
Gold(I)-Catalyzed Direct Alkyne Hydroarylation in Ionic Liquids: Mechanistic Insights


1
Department of Chemical Sciences, University of Padua, Via Marzolo 1, I-35131 Padua, Italy
2
Consortium for Chemical Reactivities and Catalysis (CIRCC), c/o University of Padua, Via Marzolo 1, I-35131 Padua, Italy
*
Author to whom correspondence should be addressed.
Catalysts 2023, 13(5), 822; https://doi.org/10.3390/catal13050822
Submission received: 11 April 2023
/
Revised: 27 April 2023
/
Accepted: 27 April 2023
/
Published: 29 April 2023
(This article belongs to the Collection Gold Catalysts)
Abstract
:The factors determining the catalytic performance of a recently discovered system for direct intermolecular alkyne hydroarylations, comprising a gold(I) complex of general formula LAuX (L = neutral ligand; X = weakly coordinating counteranion) and an ionic liquid as solvent, have been investigated. In particular, the effect of the ionic liquid anion, cation, and of the electronic and steric properties of the neutral ligand L have been considered. The results of the investigation shed some light in particular on the role of the anion, which is multiform and can potentially influence the catalytic performance in many respects, and on the role of the neutral ligand, which should be a relatively poor electron donor but not so much as to compromise the stability of the catalyst. Knowledge of the factors affecting catalytic performance will speed up the optimization of this catalytic system, in the case of direct alkyne hydroarylations with different substrates, and will also facilitate the extension of its application to other reactions.
1. Introduction
Homogeneous catalysis by gold complexes has undergone tremendous development in the course of the 21st century [1,2,3,4,5,6]. In particular, gold(I) complexes of general stoichiometry L-Au-X, where L is a neutral, soft ligand and X a weakly coordinating counteranion, have emerged as extremely useful, unique catalysts for several applications, mainly involving activation of multiple C-C bonds [3,4,5,6]. It was quite early recognized that the steric and electronic properties of the ligand L can significantly influence the performance of the catalyst [7], and the impact of the ligand on gold(I) catalysis has been comprehensively described with in-depth kinetic and mechanistic studies, aimed at understanding the ligand effects on the different steps of a catalytic cycle [7,8,9]. On the other hand, the role of the weakly coordinating counteranion X, although evidenced in earlier work by, e.g., Zhang and Widenhoefer [10], was initially quite neglected. Indeed, such anionic ligands were widely believed to bear a minor role in these processes, since they generally have to be substituted by the unsaturated substrate in the coordination sphere of gold in order to start the catalytic cycle; consequently, X was expected to act, at best, as a poison for the gold catalyst. However, it was later highlighted that the anionic ligand can influence the catalytic activity [11] and the regio- [12] and even the stereoselectivity [13] in gold-catalyzed reactions, taking up a prominent role in the catalytic process [14,15,16]. The effect exerted by the L and X ligands though, cannot be generalized to any given reaction, since it depends on the one hand on the nature of the rate-determining reaction step and on the other hand on the kind of cooperative effect that X can exert in the catalytic process itself [17]. Further to this, the effects of L and X are mutually related, hence they cannot be treated independently from one another [18,19].
Some time ago, we reported on the use of gold(I) complexes in ionic liquids (ILs) as particularly efficient catalysts for direct alkyne hydroarylation reactions [20]. Use of ILs as solvents for this catalytic system clearly represents a particular case of study, since the LAuX catalyst finds itself in the presence of an excess of the IL anion (and countercation) that bears a decisive influence on the catalytic process. Indeed, we have previously shown that this effect can be rather dramatic and may lead to anything from a drastic increase in catalytic efficiency, compared to common organic solvents, down to a complete shutdown in the catalytic activity [20]. In this communication, we try to rationalize the effect of the IL in conjunction with that of the neutral ligand L, in the context of the catalysis of a model intermolecular direct hydroarylation reaction, in order to shed some light on their importance and on the degree of control on the reaction that can be achieved through the optimization of these parameters.
2. Results
2.1. The Ionic Liquid Effect
The first aim of this investigation is to achieve a better understanding of how different IL anions (and cations) affect the catalytic activity of gold(I) catalysts in direct alkyne hydroarylation reactions.
As outlined in the Introduction, the roles of the neutral ligand L and of the counteranion X (in our case, the IL counteranion) can be crucial in gold(I) complex catalysis. Whereas the steric and electronic properties of the neutral ligand L can be parametrized using well established descriptors for their steric (e.g., Tolman cone angle [21], % buried volume [22]) and electronic (e.g., Tolman electronic parameter [23]) features, it is more difficult to individuate analogous descriptors for the anionic ligand/counteranion. Hammond and co-workers proposed an interpretation of the counteranion effect in cationic gold catalysis in terms of a “gold affinity index” (GAI, dependent on the size and charge distribution of the ion) and a “hydrogen bonding basicity index” (HBI, a measure of the capability of the anion to act as a hydrogen bond acceptor) [24]; the latter parameter bears a particular importance in reactions that involve an “active hydrogen”, e.g., hydrofunctionalizations of unsaturated organic compounds in which the added H-Nu nucleophile bears a hydrogen capable of forming strong hydrogen bonds. However, Hammond and co-workers had to conclude that, due to the multiple complex interactions that the ion may build during a reaction cycle, the anion effect on the catalytic performance cannot be invariably predicted using these two parameters. Zuccaccia and co-workers considered instead the influence of the coordinating ability, the Brønsted basicity, and also the geometry of the counteranion on all steps of standard alkyne hydroalkoxylation reactions; they identified anions with intermediate coordinating ability and basicity as the most efficient ones [25].
All these remarkable previous studies were conducted in organic solvents, in which the gold complex counteranion X was the only anionic species and was present in stoichiometric amounts. There is no precedent to look at concerning the same kind of studies in ILs. Moreover, no mechanistic studies concerning the role of the counteranion in gold-catalyzed direct alkyne hydroarylation reactions have appeared in the literature so far, apart from our preliminary investigation in ILs [20] and the extensive anion screening conducted by the group of Hashmi on a set of reactions also comprising a standard intramolecular alkyne hydroarylation [18,19]. Therefore, at the start of our study we were able to make only hypotheses about the anion effect in our system.
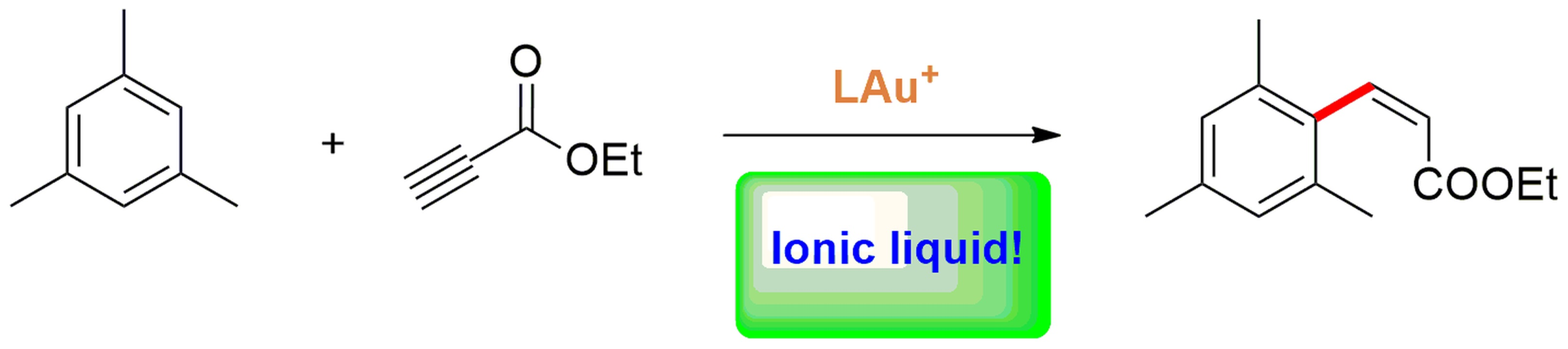
We took as a starting point the general mechanism for gold(I)-catalyzed alkyne hydrofunctionalization reactions, outlined in Figure 1 [26]. As can be inferred from the figure, the IL solvent as a whole can influence the catalytic cycle by solvating and stabilizing the charged intermediates A and B in the reaction cycle; further to this, the IL anion (highlighted in green in the figure) competes with the alkyne substrate for coordination at Au, which may result in catalyst poisoning but also in its stabilization in the resting state. Finally, the anion can also activate and orient the attack of the nucleophile HNu (if the nucleophile bears an active hydrogen) and assist the proton transfer from Nu to the Au-bound carbon atom.
