
Methanation of CO2 over High Surface Nickel/Aluminates Compounds Prepared by a Self-Generated Carbon Template


Abstract
:1. Introduction
2. Results
2.1. Textural Data and XRD of Calcined Samples
2.2. Morphological and Micrographs of Calcined Samples (SEM/TEM)
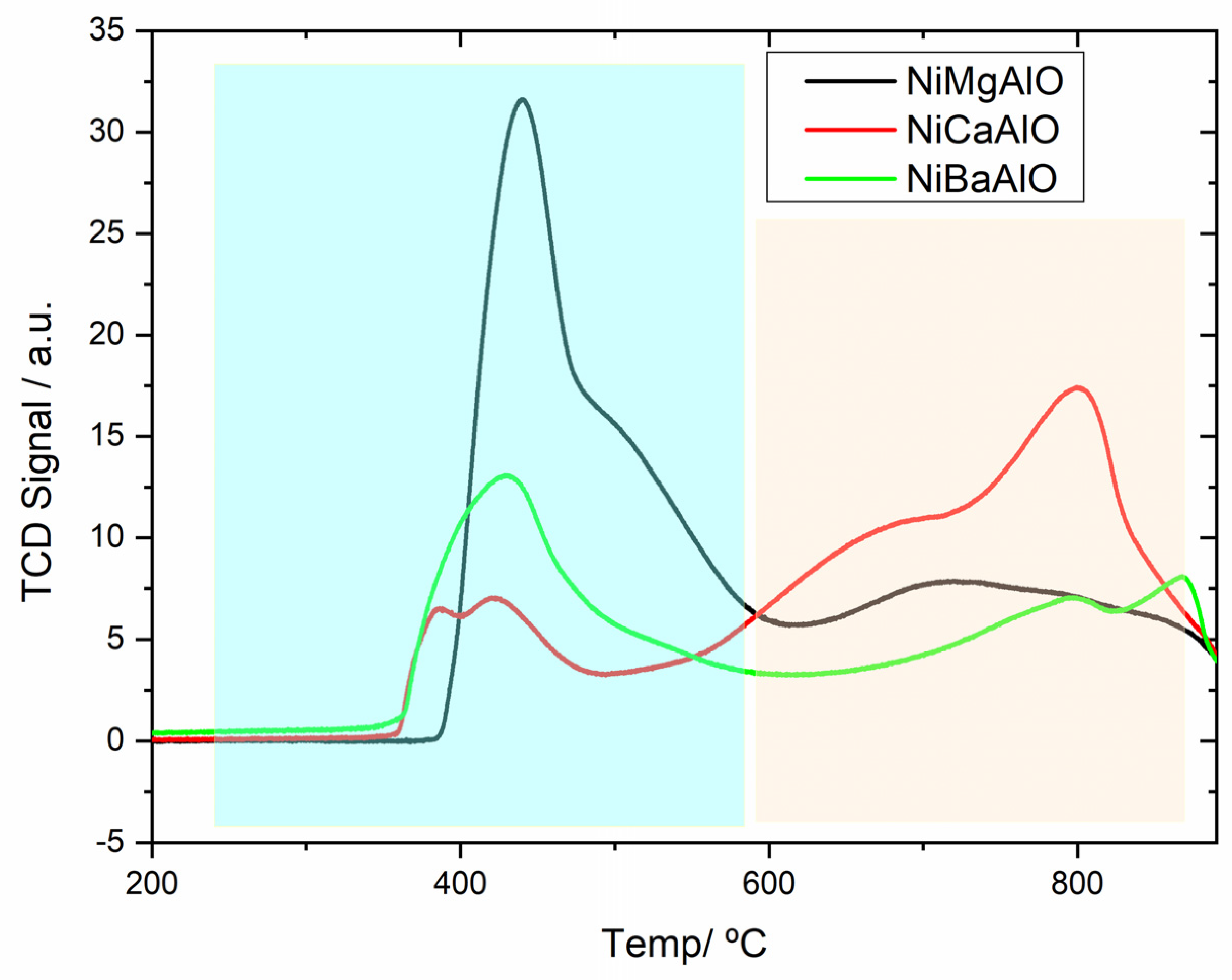
2.3. Temperature-Programmed Reduction (TPR)
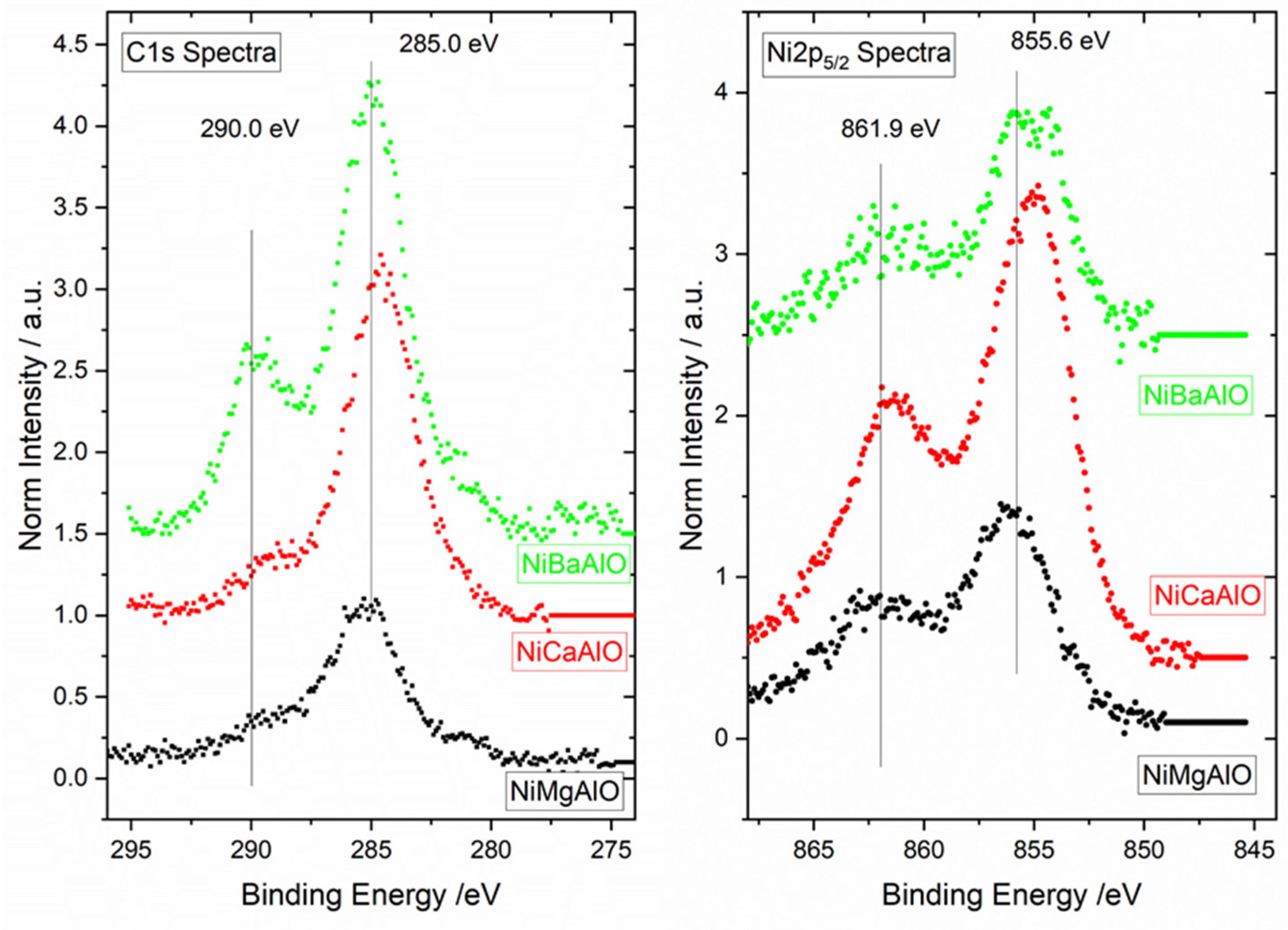
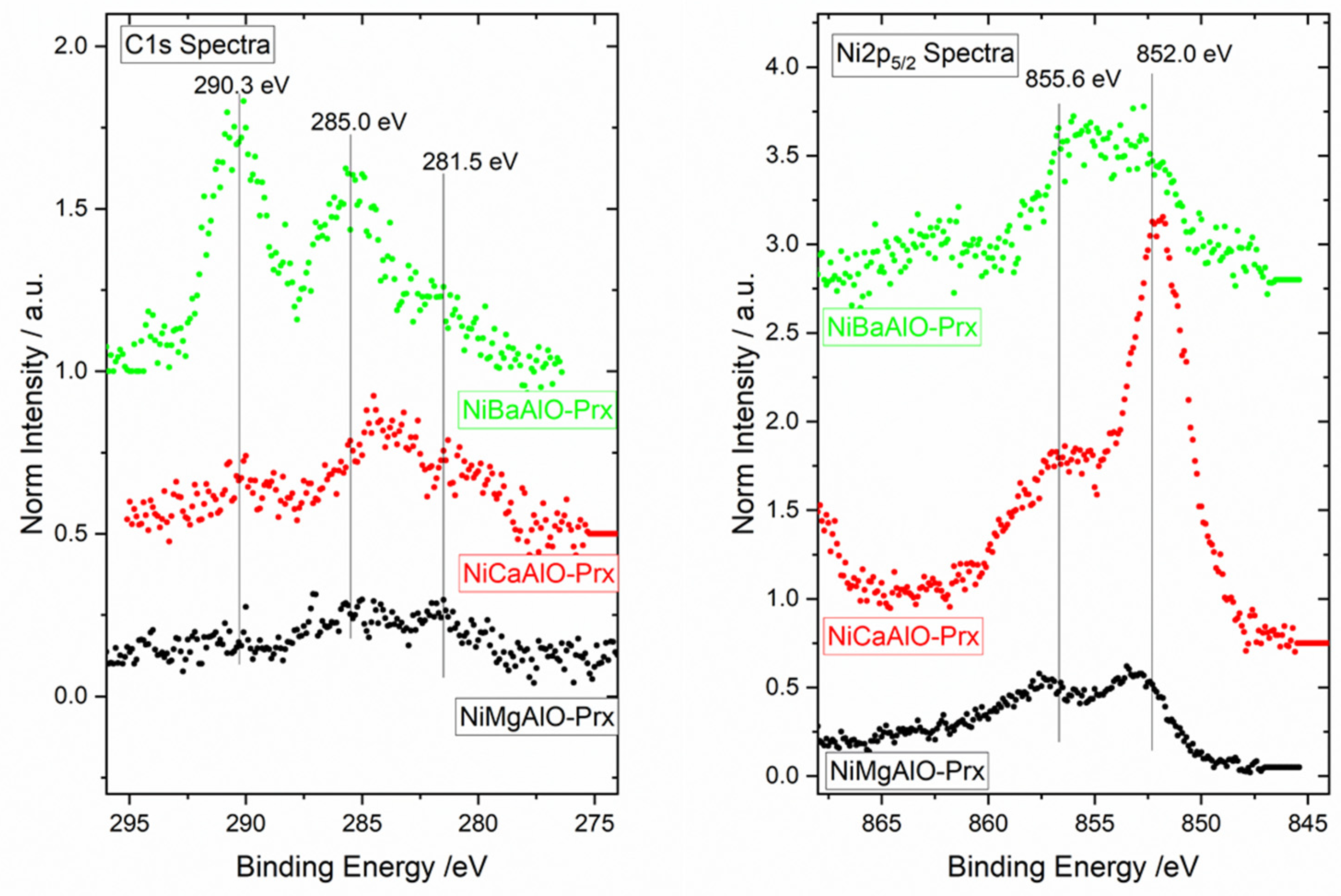
2.4. X-ray Photoelectron Spectroscopy (XPS) of Calcined Samples
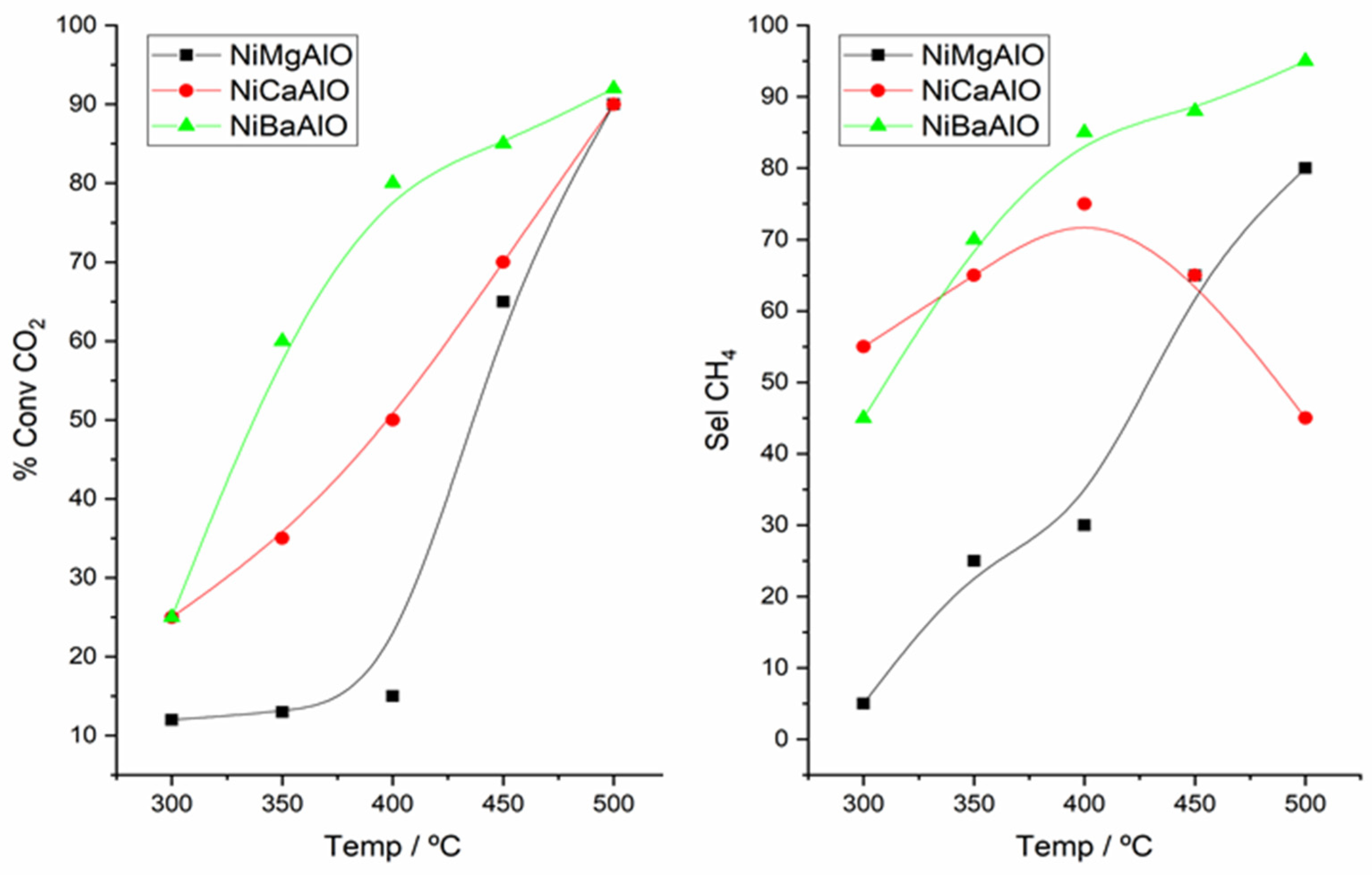
