported on the nature of tungsten species formed on Al2O3 support with the progressive increment of tungsten surface density. There is a consensus that at low surface coverage (<5 W/nm2), tetrahedrally coordinated WO42- species coexist with octahedrally coordinated polymeric ones, being amorphous or finely dispersed on the support. Polytungstates dominate when monolayer coverage is achieved, whereas above monolayer coverage, WO3 crystallites are additionally formed on top of the monolayer, in which their size increases upon further increases in the W surface density [41,49]. In the present study, the Raman results confirmed that the monolayer coverage was attained already at a 5.1 W/nm2 surface density corresponding to 28WO3/Al2O3, since the characteristic bands of crystalline WO3 were present. These findings are consistent with the XRD results shown in Figure 2, where bulk WO3 was detected at tungsten surface densities equal to or higher than 5.1 W/nm2.
2.1.2. XPS
The determination of the surface composition of the WO3/Al2O3 catalysts was conducted via XPS measurements (Figure 4). According to the XP survey scan of 37WO3/Al2O3 shown in Figure 4a, the sample consists of Al, W, O and C. Similar spectra were recorded for all studied catalysts. Figure 4b presents the W4f XP spectra of 10, 19, 37 and 44WO3/Al2O3 catalysts. W4f XP spectra consist of two main peaks centered at 35.8 ± 0.1 and 37.8 ± 0.1 eV with a spin-orbit splitting of 2 eV, assigned to W4f5/2 and W4f7/2, respectively. These values are characteristic of the hexavalent oxidation state of tungsten (W6+) and subsequent deconvolution of the spectra confirmed that W6+ was the only oxidation state of tungsten encountered on the examined samples [52,53,54]. Figure 4c exhibits the Al2p spectra of the supported catalysts. Al2p spectra comprise a main peak centered at 74.3 ± 0.1 eV, whereas no alterations were observed upon increase in tungsten content of the samples [52,53]. The O1s core level spectra of the examined catalysts consist of a main peak centered at 531 ± 0.1 eV, attributed to lattice oxygen species [55]. The binding energy values of W4f, Al2p and O1s peaks, corrected using the C1s peak at 284.5 eV, are listed in Table 2.
The bulk and surface compositions of the catalysts determined via XRF and XPS, respectively, expressed as the atomic ratio of tungsten to aluminum (W/Al), are illustrated in Figure 5. It is apparent that at low tungsten coverage, namely, 10WO3/Al2O3, the surface W/Al atomic ratio was higher than the nominal one, i.e., the W/Al atomic ratio determined via XPS was 0.04, whereas the nominal one was 0.02. This deviation can be attributed to the high dispersion of tungsten species on the surface of the support; thus, there is a tungsten enrichment of the surface leading to a higher W/Al atomic ratio. This can also be deduced from the absence of WO3 reflections and the characteristic bands in XRD and Raman results, respectively. Focusing on the 19WO3/Al2O3 catalyst, the bulk and surface composition were in complete agreement. For the catalysts with higher surface coverages, 37 and 44WO3/Al2O3, the surface W/Al ratios were lower than the nominal ones, and it was observed that upon increase in tungsten content, this deviation was becoming more noticeable. This can be explained by the fact that the monolayer coverage was well exceeded on these catalysts, leading to the formation of bulk WO3, growth of larger crystallites and, therefore, a decrease in WO3 dispersion as tungsten loading increased. It should be noted that XPS is a surface-sensitive spectroscopic technique with an analysis depth of ~4–5 nm; hence, if larger crystallites are present on the catalyst surface, they will not be taken into account at sizes above 5 nm.
2.1.3. Acidity of the Catalysts
The total acidity of WO3/Al2O3 catalysts was investigated by means of NH3-TPD. The pretreated under He flow catalysts were exposed to a 1.1% NH3/He/Ar stream up to equilibration of the adsorption process. Ar was used as an inert tracer, since it does not adsorb on the catalyst’s surface. The physically adsorbed NH3 at room temperature was flushed away by subsequent He flow through the TPD cell followed by the TPD run in the temperature region 30–650 °C. NH3 desorbs molecularly, since it was the only species detected in the effluent. The NH3-TPD profiles of the supported WO3/Al2O3 catalysts are delineated in Figure 6a. The corresponding profile of γ-Al2O3 was also included for comparison. All TPD profiles consist of a main peak centered at 105–115 °C, which was extended up to 650 °C. The broad TPD profiles were indicative of the existence of both weak and intermediate acid sites on the surface of the catalysts. The addition of tungsten oxo-species did not induce any alterations regarding the strength of acid sites. The amount of NH3 desorbed (in μmol gcat−1) during TPD as a function of the tungsten surface density is depicted in Figure 6b (left axis). At low surface coverages up to 2.8 W/nm2, the amount of NH3 desorbed and, consequently, the number of acid sites remained practically unchanged. At higher tungsten surface densities, the number of acid sites gradually decreased, which was related to the lower specific surface areas of the tungsten-containing samples. Since the catalysts significantly differed in terms of specific surface area, which plays a crucial role in the adsorption of NH3 and, as a consequence, in the determination of the catalyst acidity, this factor can be eliminated by expressing the acidity on a surface area basis. Hence, the concentration of acid sites of the supported catalysts as a function of the tungsten surface density is demonstrated in Figure 6b (right axis). It is apparent that upon an increase in the tungsten surface coverage, a gradual increase in the density of acid sites was observed. The values of the amount and concentration of acid sites are listed in Table 1. Our results at low W surface densities are in accordance with those reported by Soled et al. [56]. They investigated the acidity of WO3/γ-Al2O3 (10% wt.) employing NH3-TPD, IR-pyridine and high-temperature gravimetric titration of amines and concluded that the addition of tungsten at a low loading (10%) did not significantly alter the number and density of total acid sites between γ-Al2O3 and WO3/Al2O3 catalysts but led to the formation of Brønsted at the expense of Lewis acid sites, whereas Al2O3 possessed only Lewis ones. The monotonic increase in Brønsted acid sites upon increase in tungsten surface density at submonolayer coverage (up to 4.1 W/nm2) was also observed by Chen et al. [57]. They found a direct correlation between the catalytic activity in 2-propanol reaction and the evolution of Brønsted acid sites, which were mainly linked with the formation of polytungstate species over WOx/Al2O3 catalysts. Zhang et al. investigated the acidity of 0–30% wt. WO3/Al2O3 catalysts employing the colorimetric titration and inverse-gas chromatography techniques and concluded that in line with previous studies, Brønsted acid sites are introduced upon addition of tungsten, while the ratio of Brønsted to Lewis acid sites is systematically increasing even at high tungsten surface densities. They proposed that the cause of the Brønsted site formation is the existence of terminal oxygen atoms that lead to charge delocalization in the support [58].
For the determination of Brønsted acid sites, 2,6-di-tert-butylpyridine was employed as a probe molecule. 2,6-Di-tert-butylpyridine is an alkylated pyridine with bulky tert-butyl substituents serving as hindered base that selectively adsorbs on Brønsted acid sites, forming the corresponding pyridinium ion, whereas the Lewis acid sites are inaccessible due to the steric hindrance phenomena. Many research groups have used 2,6-di-tert-butylpyridine for the discrimination between Brønsted and Lewis acid sites or the determination of Brønsted acidity on the external surface of zeolites, since the bulky substituted pyridine cannot enter into the smaller pores of the studied zeolites [37,43,59,60]. Table 1 summarizes the 2,6-di-tert-butylpyridine uptake over selected WO3/Al2O3 catalysts determined via adsorption experiments at 120 °C followed by desorption of weakly adsorbed molecules at the same temperature. The uptake and consequently the amount of Brønsted acid sites follow the order 37WO3/Al2O3 > 44WO3/Al2O3 > 10WO3/Al2O3 > Al2O3. Tungsten addition results in the development of Brønsted acid sites, which increase up to 7.4 W/nm2 in surface coverage while further increases in tungsten content lead to a decrease in Brønsted acid sites, probably linked to the formation of larger WO3 crystallites. Macht et al. reported on the titration of Brønsted acid sites of WOx/Al2O3 catalysts using 2,6-di-tert-butylpyridine during the catalytic tests in 2-butanol dehydration. They concluded that the density of Brønsted acid sites reached a maximum at above monolayer coverage (9.8 W/nm2), whereas further increases in tungsten loading led to the diminution of the concentration of Brønsted acid sites [37]. The impact of tungsten addition on various supports on the formation of Brønsted acid sites has been reported by many research groups employing various methods [36,38,39,42,61].
