
Catalytic Reduction of N2O by CO on Single-Atom Catalysts Au/C2N and Cu/C2N: A First-Principles Study

1
Institute of Functional Material Chemistry, Faculty of Chemistry, National & Local United Engineering Lab for Power Battery, Northeast Normal University, Renmin Road 5268, Changchun 130024, China
2
School of Chemical and Biomolecular Science, Southern Illinois University Carbondale, Carbondale, IL 62901, USA
*
Author to whom correspondence should be addressed.
Catalysts 2023, 13(3), 578; https://doi.org/10.3390/catal13030578
Submission received: 18 January 2023
/
Revised: 2 March 2023
/
Accepted: 11 March 2023
/
Published: 13 March 2023
(This article belongs to the Special Issue Advanced Opportunities and Insights on Novel Catalysts for Electrocatalytic Energy Conversion)
Abstract
:The catalytic conversion of greenhouse gases, such as N2O, is a promising way to mitigate global warming. In this work, density functional theory (DFT) studies were performed to study N2O reduction by CO over single-atom catalysts (SACs) and compare the performance of noble (Au/C2N) and non-noble (Cu/C2N) SACs. The computational results indicated that catalytic N2O reduction on both catalysts occurs via two mechanisms: (I) the N2O adsorption mechanism—starting from the adsorption on the catalysts, N2O decomposes to a N2 molecule and O-M/C2N intermediate, and then CO reacts with O atom on the O-M/C2N intermediate to form CO2; and (II) the CO adsorption mechanism—CO and N2O are adsorbed on the catalyst successively, and then a synergistic reaction occurs to produce N2 and CO2 directly. The computational results show that mechanism I exhibits an obvious superiority over mechanism II for both catalysts due to the lower activation enthalpy. The activation enthalpies of the rate-determining step in mechanism I are 1.10 and 1.26 eV on Au/C2N and Cu/C2N, respectively. These results imply that Cu/C2N, an abundant-earth SAC, can be as active as expensive Au/C2N. Herein, our research provides a theoretical foundation for the catalytic reduction of N2O and broadens the application of non-noble-metal SACs.
1. Introduction
Nitrous oxide (N2O) emission comes from microbial nitrification and the denitrification of solids and waters [1], biomass burning, fossil-fuel combustion through industrial processes, and selective catalytic reduction of NOx [1,2]. Along with carbon dioxide, N2O is one of the main contributors to the greenhouse effect. The accumulation of N2O gas in the atmosphere is associated with ozone depletion, global warming, and acid rain [3,4,5]. Furthermore, N2O has an atmospheric lifetime that is approximate 300-times longer than CO2 [6]. Therefore, considerable efforts have been made to regulate N2O emission and conversion to prevent and alleviate its environmental impact.
It is commonly known that the catalytic reduction of N2O by carbon monoxide (CO) is one of the most promising methods to mitigate N2O emissions [7,8,9,10,11]. Not only is CO inexpensive and easily accessible, but the use of CO, a toxic air pollutant, further limits harmful gas emission. Nevertheless, N2O reduction by CO has a high activation temperature. Indeed, an experimental study carried out by Loirat et al. found that N2O reduction by CO has an activation energy as high as 193 ± 8 kJ/mol (about 2.0 eV) between 1076 and 1228 K [12]; thus, the reaction needs efficient catalysts to lower the operating temperature. Precious-metal catalysts such as Au, Pt, and Pd have been extensively studied for N2O reduction [13,14,15,16,17]. However, their high cost, scarcity, and high reaction temperatures limit their application on a larger scale. Consequently, a search for more affordable alternatives, with higher activity, has been the focus in recent years [18,19,20,21,22,23,24,25].
In 2011, Zhang et al. created a new class of catalysts called single-atom catalysts (SAC). The synthesized SAC platinum demonstrated excellent catalytic performance for CO oxidation [26]. Due to the excellent stability, high catalytic activity and remarkable atomic efficiency, SCAs have attracted considerable researcher attention in recent years [27,28,29,30]. Some SACs proved to be highly active for N2O reduction [14,18,23,31,32,33,34]. Density functional theory (DFT) studies showed that Pt-doped graphene [14], an Fe-embedded graphene SAC [23], the boron nitride nanotube-supported Cr SAC [35] and single-atom Ge-embedded graphene are effective catalyst for N2O reduction [36]. Furthermore, non-metal SACs, such as Si-doped graphene, also showed comparable catalytic activity to transition-metal-embedded graphene [18].
In 2017, Mahmood et al. synthesized a novel two-dimensional layered C2N with high thermal and structural stability [37]. This C2N monolayer proved to be a promising SAC support based on its uniformly distributed N6 cavity, which is a suitable anchoring site for a single metal atom. Furthermore, many studies have shown that a single metal atom embedded in a C2N monolayer exhibits excellent catalytic activity in many important reactions, such as CO oxidation, CO2 reduction, N2 reduction, oxygen reduction, and hydrogen evolution reactions as well [38,39,40]. In our previous work, we investigated the reduction of CO2 to formic acid over Cu/C2N SAC [41]. Our results suggested that Cu/C2N can effectively catalyze CO2 reduction with high selectivity at room temperature.
In the present work, we aim to explore the catalytic performance of Au/C2N and Cu/C2N for N2O reduction using DFT. Additionally, the activity of noble and abundant earth metal SACs was evaluated and compared. Our results showed that both Au/C2N and Cu/C2N are highly active and exhibit comparable catalytic efficiency. Therefore, abundant-earth-metal SACs proved to be promising catalysts for N2O reduction. Our study provides theoretical information for the prospects of affordable metal SACs in catalytic reactions and broadens their field of application.
2. Results and Discussion
2.1. Geometric and Electronic Properties of Cu/C2N and Au/C2N
Studies have shown that Au and Cu atoms can be embedded in the N6 cavity of the C2N monolayer [42,43]. In our previous work, molecular dynamics simulation showed that Cu/C2N has high thermal stability [41]. Therefore, stable M/C2N can be formed and used as a catalyst. Due to differences in the atomic radius, the Au and Cu atoms anchor on the C2N sheet differently, as shown in Figure 1. The Au atom preferably bonds in the center of the N6 cavity, with an average Au-N bond length of 2.752 Å in the case of the Cu atom, which has a smaller atomic radius than Au (1.28 vs. 1.79Å). In contrast, Cu most favorably bonds to two N atoms of the N6 cavity, with a bond length of 2.101 Å. The doping energies for Au/C2N and Cu/C2N are −3.88 eV and −4.19 eV, respectively.
To better understand the catalytic potential of both SACs, Mulliken charge analysis was performed. In Au/C2N, we found that Au carries 0.548 e charge, while each six surrounding N atoms carry a charge of −0.250 e. In the Cu/C2N case, the charge of Cu is 0.622 e, the Cu two closest N neighbors have a charge of −0.350 e each, while the other four N atoms have a lower charge density of −0.216 e each. The electron transfer between the metal atom and the C2N sheet indicates metal–N bonding.
The calculated frontier molecular orbitals of Au/C2N and Cu/C2N show that the HOMOs are mainly formed by the d orbitals of the single metal (Figures S1 and S2). This suggests that the metal atoms can donate electrons to N2O and, thus, activate the N2O reduction reaction.
2.2. Mechanism of Catalytic N2O Reduction
Our MEP calculations show that N2O reduction by CO can proceed via the two following mechanisms on Au/C2N and Cu/C2N:
Mechanism I:
N2O + M/C2N → N2 + O-M/C2N
CO + O-M/C2N → CO2 + M/C2N
Mechanism II:
CO + M/C2N → CO-M/C2N
N2O + CO-M/C2N → N2O-CO-M/C2N
N2O-CO-M/C2N → N2 + CO2 + M/C2N
2.2.1. Mechanism I: N2O Adsorption Mechanism
Au/C2N-Catalyzed N2O Reduction
In mechanism I: a N2O molecule is first adsorbed on the catalyst. Then, an O atom is abstracted by catalysts, leading to the release of a N2 molecule. The O-M/C2N intermediate then reacts with a CO molecule to produce CO2 (Equations (1) and (2)). The structure diagram and energy profile of reaction mechanism I on Au/C2N are shown in Figure 2 and Figure 3. Table 1 lists the relative energies, enthalpies and free energies with zero-point vibrational correction for all the species in the N2 reduction catalyzed by Au/C2N and Cu/C2N. The enthalpy values were used to discuss the energy changes in the reaction systems in the following discussion. The Mulliken charge analysis is shown in Table 2.