2.5. Catalytic CO2 Hydrogenation
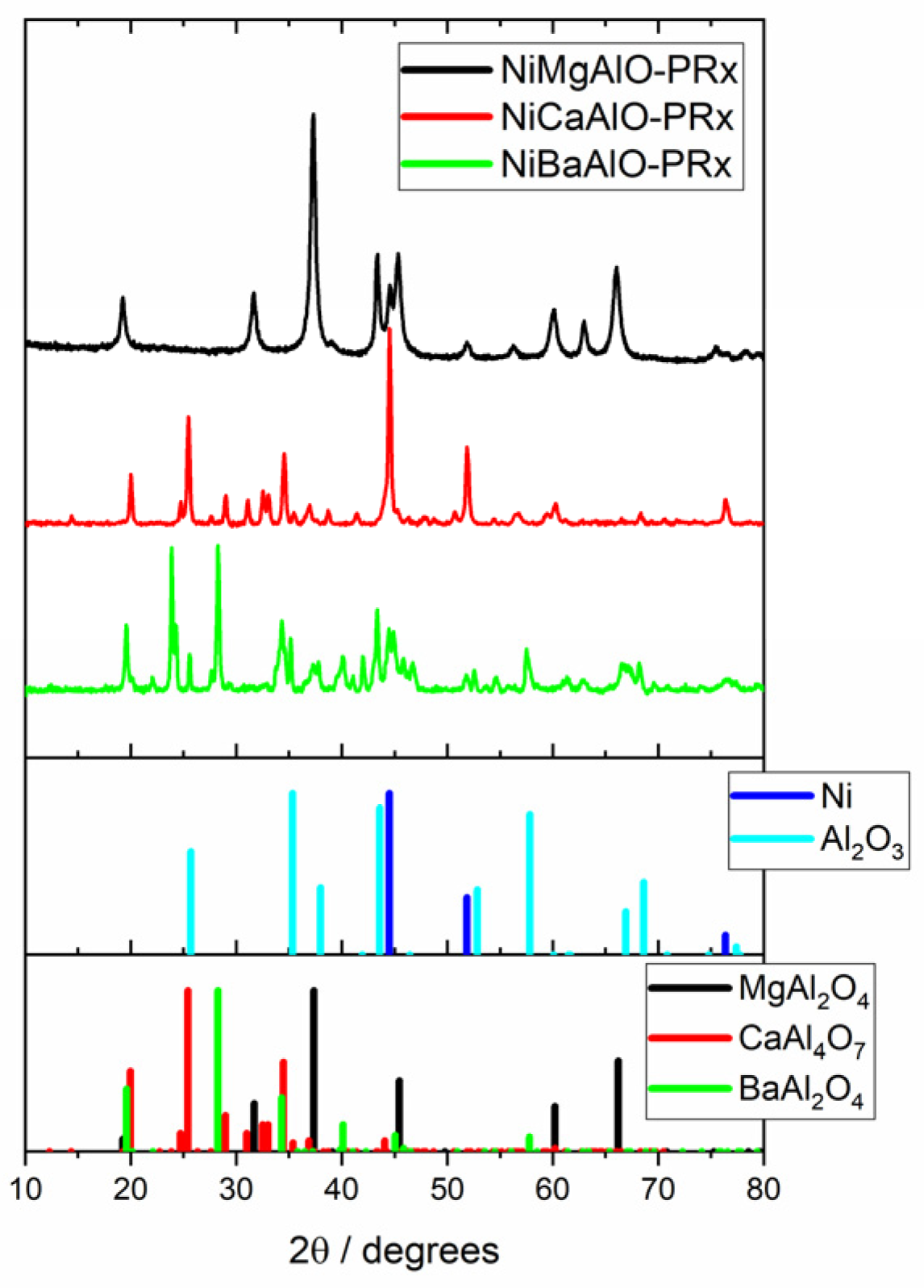
2.6. Textural Data and XRD of Post-Reaction Samples
2.7. SEM/TEM of Post-Reaction Samples
2.8. “In Situ” XPS Analysis of Post-Reaction Samples
3. Discussion
4. Materials and Methods
4.1. Preparation of the Catalysts
4.2. Catalytic Test
4.3. N2-Physisorption
4.4. SEM/TEM
4.5. XRD
4.6. TPR
4.7. XPS
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Energy Agency. World Energy Outlook; OECD/IEA: Paris, France, 1995. [Google Scholar]
- BP Energy Outlook; 2019 Edition; BP: London, UK, 2019.