2.2. Catalytic Evaluation in DME Hydrolysis
The supported WO3/Al2O3 catalysts were tested in DME hydrolysis reaction in the temperature range of 140–300 °C with W/F = 0.5 g s cm−3. The variation in the conversion as a function of the reaction temperature over the xWO3/Al2O3 catalysts is presented in Figure 7. DME hydrolysis is a thermodynamically limited reaction; hence, the equilibrium conversion was calculated taking into account the composition of the feed, i.e., 0.09% DME and 3% H2O vapor, and it is included in Figure 7. In the temperature range studied, CH3OH was the only product detected. γ-Al2O3 is the sample with the poorest catalytic performance, where the production of CH3OH commenced above 220 °C and reached the equilibrium value of 300 °C. Tungsten-containing catalysts demonstrated substantially improved catalytic activity. Upon increases in tungsten content, the activity of the catalysts towards DME hydrolysis gradually increased up to 37% wt. WO3/Al2O3, whereas higher tungsten loading (44WO3/Al2O3) led to inferior activity. The best-performing catalyst was already active at 150 °C and reached equilibrium at 250 °C (XDME = 50%). By comparison, the temperature at which 10% conversion was achieved (T10) over γ-Al2O3 and 37WO3/Al2O3 was 243 and 183 °C, respectively, which is a significant shift by 60 °C to lower temperatures.
Figure 8 presents the reaction rates of the WO3/Al2O3 catalysts fresh and poisoned with 2,6-di-tert-butylpyridine, measured at 200 °C (left axis) and the 2,6-di-tert-butylpyridine uptake (right axis) as a function of tungsten surface density. The reaction rates are expressed on a unit mass basis, and they are measured under differential conditions (XDME ≤ 20%). The catalytic activity in terms of reaction rates followed the order: 7.4 W/nm2 > 10.1 W/nm2 > 5.1 W/nm2 > 2.8 W/nm2 > 1.2 W/nm2 >> γ-Al2O3. A direct comparison between 37WO3/Al2O3 and γ-Al2O3 cannot be made, since there was no measurable reaction rate at 200 °C for γ-Al2O3, whereas at 225 °C, where γ-Al2O3 exhibited catalytic activity, the conditions were not differential for the 37WO3/Al2O3 catalyst (XDME = 35%). Thus, the comparison can be made by employing the sample with the lowest tungsten content, viz., 10WO3/Al2O3. Table 3 summarizes the reaction rates, the specific reaction rates and T10 values of the tested samples. The highest reaction rate per catalyst weight achieved by 37WO3/Al2O3 (15.74 × 10−3 μmol gcat−1 s−1) was 19 times higher than the corresponding one of 10WO3/Al2O3 (0.82 × 10−3 μmol gcat−1 s−1), while in terms of WO3 weight the former exhibited fivefold higher activity than 10WO3/Al2O3. Regarding the reaction rates expressed per WO3 weight, it can be concluded that the catalytic activity increases up to 7.4 W/nm2 where tungsten-oxo species and WO3 crystallites (where present) developed beneficially for the hydrolysis reaction, whereas further addition of tungsten led to the formation of larger crystallites that were less active or even inert towards DME hydrolysis. The superiority of 37WO3/Al2O3 was even more pronounced in terms of intrinsic activity, which was 34 times higher than the specific reaction rate measured at 200 °C over 10WO3/Al2O3. This difference was intensified by the fact that 37WO3/Al2O3 had almost half the specific surface area of 10WO3/Al2O3, yet was significantly more active towards DME hydrolysis. At this point, it should be mentioned that the expression of the reaction rates per specific surface area was not completely appropriate for the supported catalysts, since the activity of the catalyst was mainly attributed to the supported phase and not to the entire exposed surface.
In any case, the activity of the supported WO3/Al2O3 catalysts exhibited a volcanic trend in DME hydrolysis, and the best-performing catalyst was the one with 7.4 W/nm2. The same trend was found for the 2,6-di-tert-butlypyridine uptake as shown in Figure 8 (right axis). As previously discussed (Section 2.1.3), 2,6-di-tert-butylpyridine uptake was directly associated with the number of Brønsted acid sites formed on the surface of the catalysts; thus, it can be concluded that DME hydrolysis occurred primarily on the Brønsted acid sites of WO3/Al2O3 catalysts. To further confirm the role of Brønsted acid sites on the catalytic activity of the tungsten-containing catalysts, DME hydrolysis rates were also measured after blocking the Brønsted acid sites of the best-performing samples, namely, 37WO3/Al2O3 and 44WO3/Al2O3, with 2,6-di-tert-butylpyridine (Figure 8, left axis). Prior to catalytic tests at 200 °C, 2,6-di-tert-butylpyridine was adsorbed on the samples at 120 °C overnight followed by the desorption of weakly bound molecules at the same temperature. Indeed, the reaction rates obtained over poisoned 37WO3/Al2O3 and 44WO3/Al2O3 were 0.39 × 10−3 and 0.30 × 10−3 μmol gcat−1 s−1 compared to 15.74 × 10−3 and 10.92 × 10−3 μmol gcat−1 s−1 of the fresh samples. Therefore, even though the reaction took place at 200 °C, Brønsted acid sites were blocked by a part of 2,6-di-tert-butylpyridine that remained strongly chemisorbed on them, resulting in a dramatic decrease in the reaction rates by 36–40 times. Thus, it can be concluded that DME hydrolysis primarily occurs on Brønsted acid sites, whereas the residual reaction rates measured may be attributed to Lewis acid sites on WOx domains that remained unaffected by the addition of 2,6-di-tert-butylpyridine [37].