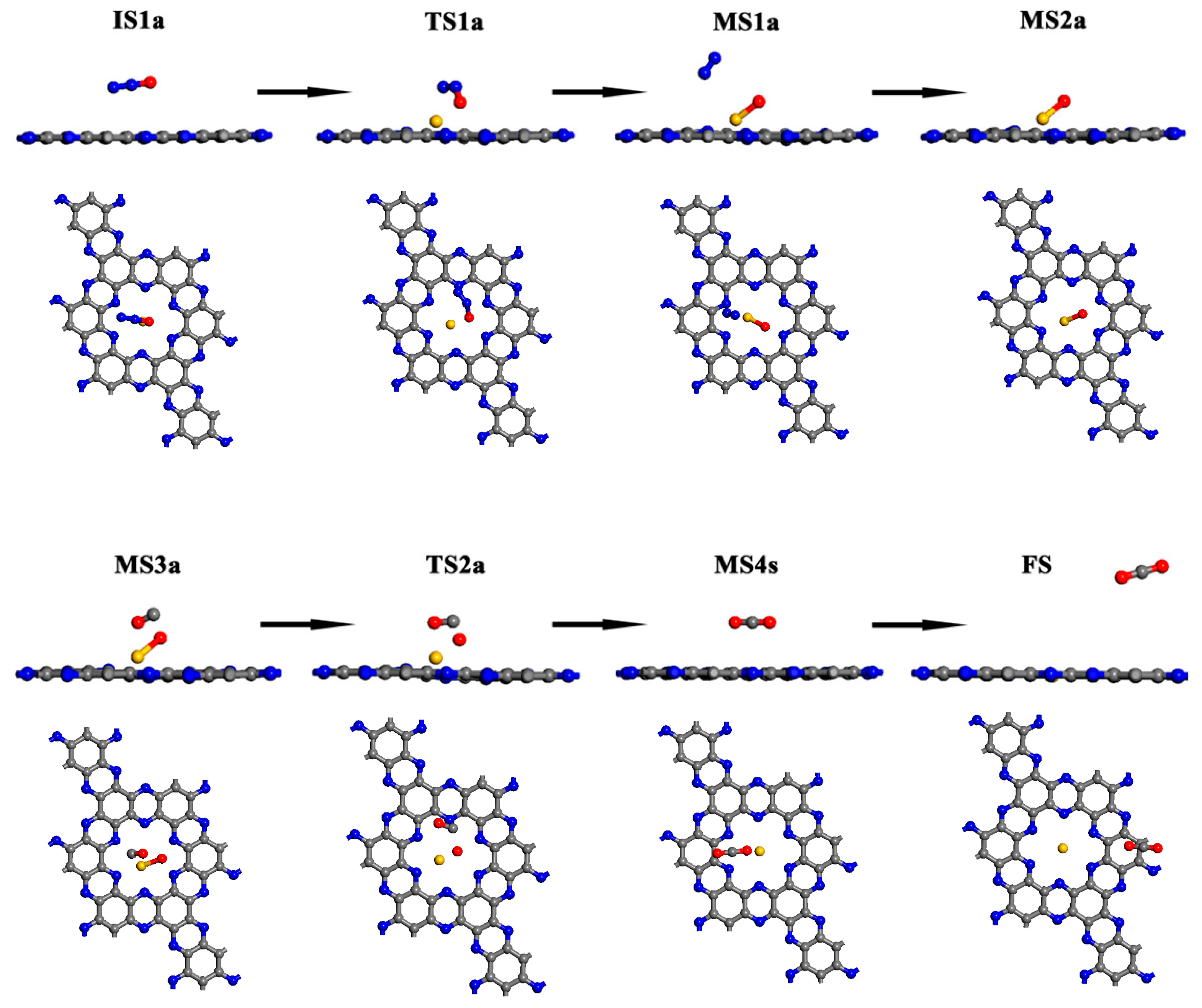
Mechanism I is initiated by the adsorption of N2O on the Au/C2N (IS1a). The adsorption enthalpy is −0.06 eV and the distance between the Au and O atoms is 3.010 Å. Compared with isolated systems, Au and O atoms are slightly charged while N2O is nearly neutral in IS1a, which is consistent with no overlap between the wave functions of the O atom and Au atom near the Fermi level. The small adsorption enthalpy, long interacting distance, and slight charge transfer suggest that N2O is only physisorbed on Au/C2N. The FMOs in Figure 4 show no overlap between N2O and Au/C2N, further confirming the absence of a chemical bond between the O and Au atoms. As the O atom in N2O approaches the Au atom, the N–O bond elongates from 1.198 Å to 1.604 Å, and the distance between Au and O shortens to 2.024 Å in TS1a, leading to an activation enthalpy of 1.10 eV. The negative charge of the O atom increases from −0.291 e to −0.466 e, and the positive charge of the Au atom rises from 0.551 e to 0.705 e. Thus, the catalyst donates electrons to the N2O molecule, which lengthens the N(a)-O bond (TS1a). The increased charge transfer from the Au to the O atom facilitates the formation of the O–Cu bond and the dissociation of the N–O bond, resulting in the formation of an O–Au/C2N intermediate (MS1a). The frontier molecular orbitals (FMO) plots and FMO charge density plots in this step are shown in Figure 4 and Figure S5. From the orbital analysis and PDOS in Figure 5b, there is not only an orbital overlap between N2O and Au/C2N but also a significant hybridization of the 4d orbital of Au and the 2p orbital of O(a) in TS1a. Both these results confirm the bonding between N2O and Au in the catalyst. The IS1a → TS1a → MS1a step has the highest enthalpy barrier, making it the rate-determining step for mechanism I.
From TS1a to MS1a, the negative charge on the O atom changes from −0.466 e to −0.478 e, while the positive charge on the Au atom increases from 0.705 e to 0.734 e. The decrease in charge transfer between the N and O atoms leads to the dissociation of the N(a)–O bond and the release of N2. It can also be proved by the disappearance of hybridizations between the N(a) 2p and O 2p orbitals in Figure 5c. Figure 4 shows that the orbital overlap between the O atom and Au in HOMO rises from IS1a to MS1a, indicating that there is an increase in electron transfer between the O and Au atom, resulting in a stronger interaction between N2O and the catalyst. From the PDOS in Figure 5c, Au 4d orbital peaks change in position, and the O 2p orbital becomes sharper near the Fermi energy level. Such changes stem from hybridization between Au 4d and O 2p, demonstrating the strong Au–O interaction. This is also consistent with the above charge and orbital analysis. In MS1a, the Au–O bond length is 2.046 Å and the N–O bond length is 3.157 Å, indicating that a normal chemical bond is formed between the O atom and Au atom, while the N–O bond is broken. This results in the formation of the O-Au/C2N (MS2a) intermediate and the release of a N2 molecule from MS1a with a low desorption enthalpy of 0.14 eV.
In MS2a, the catalyst donates −0.478 e to an O atom, and the Au-O bond is 2.144 Å. Based on MS2a, CO is adsorbed to form intermediate MS3a with an adsorption enthalpy of −0.18 eV. Due to the high negative charge of adsorbed O in MS2a, it can easily react with CO. In MS3a, the charge of the CO molecule then changes from electroneutrality to −0.008 e, and the positive charge of the Au atom decreases slightly from 0.739 e to 0.736 e. Small adsorption enthalpy and slight charge transfer are consistent with the weak overlap between O(a) 2p and C 2p orbitals in Figure 5d. Next, the O atom bonded to Au is abstracted by the CO molecule, and adsorbed CO2 is produced via the transition state TS2a, by overcoming a low enthalpy barrier of 0.28 eV. Finally, CO2 gas is released with a small desorption enthalpy of 0.28 eV. The dissociation of the Au–O bond and formation of C–O(a) bond from TS2a to MS4a can be found by the PDOS plots in Figure 5e,f. The highest activation enthalpy (TS1a) is 1.10 eV and the overall reaction in mechanism I is exothermic, releasing 3.34 eV of heat.
Cu/C2N-Catalyzed N2O Reduction
Although the two metal atoms are doped on different sites, mechanism I is the most favorable path for N2O reduction on both catalysts. The structure diagram of reaction mechanism I on the Cu/C2N catalyst is shown in Figure S3. The FMO, FMO charge density and PDOS of relevant species in the reaction are shown in Figure 6, Figure S6 and Figure 7. Firstly, N2O molecule is weakly physisorbed on Cu/C2N (IS1a) with an adsorption enthalpy of −0.01 eV. From Table 3, the positive charge of the Cu atom increases from 0.622 e to 0.639 e, and the negative charge of the O atom increases from −0.278 e to −0.306 e. This means that the charge of N2O increases by 0.023 e, indicating more charge transfer between N2O and Cu/C2N than that between N2O and Au/C2N (0.004 e). Different from Figure 5a, Figure 7a shows observable overlaps of the O 2p and Cu 4d orbitals. The difference in interaction strength between N2O and the two catalysts can be found from the adsorption enthalpy, charge transfer and PDOS. As N2O gradually approaches the Cu atom, the N–O bond stretches from 1.205 Å to 1.678 Å, while the Cu–O atom bond distance shortens to 1.792 Å. The process has an activation enthalpy of 1.26 eV (TS1a) which is slightly higher than TS1a over Au/C2N. The negative charge of the O atom increases from −0.306 e to −0.515 e, and the positive charge of the Cu atom increases from 0.508 e to 0.585 e. The charge transfer from the Cu atom to the O atom leads to Cu–O bond formation and N–O bond cleavage. The PDOS analysis in Figure 7b also shows the overlaps of O 2p and Cu 3d at a lower energy and weaker hybridization peak between O and N(a) than that in Figure 5b, which implies the formation of Au–O chemical binding and the dissociation of the N–O bond and TS1a on the Cu/C2N catalyst is more like a product and may need a higher activation enthalpy.
This oxygen abstraction leads to the intermediate MS1a where the charges on the O atom and Cu atom are slightly increased compared with TS1a. The Cu–O bond length is 1.749 Å, and there is an evident orbital overlap between the O atom and Cu, and disappearance of the overlap of O and N orbitals (Figure 7c), showing O–Cu bond formation and C–O bond dissociation. The weak N–O bond results in facile N2 desorption (0.26 eV), and the formation of intermediate O-Cu/N2C (MS2a).