In our study, we considered in situ activated catalysts, i.e., we employed LAuCl complexes in which the strongly coordinating chloride is substituted in situ with a less coordinating one upon metathesis with a proper silver salt (AgSbF6). Irrespective of the employed silver salt, it can be assumed that, due to the mass action law, the actual counteranion of the catalytically competent LAu+ species will be the counteranion of the IL, which in our system is present in 250-fold excess with respect to gold. Indeed, we have recorded the same catalytic performances in a given IL using different silver salts as activators, whereas the starting LAuCl complexes are instead completely inactive in the reaction if a silver salt activator is not added. As a further notice, when a silver salt activator was employed, the resulting insoluble AgCl coproduct was not removed from the reaction mixture, as it has been demonstrated to be inert under these reaction conditions. Indeed, we have previously verified [20] that catalytically competent species in direct alkyne hydroarylation reactions formed in situ in ILs upon silver salt metathesis performed similarly to preformed catalysts with weakly coordinating counteranions (no “silver effect” [14] present).
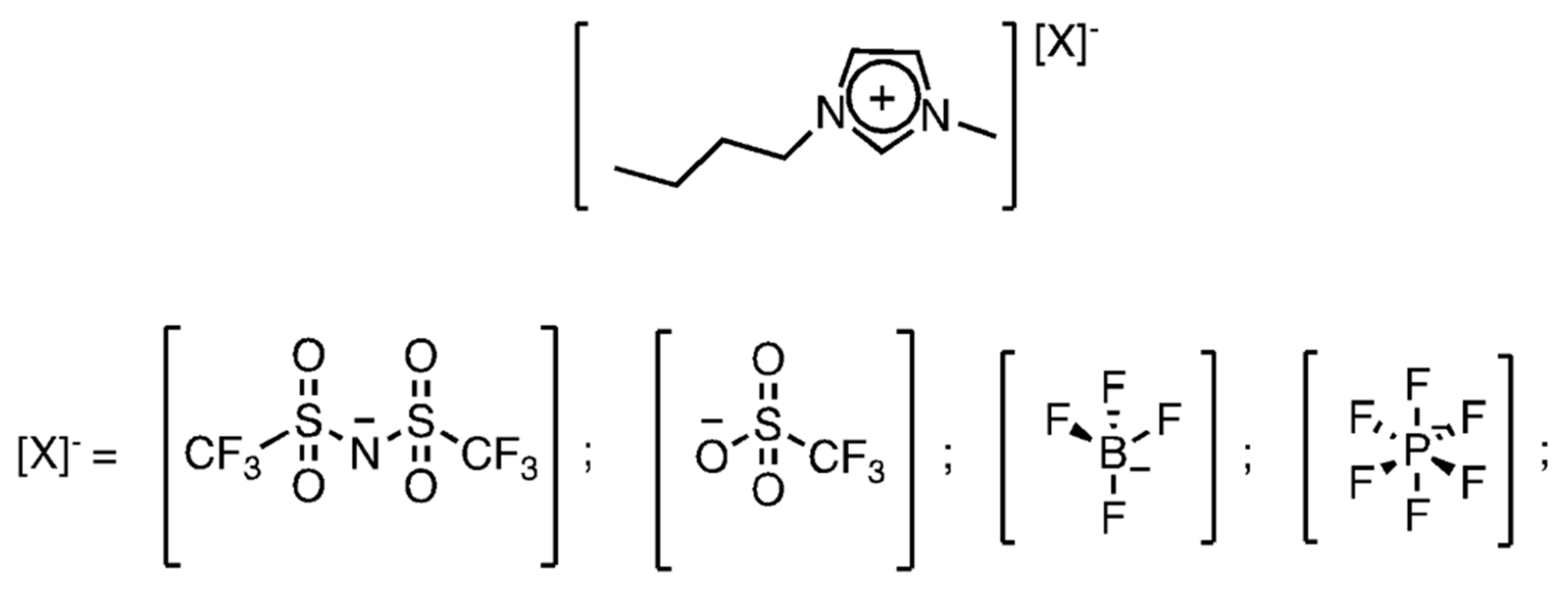
We screened a series of four ILs having 1-butyl-3-methylimidazolium (BMIM+) as the cation and different anions, namely hexafluorophosphate (PF6−), tetrafluoroborate (BF4−). bis(trifluoromethylsulfonyl)imide (NTf2−) and trifluoromethanesulfonate (OTf−) (Scheme 1). Relevant descriptors for the various employed anions (also including SbF6−, that we employ as weakly coordinating counteranion for gold in the activation process by metathesis) are listed in Table 1. Besides GAI and HBI, we have also included data for estimating the Brønsted basicity of the more basic anions, experimentally determined as the pKa of the conjugated acids in 1,2-dichloroethane [27]. Such data are not available for the conjugated acids of PF6− and SbF6−; nevertheless, the available experimental values match the HBI data.
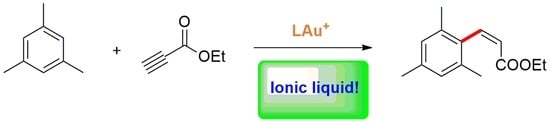
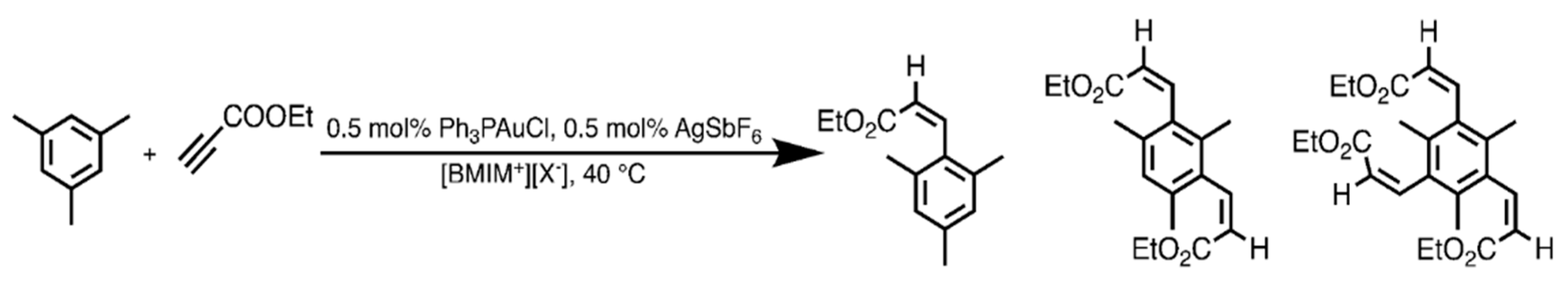
We addressed the hydroarylation of ethyl propiolate with mesitylene at 40 °C as a target reaction, using conventional chloro(triphenylphosphine)gold(I) as precatalyst and AgSbF6 as activator (Scheme 2). The outcomes of the reactions are reported in Table 2; products of formal trans-hydroarylation of the triple bond are invariably formed, in accordance with the current mechanistic understanding of gold-catalyzed alkyne hydroarylation reactions [28], which conforms to the general reaction mechanism outlined in Figure 1. Catalytic performance is monitored in terms of alkyne conversion, since the same mesitylene substrate can hydroarylate up to three alkyne molecules (the reaction chemoselectivity issue is treated below).