Since there are no reports in DME hydrolysis over supported WOx catalysts and because of the principle of microscopic reversibility, we took into consideration the literature concerning alcohol dehydration reactions. Suwannapichat et al. investigated the direct synthesis of DME from CO2 hydrogenation over hybrid CuO/ZnO/ZrO2 and WOx/Al2O3 catalysts. Methanol dehydration took place over tungsten-supported catalysts varying the tungsten loading and the pore size of the support. In accordance with our results, they also observed a volcanic trend in DME production as a function of tungsten surface density, while the maximum value was obtained above monolayer coverage at 8 W/nm2 [41]. The optimization of the catalytic activity in alcohol dehydration above the theoretical monolayer coverage over WOx-supported catalysts (i.e., WOx/ZrO2 and WOx/TiO2) was reported by various research groups [37,39,40,62]. It is widely accepted that CH3OH dehydration as well as the reverse reaction, DME hydrolysis, are acid-catalyzed reactions; hence, the acidity of the catalysts plays a crucial role in the overall process. However, based on the NH3-TPD results (Figure 6b), it can be seen that the most active catalysts in DME hydrolysis exhibited lower total acidity (per catalyst weight). Adsorption of NH3 is a measure of the total acidity of the catalysts, both Brønsted and Lewis; thus, no direct correlation could be made between the total acidity and the catalytic activity. Therefore, the type and strength rather than the number of the acid sites are the key factors that drive the catalytic activity. Indeed, it is generally agreed that mainly Brønsted acid sites are responsible for the higher reaction rates obtained in DME hydrolysis and CH3OH dehydration [5,39,63,64,65,66]. γ-Al2O3 that possesses only Lewis acid sites has low activity, as water competitively adsorbs on the same sites with methanol leading to their blockage [64,67]. Macht et al. concluded that the enhanced catalytic activity in 2-butanol dehydration over WO3/Al2O3 and WO3/ZrO2 is directly correlated with the development Brønsted acidity. Lewis acid sites were constantly diminishing upon tungsten addition on Al2O3. The dehydration rates increased upon tungsten surface density increment, reaching the maximum values at 9–10 W/nm2. After the titration of the Brønsted acid sites with 2,6-di-tert-butylpyridine, the dehydration rates obtained dramatically decreased. The residual reaction rates remained constant upon increase in tungsten surface density up to 10 W/nm2, while at even higher surface densities, where large WO3 crystallites were formed, they diminished further. On the other hand, the blocking of acid sites with pyridine, which coordinated with both Brønsted and Lewis acid sites, led to the complete loss of the dehydration activity of the catalysts [37]. The results obtained in the present work regarding the enhanced catalytic activity of the WO3/Al2O3 catalysts in DME hydrolysis could, therefore, be directly associated with the development of Brønsted acid sites upon tungsten addition on Al2O3 support. A DME molecule adsorbs on a Brønsted acid site forming a dimethyloxonium ion ((CH3)2OH+), whereas its subsequent hydration leads to the formation of methanol.
3. Materials and Methods
3.1. Catalysts Synthesis
WO3/γ-Al2O3 samples with tungsten (VI) oxide loading in the range 0–44% w/w were prepared via the incipient wetness impregnation method. Prior to impregnation, the γ-Al2O3 (ALCOA DD-2) support was crushed, sieved (90 < dp < 180 μm) and calcined at 500 °C for 4 h at a heating rate of 5 °C min−1. The desired amount of ammonium metatungstate hydrate ((NH4)6W12O39·xH2O, Sigma Aldrich, Steinheim, Germany), depending on the final percentage of WO3 on the catalyst, was dissolved in triple-distilled water followed by the impregnation of the support. The suspension was left for equilibration under rotation for 1 h at room temperature and atmospheric pressure, while subsequent evaporation under vacuum at 50 °C led to a slurry which was left to dry at room temperature overnight. The samples were further dried by gradually increasing the temperature from 50 to 120 °C with a heating rate of 10 °C h−1 and, finally, they were calcined at 500 °C with a heating rate of 5 °C min−1 for 5 h. The resulting catalysts were nominated as xWO3/Al2O3 (where x refers to the WO3 loading by weight percent, i.e., 0, 10, 19, 28, 37, and 44%).
3.2. Catalyst Characterization
The specific surface area (SBET) and porosity of the catalysts were determined from the N2 sorption-isotherms at −196 °C, using a Quantachrome Autosorb IQ-C-MP apparatus. Prior to measurements, the samples were degassed at 150 °C in vacuum for 2 h. N2 adsorption isotherms were measured at relative pressures (P/P0, where P is the actual pressure of N2, and P0 is the saturation pressure of N2 at −196 °C) in the range of 0.015 to 0.995. The specific surface area was calculated in the range 0.015 < P/P0 < 0.35, total pore volume at P/P0 = 0.995 and pore size distribution (PSD) using the Barret–Joyner–Halenda (BJH) model using the desorption branches of the N2 isotherms.
The XRD diffractograms of the catalysts were recorded using a Bruker D8 Advance X-ray powder diffractometer operated at 40 mA and 40 kV employing a Ni-filtered Cu Ka radiation (λ = 1.5418 Å) in the range of 20° < 2θ < 80° with a scan step of 0.02° and a scan speed of 1.5 s/step.
Raman spectra were recorded using a T64000 micro-Raman spectrometer (Horiba Jobin Yvon, Kyoto, Japan) equipped with a 514.5 nm laser line as the excitation source. The spot size was ~2–3 μm while a microscope objective of 50× magnification was employed. The spectra were corrected taking into account the 520 cm−1 Raman band of crystalline Si.
The composition of the catalysts was determined via X-ray fluorescence spectroscopy (XRF) using a S2 PicofoxTM Bruker Nano X-Ray Fluorescence spectrometer equipped with a metal–ceramic air-cooled molybdenum (Mo) target X-ray tube operating at 50 kV and 0.6 mA.
X-ray photoelectron spectroscopy (XPS) was performed in a UHV chamber (5 × 10−10 mbar) equipped with a SPECS Phoibos 100-1D-DLD hemispherical electron analyzer and a non-monochromatized dual-anode Mg/Al X-ray source. Samples were excited using MgKα at 1253.6 eV photon energy, and the pass energy of the analyzer was 10 eV. Prior to XPS measurements, the powdered samples were pressed and pelletized, whereas the diameter of the analyzed area spot was 7 × 15 mm. The spectra were recorded and analyzed using the commercial software, SpecsLab Prodigy (Specs GmbH, Berlin, Germany). The binding energy at 284.5 eV corresponding to C 1s of contaminant carbon was used as reference for the calculation of the binding energies.
The total acidity of the catalysts was determined via temperature-programmed desorption of ammonia (NH3-TPD) under atmospheric pressure in a fixed-bed reactor. The system consisted of two independent gas lines equipped with mass flow controllers (Aera GmbH, Kirchheim, Germany) and a mass spectrometer (Omnistar/Pfeiffer Vacuum, Asslar, Germany) for online monitoring of effluent gases. During NH3 adsorption at room temperature, a gas mixture (30 cm3 min−1) containing 1.1% NH3, 1% Ar as inert tracer and He as balance was fed into the TPD cell (100 mg of catalyst) until saturation was attained, marked by the stabilization of signals in the mass spectrometer. Then, the excess of NH3 was purged with He until all signals met their baselines. The subsequent TPD run was conducted under He flow (30 cm3 min−1) from 30 to 650 °C with a heating rate of 10 °C min−1. Prior to NH3 adsorption, the samples were preheated under He flow (30 cm3 min−1) up to 500 °C with a heating rate of 10 °C min−1 and soaked at 500 °C for 15 min followed by cooling down to room temperature under the same flow. The following fragments were recorded in the mass spectrometer during TPD experiments: m/z = 15, 16 (CH4), 17 (NH3), 18 (H2O), 28 (N2), 44 (CO2), 32 (O2), 40 (Ar) and 2 (H2).
The 2,6-di-tert-butylpyridine (>98%, Merck-Schuchardt, Hohenbrunn, Germany) was used as probe molecule for the determination of Brønsted acid sites of WO3/Al2O3 catalysts via a static gravimetric method. In brief, vapors of 2,6-di-tert-butylpyridine were left to adsorb on the powdered samples (dried at 150 °C overnight) enclosed in a sealed glass vessel at 120 °C overnight with a subsequent desorption at the same temperature to remove the weakly bound 2,6-di-tert-butylpyridine. After the adsorption and desorption process, the weight gain and loss, respectively, were measured. The 2,6-di-tert-butylpyridine uptake is referred to the remaining adsorbed amount of 2,6-di-tert-butylpyridine after the desorption step.
