
Enhancing the Activity of Cu-MOR by Water for Oxidation of Methane to Methanol

1
Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
2
State Key Laboratory of Fine Chemicals, The Pennsylvania State University-Dalian University of Technology (PSU-DUT) Joint Center for Energy Research, School of Chemical Engineering, Dalian University of Technology, Dalian 116023, China
3
School of chemical engineering, University of Chinese Academy of Sciences, Beijing 100039, China
*
Authors to whom correspondence should be addressed.
Catalysts 2023, 13(7), 1066; https://doi.org/10.3390/catal13071066
Submission received: 27 May 2023
/
Revised: 27 June 2023
/
Accepted: 1 July 2023
/
Published: 3 July 2023
(This article belongs to the Special Issue Heterogeneous Catalysis for Clean Energy Production and Carbon Dioxide Utilization)
Abstract
:As clean energy, methane has huge reserves and great development potential in the future. Copper zeolites are efficient in the oxidation of methane to methanol. Water has been confirmed as a source of oxygen to regenerate the copper-zeolite active sites to enable selective anaerobic oxidation of methane to methanol. In this work, we report that the methanol yield increased from 36 μmol/g (Cu-MOR1) to 92 μmol/g (Cu-MOR1-water) as a result of water enhancing the activity of copper ion-exchange mordenite catalyst. We show for the first time that water could convert inactive copper species into active copper species during catalyst activation. A combination of the XPS, FTIR, and NMR results indicates that water dissociates and then converts ZCuIIZ into ZCuII(OH) (where Z indicates framework O (Ofw) bonded to one isolated Al in a framework T-site, i.e., 1Al) and simultaneously produces a Brönsted acid site during catalyst activation. This finding can be used to tune the state of copper species and design highly active copper-zeolite catalysts for methane oxidation to methanol.

1. Introduction
Methane is the main component of natural gas and natural gas hydrate, being clean and easy to distribute, and the reserves of natural gas hydrate are huge [1,2]. In recent years, the development of shale gas technology has made methane (CH4) inexpensive and accessible.
Currently, the mainly utilization of methane is combustion for power generation and heating [3]. There are limited industrial routes for converting methane into fuels and chemicals. Methane could be converted to chemicals via both direct and indirect routes. To date, methane is mainly converted to chemicals and fuels through indirect processes. For the indirect process, methane is first converted to syngas (H2 and CO) through a steam reforming process and then the syngas is converted to chemicals such as methanol and gasoline. The steam reforming process operates at high temperatures (700–1000 °C) and pressures (15–40 atm) and is an energy-intensive process, making direct conversion an attractive alternative [3]. The direct conversion of methane to chemicals has been a very active topic in the past few decades. Studies on the direct conversion of methane to chemicals have mainly focused on three processes: oxidative coupling of methane to C2+ carbon species, nonoxidative conversion of methane to aromatics, and partial oxidation of methane to methanol [4,5,6,7,8,9,10]. Oxidative coupling of methane usually requires activation of methane at high temperatures. However, high temperature favors the complete oxidation of methane and C2 (C2H4 and C2H6). The process currently faces two important challenges. The first is catalyst selectivity and the second is catalyst deactivation. To date, despite extensive research, no specific catalyst has fully met the practical industrial and economic requirements. The advantage of the nonoxidative conversion of methane to aromatics is that it prevents irreversible overoxidation, which leads to thermodynamically stable undesired products, such as CO2 and H2O. The nonoxidative conversion of methane to aromatics faces similar challenges to the oxidative coupling of methane. The reaction process requires high temperatures, but the catalyst will deactivate rapidly at high temperatures. Unlike the oxidative coupling of the methane process and the anaerobic conversion of the methane to aromatics process, the methane oxidation to methanol process does not a require high temperature. The peroxidation of the product is inhibited at low temperatures, and the product selectivity is higher than that of the high-temperature process.
The direct, low-temperature conversion of methane to transportable liquid methanol is an ideal alternative pathway for the transportation and utilization of methane. Fe, Ni ion-exchanged zeolites have been shown to convert methane to methanol, but are less active than copper ion-exchanged zeolites [11,12]. Copper ion-exchanged zeolites have shown excellent performance in the selective oxidation of methane to methanol [13], at relatively mild temperatures (typically 125–200 °C) using O2 as an oxygen source.