In MS3a, CO is adsorbed on the catalyst along with MS2a. CO adsorption has a weak adsorption enthalpy of −0.26 eV. The adsorption is evidenced by a new C–O bond formation and Cu–O bond cleavage. This step results in the formation of adsorbed CO2 on Cu/C2N (MS4a) with an enthalpy barrier of 0.41 eV (TS2a). Finally, CO2 gas is released with a desorption enthalpy of 0.61 eV.
2.2.2. Mechanism II: CO Adsorption Mechanism
In mechanism II, CO and N2O molecules are adsorbed on the catalysts, and react through a synergistic step as shown in Equations (3)–(5).
Au/C2N-Catalyzed N2O Reduction
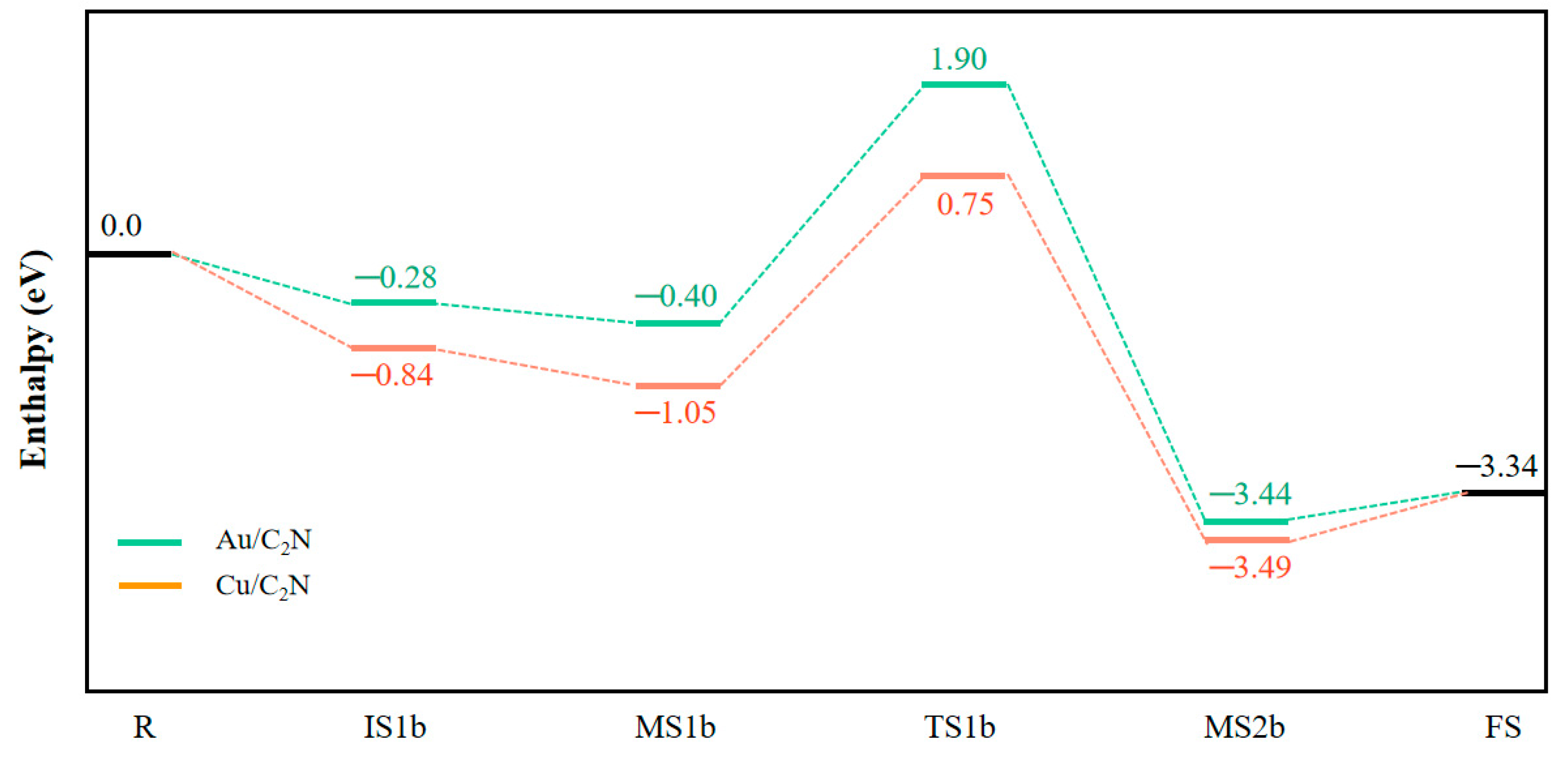
The schematic diagram and energy profile of mechanism II are shown in Figure 8 and Figure 9. Here, CO first adsorbs onto Au/C2N with an adsorption enthalpy of −0.28 eV (IS1b). The C-Au atom distance is as long as 2.426 Å and is accompanied by a small charge transfer of 0.046 e from CO to the catalyst. Then, the N2O molecule also adsorbs on the catalyst, resulting in a co-adsorption state (MS1b), with an adsorption enthalpy of −0.40 eV. In MS1b, the Au–C bond length is 2.419 Å, and the distance between C and O atoms in N2O is 3.335 Å. A negligible charge transfer is also found between the reactants and the catalyst. On the geometry analysis, adsorption enthalpy, and charge transfer, the interaction between reactants and catalyst is weak in both IS1b and MS1b.
As both N2O and CO are in proximity to each other, they can react. The N–O bond in N2O extends from 1.198 Å to 2.113 Å and the distance between the C in the CO and O atom in N2O is 2.023 Å in TS1b. This is considered a loose transition state, initiating the oxygen abstraction of N2O by CO. This step has an activation enthalpy of 2.30 eV, leading to the formation of N2 and CO2, which are subsequently released with a small desorption enthalpy of 0.10 eV.
Cu/C2N-Catalyzed N2O Reduction
Similar to the initial step on Au/C2N (Figure S4 and Figure 8), the CO molecule is adsorbed first on Cu/C2N (IS1b). The adsorption enthalpy (−0.84 eV) is, in contrast, more exothermic than on Au/C2N (−0.28 eV). In IS1b, the length of the Cu–C bond is 1.834 Å and the charge transfer from CO to the catalyst is much higher (0.053 e). Then, as the N2O molecule is physisorbed, a co-adsorption state (MS1b) is formed, and the total adsorption enthalpy is −1.05 eV. The adsorption energy mainly originates from the interaction between CO and the catalyst, since the charge transfer between N2O and Cu/C2N is negligible. Subsequently, the N–O bond extends from 1.199 Å to 1.592 Å in TS1b. As the O atom in N2O approaches the C atom of CO, a stable state is reached with a N2O–CO distance of 2.042 Å. For both reactants to react, an enthalpy barrier of 1.80 eV has to be overcome to form TS1b, a more reactant-like TS. Although significant, this activation enthalpy is lower than on Au/C2N (2.30 eV), where the TS is more product-like. Then, the N–O bond in N2O is broken and the O atom is abstracted by C in CO to form adsorbed CO2 in MS2b with a newly formed C–O bond length of 1.205 Å. Finally, N2 and CO2 are released with a desorption enthalpy of 0.15 eV, and the catalyst is regenerated and available for a new reaction cycle.
In summary, the homology of Au and Cu elements results in a similar N2O + CO reaction mechanism. By comparing the highest activation enthalpy of the two reaction mechanisms, we found that mechanism I is the most favorable route on both Au/C2N and Cu/C2N for N2 reduction. In mechanism I, N2O activation is the rate-determining step, i.e., IS1a → TS1a → MS1a. The calculated activation enthalpy are 1.10 eV and 1.26 eV on Au/C2N and Cu/C2N, respectively, which demonstrates that non-precious-metal SAC can exhibit a high catalytic activity comparable to a noble-metal SAC. It is worth noting that the calculated activation enthalpies are much lower than experimental values without any catalyst (about 2.0 eV) [12].
3. Computational Methods
All calculations, from the geometric optimizations to electronic properties, were performed in DMol3 package [44] using the spin-polarized Perdew-Burke-Ernzerhof (PBE) functional [45]. The double numerical basis set with polarization function (DNP) and the DFT semi-core pseudopotentials (DSPPs) were employed [46]. As the generalized gradient approximation (GGA) method is insufficient in describing weak interaction, dispersion correction was added to the system [47]. Transition state (TS) correction was also employed to evaluate dispersion interactions [48]. The global orbital cutoff was set to fine quality: 4.5 Å and 4.4 Å for Au and Cu, respectively. The sampling of the Brillouin zone integration was carried out with 3 × 3 × 1 k-points for the C2N monolayer [49]. To avoid C2N monolayer interaction with its periodic image, the cell length along the z-direction was set to 15Å. For all optimization calculations, the cutoff energy and force were set to 1.0 × 10−5 Ha and 2.0 × 10−3 Ha/Å, respectively, and the SCF convergence was set to 1.0 × 10−6. Au- and Cu- embedded C2N monolayers were modeled with a C2N (2 × 2) supercell. To study the minimum-energy pathway (MEP) for N2O reduction, the linear synchronous transit (LST), and quadratic synchronous transit (QST) methods were used for transition state (TS) search [50,51]. The vibrational frequencies for each obtained structure along the MEP were calculated to ensure that every TS had a single imaginary frequency.