3.3. Catalyst Evaluation Tests
The catalytic performance of WO3/γ-Al2O3 samples in DME hydrolysis was carried out in a conventional quartz fixed-bed flow reactor at atmospheric pressure whereas the reaction temperature was in the range of 140–300 °C. In a typical experiment, 0.5 g of catalyst (crushed and sieved to particle size 90 < dp < 180 μm) was introduced in the reactor and heated under He (30 cm3 min−1) at 300 °C for 1 h. Then, the catalyst was exposed to a reactant feed (60 cm3 min−1) containing 900 ppm DME (3.5% DME/He, home-made mixture in gas cylinder using DME, ≥99.9% (GC), Sigma Aldrich, Steinheim, Germany), ~3% H2O and He (99.999%, Linde Hellas, Athens, Greece) as balance (W/F = 0.5 g s cm−3). H2O vapor was introduced by flowing He through a saturator containing triple-distilled water at room temperature. The reactant and product composition were analyzed online by a gas chromatograph (Shimadzu GC-14B) equipped with a flame ionization detector (FID). The employed chromatographic column was 0.19% Picric acid/Graphpac-CC 80/100, 7′ × 1/8″ × 0.085″/SS.
The measurements of the reaction rates over the poisoned catalysts with 2,6-di-tert-butylpyridine took place at 200 °C with no alterations regarding the reactant feed or the W/F. Prior to the experiment, the sample was exposed to 2,6-di-tert-butylpyridine vapors in a sealed vessel at 120 °C overnight followed by the removal of excess 2,6-di-tert-butylpyridine at the same temperature. Then, the catalyst was loaded in the reactor, and the feed stream passed though the reactor at 200 °C. The reaction rate was measured after steady state was achieved (~1 h).
The conversion (X) of DME was calculated using the following equation:
where , are the feed and the outlet concentration of DME and CH3OH, respectively.
For the calculation of the reaction rates on a unit catalyst weight (, in μmol gcat−1 s−1) the following equation was used:
where: FDME is the molar flow of DME (in μmol s−1), XDME is the conversion of DME to CH3OH and W is the catalyst weight (in g).
For the calculation of the reaction rates per WO3 weight (, in μmol gWO3−1 s−1) the following equation was used:
where rW is the reaction rate on a unit catalyst weight of DME hydrolysis (in μmol gcat−1 s−1), and w is the WO3 weight calculated from the WO3 content of the catalyst (in g).
For the calculation of the specific reaction rates (r, in μmol m−2 s−1), the following equation was used:
where rW is the reaction rate on a unit catalyst weight of DME hydrolysis (in μmol gcat−1 s−1), and SSA is the specific surface area of the catalysts (in m2 gcat−1).
4. Conclusions
DME hydrolysis is the rate-determining step in steam reforming of DME, which can be employed in the production of high-quality reformate gas as feed for fuel cells. Hence, the improvement of DME hydrolysis reaction would lead to the enhanced efficiency of the overall DME steam reforming process. Acidic Al2O3 and zeolites were mainly used to catalyze the reaction, with the former being inactive beyond 250 °C and the latter being active but not stable due to the coke formation resulting from the simultaneous occurrence of side reactions such as an MTG reaction. Therefore, the modification of γ-Al2O3 focusing on the development of additional acid sites would lead to enhanced catalytic activity at a lower temperature and, at the same time, its high stability could be retained. WO3/Al2O3-supported catalysts with various WO3 loadings (0–44% wt.), prepared by the incipient wetness impregnation method, were investigated in DME hydrolysis to CH3OH. According to XRD and Raman results, amorphous and well-dispersed mono- and polytungstate species prevailed on the surface of the catalysts at low tungsten surface densities (<5.1 W/nm2), whereas monolayer coverage was exceeded at higher surface densities (≥5.1 W/nm2) indicated by the formation of WO3 crystallites. XPS measurements further validated the fine dispersion of tungsten oxo-species at low surface coverage, whereas W6+ was the only oxidation state encountered on the samples. The concentration of acid sites determined via NH3-TPD experiments as a function of tungsten loading exhibited a monotonic increase, although the amount of total acid sites per catalyst unit weight decreased due to the specific surface area loss of the samples upon augmentation of tungsten content. A volcanic trend was observed between the hydrolysis rates and the tungsten surface density. The number of Brønsted acid sites, determined via the selective adsorption of 2,6-di-tert-butylpyridine as a function of tungsten surface density, followed the same trend. The most active catalyst was 37WO3/Al2O3 with a tungsten surface density of 7.4 W/nm2, which is above the theoretical monolayer coverage. Blocking of Brønsted acid sites with 2,6-di-tert-butylpyridine led to a dramatic decrease in hydrolysis rates by 40 times. The enhanced catalytic activity was attributed to the development of Brønsted acid sites at the expense of Lewis ones upon tungsten addition. Therefore, the type and strength rather than the number of acid sites have a key role in DME hydrolysis reaction.
Author Contributions
M.S., Methodology, validation, formal analysis, investigation, data curation, and writing—original draft preparation; T.I., Conceptualization, methodology, validation, formal analysis, investigation, resources, writing—review and editing, supervision, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation under the call RESEARCH–CREATE–INNOVATE (project code: T1EDK-01704).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors would like to thank L. Sygellou for her assistance with the XPS measurements and Paraskevopoulou for the donation of the 2,6-di-tert-butylpyridine reagent.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Namuangruk, S.; Meeprasert, J.; Khemthong, P.; Faungnawakij, K. A Combined Experimental and Theoretical Study on the Hydrolysis of Dimethyl Ether over H-ZSM-5. J. Phys. Chem. C 2011, 115, 11649–11656. [Google Scholar] [CrossRef]
- Hirunsit, P.; Faungnawakij, K.; Namuangruk, S.; Luadthong, C. Catalytic Behavior and Surface Species Investigation over γ-Al2O3 in Dimethyl Ether Hydrolysis. Appl. Catal. Gen. 2013, 460–461, 99–105. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical Recycling of Carbon Dioxide to Methanol and Dimethyl Ether: From Greenhouse Gas to Renewable, Environmentally Carbon Neutral Fuels and Synthetic Hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Faungnawakij, K.; Kikuchi, R.; Matsui, T.; Fukunaga, T.; Eguchi, K. A Comparative Study of Solid Acids in Hydrolysis and Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2007, 333, 114–121. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Ott, K.C.; Borup, R.L.; Greene, H.L. Role of Acidity on the Hydrolysis of Dimethyl Ether (DME) to Methanol. Appl. Catal. B Environ. 2005, 61, 281–287. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl Ether (DME) as an Alternative Fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Basu, A.; Gradassi, M.J.; Masin, J.G. Dimethyl Ether: A Fuel for the 21st Century. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1997; Volume 107, pp. 117–125. ISBN 978-0-444-82352-6. [Google Scholar]