These results show that the effect of the nature of the anion is very significant and that NTf2−, i.e., the anion exhibiting the highest GAI and the lowest HBI, is by far the most effective anion. The effect of the anion under these reaction conditions is even more drastic compared to previous preliminary tests reported by us on the same reaction but at higher temperature (60 °C), in which also the IL with the PF6− anion enabled moderate activity [20]. The observed trend is in contrast to the one recorded by Zuccaccia et al., who investigated alkyne hydroalkoxylation reactions (i.e., a reaction with “active hydrogen”) and found anions of intermediate coordinating ability and hydrogen bonding basicity to be the best compromise [25]. However, the observed trend is also not consistent with the quantitative analysis of the counterion effect made by Hammond and co-workers [24]. In their view, since the direct hydroarylation reaction is not a reaction involving an “active hydrogen”, such as the hydroalkoxylation mentioned before, weakly coordinating counterions are expected to deliver the highest reactivity, provided they sufficiently stabilize the catalyst. In our case, the catalytic system showed no sign of decomposition, at least for the first few hours of reaction, but nevertheless, catalytic activity was recorded only with NTf2−. We hypothesized that such an inconsistency was indeed due to the fact that we were conducting the tests in an IL solvent, i.e., under conditions in which the anion is present in great excess with respect to the catalyst and greatly contributes to the liquid phase in which the reaction takes place. Consequently, under these conditions the observed difference in reactivity could depend on a property of the employed IL as a whole, and not simply of the IL anion.
In addition to stabilizing the cationic reaction intermediates due to coulombic interactions, the IL is also expected to solubilize the reagents, in order for the reaction to proceed efficiently. Indeed, we have observed that in our system the reagents are only partially soluble in the IL solvent, hence their solubility may play an important role in determining the observed catalytic activity. In order to determine a parameter with which to express the capability of the IL solvent to dissolve organic substrates, it is helpful to consider the Hildebrand solubility parameter, δ. When two substances have similar intermolecular interactions and, as a consequence, their Hildebrand solubility parameters are similar, they are likely to be miscible. Lee and Marciniak proved that the solubility parameter for a series of ILs with BMIM as the cation and variable anions increases in the following order: [OTf]− < [NTf2 ]− < [PF6]− < [BF4]− (Table 3) [29,30]. Considering that less polar species (such as the organic species used in our system) feature a low δ, it is reasonable that [NTf2]−, which provides a relatively low solubility parameter, favors the reaction. The IL with the [NTf2] also exhibits the lowest viscosity among all the tested ILs [31] (Table 3), which also points towards a higher reaction efficiency.
However, whereas the reported data point towards low lipophilicity and high viscosity as the cause of the inactivity of the catalytic system in ILs such as [BMIM][BF4] or [BMIM][PF6], they appear insufficient to explain the observed differences in the behavior of the other ILs, i.e., [BMIM][NTf2] and [BMIM][OTf]. The latter, in fact, exhibits even higher lipophilicity, only marginally higher viscosity, and anion properties that should still enable efficient catalysis. While additional tests are needed in order to provide experimental support for alternative explanations, we tend to invoke a recently highlighted property of ILs as being responsible for these differences. In particular, during the last decade several authors have pointed out that ILs are often microheterogeneous and tend to resemble, in their structure, concentrated microemulsions rather than conventional molecular solvents, since more polar and less polar functionalities do organize themselves into microdomains with different properties [32,33,34]. This heterogeneity has been experimentally verified in several cases, including imidazolium-based ILs: in particular, ILs involving the BMIM cation invariably exhibit such a microphase separation [35]. This microheterogeneous nature, which is often maintained also upon dilution with conventional liquid reagents/solvents [36], has obvious consequences in the preferential solvation of solutes by either of the two domains, and consequently on reactions carried out in these solvents: very recently, an example has been highlighted, in which changing from a [BMIM][NTf2] to a [BMIM][OTf], as in our case, leads to a complete shutdown of reactivity, which has been explained in terms of the different properties of the microdomains of the IL in the two cases [37]: OTf−, which is a stronger hydrogen bond acceptor compared to NTf2− (Table 1), engages in stronger hydrogen bond interactions with the BMIM cation (which acts as an H-bond donor through its C-H moiety in the 2-position) producing polar microdomains with different properties. This influences the partition of the substrate between polar and apolar microdomains and consequently its reactivity. In our system, a similar situation could arise, with different partitions of catalyst and reagent(s) between the microdomains, allowing the reaction in the case of NTf2− but preventing it in others.
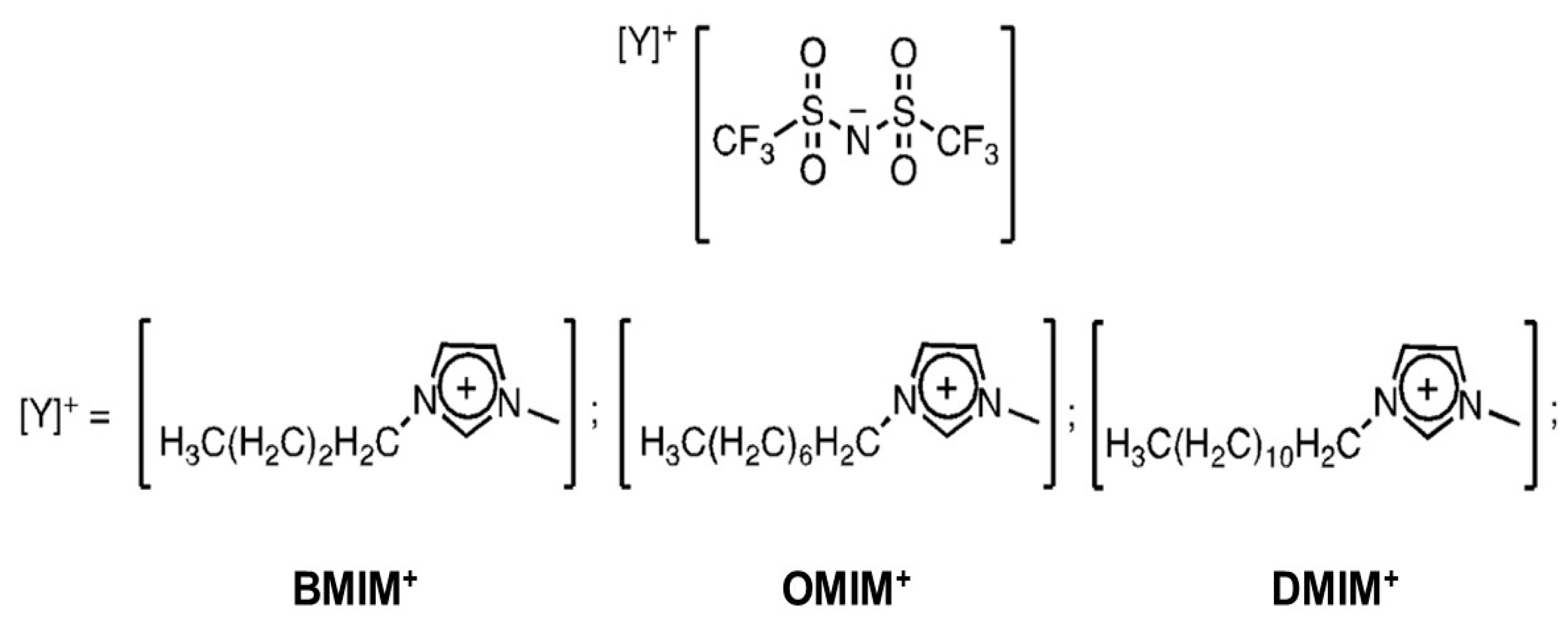
A second screening of the employed IL solvent focused on the cation effect. We have previously determined that methylation of the BMMIM cation in the 2-position results in no effect on the catalytic performance of the system when NTf2− is the counteranion. Similar to the previous investigation, we carried out the same reaction reported in Scheme 2 in a series of three ILs composed of NTf2− as the anion and of cations with decreasing hydrophilicity (Scheme 3). More precisely, all of the three cations had the same imidazolium-based structure but different aliphatic chain length substituents. Indeed, it was found that increasing the chain length of alkyl substituents of the ions of ILs leads to higher lipophilicity of the solvent, as determined by the Hildebrand solubility parameter [29,30]. Further to this, increasing the chain length also results in an enhancement of the microphase separation within the IL [35]. The results are reported in Table 4.
It can be appreciated that the catalytic activity is almost independent from the nature of the employed IL cation, as should actually be the case if the reaction takes place in the polar microdomains of the IL, where the cationic gold(I) catalyst is presumably located.