- Höök, M.; Tang, X. Depletion of fossil fuels and anthropogenic climate change—A review. Energy Policy 2013, 52, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Meylan, F.D.; Moreau, V.; Erkman, S. Material constraints related to storage of future European renewable electricity surpluses with CO2 methanation. Energy Policy 2016, 94, 366–376. [Google Scholar] [CrossRef]
- Reiter, G.; Lindorfer, J. Evaluating CO2 sources for power-to-gas applications–A case study for Austria. J. CO2 Util. 2015, 10, 40–49. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation–from fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Schiebahn, S.; Grube, T.; Robinius, M.; Tietze, V.; Kumar, B.; Stolten, D. Power to gas: Technological overview, systems analysis and economic assessment for a case study in Germany. Int. J. Hydrogen Energy 2015, 40, 4285–4294. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 Methanation: The Effect of Catalysts and Reaction Conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Bengaouer, A.; Bedel, L. 20. CO2 hydrogenation to methane. In Volume 2 Transformations; De Gruyter: Berlin, Germany, 2019; pp. 385–412. [Google Scholar]
- Sterner, M.; Specht, M. Power-to-Gas and Power-to-X—The History and Results of Developing a New Storage Concept. Energies 2021, 14, 6594. [Google Scholar] [CrossRef]
- Li, K.; An, X.; Park, K.H.; Khraisheh, M.; Tang, J. A critical review of CO2 photoconversion: Catalysts and reactors. Catal. Today 2014, 224, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ghoussoub, M.; Wang, H.; Shao, Y.; Sun, W.; Tountas, A.A.; Wood, T.E.; Li, H.; Loh, J.Y.Y.; Dong, Y.; et al. Photocatalytic Hydrogenation of Carbon Dioxide with High Selectivity to Methanol at Atmospheric Pressure. Joule 2018, 2, 1369–1381. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, J.; Li, Y.; Wang, C. Selective photocatalytic CO2 reduction to CH4 over Pt/In2O3: Significant role of hydrogen adatom. Appl. Catal. B Environ. 2018, 226, 544–553. [Google Scholar] [CrossRef]
- Yaashikaa, P.R.; Kumar, P.S.; Varjani, S.J.; Saravanan, A. A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J. CO2 Util. 2019, 33, 131–147. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.-H.; Qi, M.-Y.; Tang, Z.-R.; Xu, Y.-J. Boosting the activity and stability of Ag-Cu2O/ZnO nanorods for photocatalytic CO2 reduction. Appl. Catal. B Environ. 2019, 268, 118380. [Google Scholar] [CrossRef]
- Rozzi, E.; Minuto, F.; Lanzini, A.; Leone, P. Green synthetic fuels: Renewable routes for the conversion of non-fossil feedstocks into gaseous fuels and their end uses. Energies 2020, 13, 420. [Google Scholar] [CrossRef] [Green Version]
- Karelovic, A.; Ruiz, P. Mechanistic study of low temperature CO2 methanation over Rh/TiO2 catalysts. J. Catal. 2013, 301, 141–153. [Google Scholar] [CrossRef]
- Petala, A.; Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: I. Effect of alkali additives on catalytic activity and selectivity. Appl. Catal. B Environ. 2018, 224, 919–927. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Duan, H.; Hou, B.; Lin, Q.; Liu, X.; Pan, X.; Pei, G.; Geng, H.; Huang, Y. Influence of pretreatment tem-perature on catalytic performance of rutile TiO2-supported ruthenium catalyst in CO2 methanation. J. Catal. 2016, 333, 227–237. [Google Scholar] [CrossRef]
- Panagiotopoulou, P. Hydrogenation of CO2 over supported noble metal catalysts. Appl. Catal. A Gen. 2017, 542, 63–70. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Özdemir, H.; Öksüzömer, M.F.; Gürkaynak, M.A. Preparation and characterization of Ni based catalysts for the catalytic partial oxidation of methane: Effect of support basicity on H2/CO ratio and carbon deposition. Int. J. Hydrogen Energy 2010, 35, 12147–12160. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.Y.; Li, H.; Yu, Z. Active and stable Ni based catalysts and processes for biogas upgrading: The effect of temperature and initial methane concentration on CO2 methanation. Appl. Energy 2018, 227, 206–212. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. Bimetallic Ni-Based Catalysts for CO2 Methanation: A Review. Nanomaterials 2020, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, S.; Zhu, X.; Li, C.; Shan, H. Dehydrogenation versus hydrogenolysis in the reaction of light alkanes over Ni-based catalysts. J. Ind. Eng. Chem. 2020, 86, 1–12. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous Catalysis on Atomically Dispersed Supported Metals: CO2 Reduction on Multifunctional Pd Catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Wu, Z.; Ge, S.; Zhang, M.; Li, W.; Tao, K. Synthesis of nickel nanoparticles supported on metal oxides using electroless plating: Controlling the dispersion and size of nickel nanoparticles. J. Colloid Interface Sci. 2009, 330, 359–366. [Google Scholar] [CrossRef]