- Liu, H.; Iglesia, E. Selective One-Step Synthesis of Dimethoxymethane via Methanol or Dimethyl Ether Oxidation on H3+nVnMo12-nPO40 Keggin Structures. J. Phys. Chem. B 2003, 107, 10840–10847. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Tan, Y.; Yang, C.; Han, Y. MnCl2 Modified H4SiW12O40/SiO2 Catalysts for Catalytic Oxidation of Dimethy Ether to Dimethoxymethane. J. Mol. Catal. Chem. 2007, 263, 149–155. [Google Scholar] [CrossRef]
- Cheung, P.; Bhan, A.; Sunley, G.; Law, D.; Iglesia, E. Site Requirements and Elementary Steps in Dimethyl Ether Carbonylation Catalyzed by Acidic Zeolites. J. Catal. 2007, 245, 110–123. [Google Scholar] [CrossRef]
- Zhou, W.; Kang, J.; Cheng, K.; He, S.; Shi, J.; Zhou, C.; Zhang, Q.; Chen, J.; Peng, L.; Chen, M.; et al. Direct Conversion of Syngas into Methyl Acetate, Ethanol, and Ethylene by Relay Catalysis via the Intermediate Dimethyl Ether. Angew. Chem. Int. Ed. 2018, 57, 12012–12016. [Google Scholar] [CrossRef]
- Liu, H.; Cheung, P.; Iglesia, E. Zirconia-Supported MoOxCatalysts for the Selective Oxidation of Dimethyl Ether to Formaldehyde: Structure, Redox Properties, and Reaction Pathways. J. Phys. Chem. B 2003, 107, 4118–4127. [Google Scholar] [CrossRef] [Green Version]
- Peláez, R.; Marín, P.; Ordóñez, S. Synthesis of Formaldehyde from Dimethyl Ether on Alumina-Supported Molybdenum Oxide Catalyst. Appl. Catal. Gen. 2016, 527, 137–145. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for Methanol Steam Reforming—A Review. Appl. Catal. B Environ. 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Papavasiliou, J.; Avgouropoulos, G.; Ioannides, T. Steam Reforming of Methanol over Copper–Manganese Spinel Oxide Catalysts. Catal. Commun. 2005, 6, 497–501. [Google Scholar] [CrossRef]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S.; Tsunoda, T.; Ishii, T.; Kumagai, M. Highly Active Copper/Ceria Catalysts for Steam Reforming of Methanol. Appl. Catal. Gen. 2002, 223, 137–145. [Google Scholar] [CrossRef]
- Ranganathan, E.S.; Bej, S.K.; Thompson, L.T. Methanol Steam Reforming over Pd/ZnO and Pd/CeO2 Catalysts. Appl. Catal. Gen. 2005, 289, 153–162. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kikuchi, R.; Takeguchi, T.; Eguchi, K. Steam Reforming of Dimethyl Ether over Composite Catalysts of γ-Al2O3 and Cu-Based Spinel. Appl. Catal. B Environ. 2005, 57, 211–222. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Tanaka, Y.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Hydrogen Production from Dimethyl Ether Steam Reforming over Composite Catalysts of Copper Ferrite Spinel and Alumina. Appl. Catal. B Environ. 2007, 74, 144–151. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Cu-Based Spinel Catalysts CuB2O4 (B=Fe, Mn, Cr, Ga, Al, Fe0.75Mn0.25) for Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2008, 341, 139–145. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Crystal Structure and Surface Species of CuFe2O4 Spinel Catalysts in Steam Reforming of Dimethyl Ether. Appl. Catal. B Environ. 2009, 92, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Pan, X.; Lin, R.; Kou, S.; Zou, W.; Ma, J.-X. Steam Reforming of Dimethyl Ether over Cu–Ni/γ-Al2O3 Bi-Functional Catalyst Prepared by Deposition–Precipitation Method. Int. J. Hydrogen Energy 2010, 35, 4060–4068. [Google Scholar] [CrossRef]
- Takeishi, K.; Akaike, Y. Hydrogen Production by Dimethyl Ether Steam Reforming over Copper Alumina Catalysts Prepared Using the Sol–Gel Method. Appl. Catal. Gen. 2016, 510, 20–26. [Google Scholar] [CrossRef]
- Shimoda, N.; Faungnawakij, K.; Kikuchi, R.; Fukunaga, T.; Eguchi, K. Catalytic Performance Enhancement by Heat Treatment of CuFe2O4 Spinel and γ-Alumina Composite Catalysts for Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2009, 365, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, T.; Matsuoka, H.; Shishido, T.; Li, D.; Tian, Y.; Sano, T.; Takehira, K. Steam Reforming of Dimethyl Ether over ZSM-5 Coupled with Cu/ZnO/Al2O3 Catalyst Prepared by Homogeneous Precipitation. Appl. Catal. Gen. 2006, 308, 82–90. [Google Scholar] [CrossRef]
- Shimoda, N.; Faungnawakij, K.; Kikuchi, R.; Eguchi, K. A Study of Various Zeolites and CuFe2O4 Spinel Composite Catalysts in Steam Reforming and Hydrolysis of Dimethyl Ether. Int. J. Hydrogen Energy 2011, 36, 1433–1441. [Google Scholar] [CrossRef]
- Ereña, J.; Vicente, J.; Aguayo, A.T.; Gayubo, A.G.; Olazar, M.; Bilbao, J. Effect of Combining Metallic and Acid Functions in CZA/HZSM-5 Desilicated Zeolite Catalysts on the DME Steam Reforming in a Fluidized Bed. Int. J. Hydrogen Energy 2013, 38, 10019–10028. [Google Scholar] [CrossRef]
- Feng, D.; Zuo, Y.; Wang, D.; Wang, J. Steam Reforming of Dimethyl Ether over Coupled Catalysts of CuO-ZnO-Al2O3-ZrO2 and Solid-Acid Catalyst. Chin. J. Chem. Eng. 2009, 17, 64–71. [Google Scholar] [CrossRef]
- Fukunaga, T.; Ryumon, N.; Shimazu, S. The Influence of Metals and Acidic Oxide Species on the Steam Reforming of Dimethyl Ether (DME). Appl. Catal. Gen. 2008, 348, 193–200. [Google Scholar] [CrossRef]
- Matsumoto, T.; Nishiguchi, T.; Kanai, H.; Utani, K.; Matsumura, Y.; Imamura, S. Steam Reforming of Dimethyl Ether over H-Mordenite-Cu/CeO2 Catalysts. Appl. Catal. Gen. 2004, 276, 267–273. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Ott, K.C.; Borup, R.L.; Greene, H.L. Generating Hydrogen-Rich Fuel-Cell Feeds from Dimethyl Ether (DME) Using Cu/Zn Supported on Various Solid-Acid Substrates. Appl. Catal. Gen. 2006, 309, 210–223. [Google Scholar] [CrossRef]
- Nishiguchi, T.; Oka, K.; Matsumoto, T.; Kanai, H.; Utani, K.; Imamura, S. Durability of WO3/ZrO2–CuO/CeO2 Catalysts for Steam Reforming of Dimethyl Ether. Appl. Catal. Gen. 2006, 301, 66–74. [Google Scholar] [CrossRef]
- Galvita, V.V.; Semin, G.L.; Belyaev, V.D.; Yurieva, T.M.; Sobyanin, V.A. Production of Hydrogen from Dimethyl Ether. Appl. Catal. Gen. 2001, 216, 85–90. [Google Scholar] [CrossRef]
- Busca, G. The Surface Acidity of Solid Oxides and Its Characterization by IR Spectroscopic Methods. An Attempt at Systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Can, F.; Courtois, X.; Duprez, D. Tungsten-Based Catalysts for Environmental Applications. Catalysts 2021, 11, 703. [Google Scholar] [CrossRef]