There are two types of CuII ions in the copper ion-exchange zeolite catalyst: ZCuII(OH) and ZCuIIZ (where Z indicates framework O (Ofw) bonded to one isolated Al in a framework T-site, i.e., 1Al), but only ZCuII(OH) can be converted into active sites [14]. Therefore, maximizing the amount of ZCuII(OH) by adjusting the ion exchange pH (5.2–5.7) is essential to the synthesis of highly-active catalysts [14,15]. Several copper-oxygen species have been proposed as active sites for the oxidation of methane to methanol, including ZCuII(OH) [16], ZCu2O2Z [13], ZCu2OZ [17,18], ZCu3O3Z [14], and ZCu2AlO3Z [19]. Methane is first activated by forming a complex intermediate with an active CuII species, followed by a reaction of the intermediate with a water molecule that facilitates methanol formation and desorption [13]. In addition, Bokhoven et al. [20] found that water can be used as an oxygen source to regenerate active sites under anaerobic conditions. Koishybay et al. [21] demonstrate that water is the main source of oxygen present in the methanol produced in the partial oxidation of methane to methanol over Cu-SSZ-13 in a continuous flow reactor.
Water has been confirmed to be involved in the formation of methanol from methane oxidation, but the effect of water on the generation of active sites has not been reported. In this work, we investigated the role of water during the generation of active sites and its effect on the catalyst activity for methane oxidation to methanol.
2. Results and Discussion
The copper-exchanged mordenite catalysts were synthesized via aqueous ion exchange of a parent Na-MOR (SiO2/Al2O3 = 8.73 and 15.5). The copper loading was 5.3%, corresponding to 834 μmol Cu/g on the sample having SiO2/Al2O3 = 8.73. The sample with SiO2/Al2O3 = 15.5 has a copper loading of 3.39%, corresponding to 533 μmol Cu/g. Detailed bulk catalyst preparation and evaluation are given in the Section 3. The samples are named Cu-MOR and Cu-MOR-water according to the pretreatment conditions. Cu-MOR refers to samples that were not subjected to additional treatment after calcination in the air at 550 °C for 6 h. Cu-MOR-water refers to samples that adsorbed water at room temperature after the initial calcination in air at 550 °C for 6 h; the samples that adsorbed excess water were further dried at 110 °C for 24 h. Prior to evaluation, all samples were recalcined in dry air at 450 °C for 2 h before cooling to 200 °C for the reaction.
Table 1 shows the effect of adsorbed water after the initial calcination on the activity of Cu-MOR samples. The Cu-MOR1 catalyst catalyzed the oxidation of methane with a methanol yield of 36 μmol/g (Table 1, entry 1), but the Cu-MOR1-water catalyst gave a methanol yield of 92 μmol/g (Table 1, entry 2), corresponding to an efficiency of 0.17 mol CH3OH/mol Cu. It is noted from the literature that catalysts prepared using copper acetate typically resulted in much higher methanol production efficiency than using copper nitrate [13,14,20]. In this work, Cu(NO3)2 is used. The efficiency reached 0.17 mol CH3OH/mol Cu which is similar to the previous work. The methanol yields of Cu-MOR1-water reacted at 200 °C for 8 h and 4 h were 92 μmol/g and 90 μmol/g, respectively (Table 1, entry 3,4). The methanol yields of Cu-MOR1-water reacted at 150 and 175 °C were 26 μmol/g and 46 μmol/g, respectively (Table 1, entry 5,6). The reaction temperature strongly affects the catalyst activity, probably due to the fact that methane is very inert, so the reaction requires a relatively high temperature. To further confirm this result, another Cu-MOR2 catalyst with SiO2/Al2O3 = 8.73 and Cu/Al = 0.38 was prepared. The methanol yield of Cu-MOR2 is 50 μmol/g (Table 1, entry 7), but the Cu-MOR2-water gave a methanol yield of 128 μmol/g (Table 1, entry 8). In addition, the gas is collected using a gas bag after the reaction and analyzed by a GC with a TCD detector. However, none of the samples detected CO and CO2, which may be due to their low levels below the detection limit.
The states of copper species in Cu-MOR and Cu-MOR-water were characterized by H2-TPR and TEM. H2-TPR curves typically show three H2 consumption signatures in the temperature range from 100 to 550 °C (Figure S1). The low-temperature peaks at 193 and 199 °C represent the reduction in CuII to CuI [22,23]. The peaks at 265 and 251 °C represent the reduction in CuO [24,25,26]. The high-temperature peaks at 316 and 438 °C represent the reduction in CuI to Cu0 [22,23]. The H2-TPR results are consistent with the TEM results. TEM images showed dense nanoparticles on the outer surface of mordenite (Figure S2), and the size of the nanoparticles was below 3 nm. The TEM and H2-TPR results indicated the presence of various copper species, including CuO and Cu ions in Cu-MOR and Cu-MOR-water.