2.2. The Ligand Effect
As soon as the IL counteranion imparting the highest catalytic activity to the system was identified, a further optimization of the neutral ligand L at gold was conducted. Indeed, the neutral ligand plays a significant role in tuning the reactivity of gold(I) catalysis: in particular, its electron donating ability modulates the acidic character of the metal fragment in the catalytic cycle and affects the stability of the postulated intermediates [9]. On this topic, Wang et al. conducted a systematic ligand-effect investigation on each of the three major stages in the catalytic event (Figure 1) [8]: external nucleophilic attack on the coordinated alkyne, protodeauration of the vinylic intermediate, and decay of the active gold catalyst. They concluded that the ligand effect is not easily predictable, but they were nevertheless able to identify the most efficient class of ligands in each step. Through establishing a structure–activity relationship between the ligand and the kinetics of each stage in the catalytic cycle, they observed that when the rate-determining step (RDS) is the nucleophilic attack, less-electron-donating ligands, that promote electrophilic activation of the substrate by rendering the gold center electron-poorer, accelerate the overall reaction. An example is the intermolecular hydroamination reported by Toste and co-workers [38]. Instead, more-electron-donating ligands accelerate the reaction when protodeauration is the RDS.
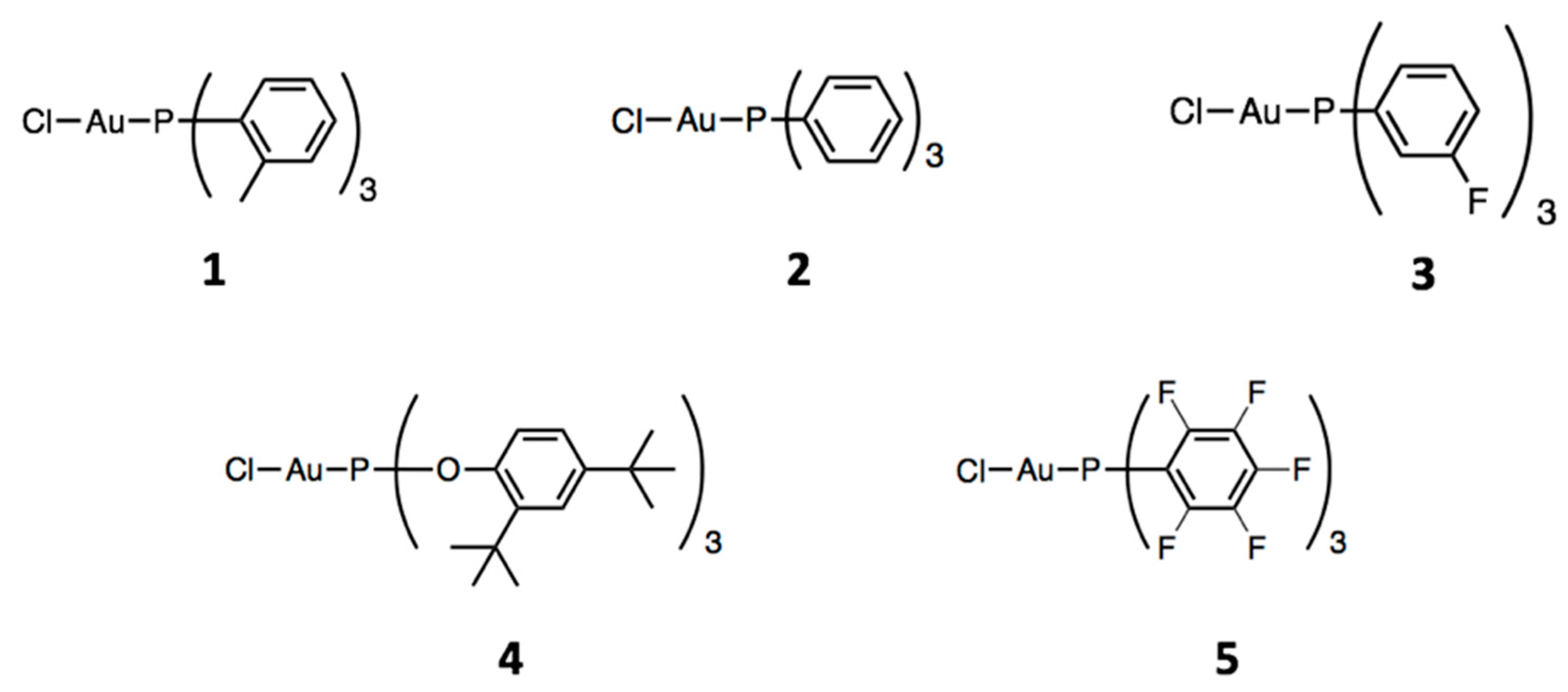
Because of the small amount of investigations on gold(I)-catalyzed intermolecular hydroarylation reactions with simple arenes, we embarked on a systematic study on the ligand effect in gold(I) complexes catalysts in this type of reaction using ILs as non-innocent solvents. The standard reaction between mesitylene and ethyl propiolate (Scheme 2) in [BMIM][NTf2] was taken as the benchmark for ligand evaluation. We selected five Au(I) complexes with phosphane- and phosphite-based ligands of different electron-donating abilities because, as reported by Wang et al., this is the ligand parameter which mainly influences the catalytic activity. The chosen complexes are reported in Scheme 4.
Like in the previous analysis, the precatalysts were activated in situ with the assistance of AgSbF6. Complexes 2 and 4 were commercial, the others were synthesized following the procedure reported in Section 3. All of the synthesized complexes were analyzed by 1H NMR, 31P NMR, and elemental analysis techniques.
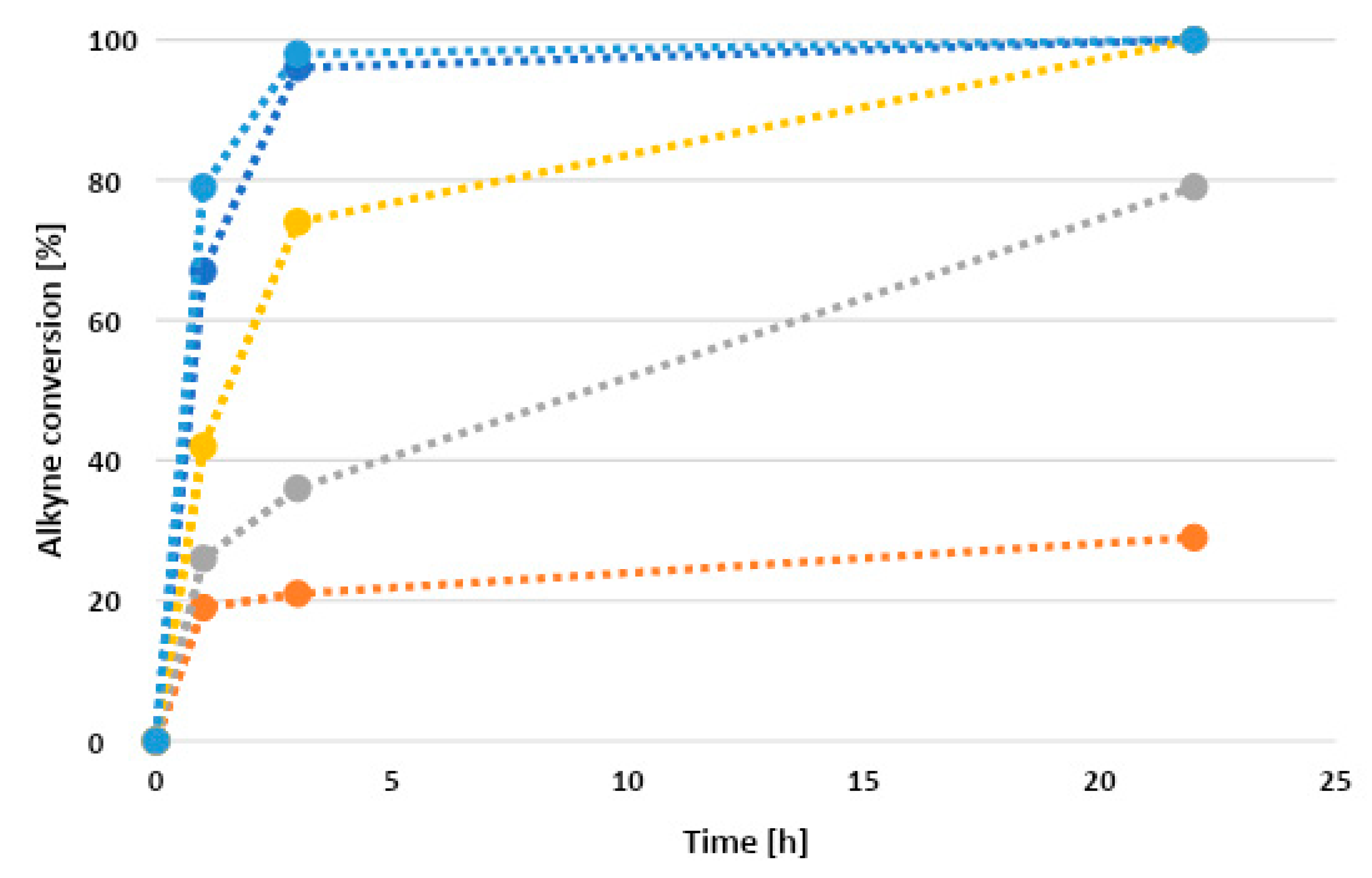
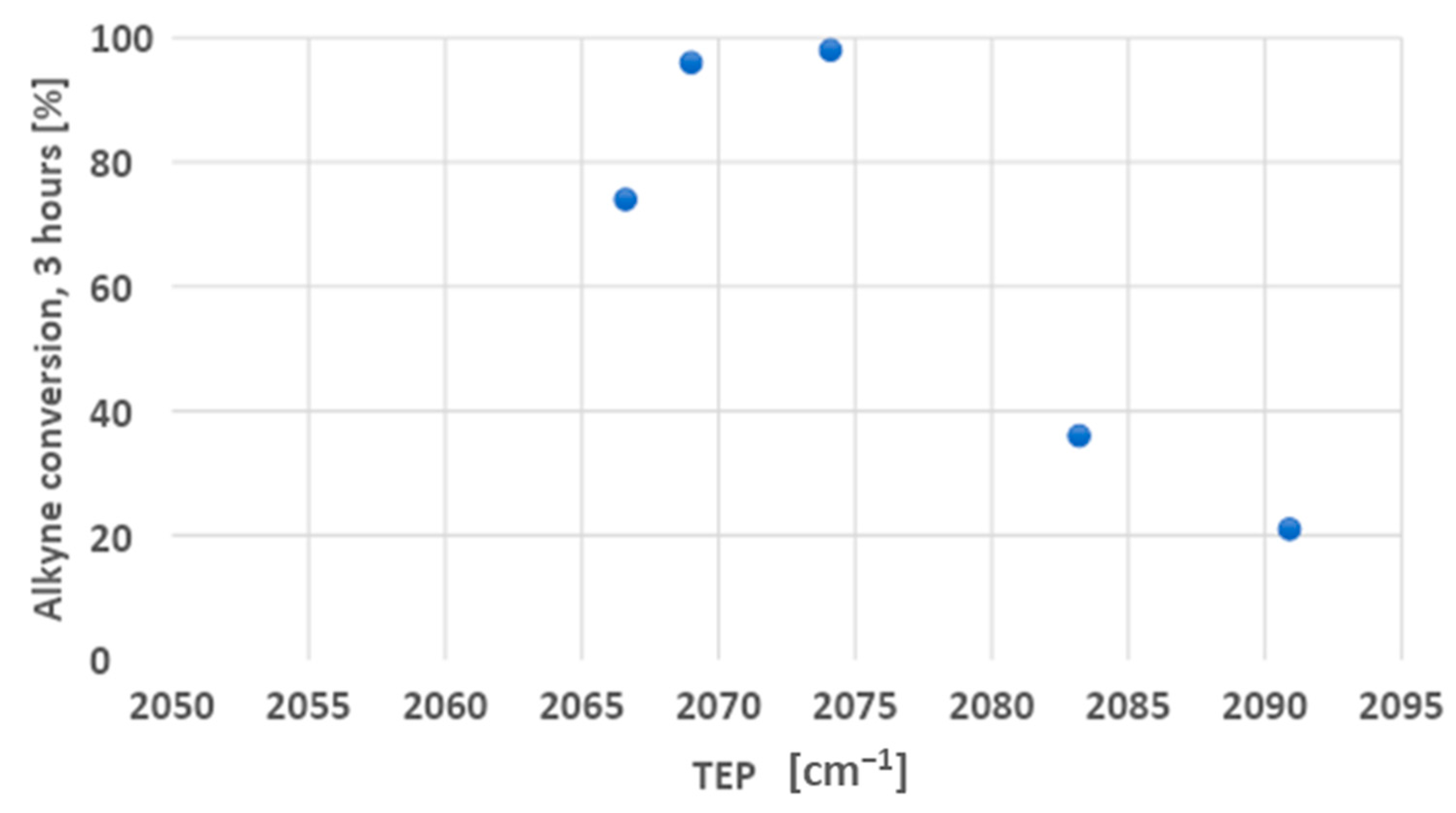
We used the Tolman electronic parameter (TEP) to define the net electron-donating power of the ligands [23]. The complexes reported in Scheme 4 were specifically selected because they cover a relatively wide range of TEP values (from 2067 to 2091 cm−1). The order of the complexes on the basis of the TEP is: 1 < 2 < 3 < 4 < 5. The kinetic profiles of the hydroarylation reaction of ethyl propiolate with mesitylene catalyzed by different Au(I) complexes are plotted in Figure 2. Dashed lines are drawn to guide the eye.