The adsorption enthalpy (Hads) was defined as follows:
where Htotal, HM/C2N, and Hadsorbate represent the enthalpies of the gas molecules adsorbed on the catalyst, M/C2N, and gas molecules, respectively. The more negative the Eads value, the higher the stability of the gas molecules on the catalyst.
Hads = Htotal − (HM/C2N + Hadsorbate)
The activation enthalpy (Ha), i.e., enthalpy barrier, which is one of the most important factors to evaluate the catalytic activity, was defined as follows:
where HTS and HIM1 represent the enthalpies of the transition state and intermediate IM1 in the reaction step of IM1 → TS → IM2.
Ha = HTS − HIM1
4. Conclusions
Herein, the catalytic reduction of N2O by CO on the SACs Au/C2N and Cu/C2N was investigated using DFT. The reaction can proceed through two mechanisms: Mechanism I starts with the N2O adsorption on the catalyst, followed by its decomposition into N2 gas and O-M/C2N intermediate. The adsorbed oxygen species then reacts with CO to produce CO2. The highest activation enthalpy on Au/C2N and Cu/C2N are 1.10 eV and 1.26 eV, respectively. In mechanism II, both CO and N2O are co-adsorbed on the catalyst, and in a one-step synergistic reaction, they react to produce N2 and CO2 gas. The activation enthalpies on Au/C2N and Cu/C2N are 1.80 eV and 2.30 eV, respectively. Our results show that mechanism I is noticeably more favorable on both catalysts, rendering it the dominant path. Based on these results, abundant-earth-metal SACs can demonstrate catalytic efficiency comparable to that of a noble-metal SAC for N2O reduction. Our work provides a theoretical basis for the catalytic reduction of N2O and broadens the application of non-noble-metal single-atom catalysts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13030578/s1, Figure S1: The schematic diagram of frontier molecular orbitals for catalyst Au/C2N; Figure S2: The schematic diagram of frontier molecular orbitals for catalyst Cu/C2N; Figure S3: The schematic diagram of all species along the MEP for the N2O reduction on the Cu/C2N monolayer via mechanism I; Figure S4: The frontier molecular orbitals (FMOs) for rate-determining step IS1a → TS1a → MS1a in N2O reduction mechanism I on Cu/C2N; Figure S5: The HOMO and LUMO charge-density plots for IS1a, TS1a and MS1a on Au/C2N; Figure S6: The HOMO and LUMO charge-density plots for IS1a, TS1a and MS1a on Cu/C2N; Table S1: The Mulliken charge of species involved in N2O reduction mechanism II.
Author Contributions
Conceptualization, H.S.; methodology, S.S.; formal analysis, S.S., J.M., Z.L. and H.S.; investigation, S.S. and J.M.; writing—original draft preparation, J.M. and S.S.; writing—review and editing, D.H., Z.L. and H.S.; supervision, H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Acknowledgments
The authors greatly thank the reviewers for their valuable comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rockmann, T.; Kaiser, J.; Brenninkmeijer, C.A.M. The isotopic fifingerprint of the pre-industrial and the anthropogenic N2O source. Atmos Chem. Phys. Discuss. 2002, 2, 2021–2043. [Google Scholar]
- Zhang, X.X.; Wang, X.P.; Zhao, X.J.; Xu, Y.Z.; Zhang, F.F. An investigation on N2O formation route over Pt/Hy in H2-SCR. Chem. Eng. J. 2014, 252, 288–297. [Google Scholar] [CrossRef]
- Duce, R.A.; Laroche, J.; Altieri, K.; Arrigo, K.R.; Baker, A.R.; Capone, D.G.; Cornell, S.; Dentener, F.; Galloway, J.; Ganeshram, R.S.; et al. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 2008, 320, 893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 2009, 326, 123. [Google Scholar] [CrossRef] [Green Version]
- Dameris, M. Depletion of the ozone layer in the 21st century, Angew. Chem. Int. Edit. 2010, 49, 489–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montzka, S.A.; Dlugokencky, E.J.; Butler, J.H. Non-CO2 greenhouse gases and climate change. Nature 2011, 476, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Liu, Y.Q.; Liu, Y.; Lian, D.X.; Chen, M.; Ji, Y.J.; Xing, L.W.; Wu, K.; Liu, S.M. Recent advances of Cu-based catalysts for NO reduction by CO under O2-containing conditions. Catalysts 2022, 12, 1402. [Google Scholar] [CrossRef]
- Blagojevic, V.; Orlova, G.; Bohme, D.K. O-atom transport catalysis by atomic cations in the gas phase: reduction of N2O by CO. J. Am. Chem. Soc. 2005, 127, 3545–3555. [Google Scholar] [CrossRef]
- Saramok, M.; Szymaszek, A.; Inger, M.; Antoniak-Jurak, K.; Samojeden, B.; Motak, M. Modified zeolite catalyst for a NOx selective catalytic reduction process in nitric acid plants. Catalysts 2021, 11, 450. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, Z.; Guo, W.; Zhang, L.; Zhang, F.; Zhu, H.; Shan, H. Theoretical investigation of the gas-phase Mn+- and Co+-catalyzed oxidation of benzene by N2O. Phys. Chem. Chem. Phys. 2009, 11, 4219–4229. [Google Scholar] [CrossRef]
- Kartha, K.K.; Pai, M.R.; Banerjee, A.M.; Pai, R.V.; Meena, S.S.; Bharadwaj, S.R. Modifified surface and bulk properties of Fe-substituted lanthanum titanates enhances catalytic activity for CO + N2O reaction. J. Mol. Catal. A Chem. 2011, 335, 158–168. [Google Scholar] [CrossRef]
- Loirat, H.; Caralp, F.; Destriau, M.; Lesclaux, R. Oxidation of CO by N2O between 1076 and 1228 K: Determination of the rate constant of the exchange reaction. J. Phys. Chem. 1987, 91, 6538–6542. [Google Scholar] [CrossRef]
- Balaj, O.P.; Balteanu, I.; Roßteuscher, T.T.J.; Beyer, M.K.; Bondybey, V.E. Catalytic oxidation of CO with N2O on gas-phase platinum clusters. Angew. Chem. Int. Edit. 2004, 43, 6519–6522. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Wang, Y.; Wang, Q. Theoretical investigation for the reaction of N2O with CO catalyzed by Pt-graphene. Struct. Chem. 2017, 28, 1679–1685. [Google Scholar] [CrossRef]
- Elizundia, U.; Duraiswami, D.; Pereda-Ayo, B.; López-Fonseca, R.; González-Velasco, J.R. Controlling the selectivity to N2O over Pt/Ba/Al2O3 NOX storage/reduction catalysts. Catal. Today 2011, 176, 324–327. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Yu, Z.; Liu, C. Mechanism of N2O formation during NO reduction on the Au(111) Surface. J. Phys. Chem. C 2010, 114, 2711–2716. [Google Scholar] [CrossRef]
- Wei, X.; Yang, X.-F.; Wang, A.-Q.; Li, L.; Liu, X.-Y.; Zhang, T.; Mou, C.-Y.; Li, J. Bimetallic Au-Pd alloy catalysts for N2O decomposition: Effects of surface structures on catalytic activity. J. Phys. Chem. C 2012, 116, 6222–6232. [Google Scholar] [CrossRef]
- Gholizadeh, R.; Yu, Y.-X. N2O+CO reaction over Si- and Se-doped graphenes: An ab initio DFT study. Appl. Surf. Sci. 2015, 357, 1187–1195. [Google Scholar] [CrossRef]
- Lambeets, S.V.; Barroo, C.; Owczarek, S.; Jacobs, L.; Genty, E.; Gilis, N.; de Bocarmé, T.V. Adsorption and hydrogenation of CO2 on Rh nanosized crystals: Demonstration of the role of interfacet oxygen spillover and comparative studies with O2, N2O, and CO. J. Phys. Chem. C 2017, 121, 16238–16249. [Google Scholar] [CrossRef]