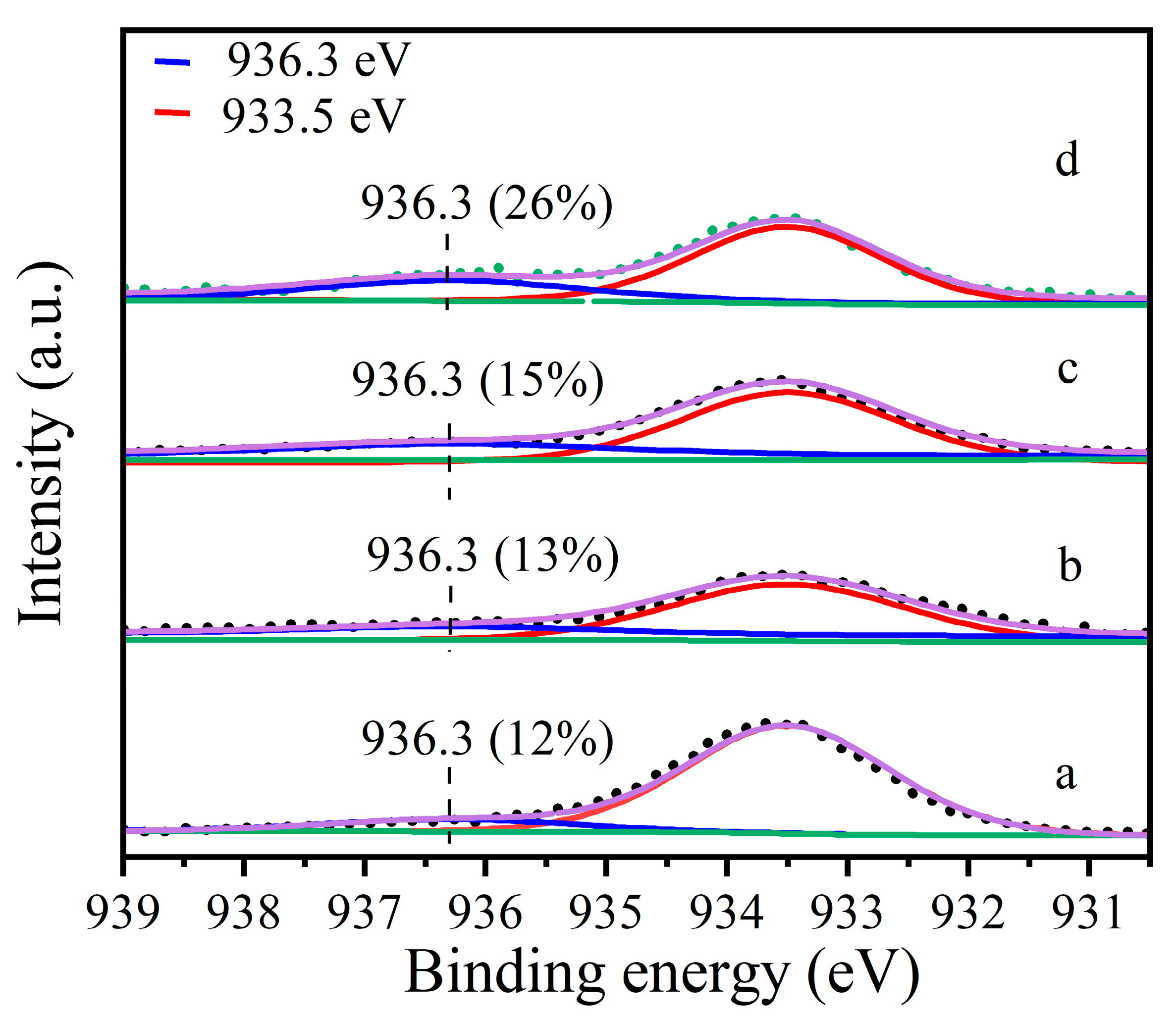
To understand the role of water in the treatment, the sample Cu-MOR1 (SiO2/Al2O3 = 15.5) was further characterized at various conditions corresponding to the evaluations. XPS was used to compare the Cu oxidation states at various treatment conditions. The peak at 933.5 eV (Figure 1) is assigned to CuII, mostly existing as ZCuIIZ or CuO [25,27,28,29]. The peak at 936.3 eV (Figure 1) is assigned to Cu(II) species bound to extra-framework oxygen species active in the methane to methanol reaction [30], including mono(μ-oxo) dicopper, bis(μ-oxo) dicopper, tricopper species, and ZCuII(OH). The ratio of different valence species can be quantified by XPS peak fitting [28,31,32,33]. The proportion of copper species to total copper species was calculated based on the ratio of the peak areas. The fraction of 936.3 eV species evaluated by the fitting of Figure 1a is 12% in Cu-MOR1. After calcination at 450 °C for 2 h, the proportion in Cu-MOR1 increased from 12% to 15% (Figure 1c). While the fraction of 936.3 eV species evaluated by the fitting of Figure 1b is 13% in Cu-MOR1-water. After calcination at 450 °C for 2 h, the proportion in Cu-MOR1-water increased from 13% to 26% (Figure 1d). After calcination at 450 °C for 2 h, the active copper content of Cu-MOR1-water (Figure 1d) is 11% higher than that of Cu-MOR1 (Figure 1c). This is consistent with the reaction results that Cu-MOR1-water is more active than Cu-MOR1 after calcination at 450 °C for 2 h. If only ZCuII(OH) could be converted into active sites, the proportion of the 936.3 eV species should be consistent before and after calcined at 450 °C for 2 h. After calcination at 450 °C for 2 h, the copper species at 936.3 eV in Cu-MOR1-water increased from 13% (Figure 1b) to 26% (Figure 1d), suggesting that the water has converted the inactive copper species (ZCuIIZ or CuO) into active copper species (CuII species bound to extra-framework oxygen species) during the activation process.