- Macht, J.; Baertsch, C.D.; May-Lozano, M.; Soled, S.L.; Wang, Y.; Iglesia, E. Support Effects on Brønsted Acid Site Densities and Alcohol Dehydration Turnover Rates on Tungsten Oxide Domains. J. Catal. 2004, 227, 479–491. [Google Scholar] [CrossRef]
- Hong, E.; Sim, H.-I.; Shin, C.-H. The Effect of Brønsted Acidity of WO3 /ZrO2 Catalysts in Dehydration Reactions of C3 and C4 Alcohols. Chem. Eng. J. 2016, 292, 156–162. [Google Scholar] [CrossRef]
- Ladera, R.; Finocchio, E.; Rojas, S.; Busca, G.; Fierro, J.L.G.; Ojeda, M. Supported WOx-Based Catalysts for Methanol Dehydration to Dimethyl Ether. Fuel 2013, 113, 1–9. [Google Scholar] [CrossRef]
- Witoon, T.; Kidkhunthod, P.; Chareonpanich, M.; Limtrakul, J. Direct Synthesis of Dimethyl Ether from CO2 and H2 over Novel Bifunctional Catalysts Containing CuO-ZnO-ZrO2 Catalyst Admixed with WOx/ZrO2 Catalysts. Chem. Eng. J. 2018, 348, 713–722. [Google Scholar] [CrossRef]
- Suwannapichat, Y.; Numpilai, T.; Chanlek, N.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J.; Witoon, T. Direct Synthesis of Dimethyl Ether from CO2 Hydrogenation over Novel Hybrid Catalysts Containing a Cu ZnO ZrO2 Catalyst Admixed with WOx/Al2O3 Catalysts: Effects of Pore Size of Al2O3 Support and W Loading Content. Energy Convers. Manag. 2018, 159, 20–29. [Google Scholar] [CrossRef]
- Pfriem, N.; Liu, Y.; Zahn, F.; Shi, H.; Haller, G.L.; Lercher, J.A. Impact of the Local Concentration of Hydronium Ions at Tungstate Surfaces for Acid-Catalyzed Alcohol Dehydration. J. Am. Chem. Soc. 2021, 143, 20133–20143. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, C.D.; Komala, K.T.; Chua, Y.-H.; Iglesia, E. Genesis of Brønsted Acid Sites during Dehydration of 2-Butanol on Tungsten Oxide Catalysts. J. Catal. 2002, 205, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Yi, L.; Deng, R.; You, K.; Song, J.; Jian, J.; Liu, P.; Ai, Q.; Luo, H. Supported WO3/γ-Al2O3 as Bifunctional Catalyst for Liquid-Phase Highly Selective Oxidation of Cyclohexylamine to Cyclohexanone Oxime under Solvent-Free Conditions. Mol. Catal. 2019, 475, 110494. [Google Scholar] [CrossRef]
- Said, A.E.-A.A.; Abd El-Wahab, M.M.M.; Abd El-Aal, M. Catalytic Dehydration of Methanol to Dimethyl Ether over Nanosized WO3/Al2O3 System under Inert and Oxidative Atmosphere. Mon. Chem. Chem. Mon. 2016, 147, 1507–1516. [Google Scholar] [CrossRef]
- Wachs, I.E. Raman and IR Studies of Surface Metal Oxide Species on Oxide Supports: Supported Metal Oxide Catalysts. Catal. Today 1996, 27, 437–455. [Google Scholar] [CrossRef]
- Ross-Medgaarden, E.I.; Wachs, I.E. Structural Determination of Bulk and Surface Tungsten Oxides with UV−vis Diffuse Reflectance Spectroscopy and Raman Spectroscopy. J. Phys. Chem. C 2007, 111, 15089–15099. [Google Scholar] [CrossRef]
- Chan, S. Laser Raman Characterization of Tungsten Oxide Supported on Alumina: Influence of Calcination Temperatures. J. Catal. 1985, 92, 1–10. [Google Scholar] [CrossRef]
- Salvati, L.; Makovsky, L.E.; Stencel, J.M.; Brown, F.R.; Hercules, D.M. Surface Spectroscopic Study of Tungsten-Alumina Catalysts Using x-Ray Photoelectron, Ion Scattering, and Raman Spectroscopies. J. Phys. Chem. 1981, 85, 3700–3707. [Google Scholar] [CrossRef]
- Martín, C.; Solana, G.; Malet, P.; Rives, V. Nb2O5-Supported WO3: A Comparative Study with WO3/Al2O3. Catal. Today 2003, 78, 365–376. [Google Scholar] [CrossRef]
- Vermaire, D. The Preparation of WO3/TiO2 and WO3/Al2O3 and Characterization by Temperature-Programmed Reduction. J. Catal. 1989, 116, 309–317. [Google Scholar] [CrossRef]
- Grunert, W. Reduction Behavior and Metathesis Activity of WO3/Al2O3 Catalysts I. An XPS Investigation of WO3/Al2O3 Catalysts. J. Catal. 1987, 107, 522–534. [Google Scholar] [CrossRef]
- Rodríguez-Ramos, I.; Guerrero-Ruiz, A.; Homs, N.; de la Piscina, P.R.; Fierro, J.L.G. Reactions of Propene on Supported Molybdenum and Tungsten Oxides. J. Mol. Catal. Chem. 1995, 95, 147–154. [Google Scholar] [CrossRef]
- Wang, H.; Wu, Y.; Liu, Z.; He, L.; Yao, Z.; Zhao, W. Deposition of WO3 on Al2O3 via a Microwave Hydrothermal Method to Prepare Highly Dispersed W/Al2O3 Hydrodesulfurization Catalyst. Fuel 2014, 136, 185–193. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Zheng, H.; Wan, F.; Yu, F.; Zhang, X.; Liu, Y. Surface Oxygen Vacancies on WO3 Contributed to Enhanced Photothermo-Synergistic Effect. Appl. Surf. Sci. 2017, 391, 654–661. [Google Scholar] [CrossRef]
- Soled, S. Comparison of the Acidities of WO3/Al2O3 and Ultrastable Faujasite Catalysts. J. Catal. 1988, 111, 286–295. [Google Scholar] [CrossRef]
- Chen, X.; Clet, G.; Thomas, K.; Houalla, M. Correlation between Structure, Acidity and Catalytic Performance of WOx/Al2O3 Catalysts. J. Catal. 2010, 273, 236–244. [Google Scholar] [CrossRef]
- Zhang, R.; Jagiello, J.; Hu, J.F.; Huang, Z.-Q.; Schwarz, J.A.; Datye, A. Effect of WO3 Loading on the Surface Acidity of WO3/Al2O3 Composite Oxides. Appl. Catal. Gen. 1992, 84, 123–139. [Google Scholar] [CrossRef]
- Rorrer, J.; He, Y.; Toste, F.D.; Bell, A.T. Mechanism and Kinetics of 1-Dodecanol Etherification over Tungstated Zirconia. J. Catal. 2017, 354, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Góra-Marek, K.; Tarach, K.; Choi, M. 2,6-Di- Tert- Butylpyridine Sorption Approach to Quantify the External Acidity in Hierarchical Zeolites. J. Phys. Chem. C 2014, 118, 12266–12274. [Google Scholar] [CrossRef]
- Kitano, T.; Hayashi, T.; Uesaka, T.; Shishido, T.; Teramura, K.; Tanaka, T. Effect of High-Temperature Calcination on the Generation of Brønsted Acid Sites on WO3/Al2O3. ChemCatChem 2014, 6, 2011–2020. [Google Scholar] [CrossRef]
- Rossmedgaarden, E.; Knowles, W.; Kim, T.; Wong, M.; Zhou, W.; Kiely, C.; Wachs, I. New Insights into the Nature of the Acidic Catalytic Active Sites Present in ZrO2-Supported Tungsten Oxide Catalysts. J. Catal. 2008, 256, 108–125. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, T.; Chen, J.; Auroux, A.; Shen, J. Surface Acidity and the Dehydration of Methanol to Dimethyl Ether. Thermochim. Acta 2005, 434, 22–26. [Google Scholar] [CrossRef]
- Xu, M.; Lunsford, J.H.; Goodman, D.W.; Bhattacharyya, A. Synthesis of Dimethyl Ether (DME) from Methanol over Solid-Acid Catalysts. Appl. Catal. Gen. 1997, 149, 289–301. [Google Scholar] [CrossRef]
- Sun, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Catalysis Chemistry of Dimethyl Ether Synthesis. ACS Catal. 2014, 4, 3346–3356. [Google Scholar] [CrossRef]
- Yaripour, F.; Baghaei, F.; Schmidt, I.; Perregaard, J. Catalytic Dehydration of Methanol to Dimethyl Ether (DME) over Solid-Acid Catalysts. Catal. Commun. 2005, 6, 147–152. [Google Scholar] [CrossRef]
- Sabour, B.; Peyrovi, M.H.; Hamoule, T.; Rashidzadeh, M. Catalytic Dehydration of Methanol to Dimethyl Ether (DME) over Al-HMS Catalysts. J. Ind. Eng. Chem. 2014, 20, 222–227. [Google Scholar] [CrossRef]
Figure 1.