On the basis of the kinetic profiles reported above, it is clear that complexes with an intermediate TEP (2 and 3) were the most efficient, achieving 96% and 98% alkyne conversion after only 3 h reaction time. With the more electron-rich phosphane (1) the conversion was lower (74% after the same time). The conversion lowered again to 36% and 21% with the even less electron-donating ligands (4 and 5). Figure 3 illustrates the correlation between the ligands’ TEP and % conversion after 3 h reaction time.
The bell-shaped dependence of the reported alkyne conversions from the TEP of the ligand demonstrates that the correlation between these two parameters is not simple. We suppose that such a dependence may be the result of the interplay between the ligand’s electron-donating capability and its stabilizing effect on the gold catalyst. Indeed, according to Hashmi and co-workers (who analyzed intramolecular hydroarylation reactions) [18], the catalytic activity seems to inversely correlate with the ligand’s electron-donating power in this reaction. These results are consistent with a scenario in which the actual RDS is the nucleophilic attack of the arene on the coordinated alkyne, which is favored by a higher electrophilicity of the gold(I) center [8]. In the case of an intermolecular hydroarylation reaction, it is even more probable that this step is the actual RDS. Nevertheless, a decreasing electron-donating ability of the ligand undermines the stability of the active gold complex, thus lowering the overall catalytic efficiency. Consequently, in reactions with 1, 2, and 3 as precatalysts, the rate of gold(I) complex decomposition does not seem to significantly affect the catalytic efficiency, while with 4 and 5 instead, the effects are drastic. This may also explain why the alkyne conversion with 5 was far from quantitative, even after several hours. In addition, the fast catalyst decomposition is consistent with the darkening of the reaction mixtures that can be observed already at short reaction times with 4 and 5.
In addition to the electronic effect of the ligand, which we anticipated to significantly influence the catalytic activity of the complex, we also checked whether steric effects could play a role in the reaction outcome. In particular, we were looking at the reaction chemoselectivity with substrates such as mesitylene, with which multiple hydroarylation reactions are possible on the same aromatic substrate. We chose the set of previously employed complexes 1–4, plus a complex 9, with the biphenyl-derived JackiePhos ligand, as an example of a more sterically hindered complex with similar electron-donating capability. We carried out the same reaction reported in Scheme 2 and checked the product selectivity of the selected catalysts after 22 h, i.e., a reaction time at which complete alkyne conversion is achieved in most cases. The results are reported in Table 5.