In addition, the copper loading was 3.39%, corresponding to 533 μmol Cu/g on the Cu-MOR1. According to the XPS result, the active copper content of Cu-MOR1-water (Figure 1d) is 11% higher than that of Cu-MOR1 (Figure 1c) after calcination at 450 °C for 2 h, which means that the active copper content of Cu-MOR1-water is 59 μmol/g higher than that of Cu-MOR1. The methanol yield of Cu-MOR1-water is 56 μmol/g higher than Cu-MOR1. It means that the addition of 59 μmol/g of active copper species produced 56 μmol/g of methanol, equivalent to each active copper species converting one molecule of methane to one molecule of methanol. The Cu efficiency was larger than the maximum Cu efficiency when the active site was ZCu2O2Z [13], ZCu2OZ [17,18], ZCu3O3Z [14], and ZCu2AlO3Z [19]. However, the Cu efficiency is the same as the theoretical copper efficiency when the active site is ZCuII(OH), suggesting that the active site is ZCuII(OH).
To further confirm this result, another Cu-MOR catalyst with SiO2/Al2O3 = 8.73 and Cu/Al = 0.38 was also characterized by XPS. The fraction of 936.3 eV species, evaluated by the fitting of Figure S3a, is 24% in Cu-MOR2. After calcination at 450 °C for 2 h, the proportion in Cu-MOR2 increased to 32% (Figure S3c). While the fraction of 936.3 eV species, evaluated by the fitting of Figure S3b is 27% in Cu-MOR2-water. After calcination at 450 °C for 2 h, the proportion in Cu-MOR2-water increased to 44% (Figure S3d). Which is much larger than the Cu-MOR2. This result is in agreement with the Cu-MOR1 catalyst, which has a SiO2/Al2O3 = 15.5.
We hypothesize that the increase in active copper species was due to the hydrolysis of hydrated CuII ions. During activation ZCuII(H2O)nZ gradual dehydration leads to water dissociation to give ZCuII(OH) and Brönsted acid site (ZH) [34,35,36] as follows:
2ZCuII(H2O)n → ZCuII(OH) + ZH + (n − 1)H2O
FTIR has been used to monitor the change of the Brönsted acid site. The results of FTIR characterization are shown in Figure 2. As shown in Figure 2, the characteristic peaks at 3608 and 3594 cm−1 corresponded to zeolite Brönsted acid (ZH) [14]. The peak at 3608 cm−1 was assigned to the vibration of the O-H bond which is associated with the Brönsted acid located in the main pore of mordenite; the peak at 3594 cm−1 was assigned to the vibration of the O-H bond, which is associated with the Brönsted acid located in the side cage of the mordenite [14]. The characteristic peak at 3650 cm−1 corresponds to CuII(OH) [36,37]. Compared with Cu-MOR1, the peaks at 3650, 3608, and 3594 cm−1 of Cu-MOR1-water were simultaneously significantly increased. FTIR results demonstrate that during the activation process, the water dissociation converts hydrated CuII ions to CuII(OH) and simultaneously produces the Brönsted acid site, Equation (1). In addition, the FTIR result is consistent with the XPS and reaction results, the amount of active site ZCuII(OH) in Cu-MOR1-water was greater compared with Cu-MOR1 (Figure 1) after calcination at 450 °C for 2 h.
To further confirm this result, another Cu-MOR catalyst with SiO2/Al2O3 = 8.73 and Cu/Al = 0.38 was also characterized by FTIR (Figure S4). Compared with Cu-MOR2, the peaks at 3650, 3608, and 3594 cm−1 of Cu-MOR2-water were simultaneously significantly increased. The trend is similar to that of Cu-MOR1 (SiO2/Al2O3 = 15.5).
The formation of Brönsted acid sites was further confirmed by pyridine-IR analysis. Figure S5 shows three peaks at 1544, 1450, and 1490 cm−1, which are assigned the adsorption of pyridine on Brönsted acid sites, Lewis acid sites, and the overlapping of pyridine adsorption on Lewis and Brönsted acid sites [38], respectively. After calcination at 450 °C for 2 h, the amount of Brönsted acid sites in Cu-MOR1-water was higher than that of Cu-MOR1 (Figure S5), indicating that the water promoted the formation of more Brönsted acid sites.