N2 adsorption–desorption isotherms (a) and pore size distribution (b) of WO3/Al2O3 catalysts. Isotherms in (a) are shifted vertically for clarity.
Figure 1.
N2 adsorption–desorption isotherms (a) and pore size distribution (b) of WO3/Al2O3 catalysts. Isotherms in (a) are shifted vertically for clarity.

Figure 2.
X-ray diffractograms of WO3/γ-Al2O3 catalysts.

Figure 3.
Raman spectra of WO3/γ-Al2O3 catalysts.

Figure 4.
(a) Survey spectra of 37WO3/Al2O3 catalyst; (b) W4f XP spectra; (c) Al2p XP spectra; (d) O1s spectra of WO3/Al2O3 catalysts.
Figure 4.
(a) Survey spectra of 37WO3/Al2O3 catalyst; (b) W4f XP spectra; (c) Al2p XP spectra; (d) O1s spectra of WO3/Al2O3 catalysts.
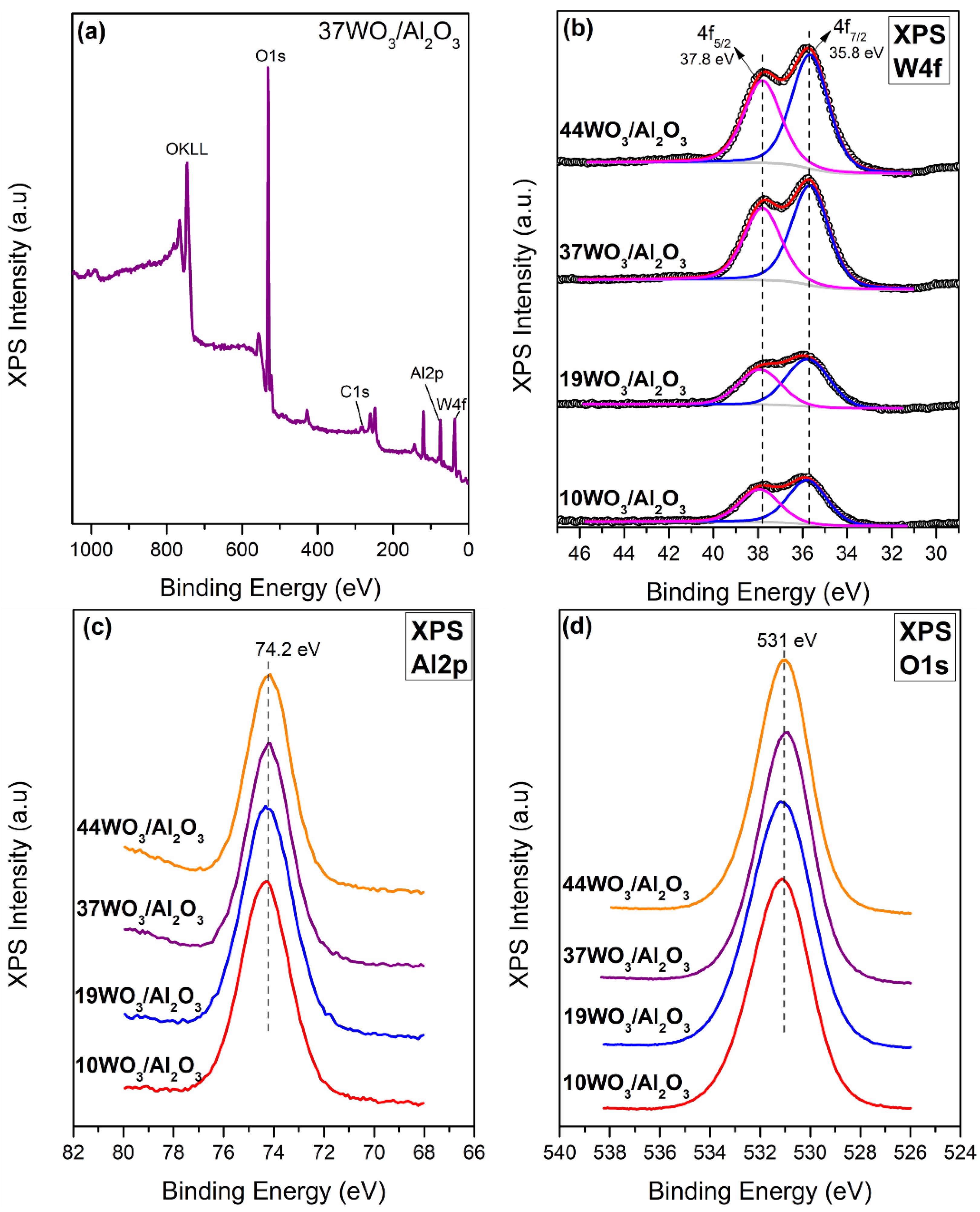
Figure 5.
Surface versus bulk W/Al atomic ratios determined via XPS and XRF of WO3/Al2O3 catalysts.

Figure 6.
(a) NH3-TPD profiles and (b) number (left axis) and concentration (right axis) of total acid sites of the WO3/Al2O3 catalysts.
Figure 6.
(a) NH3-TPD profiles and (b) number (left axis) and concentration (right axis) of total acid sites of the WO3/Al2O3 catalysts.
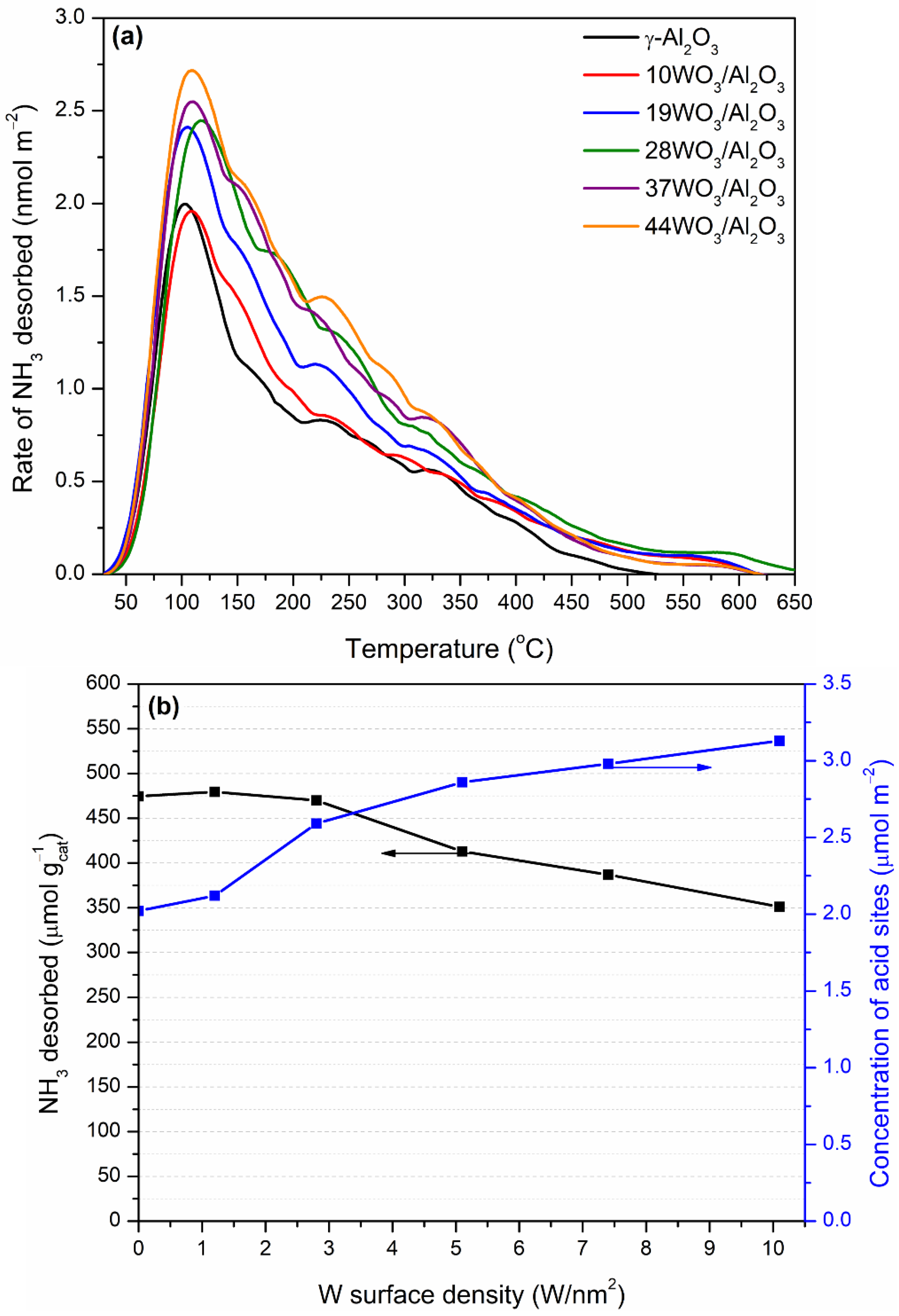
Figure 7.