- Kokalj, A.; Matsushima, T. A density-functional theory study of the interaction of N2O with Rh(110). J. Chem. Phys. 2005, 122, 034708. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.-F.; Pérez-Ramírez, J.; Ample, F.; Ricart, J.M. Theoretical studies of N2O adsorption and reactivity to N2 and NO on Rh(111). J. Phys. Chem. B 2004, 108, 17921–17927. [Google Scholar] [CrossRef]
- Song, E.H.; Yan, J.M.; Lian, J.S.; Jiang, Q. External electric field catalyzed N2O decomposition on Mn-embedded graphene. J. Phys. Chem. C 2012, 116, 20342–20348. [Google Scholar] [CrossRef]
- Wannakao, S.; Nongnual, T.; Khongpracha, P.; Maihom, T.; Limtrakul, J. Reaction mechanisms for co catalytic oxidation by N2O on Fe-embedded graphene. J. Phys. Chem. C 2012, 116, 16992–16998. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Sharifi, F.; Nematollahi, P. Al- or Si-decorated graphene oxide: A favorable metal-free catalyst for the N2O reduction. Appl. Surf. Sci. 2016, 387, 454–460. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F.; Nematollahi, P. Oxidation of CO by N2O over Al- and Ti-doped graphene: A comparative study. RSC Adv. 2016, 6, 64832–64840. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of co oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Tosoni, S.; Pacchioni, G. Role of support in tuning the properties of single atom catalysts: Cu, Ag, Au, Ni, Pd, and Pt adsorption on SiO2/Ru, SiO2/Pt, and SiO2/Si ultrathin films. J. Chem. Phys. 2021, 154, 134706. [Google Scholar] [CrossRef]
- Tosoni, S.; Liberto, G.D.; Matanovic, I.; Pacchioni, G. Modelling single atom catalysts for water splitting and fuel cells: A tutorial review. J. Power Sour. 2023, 556, 232492. [Google Scholar] [CrossRef]
- Liberto, G.D.; Tosoni, S.; Cipriano, L.A.; Pacchioni, G. A few questions about single-atom catalysts: When modeling helps. Acc. Mater. Res. 2023, 3, 986–995. [Google Scholar] [CrossRef]
- Cipriano, L.A.; Liberto, G.D.; Pacchioni, G. Superoxo and peroxo complexes on single-atom catalysts: Impact on the oxygen evolution reaction. ACS Catal. 2022, 12, 11682–11691. [Google Scholar] [CrossRef]
- Zhang, L.L.; Chen, X.M.; Liu, C.G. Reduction of N2O by CO via Mars-van Krevelen mechanism over phosphotungstic acid supported single-atom catalysts: A density functional theory study. Inorg. Chem. 2019, 58, 5221–5229. [Google Scholar] [CrossRef] [PubMed]
- Anchalee, J.; Supawadee, N.; Phornphimon, M.; Masahiro, E. Silicon-coordinated nitrogen-doped graphene as a promising metal-free catalyst for N2O reduction by CO: A theoretical study. RSC Adv. 2018, 8, 22322–22330. [Google Scholar]
- Liu, C.G.; Sun, C.; Jiang, M.X.; Zhang, L.L.; Sun, M.J. Calculations of NO reduction with CO over a Cu1/PMA single-atom catalyst: A study of surface oxygen species, active sites, and the reaction mechanism. Phys. Chem. Chem. Phys. 2019, 21, 9975–9986. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Heidari, S. Catalytic reduction of nitrous oxide over boron-doped C3N monolayers: A DFT study. Chem. Phys. Lett. 2019, 725, 52–58. [Google Scholar] [CrossRef]
- Fan, G.H.; Wang, Q.; Xu, H.; Wang, X.H.; Tu, X.X.; Chu, X.F. Single Cr atom supported on boron nitride nanotubes for the reaction of N2O reduction by CO: A density functional theory study. Appl. Surf. Sci. 2021, 544, 148776–148782. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vessally, E. N2O + CO reaction over single Ga or Ge atom embedded graphene: A DFT study. Surf. Sci. 2017, 667, 105–111. [Google Scholar] [CrossRef]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Jeon, I.-Y.; Jung, S.-M.; Choi, H.-J.; Seo, J.-M.; Bae, S.-Y.; Sohn, S.-D.; et al. Nitrogenated holey two-dimensional structures. Nat. Commun. 2015, 6, 6486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarty, S.; Das, T.; Banerjee, P.; Thapa, R.; Das, G.P. Electron doped C2N monolayer as effiffifficient noble metal-free catalysts for CO oxidation. Appl. Surf. Sci. 2017, 418, 92–98. [Google Scholar] [CrossRef]
- Mahmood, J.; Li, F.; Kim, C.; Choi, H.-J.; Gwon, O.; Jung, S.-M.; Seo, J.-M.; Cho, S.-J.; Ju, Y.-W.; Jeong, H.Y.; et al. Fe@C2N: A highly-effiffifficient indirect-contact oxygen reduction catalyst. Nano Energy 2018, 44, 304–310. [Google Scholar] [CrossRef]
- Mahmood, J.; Li, F.; Jung, S.-M.; Okyay, M.S.; Ahmad, I.; Kim, S.-J.; Park, N.; Jeong, H.Y.; Baek, J.-B. An effiffifficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction. Nat. Nanotechnol. 2017, 12, 441–446. [Google Scholar] [CrossRef]
- Ma, J.; Gong, H.; Zhang, T.; Yu, H.; Zhang, R.; Liu, Z.; Yang, G.; Sun, H.; Tang, S.; Qiu, Y. Hydrogenation of CO2 to formic acid on the single atom catalysis Cu/C2N: A first principles study. Appl. Surf. Sci. 2019, 488, 1–9. [Google Scholar] [CrossRef]
- Ma, D.W.; Wang, Q.; Yan, X.; Zhang, X.; He, C.; Zhou, D.; Tang, Y.; Lu, Z.; Yang, Z. 3d transition metal embedded C2N monolayers as promising single-atom catalysts: A first-principles study. Carbon 2016, 105, 463–473. [Google Scholar] [CrossRef]
- He, B.L.; Shen, J.S.; Tian, Z.X. Iron-embedded C2N monolayer: A promising low-cost and high-activity single-atom catalyst for CO oxidation. Phys. Chem. Chem. Phys. 2016, 18, 24261–24269. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756. [Google Scholar] [CrossRef]
- Perdew, J.P.; Yue, W. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8800–8802. [Google Scholar] [CrossRef] [PubMed]
- Basiuk, V.A.; Henao-Holguín, L.V. Dispersion-Corrected Density Functional Theory Calculations of meso-Tetraphenylporphine-C60 Complex by Using DMol3 Module. J. Comput. Theor. Nanosci. 2014, 11, 1609–1615. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [Green Version]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
Figure 1.
The optimized structures of the two catalysts: (a) Au/C2N and (b) Cu/C2N. (Blue, grey, yellow and orange balls represent N, C, Au, and Cu atoms, respectively). Number 1 and 2 in (b) represent the labels of the two N atoms.
Figure 1.
The optimized structures of the two catalysts: (a) Au/C2N and (b) Cu/C2N. (Blue, grey, yellow and orange balls represent N, C, Au, and Cu atoms, respectively). Number 1 and 2 in (b) represent the labels of the two N atoms.

Figure 2.
The schematic diagram of all species along the MEP for the N2O reduction on the Au/C2N monolayer via mechanism I. (Blue, grey, yellow and red balls represent N, C, Au and O atoms, respectively).
Figure 2.
The schematic diagram of all species along the MEP for the N2O reduction on the Au/C2N monolayer via mechanism I. (Blue, grey, yellow and red balls represent N, C, Au and O atoms, respectively).
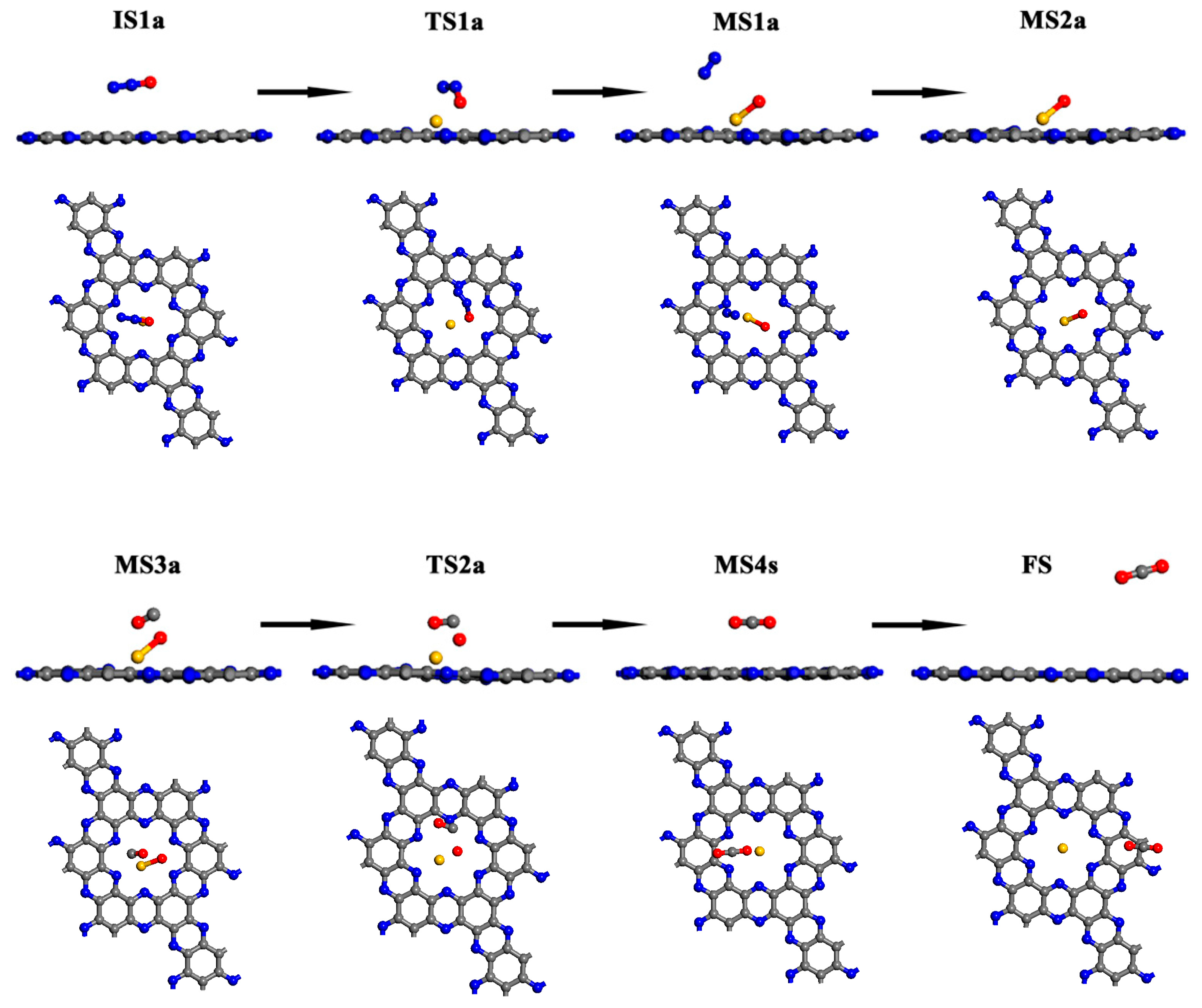
Figure 3.