According to Xue et al., the removal of framework aluminum in the main channels (12-MR) would result in a decrease in the intensity of the band at 3610 cm−1 [39]. Meanwhile, the removal of the frame aluminum creates extra-framework aluminum. NMR is an efficient technique for distinguishing extra-framework aluminum from framework aluminum. Figure 3 shows 27Al NMR spectra of Cu-MOR1 and Cu-MOR1-water after calcination at 450 °C for 2 h. All spectra contain only one peak at 55 ppm, which is due to tetrahedral coordinated Al sites in the framework. However, no extra framework aluminum associated with the peak around 0 ppm is visible [40]. Based on the NMR results, we could exclude the possibility of zeolite dealumination. The NMR results further confirm the increase in the Brönsted acid peak (Figure 2) is due to the water dissociation.
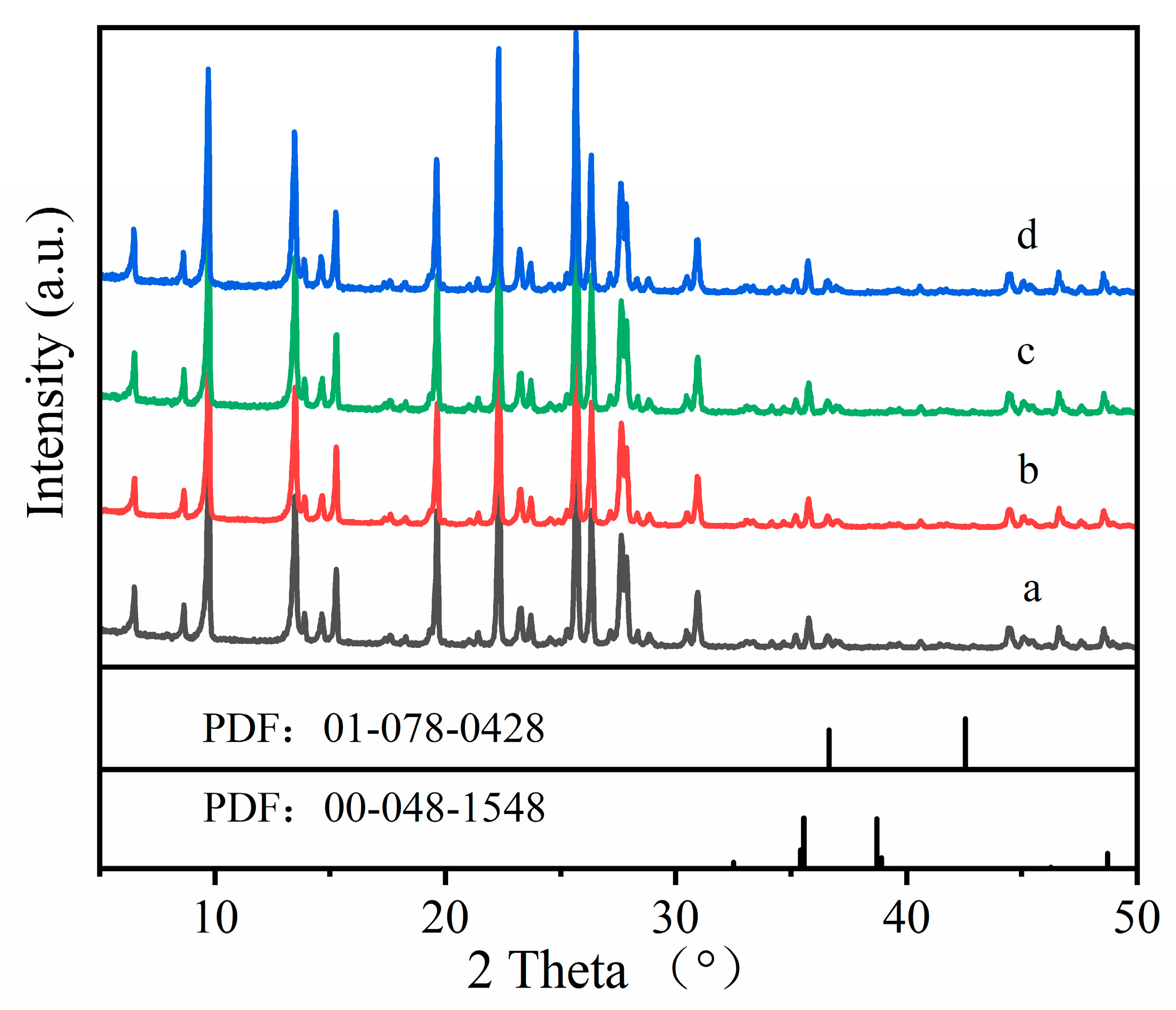
The 27Al NMR results are consistent with the XRD results. Figure 4 shows the XRD patterns of Cu-MOR1 and Cu-MOR1-water. The diffraction peaks of Cu-MOR1 and Cu-MOR1-water before and after calcination at 450 °C for 2 h were identical, indicating that calcination at 450 °C for 2 h did not significantly change the structure of the samples. This feature is expected because the water vapor dealumination of zeolites generally requires high temperatures [41]. There are two kinds of CuO phases (Figure 4): monoclinic CuO (PDF: 00-048-1548) and cubic CuO (PDF: 01-078-0428). Compared with standard XRD of CuO, the Cu-MOR1-water and Cu-MOR1 has no characteristic peak associated with CuO, suggesting that the copper species are highly dispersed in the Cu-MOR1 and Cu-MOR1-water after calcination at 450 °C for 2 h and did not form large CuO nanoparticles.