- Azancot, L.; Bobadilla, L.F.; Santos, J.L.; Córdoba, J.M.; Centeno, M.A.; Odriozola, J.A. Influence of the preparation method in the metal-support interaction and reducibility of Ni-Mg-Al based catalysts for methane steam reforming. Int. J. Hydrogen Energy 2019, 44, 19827–19840. [Google Scholar] [CrossRef]
- Damyanova, S.; Pawelec, B.; Palcheva, R.; Karakirova, Y.; Sanchez, M.C.; Tyuliev, G.; Gaigneaux, E.; Fierro, J. Structure and surface properties of ceria-modified Ni-based catalysts for hydrogen production. Appl. Catal. B Environ. 2018, 225, 340–353. [Google Scholar] [CrossRef]
- Ho, P.; de Luna, G.S.; Angelucci, S.; Canciani, A.; Jones, W.; Decarolis, D.; Ospitali, F.; Aguado, E.; Rodríguez-Castellón, E.; Fornasari, G.; et al. Understanding structure-activity relationships in highly active La promoted Ni catalysts for CO2 methanation. Appl. Catal. B Environ. 2020, 278, 119256. [Google Scholar] [CrossRef]
- Mebrahtu, C.; Krebs, F.; Perathoner, S.; Abate, S.; Centi, G.; Palkovits, R. Hydrotalcite based Ni–Fe/(Mg, Al)Ox catalysts for CO2 methanation–tailoring Fe content for improved CO dissociation, basicity, and particle size. Catal. Sci. Technol. 2018, 8, 1016–1027. [Google Scholar] [CrossRef]
- Ren, J.; Mebrahtu, C.; Palkovits, R. Ni-based catalysts supported on Mg–Al hydrotalcites with different morphologies for CO2 methanation: Exploring the effect of metal–support interaction. Catal. Sci. Technol. 2020, 10, 1902–1913. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential Role of the Support for Nickel-Based CO2 Methanation Catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Siakavelas, G.; Charisiou, N.; AlKhoori, S.; AlKhoori, A.; Sebastian, V.; Hinder, S.; Baker, M.; Yentekakis, I.; Polychronopoulou, K.; Goula, M. Highly selective and stable nickel catalysts supported on ceria promoted with Sm2O3, Pr2O3 and MgO for the CO2 methanation reaction. Appl. Catal. B Environ. 2020, 282, 119562. [Google Scholar] [CrossRef]
- Sterk, E.B.; Nieuwelink, A.-E.; Monai, M.; Louwen, J.N.; Vogt, E.T.C.; Filot, I.A.W.; Weckhuysen, B.M. Structure Sensitivity of CO2 Conversion over Nickel Metal Nanoparticles Explained by Micro-Kinetics Simulations. JACS Au 2022, 2, 2714–2730. [Google Scholar] [CrossRef]
- Ortega-Liebana, M.C.; Hueso, J.L.; Ferdousi, S.; Arenal, R.; Irusta, S.; Yeung, K.L.; Santamaria, J. Extraordinary sensitizing effect of co-doped carbon nanodots derived from mate herb: Application to enhanced photocatalytic degradation of chlorinated wastewater compounds under visible light. Appl. Catal. B Environ. 2017, 218, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Gerlak, C.A.; Liu, C.; Llorca, J.; Yao, S.; Rui, N.; Zhang, F.; Liu, Z.; Zhang, S.; Deng, K.; et al. Effect of Ni particle size on the production of renewable methane from CO2 over Ni/CeO2 catalyst. J. Energy Chem. 2021, 61, 602–611. [Google Scholar] [CrossRef]
- Marconi, E.; Tuti, S.; Luisetto, I. Structure-Sensitivity of CO2 Methanation over Nanostructured Ni Supported on CeO2 Nanorods. Catalysts 2019, 9, 375. [Google Scholar] [CrossRef] [Green Version]
- Munnik, P.; Velthoen, M.E.Z.; de Jongh, P.E.; de Jong, K.P.; Gommes, C.J. Nanoparticle Growth in Supported Nickel Catalysts during Methanation Reaction-Larger is Better. Angew. Chem. 2014, 126, 9647–9651. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Binas, V.; Marnellos, G.E.; Konsolakis, M. Deciphering the role of Ni particle size and nickel-ceria interfacial perimeter in the low-temperature CO2 methanation reaction over remarkably active Ni/CeO2 nanorods. Appl. Catal. B Environ. 2021, 297, 120401. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; Moioli, E.; Chen, W.; Zhang, M.; Terlingen, B.J.; Zijlstra, B.; Filot, I.; Züttel, A.; Pidko, E.; Hensen, E.J. Efficient Base-Metal NiMn/TiO2 Catalyst for CO2 Methanation. ACS Catal. 2019, 9, 7823–7839. [Google Scholar] [CrossRef]