Conversion of DME to CH3OH as a function of reaction temperature over WO3/Al2O3 catalysts. The equilibrium conversion is also included. Reaction conditions: 0.09% DME, 3% H2O, W/F = 0.5 g s cm−3.
Figure 7.
Conversion of DME to CH3OH as a function of reaction temperature over WO3/Al2O3 catalysts. The equilibrium conversion is also included. Reaction conditions: 0.09% DME, 3% H2O, W/F = 0.5 g s cm−3.

Figure 8.
Variation in the reaction rates (in μmol gcat−1 s−1) of DME hydrolysis of fresh WO3/Al2O3 catalysts (solid square symbols, left axis) and poisoned with 2,6-di-tert-butylpyridine prior to catalytic tests (open square symbols, left axis) and 2,6-di-ter-butylpyridine uptake (right axis) with the tungsten surface density. Reaction conditions: 0.09% DME, 3% H2O, reaction temperature = 200 °C, W/F = 0.5 g s cm−3.
Figure 8.
Variation in the reaction rates (in μmol gcat−1 s−1) of DME hydrolysis of fresh WO3/Al2O3 catalysts (solid square symbols, left axis) and poisoned with 2,6-di-tert-butylpyridine prior to catalytic tests (open square symbols, left axis) and 2,6-di-ter-butylpyridine uptake (right axis) with the tungsten surface density. Reaction conditions: 0.09% DME, 3% H2O, reaction temperature = 200 °C, W/F = 0.5 g s cm−3.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Tungsten surface density, textural properties and number of acid sites, total and Brønsted, determined via NH3-TPD and 2,6-di-tert-butylpyridine adsorption experiments, respectively.
Table 1.
Tungsten surface density, textural properties and number of acid sites, total and Brønsted, determined via NH3-TPD and 2,6-di-tert-butylpyridine adsorption experiments, respectively.
| Catalyst | W Surface Density (W/nm2) | SBET (m2 g−1) | Vp (cm3 g−1) | DP (nm) | Total Acid Sites (μmol gcat−1) | Acid Sites Density (μmol m−2) | Brønsted Acid Sites (μmol gcat−1) |
|---|---|---|---|---|---|---|---|
| γ-Al2O3 | 0.0 | 235 | 0.49 | 4.11 | 475 | 2.02 | 16 |
| 10WO3/Al2O3 | 1.2 | 226 | 0.39 | 3.66 | 480 | 2.12 | 57 |
| 19WO3/Al2O3 | 2.8 | 182 | 0.36 | 3.67 | 470 | 2.59 | n.d |
| 28WO3/Al2O3 | 5.1 | 144 | 0.30 | 3.65 | 413 | 2.86 | n.d |
| 37WO3/Al2O3 | 7.4 | 130 | 0.26 | 3.66 | 387 | 2.98 | 139 |
| 44WO3/Al2O3 | 10.1 | 112 | 0.22 | 3.70 | 351 | 3.13 | 98 |
| WO3 | 2.5 | 0.002 | 3.20 | n.d. | n.d. | n.d |
Table 2.
Bulk and surface compositions determined via XRF and XPS measurements, respectively, expressed as the atomic ratio of W/Al and the binding energy values of W4f, Al2p and O1s peaks of WO3/Al2O3 catalysts.
Table 2.
Bulk and surface compositions determined via XRF and XPS measurements, respectively, expressed as the atomic ratio of W/Al and the binding energy values of W4f, Al2p and O1s peaks of WO3/Al2O3 catalysts.
| Catalyst | W/Al Atomic Ratio | BE (eV) | ||||
|---|---|---|---|---|---|---|
| XRF | XPS | W4f | Al2p | O1s | ||
| 4f5/2 | 4f7/2 | |||||
| 10WO3/Al2O3 | 0.02 | 0.04 | 37.9 | 35.8 | 74.3 | 531.1 |
| 19WO3/Al2O3 | 0.05 | 0.05 | 37.9 | 35.8 | 74.3 | 531.1 |
| 37WO3/Al2O3 | 0.13 | 0.11 | 37.8 | 35.7 | 74.2 | 530.9 |
| 44WO3/Al2O3 | 0.17 | 0.13 | 37.8 | 35.7 | 74.2 | 531 |
Table 3.
Catalytic performance of WO3/Al2O3 in DME hydrolysis expressed as the temperature at which 10% conversion was obtained (T10) and reaction rates expressed per catalyst weight, WO3 weight and specific surface area, rW, and r, respectively.
Table 3.
Catalytic performance of WO3/Al2O3 in DME hydrolysis expressed as the temperature at which 10% conversion was obtained (T10) and reaction rates expressed per catalyst weight, WO3 weight and specific surface area, rW, and r, respectively.
| Catalyst | T10 (°C) | rW | r | |
|---|---|---|---|---|
| (×10−3 μmol gcat−1 s−1) | (×10−3 μmol gWO3−1 s−1) | (×10−5 μmol m−2 s−1) | ||
| γ-Al2O3 | 243 | 0 | n.d. | 0 |
| 10WO3/Al2O3 | 213 | 0.82 | 8.2 | 0.36 |
| 19WO3/Al2O3 | 210 | 2.02 | 10.64 | 1.11 |
| 28WO3/Al2O3 | 199 | 8.34 | 29.78 | 5.77 |
| 37WO3/Al2O3 | 192 | 15.74 | 42.54 | 12.11 |
| 44WO3/Al2O3 | 183 | 10.92 | 25.10 | 9.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Smyrnioti, M.; Ioannides, T. Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts. Catalysts 2022, 12, 396. https://doi.org/10.3390/catal12040396
AMA Style
Smyrnioti M, Ioannides T. Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts. Catalysts. 2022; 12(4):396. https://doi.org/10.3390/catal12040396
Chicago/Turabian StyleSmyrnioti, Maria, and Theophilos Ioannides. 2022. "Dimethyl Ether Hydrolysis over WO3/γ-Al2O3 Supported Catalysts" Catalysts 12, no. 4: 396. https://doi.org/10.3390/catal12040396
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.