The energy profiles along the MEP for N2O reduction via mechanism I. (R: M/C2N + N2O + CO, FS: M/C2N + N2 + CO2).
Figure 3.
The energy profiles along the MEP for N2O reduction via mechanism I. (R: M/C2N + N2O + CO, FS: M/C2N + N2 + CO2).
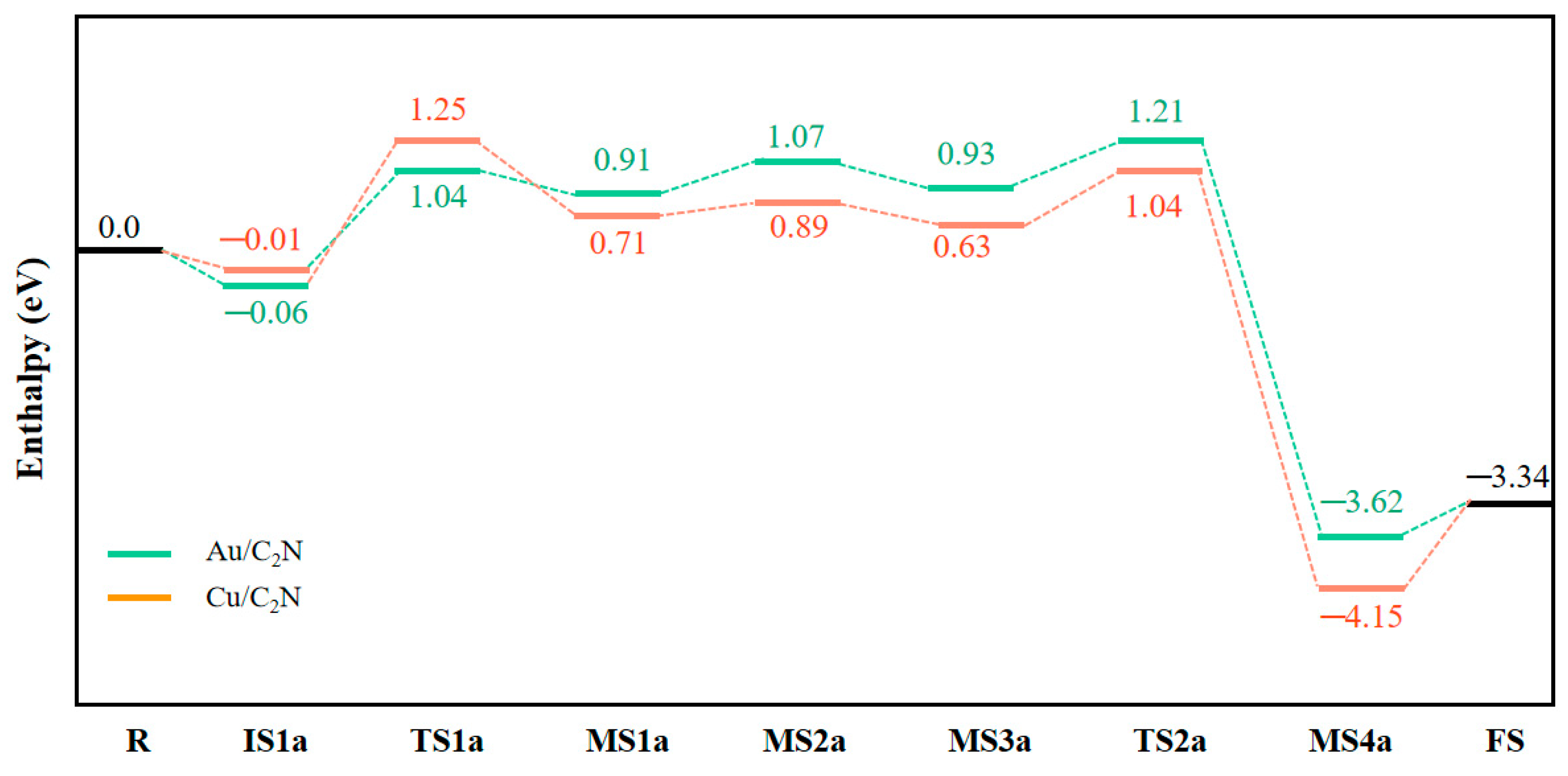
Figure 4.
The frontier molecular orbitals (FMOs) for the rate-determining step IS1a → TS1a → MS1a in the N2O reduction mechanism I on Au/C2N. (The isovalue is setting is default, 0.03 a.u.).
Figure 4.
The frontier molecular orbitals (FMOs) for the rate-determining step IS1a → TS1a → MS1a in the N2O reduction mechanism I on Au/C2N. (The isovalue is setting is default, 0.03 a.u.).
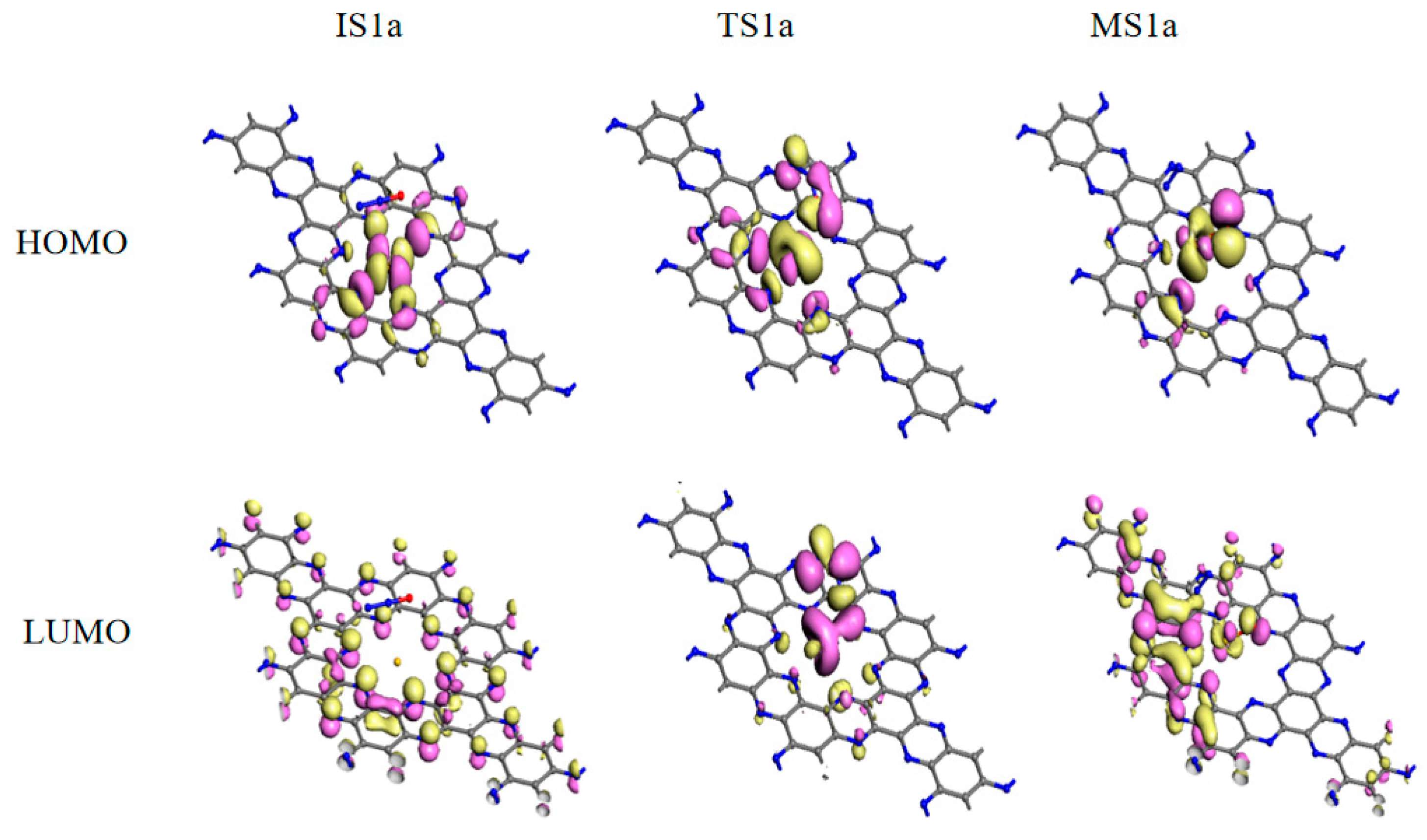
Figure 5.
The PDOS of relevant species in N2O reduction mechanism I on Au/C2N. (N(a) represents the N atom bonding with O atoms, and N(b) represents the ending N atom in N2O. O(a) and O(b) are from N2O and CO, respectively). (a) IS1a, (b) TS1a, (c) MS1a, (d) MS3a, (e) TS2a, (f) MS4a.
Figure 5.