- Wang, K.; Men, Y.; Liu, S.; Wang, J.; Li, Y.; Tang, Y.; Li, Z.; An, W.; Pan, X.; Li, L. Decoupling the size and support/metal loadings effect of Ni/SiO2 catalysts for CO2 methanation. Fuel 2021, 304, 121388. [Google Scholar] [CrossRef]
- Shen, W.; Dumesic, J.; Hill, C. Criteria for stable Ni particle size under methanation reaction conditions: Nickel transport and particle size growth via nickel carbonyl. J. Catal. 1981, 68, 152–165. [Google Scholar] [CrossRef]
- Santiago, M.; Groen, J.C.; Pérez-Ramírez, J. Carbon-templated hexaaluminates with enhanced surface area and catalytic performance. J. Catal. 2008, 257, 152–162. [Google Scholar] [CrossRef]
- Barton, T.J.; Bull, L.M.; Klemperer, W.G.; Loy, D.A.; McEnaney, B.; Misono, M.; Monson, P.A.; Pez, G.; Scherer, G.W.; Vartuli, A.J.C.; et al. Tailored Porous Materials. Chem. Mater. 1999, 11, 2633–2656. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Lou, H.; Zhao, H.; Wang, X.; Zheng, X. Novel synthesis of high surface area MgAl2O4 spinel as catalyst support. Mater. Lett. 2004, 58, 1920–1923. [Google Scholar] [CrossRef]
- Pereñiguez, R.; Gonzalez-Delacruz, V.M.; Caballero, A.; Holgado, J.P. LaNiO3 as a precursor of Ni/La2O3 for CO2 reforming of CH4: Effect of the presence of an amorphous NiO phase. Appl. Catal. B Environ. 2012, 123–124, 324–332. [Google Scholar] [CrossRef]
- Pereñíguez, R.; Gonzalez-Delacruz, V.M.; Holgado, J.P.; Caballero, A. Synthesis and characterization of a LaNiO3 perovskite as precursor for methane reforming reactions catalysts. Appl. Catal. B Environ. 2010, 93, 346–353. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Hu, Y. Interactions between Ni and La2O3 in Ni/La2O3 Catalysts Prepared Using Different Ni Precursors. J. Catal. 1996, 161, 55–61. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, A.; Caballero, A. Identification of Outer and Inner Nickel Particles in a Mesoporous Support: How the Channels Modify the Reducibility of Ni/SBA-15 Catalysts. ChemNanoMat 2016, 3, 94–97. [Google Scholar] [CrossRef]
- Bueno-Alejo, C.J.; Arca-Ramos, A.; Hueso, J.L.; Santamaria, J. LED-driven continuous flow carbon dioxide hydrogenation on a nickel-based catalyst. Catal. Today 2020, 355, 678–684. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Biju, V.; Khadar, M.A. Electronic Structure of Nanostructured Nickel Oxide Using Ni2p XPS Analysis. J. Nanoparticle Res. 2002, 4, 247–253. [Google Scholar] [CrossRef]
- McIntyre, N.S.; Johnston, D.D.; Coatsworth, L.L.; Davidson, R.D.; Brown, J.R. X-ray photoelectron spectroscopic studies of thin film oxides of cobalt and molybdenum. Surf. Interface Anal. 1990, 15, 265–272. [Google Scholar] [CrossRef]
- Espinos, J.P.; Fernandez, A.R.; Gonzalezelipe, A.; Munuera, G. Electronic Interaction of Ni particles with TiO2 and SiO2. Surf. Sci. 1991, 251, 1012–1017. [Google Scholar] [CrossRef]
- Czekaj, I.; Loviat, F.; Raimondi, F.; Wambach, J.; Biollaz, S.; Wokaun, A. Characterization of surface processes at the Ni-based catalyst during the methanation of biomass-derived synthesis gas: X-ray photoelectron spectroscopy (XPS). Appl. Catal. A Gen. 2007, 329, 68–78. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, Y.; Zhang, Y.; Wen, Y.; Zhao, J. Study on Alumina-Supported Cobalt–Nickel Oxide Catalyst for Synthesis of Acetonitrile from Ethanol. Catal. Lett. 2011, 141, 168–177. [Google Scholar] [CrossRef]
- Guczi, L.; Stefler, G.; Geszti, O.; Sajó, I.; Pászti, Z.; Tompos, A.; Schay, Z. Methane dry reforming with CO2: A study on surface carbon species. Appl. Catal. A Gen. 2010, 375, 236–246. [Google Scholar] [CrossRef]
- Wiltner, A.; Linsmeier, C. Formation of endothermic carbides on iron and nickel. Phys. Status Solidi A 2004, 201, 881–887. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Cored, J.; García-Ortiz, A.; Iborra, S.; Climent, M.J.; Liu, L.; Chuang, C.-H.; Chan, T.-S.; Escudero, C.; Concepción, P.; Corma, A. Hydrothermal Synthesis of Ruthenium Nanoparticles with a Metallic Core and a Ruthenium Carbide Shell for Low-Temperature Activation of CO2 to Methane. J. Am. Chem. Soc. 2019, 141, 19304–19311. [Google Scholar] [CrossRef]