From the data reported in Table 5, it is quite apparent that the stereoelectronic features of the ligand are able to influence the chemoselectivity of the hydroarylation to a limited extent. Furthermore, the ligand effect is not easily predictable on the basis of the usual parameters for evaluating the ligand steric and electronic properties. Indeed, whereas use of precatalyst 9 provides a slight increase in selectivity for the monosubstituted product, the opposite effect is recorded with complex 1 with tris(o-tolyl)phosphine, which is significantly bulkier than triphenylphosphine in complex 2. The electronic properties of the ligand play instead a very marginal role on the reaction chemoselectivity, as implied by the very similar results obtained with complexes 2–4.
In conclusion, the investigation on the effect of the IL on the performance of our gold(I)/IL catalytic system for direct alkyne hydroarylations has shed some light in particular on the role of the anion, which can potentially influence the catalytic performance by deactivating the catalyst, by taking up an active role in the reaction mechanism (nucleophile activation, proton transfer) and also by influencing, together with the IL cation, the solubility of the substrates and their partition in the IL microdomains. A detailed knowledge of all these factors affecting the overall catalytic performance will need further work, but will enable the rapid optimization of this catalytic system in the case of direct alkyne hydroarylations with different substrates, facilitating also the extension of its application to other reactions. The high tunability of Au(I) complexes allowed us also to investigate the effect of the ligand on the catalyst activity and selectivity in hydroarylation reactions. We found that two main factors are competing against each other: the electron-donating power of the ligand and the stability of the catalytically competent, cationic LAu+ species. The RDS seems to be the nucleophilic attack of the arene to the activated alkyne. Thus, less-electron-donating ligands render the cationic gold complex more electrophilic and consequently speed up the reaction. However, if the Au(I) center is too electron-poor, the instability of the complex becomes relevant and the ensuing decomposition shuts down the reactivity of the system. The best compromise among the tested ligands is apparently provided by tris(m-fluorophenyl)phosphine. Furthermore, the selectivity of the reaction does not seem to be very sensitive to the employed gold catalyst, as it is not significantly dependent on the steric or electronic properties of the ligand. Finally, it is useful to remark that, comparing the catalytic results obtained herein with those found in the literature, the gold(I)/IL system shows higher reactivities (especially in the case of precatalysts 2 and 3) with lower amounts of catalyst (0.5 mol% against 1–5 mol%), without arene excess, and under acid-free conditions.
3. Materials and Methods
3.1. General Remarks
All manipulations were carried out using standard Schlenk techniques under an atmosphere of argon. Commercial reagents and solvents were purchased as high-purity products and used as received. NMR spectra were recorded on Bruker Avance (Ettlingen, Germany) 300 MHz (300.1 MHz for 1H, 75.5 MHz for 13C and 121.49 MHz for 31P). The chemical shifts (δ) are reported in units of ppm relative to the residual solvent signals and multiplicities of the peaks are expressed as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants are given in Hz. Elemental analyses were carried out with a Thermo Scientific FLASH 2000 apparatus (Waltham, MA, USA).
3.2. General Procedures for Precatalyst Synthesis
The phosphine ligand (0.34 mmol) and chloro(dimethylsulfide)gold(I) (0.34 mmol) were put in a round-bottom flask which was then evacuated and filled with argon at room temperature. A volume of 10 mL of dichloromethane was then added, and the resulting solution was stirred at room temperature for 4 h. After this time, the solvent was removed by using a vacuum pump. The yield was calculated by weighing the obtained solid.
- Chloro[tri(o-tolyl)phosphine]gold(I) (1)
According to the described method, tri(o-tolyl)phosphine (120 mg, 0.34 mmol) and chloro(dimethylsulfide)gold(I) (100 mg, 0.34 mmol) were dissolved in 10 mL of dichloromethane. After 4 h, the solvent was removed to afford chloro[tri(o-tolyl)phosphine]gold(I) quantitatively (180 mg) as a white solid. 31P-NMR (121 MHz, CDCl3): δ 8.25. 1H-NMR (300 MHz, CDCl3): δ 7.47 (m, 3H, ArH), 7.36 (m, 3H, ArH), 7.20 (m, 3H, ArH), 6.92 (m, 3H, ArH), 2.68 (s, 9H, CH3). Elemental analysis % calculated for C21H21AuClP: C, 49.87; H, 4.18. Found: C, 49.59; H, 3.96.
- Chloro[tri(m-fluorophenyl)phosphine]gold(I) (3)
According to the described method, tri(m-fluorophenyl)phosphine (110 mg, 0.34 mmol) and chloro(dimethylsulfide)gold(I) (100 mg, 0.34 mmol) were dissolved in 10 mL of dichloromethane. After 4 h, the solvent was removed to afford chloro[tri(m-fluorophenyl)phosphine]gold(I) quantitatively (190 mg) as a white solid. 31P-NMR (121 MHz, CDCl3): δ 32.5. 1H-NMR (300 MHz, CDCl3): δ 7.52 (m, 1H, ArH), 7.34 (m, 2H, ArH), 7.16 (m, 1H, ArH). Elemental analysis % calculated for AuClPC18H12F3: C, 39.40; H, 2.20. Found: C, 39.32; H, 2.40.
- Chloro[tris(2,3,4,5,6-pentafluorophenyl)phosphine]gold(I) (5)
According to the described method, tris(pentafluorophenyl)phosphine (180 mg, 0.34 mmol) and chloro(dimethylsulfide)gold(I) (100 mg, 0.34 mmol) were dissolved in 10 mL of dichloromethane. After 4 h, the solvent was removed to afford chloro[tris(2,3,4,5,6-pentafluorophenyl)phosphine]gold(I) quantitatively (260 mg) as a white solid. 31P-NMR (121 MHz, CD3CN): δ = −35.4. Elemental analysis % calculated for C18AuClF15P: C, 28.28; H, 0. Found: C, 28.68; H, 0.24.
- Chloro[bis(3,5-bis(trifluoromethyl)phenyl)(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine]gold(I) (9)
According to the described method, bis(3,5-bis(trifluoromethyl)phenyl)(2′,4′,6′- triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (110 mg, 0.14 mmol) and chloro(dimethylsulfide)gold(I) (40 mg, 0.14 mmol) were dissolved in 10 mL of dichloromethane. After 4 h, the solvent was removed to afford chloro[bis(3,5-bis(trifluoromethyl)phenyl)(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine]gold(I) quantitatively (140 mg). 31P-NMR (121 MHz, CDCl3): δ 18.1. 1H-NMR (300 MHz, CDCl3): 7.97 (s, 2H, ArH), 7.86 (s, 2H, ArH), 7.82 (s, 2H, ArH), 7.31 (d, J = 9.0, 1H, ArH), 7.10 (s, 2H, ArH), 7.00 (d, J = 9.0 Hz, 1H, ArH), 3.71 (s, 3H, OCH3), 3.32 (s, 3H, OCH3), 2.99 (septet, J = 6.8 Hz, 1H, iPr), 2.33 (septet, J = 6.9 Hz, 2H, iPr), 1.37 (d, J = 6.8 Hz, 6H, iPr), 1.06 (d, J = 6.9 Hz, 6H, iPr), 0.99 (d, J = 6.9 Hz, 6H, iPr). Elemental analysis % calculated for C39H37AuClF12P: C, 45.52; H, 3.62. Found: 45.49; H, 3.75.
3.3. General Procedure for the Catalytic Hydroarylation Tests on different Gold Complexes
The gold(I) complex (0.012 mmol), AgSbF6 (4.3 mg, 0.012 mmol), and 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (0.75 mL) were placed in a Schlenk tube, which was then evacuated and filled with argon at room temperature. Ethyl propiolate (0.25 mL, 2.5 mmol) and mesitylene (0.35 mL, 2.5 mmol) were added into the Schlenk tube. The tube was placed in an oil bath thermostated at 40 °C and the reaction mixture was stirred for 22 h. Portions of the mixture (0.1 mL) were drawn off from the reaction mixture and analyzed by 1H NMR. Reaction products were identified by comparison with literature reported NMR data [20].
- Ethyl-3-mesitylpropenoate (6)
1H-NMR (300 MHz, CDCl3): δ 7.05 (d, J = 11.7 Hz, 1H, vinyl), 6.86 (s, 2H, ArH), 6.13 (d, J = 11.7 Hz, 1H, vinyl), 4.05 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.28 (s, 3H, ArCH3), 2.17 (s, 6H, ArCH3), 0.98 (t, J = 7.1 Hz, 3H, OCH2CH3).