The PDOS of relevant species in N2O reduction mechanism I on Au/C2N. (N(a) represents the N atom bonding with O atoms, and N(b) represents the ending N atom in N2O. O(a) and O(b) are from N2O and CO, respectively). (a) IS1a, (b) TS1a, (c) MS1a, (d) MS3a, (e) TS2a, (f) MS4a.

Figure 6.
The frontier molecular orbitals (FMOs) for rate-determining step IS1a → TS1a → MS1a in N2O reduction mechanism I on Cu/C2N. (The isovalue was set as default value, 0.03 a.u.).
Figure 6.
The frontier molecular orbitals (FMOs) for rate-determining step IS1a → TS1a → MS1a in N2O reduction mechanism I on Cu/C2N. (The isovalue was set as default value, 0.03 a.u.).
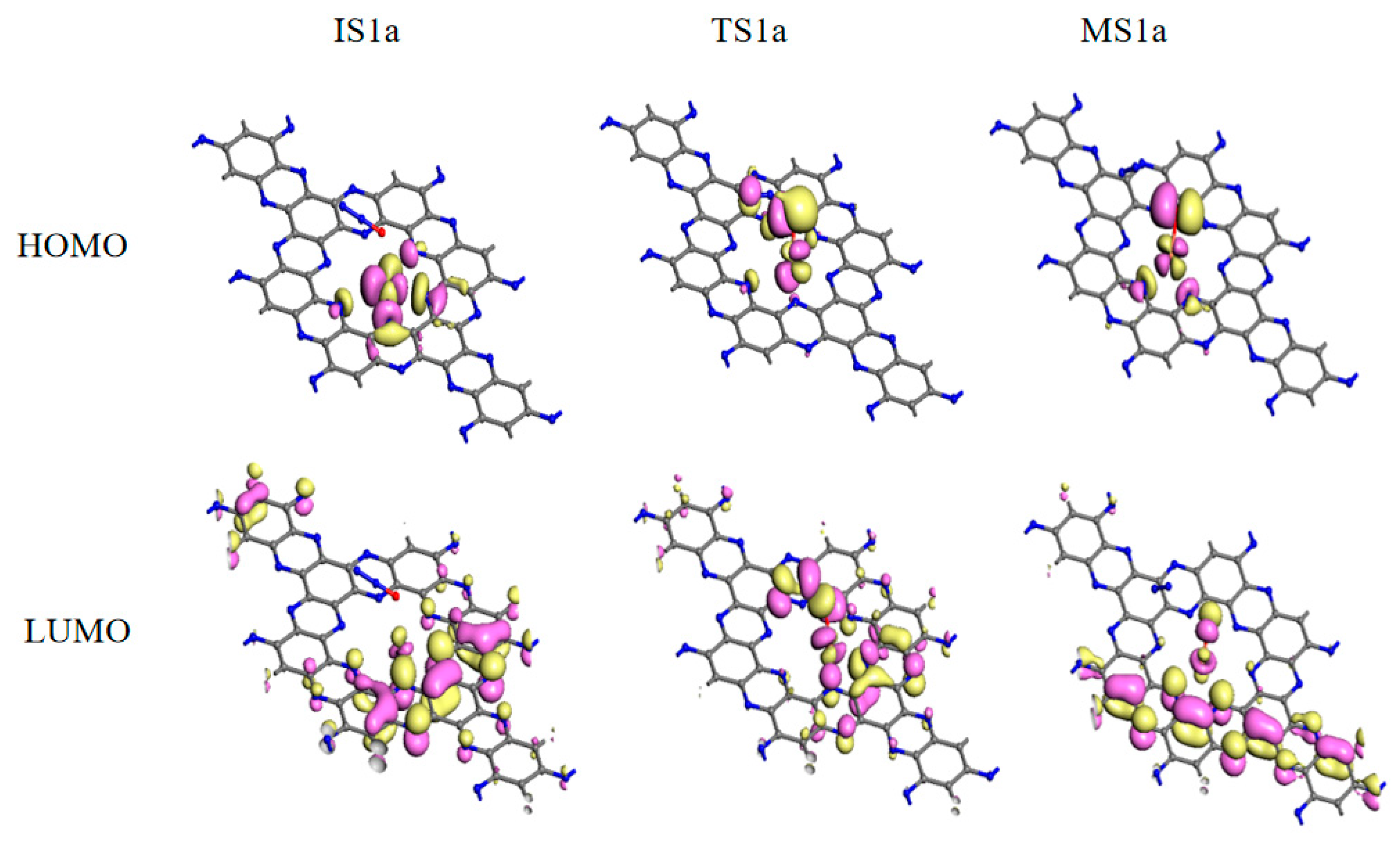
Figure 7.
The PDOS of relevant species in N2O reduction mechanism I on Cu/C2N. (N(a) represents the N atom bonding with O atoms, and N(b) represents the ending N atom in N2O. O(a) and O(b) are from N2O and CO, respectively). (a) IS1a, (b) TS1a, (c) MS1a, (d) MS3a, (e) TS2a, (f) MS4a.
Figure 7.
The PDOS of relevant species in N2O reduction mechanism I on Cu/C2N. (N(a) represents the N atom bonding with O atoms, and N(b) represents the ending N atom in N2O. O(a) and O(b) are from N2O and CO, respectively). (a) IS1a, (b) TS1a, (c) MS1a, (d) MS3a, (e) TS2a, (f) MS4a.
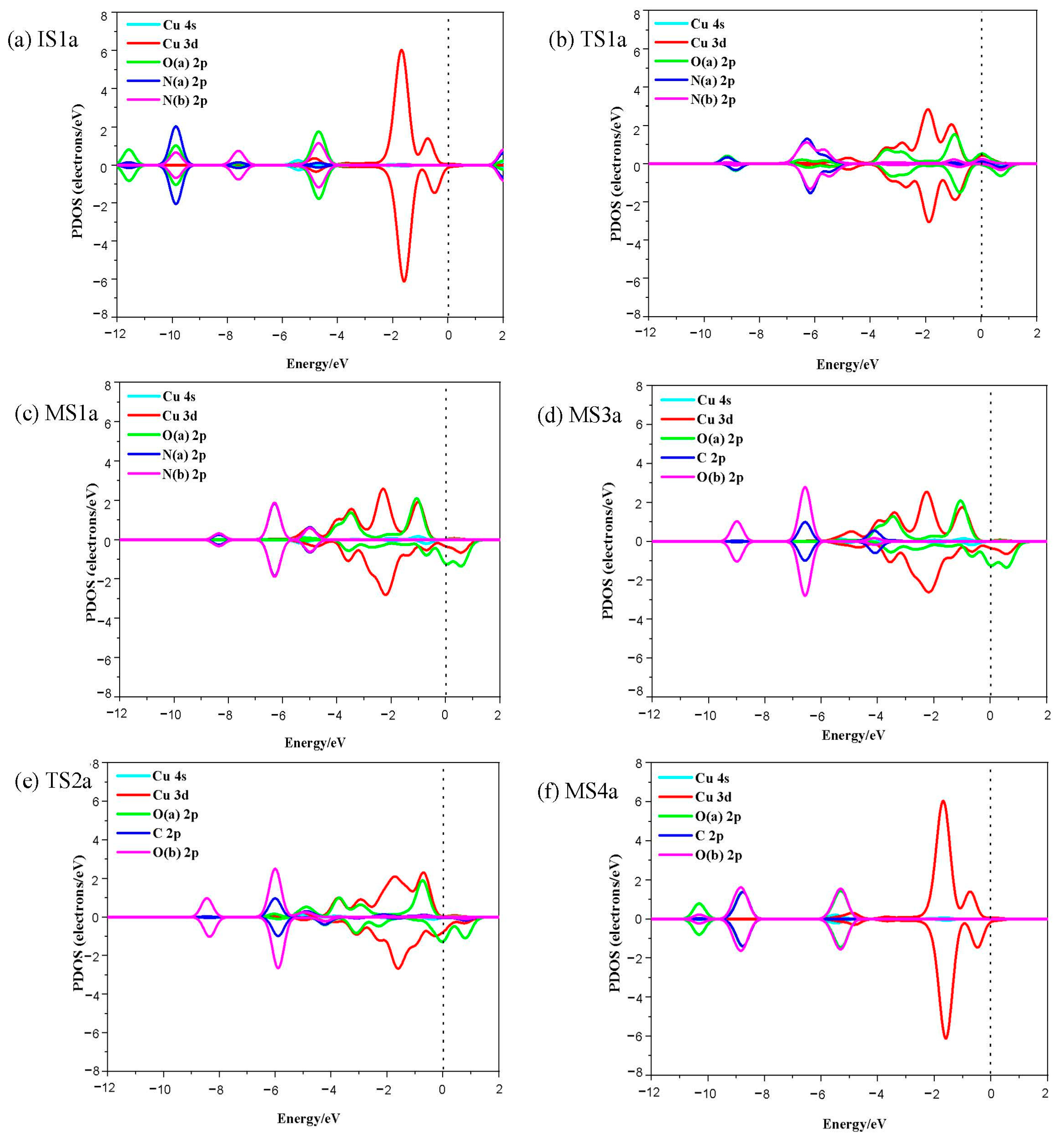
Figure 8.
The schematic diagram of all species along the MEP for the N2O reduction on the Au/C2N monolayer via mechanism II. (Blue, grey, yellow and red balls represent N, C, Au and O atoms, respectively.)