Based on the above experimental results and literature, we proposed the possible mechanism shown in Figure 5. Based on the literature, only ZCuII(OH) has been identified as active copper species, and it was recently identified as an active site for methane oxidation to methanol [16]. All the other active sites including ZCu2O2Z [13], ZCu2OZ [17,18], ZCu3O3Z [14], and ZCu2AlO3Z [19] were original from ZCuII(OH).
The amount of ZCuII(OH) in copper-zeolite determined the activity for oxidation of methane to methanol. Maximizing the amount of ZCuII(OH) by adjusting the ion exchange pH (5.2–5.7) during catalyst synthesis could maximize the catalyst activity. In summary, the catalyst synthesis condition determined the amount of ZCuII(OH) in the catalyst. Herein, we report a new ZCuII(OH) formation road. The ZCuII(H2O)nZ gradual dehydration leads to water dissociation to give ZCuII(OH) and Brönsted acid site (ZH) during the activation process [34,35,36], then ZCuII(OH) could convert methane to methanol. Water converts inactive ZCuIIZ into active site ZCuII(OH). This process was different from the work of Bokhoven et al. [20], who found that water can be used as an oxygen source to regenerate active sites under anaerobic conditions, the mechanism of it is water oxidation of two adjacent ZCuI ions to regeneration ZCuOCuZ active site. Following the work of Bokhoven et al. [20], Koishybay et al. reported the oxidation of methane to methanol using water as the sole oxidant in the absence of molecular oxygen [21]. In this work, the increased active sites come from ZcuIIZ and water. Water converts ZcuIIZ to active sites, thereby increasing the activity of the catalyst for methane oxidation to methanol. In addition, Figure 6 also shows that Cu-MOR1-water is a stable methane oxidation catalyst, and the yield of methanol remains unchanged for three consecutive utilizations.
3. Materials and Methods
3.1. Materials
NaOH, NaAlO2, and copper nitrate trihydrate (Cu(NO3)2·3H2O, 99.0–102.0%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Methanol and n-propanol were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. CH4 was purchased from Dalian special gases Co., Ltd. (Dalian, China). Silica sol was purchased from Dalian Snowchemical S&T Co., Ltd.(Dalian, China). Na-Mordenite (SiO2/Al2O3 = 15.5) was purchased from Nankai University Catalyst Co., Ltd. (Tianjin, China). Na-mordenite (SiO2/Al2O3 = 8.73) was synthesized from an aluminosilicate mixture of composition 2.25 Na2O:1 Al2O3:10 SiO2:219 H20 and digested in a stainless steel autoclave reactor at 160 °C for 36 h. The product was isolated by centrifugation, washed three times with deionized water, and dried at 110 °C for 24 h.
3.2. Catalyst Preparation
Copper-exchanged mordenite catalysts were prepared by the aqueous ion-exchange method. Typically, 10 g Na-MOR (SiO2/Al2O3 = 15.5) was ion-exchanged 3 times with 500 mL of 0.05 M Cu(NO3)2 solution at 50 °C. After each ion exchange process, the sample was filtered, washed with plenty of deionized water, and then calcined in air at 550 °C for 6 h. The samples were named Cu-MOR and Cu-MOR-water according to the drying conditions. Cu-MOR indicates that the samples were not subjected to any other treatment after calcination in the air at 550 °C for 6 h. Cu-MOR-water means that after calcined in air at 550 °C for 6h, the sample adsorbed excess water at room temperature, and then dried at 110 °C for 24 h.
3.3. Characterization of Catalysts
Phase identification was realized by XRD patterns, which were carried out on a PANalytical powder diffractometer (PANalytical B.V., Almelo, The Netherland) with Cu-Kα radiation (λ = 1.5418 Å). The XRD patterns were recorded from 5° to 40° with a scan rate of 15 °/min.
The SiO2/Al2O3 molar ratio was analyzed using X-ray fluorescence spectroscopy (PANalytical B.V., Almelo, The Netherland) on a PANalytical instrument. Cu content was determined by ICP-OES (Thermo Fisher Scientific, Waltham, MA, USA).