- Diethyl-3,3′-(2,4,6-trimethyl-1,3-phenylene)bis(propenoate) (7)
1H-NMR (300 MHz, CDCl3): 7.05 (d, J = 11.7 Hz, 2H, vinyl), 6.90 (s, 1H, ArH), 6.14 (d, J = 11.7 Hz, 2H, vinyl), 4.05 (q, J = 7.1 Hz, 4H, OCH2CH3), 2.17 (s, 6H, ArCH3), 2.07 (s, 3H, ArCH3), 0.98 (t, J = 7.1 Hz, 6H, OCH2CH3).
- Triethyl-3,3′,3′’-(2,4,6-trimethyl-1,3,5-phenylene)tris(propenoate) (8)
1H-NMR (300 MHz, CDCl3): 7.05 (d, J = 11.7 Hz, 3H, vinyl), 6.15 (d, J = 11.7 Hz, 3H, vinyl), 4.05 (q, J = 7.1 Hz, 6H, OCH2CH3), 2.07 (s, 9H, ArCH3), 0.98 (t, J = 7.1 Hz, 9H, OCH2CH3).
3.4. General Procedure for the Catalytic Hydroarylation Tests on Different Ionic Liquids
Chloro[triphenylphosphine]gold(I) (6.2 mg, 0.012 mmol), AgSbF6 (4.3 mg, 0.012 mmol), and the ionic liquid (0.75 mL) were placed in a Schlenk tube, which was then evacuated and filled with argon at room temperature. Ethyl propiolate (0.25 mL, 2.5 mmol) and mesitylene (0.35 mL, 2.5 mmol) were added into the Schlenk tube. The tube was placed in an oil bath thermostated at 40 °C and the reaction mixture was stirred for 23 h. Portions of the mixture (0.1 mL) were drawn off from the reaction mixture and analyzed by 1H NMR. Reaction products were identified by comparison with literature reported NMR data [20]. The products’ NMR data are reported in the previous paragraph.
Author Contributions
Investigation, S.B. and P.B.; validation, M.B.; supervision, writing, M.B. and A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data supporting the reported results can be found in Section 3 of this contribution.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Toste, F.D.; Michelet, V. (Eds.) Gold Catalysis: An Homogeneous Approach; Catalytic Science Series; Imperial College Press: London, UK, 2014. [Google Scholar]
- Slaughter, L.M.; Aponick, A. (Eds.) Homogeneous Gold Catalysis; Topics in Current Chemistry; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Praveen, C. Carbophilic Activation of π-Systems via Gold Coordination: Towards Regioselective Access of Intermolecular Addition Products. Coord. Chem. Rev. 2019, 392, 1–34. [Google Scholar] [CrossRef]
- Alyabyev, S.B.; Beletskaya, I.P. Gold as a Catalyst. Part I. Nucleophilic Addition to the Triple Bond. Russ. Chem. Rev. 2017, 86, 689–749. [Google Scholar] [CrossRef]
- Alyabyev, S.B.; Beletskaya, I.P. Gold as a Catalyst. Part II. Alkynes in the Reactions of Carbon–Carbon Bond Formation. Russ. Chem. Rev. 2018, 87, 984–1047. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef]
- Gorin, D.J.; Sherry, B.D.; Toste, F.D. Ligand Effects in Homogeneous Au Catalysis. Chem. Rev. 2008, 108, 3351–3378. [Google Scholar] [CrossRef]
- Wang, W.; Hammond, G.B.; Xu, B. Ligand Effects and Ligand Design in Homogeneous Gold(I) Catalysis. J. Am. Chem. Soc. 2012, 134, 5697–5705. [Google Scholar] [CrossRef]
- Obradors, C.; Echavarren, A.M. Intriguing Mechanistic Labyrinths in Gold(i) Catalysis. Chem. Commun. 2014, 50, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Widenhoefer, R.A. Regio- And Stereoselective Synthesis of Alkyl Allylic Ethers via Gold(l)-Catalyzed Intermodular Hydroalkoxylation of Allenes with Alcohols. Org. Lett. 2008, 10, 2079–2081. [Google Scholar] [CrossRef]
- Hashmi, S.A.K.; Braun, I.; Rudolph, M.; Rominger, F. The Role of Gold Acetylides as a Selectivity Trigger and the Importance of Gem-Diaurated Species in the Gold-Catalyzed Hydroarylating-Aromatization of Arene-Diynes. Organometallics 2012, 31, 644–661. [Google Scholar] [CrossRef]
- Jia, M.; Cera, G.; Perrotta, D.; Monari, M.; Bandini, M. Taming Gold(I)-Counterion Interplay in the de-Aromatization of Indoles with Allenamides. Chem. A Eur. J. 2014, 20, 9875–9878. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.L.; Eun, J.K.; Mba, M.; Toste, F.D. A Powerful Chiral Counterion Strategy for Asymmetric Transition Metal Catalysis. Science 2007, 317, 496–499. [Google Scholar] [CrossRef]
- Jia, M.; Bandini, M. Counterion Effects in Homogeneous Gold Catalysis. ACS Catal. 2015, 5, 1638–1652. [Google Scholar] [CrossRef]
- Zuccaccia, D.; Del Zotto, A.; Baratta, W. The Pivotal Role of the Counterion in Gold Catalyzed Hydration and Alkoxylation of Alkynes. Coord. Chem. Rev. 2019, 396, 103–116. [Google Scholar] [CrossRef]
- Lu, Z.; Li, T.; Mudshinge, S.R.; Xu, B.; Hammond, G.B. Optimization of Catalysts and Conditions in Gold(I) Catalysis—Counterion and Additive Effects. Chem. Rev. 2021, 121, 8452–8477. [Google Scholar] [CrossRef] [PubMed]
- Biasiolo, L.; Del Zotto, A.; Zuccaccia, D. Toward Optimizing the Performance of Homogeneous L-Au-X Catalysts through Appropriate Matching of the Ligand (L) and Counterion (X−). Organometallics 2015, 34, 1759–1765. [Google Scholar] [CrossRef]
- Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A.S.K. An Industrial Perspective on Counter Anions in Gold Catalysis: Underestimated with Respect to Ligand Effects. Adv. Synth. Catal. 2018, 360, 2493–2502. [Google Scholar] [CrossRef]
- Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A.S.K. An Industrial Perspective on Counter Anions in Gold Catalysis: On Alternative Counter Anions. Adv. Synth. Catal. 2018, 360, 3949–3959. [Google Scholar] [CrossRef]
- Baron, M.; Biffis, A. Gold(I) Complexes in Ionic Liquids: An Efficient Catalytic System for the C-H Functionalization of Arenes and Heteroarenes under Mild Conditions. Eur. J. Org. Chem. 2019, 2019, 3687–3693. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef]
- Tolman, C.A. The 16 and 18 electron rule in organometallic chemistry and homogeneous catalysis. Chem. Soc. Rev. 1972, 1, 337–353. [Google Scholar] [CrossRef]
- Lu, Z.; Han, J.; Okoromoba, O.E.; Shimizu, N.; Amii, H.; Tormena, C.F.; Hammond, G.B.; Xu, B. Predicting Counterion Effects Using a Gold Affinity Index and a Hydrogen Bonding Basicity Index. Org. Lett. 2017, 19, 5848–5851. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Belanzoni, P. Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment. ACS Catal. 2015, 5, 803–814. [Google Scholar] [CrossRef]
- Leung, C.H.; Baron, M.; Biffis, A. Gold-Catalyzed Intermolecular Alkyne Hydrofunctionalizations—Mechanistic Insights. Catalysts 2020, 10, 1210. [Google Scholar] [CrossRef]
- Kütt, A.; Rodima, T.; Saame, J.; Raamat, E.; Mäemets, V.; Kaljurand, I.; Koppel, I.A.; Garlyauskayte, R.Y.; Yagupolskii, Y.L.; Yagupolskii, L.M.; et al. Equilibrium Acidities of Superacids. J. Org. Chem. 2011, 76, 391–395. [Google Scholar] [CrossRef]