Figure 8.
The schematic diagram of all species along the MEP for the N2O reduction on the Au/C2N monolayer via mechanism II. (Blue, grey, yellow and red balls represent N, C, Au and O atoms, respectively.)
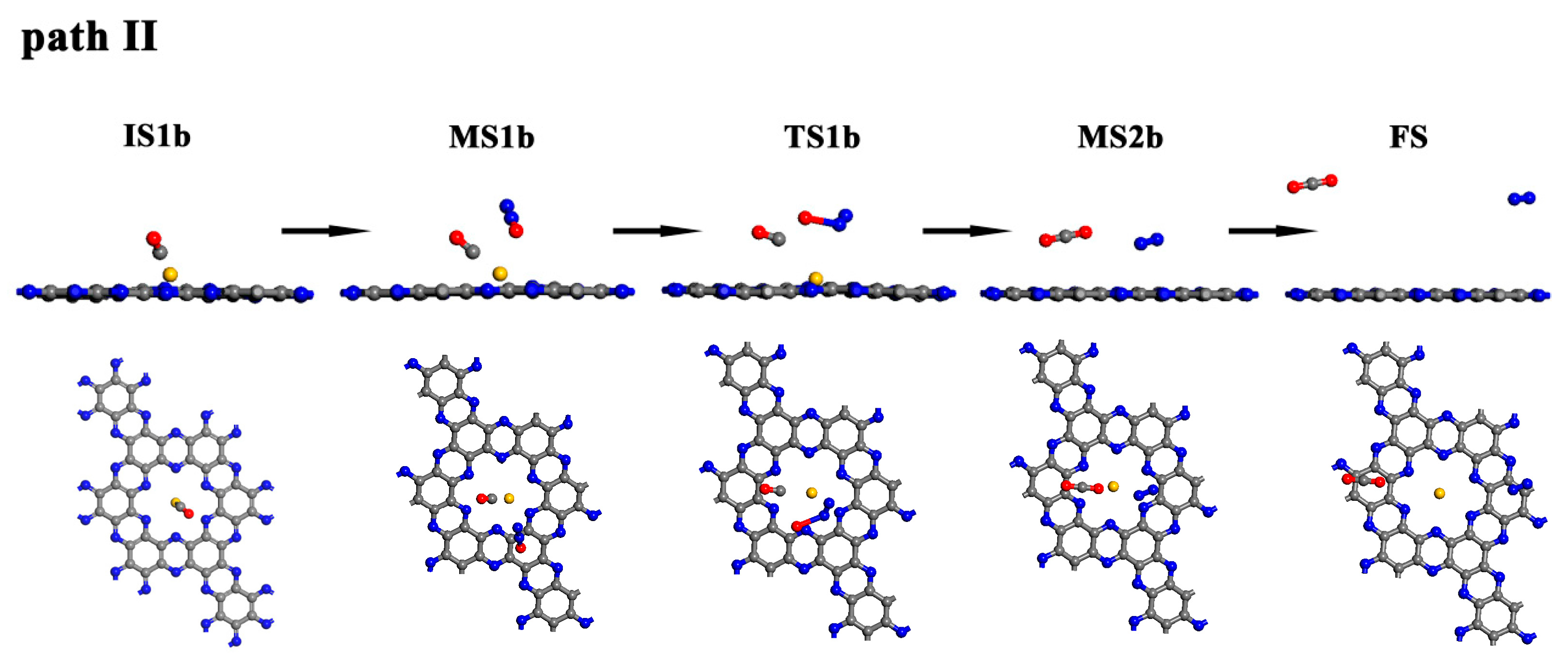
Figure 9.
Energy profiles along the MEP for the N2O reduction via mechanism II. (R: M/C2N + N2O + CO, FS: M/C2N + N2 + CO2.)
Figure 9.
Energy profiles along the MEP for the N2O reduction via mechanism II. (R: M/C2N + N2O + CO, FS: M/C2N + N2 + CO2.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Relative energy (ΔEcorr), enthalpy (ΔHcorr) and free energy (ΔGcorr) with zero-point vibrational correction for all the species in the both reactions. (unit: eV).
Table 1.
Relative energy (ΔEcorr), enthalpy (ΔHcorr) and free energy (ΔGcorr) with zero-point vibrational correction for all the species in the both reactions. (unit: eV).
| Au/C2N | Cu/C2N | |||||
|---|---|---|---|---|---|---|
| ΔEcorr | ΔHcorr | ΔGcorr | ΔEcorr | ΔHcorr | ΔGcorr | |
| R | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| IS1a | −0.05 | −0.06 | −0.09 | −0.02 | −0.01 | −0.11 |
| TS1a | 0.90 | 1.04 | 0.74 | 1.07 | 1.25 | 0.80 |
| MS1a | 0.84 | 0.91 | 0.40 | 0.63 | 0.71 | 0.20 |
| MS2a | 0.98 | 1.07 | 0.47 | 0.81 | 0.89 | 0.34 |
| MS3a | 0.83 | 0.93 | 0.60 | 0.56 | 0.63 | 0.29 |
| TS2a | 1.13 | 1.21 | 0.95 | 0.99 | 1.04 | 0.83 |
| MS4a | −3.69 | −3.62 | −3.97 | −4.13 | −4.15 | −3.80 |
| IS1b | −0.32 | −0.28 | −0.28 | −0.90 | −0.84 | −0.92 |
| MS1b | −0.43 | −0.40 | −0.14 | −1.10 | −1.05 | −0.96 |
| TS1b | 1.87 | 1.90 | 2.08 | 0.72 | 0.75 | 0.86 |
| MS2b | −3.48 | −3.44 | −3.30 | −3.58 | −3.49 | −3.54 |
| FS | −3.40 | −3.34 | −3.25 | −3.40 | −3.34 | −3.25 |
Table 2.
The Mulliken charge of species involved in Au/C2N catalytic N2O reduction mechanism I. N(a) represents the N atom bonding with O atoms, and N(b) represents the ending N atom in N2O.
Table 2.
The Mulliken charge of species involved in Au/C2N catalytic N2O reduction mechanism I. N(a) represents the N atom bonding with O atoms, and N(b) represents the ending N atom in N2O.
| Atom/ Molecule | Charges/e | |||||||
|---|---|---|---|---|---|---|---|---|
| Isolated Systems | IS1a | TS1a | MS1a | MS2a | MS3a | TS2a | MS4a | |
| Au | 0.548 | 0.551 | 0.705 | 0.734 | 0.739 | 0.736 | 0.708 | 0.561 |
| O(N2O) | −0.278 | −0.291 | −0.466 | −0.478 | −0.474 | −0.474 | −0.475 | −0.275 |
| N(a) | 0.420 | 0.435 | 0.141 | 0.013 | ||||
| N(b) | −0.143 | −0.140 | −0.112 | −0.016 | ||||
| N2O | 0 | 0.004 | −0.437 | |||||
| C | 0.08 | 0.080 | 0.102 | 0.543 | ||||
| O(CO) | −0.08 | −0.088 | −0.164 | −0.261 | ||||
| CO | 0 | −0.008 | ||||||
Table 3.
The Mulliken charge of species involved in Cu/C2N catalytic reduction of N2O reaction mechanism I. (Note that N(a) represents the N atom bonding with N and O atoms, and N(b) represents the ending N atom).
Table 3.
The Mulliken charge of species involved in Cu/C2N catalytic reduction of N2O reaction mechanism I. (Note that N(a) represents the N atom bonding with N and O atoms, and N(b) represents the ending N atom).
| Atom/ Molecule | Charges of the Systems/e | |||||||
|---|---|---|---|---|---|---|---|---|
| Isolated Systems | IS1a | TS1a | MS1a | MS2a | MS3a | TS2a | MS4a | |
| Cu | 0.622 | 0.639 | 0.785 | 0.809 | 0.807 | 0.806 | 0.725 | 0.685 |
| O(N2O) | −0.278 | −0.306 | −0.515 | −0.530 | −0.526 | −0.527 | −0.504 | −0.265 |
| N(a) | 0.420 | 0.449 | 0.148 | 0.006 | ||||
| N(b) | −0.143 | −0.120 | −0.095 | −0.006 | ||||
| N2O | 0 | 0.023 | −0.462 | |||||
| C | 0.08 | 0.080 | 0.156 | 0.538 | ||||
| O(CO) | −0.08 | −0.086 | −0.123 | −0.237 | ||||
| CO | 0 | 0.006 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Su, S.; Ma, J.; Liu, Z.; Holiharimanana, D.; Sun, H. Catalytic Reduction of N2O by CO on Single-Atom Catalysts Au/C2N and Cu/C2N: A First-Principles Study. Catalysts 2023, 13, 578. https://doi.org/10.3390/catal13030578
AMA Style
Su S, Ma J, Liu Z, Holiharimanana D, Sun H. Catalytic Reduction of N2O by CO on Single-Atom Catalysts Au/C2N and Cu/C2N: A First-Principles Study. Catalysts. 2023; 13(3):578. https://doi.org/10.3390/catal13030578
Chicago/Turabian StyleSu, Shengyang, Junmei Ma, Zhenhua Liu, Domoina Holiharimanana, and Hao Sun. 2023. "Catalytic Reduction of N2O by CO on Single-Atom Catalysts Au/C2N and Cu/C2N: A First-Principles Study" Catalysts 13, no. 3: 578. https://doi.org/10.3390/catal13030578
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.