- Muroyama, H.; Tsuda, Y.; Asakoshi, T.; Masitah, H.; Okanishi, T.; Matsui, T.; Eguchi, K. Carbon dioxide methanation over Ni catalysts supported on various metal oxides. J. Catal. 2016, 343, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.L.; Mangarella, M.C.; D’Amico, A.D.; Gallagher, J.R.; Dutzer, M.R.; Stavitski, E.; Miller, J.T.; Sievers, C. Differences in the Nature of Active Sites for Methane Dry Reforming and Methane Steam Reforming over Nickel Aluminate Catalysts. ACS Catal. 2016, 6, 5873–5886. [Google Scholar] [CrossRef]
- Malet, P.; Caballero, A. The selection of experimental conditions in temperature-programmed reduction experiments. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 2369–2375. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ICP Chemical Formula | BET Surface (m2/g) | Pore Volume (cm3/g) | Micropore Volume (cm3/g) | Pore Size (Å) |
|---|---|---|---|---|---|
| NiBaAlO | NiBa1.1Al11(O)x | 30.0 | 0.11 | 0.002 | 12–17 |
| NiMgAlO | NiMg1.0Al11(O)x | 58.0 | 0.20 | 0.005 | 14–17 |
| NiCaAlO | NiCa1.1Al11(O)x | 31.0 | 0.21 | 0.003 | 22–24 |
| NiBaAlO-AIR | NiBa1.0Al11(O)x | 7.2 | 0.02 | 1.6 × 10−4 | 11–13 |
| NiBaAlO-Carbon | NiBa1.1Al11(O)x | 15.0 | 0.03 | 6.3 × 10−4 | 13–15 |
| Crystallite Size Calculated from Scherrer Formula (nm) | |||||||
|---|---|---|---|---|---|---|---|
| NiBaAlO | NiBaAlO-PRx | NiCaAlO | NiCaAlO-PRx | NiMgAlO | NiMgAlO-PRx | NiBaAlO-Carbon NiBaAlO-AIR | |
| NiO | 23 ± 2 | -- | 31 ± 2 | -- | 24 ± 2 | -- | -- |
| Metallic Ni | -- | 20 ± 2 | -- | 30 ± 2 | -- | 20 ± 2 | -- |
| Al2O3 | 35 ± 2 | 45 ± 2 | -- | -- | -- | -- | -- |
| BaAl2O4 | 30 ± 2 | 34 ± 2 | -- | -- | -- | -- | 63 ± 2 74 ± 2 |
| BaCO3 | -- | 40 ± 2 | -- | -- | -- | -- | -- |
| CaAl4O7 | -- | -- | 24 ± 2 | 11 ± 2 | -- | -- | -- |
| (MgAl2O4) | -- | -- | -- | -- | 15 ± 2 | 12 ± 2 | -- |
| Areas and Maxima Binding Energies (eV) from XPS Zones | |||||||
|---|---|---|---|---|---|---|---|
| Al2p | C1s | Ba3d | Ca2p | Mg1s | Ni2p | O1s | |
| (B.E. eV) | (B.E. eV) | (B.E. eV) | (B.E. eV) | (B.E. eV) | (B.E. eV) | (B.E. eV) | |
| NiBaAlO | 38.9% | 13.3% | 2.5% | 0.7% | 44.6% | ||
| (74.0 ± 0.1) | (284.7 ± 0.1) | (780.2 ± 0.1) | (854.3 ± 0.1) | (531.3 ± 0.1) | |||
| NiBaAlO-PRx | 44.1% | 4.7% | 3.3% | 0.5% | 47.4% | ||
| (74.0 ± 0.1) | (290.1 ± 0.1) | (780.8 ± 0.1) | (852.7 ± 0.1) | (531.7 ± 0.1) | |||
| NiCaAlO | 43.0% | 11.2% | 1.7% | 4.5% | 39.7% | ||
| (74.0 ± 0.1) | (284.6 ± 0.1) | (347.4 ± 0.1) | (854.8 ± 0.1) | (530.4 ± 0.1) | |||
| NiCaAlO-PRx | 52.2% | 3.3% | 2.7% | 3.1% | 38.7% | ||
| (74.0 ± 0.1) | (284.5 ± 0.1) | (348.0 ± 0.1) | (851.8 ± 0.1) | (531.5 ± 0.1) | |||
| NiMgAlO | 40.6% | 6.4% | 2.1% | 1.3% | 49.7% | ||
| (74.0 ± 0.1) | (285.3 ± 0.1) | (1303.5 ± 0.1) | (856.5 ± 0.1) | (531.4 ± 0.1) | |||
| NiMgAlO-PRx | 44.1% | 1.3% | 2.1% | 1.2% | 50.6% | ||
| (74.0 ± 0.1) | (281.5 ± 0.1) | (1304.2 ± 0.1) | (853.5 ± 0.1) | (532.2 ± 0.1) | |||
| Percentage of Indicated Species for Deconvolution of C1s and Ni2p XPS Signals | |||||
|---|---|---|---|---|---|
| C1s-1% (Carbonate) | C1s-2% (Graphitic) | C1s-3% (Carbide) | Ni2p-1% (Ni2+) | Ni2p-2% (Ni0) | |
| NiBaAlO | 25.3 | 69 | 5.7 | 90.0 | 10.0 |
| NiBaAlO-PRx | 45.9 | 38.6 | 15.5 | 62.7 | 37.3 |
| NiCaAlO | 10.7 | 84.6 | 4.8 | 98.7 | 1.3 |
| NiCaAlO-PRx | 22.8 | 50.3 | 26.9 | 50.6 | 49.4 |
| NiMgAlO | 15.3 | 77 | 7.6 | 100.0 | 0.0 |
| NiMgAlO-PRx | 3.3 | 48.1 | 48.6 | 69.5 | 30.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roudane, S.; Bettahar, N.; Caballero, A.; Holgado, J.P. Methanation of CO2 over High Surface Nickel/Aluminates Compounds Prepared by a Self-Generated Carbon Template. Catalysts 2023, 13, 142. https://doi.org/10.3390/catal13010142
Roudane S, Bettahar N, Caballero A, Holgado JP. Methanation of CO2 over High Surface Nickel/Aluminates Compounds Prepared by a Self-Generated Carbon Template. Catalysts. 2023; 13(1):142. https://doi.org/10.3390/catal13010142
Chicago/Turabian StyleRoudane, Sarra, Noureddin Bettahar, Alfonso Caballero, and Juan Pedro Holgado. 2023. "Methanation of CO2 over High Surface Nickel/Aluminates Compounds Prepared by a Self-Generated Carbon Template" Catalysts 13, no. 1: 142. https://doi.org/10.3390/catal13010142
APA StyleRoudane, S., Bettahar, N., Caballero, A., & Holgado, J. P. (2023). Methanation of CO2 over High Surface Nickel/Aluminates Compounds Prepared by a Self-Generated Carbon Template. Catalysts, 13(1), 142. https://doi.org/10.3390/catal13010142