- Ghosh, T.; Chatterjee, J.; Bhakta, S. Gold-Catalyzed Hydroarylation Reactions: A Comprehensive Overview. Org. Biomol. Chem. 2022, 20, 7151–7187. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A. The Solubility Parameters of Ionic Liquids. Int. J. Mol. Sci. 2010, 11, 1973–1990. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, S.B. The Hildebrand Solubility Parameters, Cohesive Energy Densities and Internal Energies of 1-Alkyl-3-Methylimidazolium-Based Room Temperature Ionic Liquids. Chem. Commun. 2005, 23, 3469–3471. [Google Scholar] [CrossRef]
- Okoturo, O.O.; VanderNoot, T.J. Temperature Dependence of Viscosity for Room Temperature Ionic Liquids. J. Electroanal. Chem. 2004, 568, 167–181. [Google Scholar] [CrossRef]
- Russina, O.; Lo Celso, F.; Plechkova, N.; Jafta, C.J.; Appetecchi, G.B.; Triolo, A. Mesoscopic Organization in Ionic Liquids. Top. Curr. Chem. 2017, 375, 58. [Google Scholar] [CrossRef]
- Jiang, H.J.; Atkin, R.; Warr, G.G. Nanostructured Ionic Liquids and Their Solutions: Recent Advances and Emerging Challenges. Curr. Opin. Green Sustain. Chem. 2018, 12, 27–32. [Google Scholar] [CrossRef]
- Marullo, S.; D’Anna, F.; Rizzo, C.; Billeci, F. Ionic Liquids: “Normal” Solvents or Nanostructured Fluids? Org. Biomol. Chem. 2021, 19, 2076–2095. [Google Scholar] [CrossRef]
- Patra, S.; Samanta, A. Microheterogeneity of Some Imidazolium Ionic Liquids as Revealed by Fluorescence Correlation Spectroscopy and Lifetime Studies. J. Phys. Chem. B 2012, 116, 12275–12283. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, D.; Welsh, I.D.; Matthewman, E.L.; Jun, S.P.; Mckeever-Willis, M.; Gritcan, I.; Greaves, T.L.; Weber, C.C. Structural Investigations of Molecular Solutes within Nanostructured Ionic Liquids. Phys. Chem. Chem. Phys. 2020, 22, 11593–11608. [Google Scholar] [CrossRef] [PubMed]
- Matthewman, E.L.; Kapila, B.; Grant, M.L.; Weber, C.C. The Amphiphilic Nanostructure of Ionic Liquids Affects the Dehydration of Alcohols. Chem. Commun. 2022, 58, 13572–13575. [Google Scholar] [CrossRef]
- Wang, Z.J.; Benitez, D.; Tkatchouk, E.; Goddard III, W.A.; Toste, F.D. Mechanistic Study of Gold(I)-Catalyzed Intermolecular Hydroamination of Allenes. J. Am. Chem. Soc. 2010, 132, 13064–13071. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
General mechanism for gold-catalyzed alkyne hydrofunctionalization reactions.

Scheme 1.
Ionic liquids employed in the anion effect evaluation.

Scheme 2.
The hydroarylation of ethyl propiolate with mesitylene investigated in the anion effect evaluation.
Scheme 2.
The hydroarylation of ethyl propiolate with mesitylene investigated in the anion effect evaluation.

Scheme 3.
ILs employed in the cation effect evaluation.

Scheme 4.
Au(I) complexes employed as precatalysts in this study.

Figure 2.
Kinetic profiles of hydroarylation reactions obtained with Au(I) precatalysts 1 (yellow), 2 (blue), 3 (light blue), 4 (gray), and 5 (orange).
Figure 2.
Kinetic profiles of hydroarylation reactions obtained with Au(I) precatalysts 1 (yellow), 2 (blue), 3 (light blue), 4 (gray), and 5 (orange).

Figure 3.
TEP vs. alkyne conversion plot of hydroarylation reactions catalyzed by different Au(I) complexes.
Figure 3.
TEP vs. alkyne conversion plot of hydroarylation reactions catalyzed by different Au(I) complexes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Properties of the employed weakly coordinating counteranions.
| Anion | Gold Affinity Index (GAI) a | Hydrogen Bond Basicity (HBI) a | Brønsted Basicity b |
|---|---|---|---|
| SbF6− | 0 | 2.8 | - |
| PF6− | n.a. | 3.2 | - |
| BF4− | 0.5 | 5.2 | −10.3 |
| NTf2− | 2.9 | 1.0 | −11.9 |
| OTf− | 2.4 | 3.4 | −11.4 |
Table 2.
Alkyne conversion in different ILs.
| Ionic Liquid | Time (h) | Alkyne Conversion (%) |
|---|---|---|
| [BMIM][NTf2] | 1 | 67 |
| 3 | 96 | |
| 23 | 100 | |
| [BMIM][OTf] | 1 | 0 |
| 3 | 0 | |
| [BMIM][BF4] | 1 | 0 |
| 3 | <1 | |
| [BMIM][PF6] | 1 | 0 |
| 3 | 1 | |
| 23 | 4 |
Reaction conditions: 2.5 mmol ethyl propiolate, 2.5 mmol mesitylene, 0.75 mL [BMIM][X], 12.5 μmol [AuCl(PPh3)], 12.5 μmol AgSbF6, 40 °C.
Table 3.
Solubility parameters and viscosity of the studied ILs.
| Ionic Liquid | Hildebrand Solubility Parameter, δ (MPa1/2) | Viscosity (mPa·s) |
|---|---|---|
| [BMIM][BF4] | 31.6 | 92 |
| [BMIM][PF6] | 29.8 | 173 |
| [BMIM][NTf2] | 26.7 | 47 |
| [BMIM][OTf] | 25.4 | 74 |
Table 4.
Alkyne conversion in ILs with cations of different hydrophilicity.
| Ionic Liquid | Time (h) | Alkyne Conversion (%) | Hildebrand Solubility Parameter, δ (MPa1/2) |
|---|---|---|---|
| [BMIM][NTf2] | 1 | 67 | 26.7 |
| 3 | 96 | ||
| 23 | 100 | ||
| [OMIM][NTf2] | 1 | 68 | 25.0 |
| 3 | 100 | ||
| [DMIM][NTf2] | 1 | 75 | n.a. |
| 3 | 97 | ||
| 23 | 100 |
Reaction conditions: 2.5 mmol ethyl propiolate, 2.5 mmol mesitylene, 0.75 mL [BMIM][X], 12.5 μmol [AuCl(PPh3)], 12.5 μmol AgSbF6, 40 °C.
Table 5.
Chemoselectivity of various precatalysts in the hydroarylation of ethyl propiolate with mesitylene at 100% alkyne conversion.
Table 5.
Chemoselectivity of various precatalysts in the hydroarylation of ethyl propiolate with mesitylene at 100% alkyne conversion.
 | |||
| Catalyst | 6 | 7 | 8 |
| 1 | 43 | 20 | 37 |
| 2 | 57 | 22 | 21 |
| 3 | 54 | 27 | 19 |
| 4 a | 58 | 22 | 20 |
 | 62 | 22 | 16 |
| 9 | |||
Reaction conditions: 2.5 mmol ethyl propiolate, 2.5 mmol mesitylene, 0.75 mL [BMIM][NTf2], 12.5 μmol gold complex, 12.5 μmol AgSbF6, 40 °C, 22 h. a Selectivity at 80% alkyne conversion.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bonfante, S.; Bax, P.; Baron, M.; Biffis, A. Gold(I)-Catalyzed Direct Alkyne Hydroarylation in Ionic Liquids: Mechanistic Insights. Catalysts 2023, 13, 822. https://doi.org/10.3390/catal13050822
AMA Style
Bonfante S, Bax P, Baron M, Biffis A. Gold(I)-Catalyzed Direct Alkyne Hydroarylation in Ionic Liquids: Mechanistic Insights. Catalysts. 2023; 13(5):822. https://doi.org/10.3390/catal13050822
Chicago/Turabian StyleBonfante, Sara, Pietro Bax, Marco Baron, and Andrea Biffis. 2023. "Gold(I)-Catalyzed Direct Alkyne Hydroarylation in Ionic Liquids: Mechanistic Insights" Catalysts 13, no. 5: 822. https://doi.org/10.3390/catal13050822
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.