TEM analysis was performed on a Titan Themis ETEM G3 microscope (Thermo Fisher Scientific, Waltham, MA, USA).
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was used to study the difference between Cu-MOR and Cu-MOR-water after activation. The scanning range was 4000–650 cm−1 with a resolution of 4 cm−1 and a total of 64 scans/spectrum using MCT detector. The DRIFTS spectrum of Cu-MOR was collected after calcination at 450 °C for 2 h, followed by the Cu-MOR sample, it was cooled to room temperature and adsorption excess water before being dried at 110 °C for 1 h in situ to generate a Cu-MOR-water sample. Finally, the DRIFTS spectrum of Cu-MOR-water was collected after calcination at 450 °C for 2 h.
In situ pyridine-IR spectra were collected on a Nicolet™ iS50 FTIR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Firstly, Cu-MOR was calcined at 450 °C for 2 h, then the Cu-MOR sample was cooled to room temperature and excess pyridine was adsorbed under vacuum conditions. Finally, the physically adsorbed pyridine was desorbed under vacuum at 150 °C for 30 min before collecting the spectra. After collecting the spectrum of Cu-MOR, the Cu-MOR adsorbed excess water at room temperature before drying at 110 °C for 1 h in situ to generate a Cu-MOR-water sample. The pyridine-IR characterization process of Cu-MOR-water is the same as that of Cu-MOR.
Temperature-programmed reduction (TPR) profiles were obtained using a micromeritics autochem II instrument (Micromeritics, Norcross, GA, USA) fitted with a TCD detector. A total of 100 mg of the sample was heated to 650 °C (10 °C/min) in a flow of 10% H2/Ar mixture.
27Al NMR spectra of Cu-MOR activated Cu-MOR and activated Cu-MOR-water were recorded on a Bruker AVANCE NEO WB 600 M spectrometer (Buruker corporation, Billerica, MA, USA). The Cu-MOR and Cu-MOR-water were activated at 450 °C for 2 h in a flow of air to prepare samples for NMR analysis. In all cases, spectra were taken after sample equilibration with ambient water vapor to relax the lattice strain and create a more symmetric environment around the Al nucleus.
The copper species in samples were characterized by X-ray photoelectron spectroscopy (XPS) using a Thermofisher Escalab 250 Xi+ spectrometer (Miami, OK, USA) equipped with a monochromatic Al Kα X-ray source. The binding energy (BE) was calibrated by setting the measured BE of C1s to 284.8 eV. The Cu-MOR and Cu-MOR-water were first activated at 450 °C for 2 h and then transferred to a glove box to prepare samples for XPS analysis without being exposed to air.
3.4. Testing of Activity for Selective Oxidation of Methane
The activity of the sample was tested for oxidation of methane to methanol. The process of methane oxidation to methanol consists of 3 steps: (1) catalyst activation, (2) methane reaction, and (3) product extraction and quantification. The sample was activated in a quartz tube (8 mm inner diameter). The temperature of the sample was controlled by a thermocouple mounted in the center of the catalyst bed. Trace moisture in the air was removed using a dehydrator.
Typical experiment process:
- Step (1)
- A total of 0.2 g Cu-MOR was heated from ambient temperature to 450 °C at 5 °C/min and activated at 450 °C for 2 h in a flow of air (20 mL/min). The activated catalyst was cooled to room temperature at 5 °C/min in dry air.
- Step (2)
- The activated catalyst was transferred to an autoclave. The autoclave was first purged several times with CH4 and charged with 1 MPa CH4, and then heated to 200 °C for 12 h.
- Step (3)
- After the reaction, the autoclave was cooled to room temperature and excess CH4 was vented. The gas was collected using a gas bag and analyzed by a GC with a TCD detector. The catalyst was then extracted with a solution of 5% H2O/CH3CN for 2 h. Internal standard n-propanol was added to the extraction solution and the product was quantified by gas chromatography (Agilent Technologies, Santa Clara, CA, USA) on an Agilent 7890A (HP-5 column, 30 m, 0.32 mm inner diameter). The yield of methanol was calculated based on the total amount of methanol and dimethyl ether, where each dimethyl ether was calculated as two methanol.
4. Conclusions
In summary, we investigated the effect of water on the activity of Cu-MOR for the oxidation of methane to methanol. Compared with Cu-MOR1, the methanol yield of Cu-MOR1-water increased from 36 μmol/g to 92 μmol/g. The reaction results show that water can strongly affect the activity of Cu-MOR catalysts. The XPS and FTIR results indicated that the active site is ZCuII(OH). The XPS and FTIR results indicated that during the activation process, water can dissociate and then convert ZCuIIZ ions into ZCuII(OH). NMR and XRD results ruled out the dealumination of the zeolite during activation. This finding can be used to tune the state of copper species and design highly active copper-zeolite catalysts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13071066/s1, Figure S1: H2-TPR profiles: (a) Cu-MOR1; (b) Cu-MOR1-water; Figure S2: TEM images: (a) Cu-MOR1; (b) Cu-MOR1-water; Figure S3: Cu2p3/2 photoelectron spectra of (a) Cu-MOR2, (b) Cu-MOR2-water, (c) Cu-MOR2 after calcined at 450 °C for 2 h, and (d) Cu-MOR2-water after calcined at 450 °C for 2 h; Figure S4: In-situ diffuse reflectance FTIR spectra: (a) Cu-MOR2 after calcination at 450 °C; (b) Cu-MOR2-water after calcination at 450 °C; Figure S5: In-situ pyridine-IR spectra: (a) Cu-MOR1; (b) Cu-MOR1-water.
Author Contributions
X.G. (Xinwen Guo) and Z.Z.: Conceptualization, methodology, writing—review and editing, formal analysis, funding acquisition. X.G. (Xi’an Guan): methodology, validation, formal analysis, investigation, writing—original draft preparation. Y.W., X.L. and H.D.: writing—review and editing, formal analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (21932005, 22172164, and 21721004).
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dickens, G.R. The Potential Volume of Oceanic Methane Hydrates with Variable External Conditions. Org. Geochem. 2001, 32, 1179–1193. [Google Scholar] [CrossRef]
- Chong, Z.R.; Yang, S.H.B.; Babu, P.; Linga, P.; Li, X. Sen Review of Natural Gas Hydrates as an Energy Resource: Prospects and Challenges. Appl. Energy 2016, 162, 1633–1652. [Google Scholar] [CrossRef]
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Albarracín-Suazo, S.; Pagán-Torres, Y.; Nikolla, E. Advances in Methane Conversion Processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem.—Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Guan, N.; Li, L. Methane Activation and Utilization: Current Status and Future Challenges. Energy Technol. 2020, 8, 1900826. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szécsényi, À.; Hensen, E.J.M.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Zhao, G.; Drewery, M.; Mackie, J.; Oliver, T.; Kennedy, E.M.; Stockenhuber, M. The Catalyzed Conversion of Methane to Value-Added Products. Energy Technol. 2020, 8, 1900665. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment. Angew. Chem.—Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Tabor, E.; Dedecek, J.; Mlekodaj, K.; Sobalik, Z.; Andrikopoulos, P.C.; Sklenak, S. Dioxygen Dissociation over Man-Made System at Room Temperature to Form the Active α-Oxygen for Methane Oxidation. Sci. Adv. 2020, 6, eaaz9776. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Huang, W.; Nguyen, L.; Yu, Y.; Zhang, S.; Li, Y.; Frenkel, A.I.; Tao, F. Conversion of Methane to Methanol with a Bent Mono(μ-Oxo)Dinickel Anchored on the Internal Surfaces of Micropores. Langmuir 2014, 30, 8558–8569. [Google Scholar] [CrossRef] [PubMed]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-Oxo)Dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef] [Green Version]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; et al. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef]
- Heyer, A.J.; Plessers, D.; Braun, A.; Rhoda, H.M.; Bols, M.L.; Hedman, B.; Hodgson, K.O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Methane Activation by a Mononuclear Copper Active Site in the Zeolite Mordenite: Effect of Metal Nuclearity on Reactivity. J. Am. Chem. Soc. 2022, 144, 19305–19316. [Google Scholar] [CrossRef] [PubMed]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [PubMed]
- Vanelderen, P.; Snyder, B.E.R.; Tsai, M.L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Lee, M.-S.; Tao, L.; Ikuno, T.; Khare, R.; Jentys, A.; Huthwelker, T.; Borca, C.N.; Kalinko, A.; Gutiérrez, O.Y.; et al. Activity of Cu–Al–Oxo Extra-Framework Clusters for Selective Methane Oxidation on Cu-Exchanged Zeolites. JACS Au 2021, 1, 1412–1421. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef]
- Koishybay, A.; Shantz, D.F. Water Is the Oxygen Source for Methanol Produced in Partial Oxidation of Methane in a Flow Reactor over Cu-SSZ-13. J. Am. Chem. Soc. 2020, 142, 11962–11966. [Google Scholar] [CrossRef]
- Bulánek, R.; Wichterlová, B.; Sobalík, Z.; Tichý, J. Reducibility and Oxidation Activity of Cu Ions in Zeolites Effect of Cu Ion Coordination and Zeolite Framework Composition. Appl. Catal. B Environ. 2001, 31, 13–25. [Google Scholar] [CrossRef]
- Neylon, M.K.; Marshall, C.L.; Kropf, A.J. In Situ EXAFS Analysis of the Temperature-Programmed Reduction of Cu-ZSM-5. J. Am. Chem. Soc. 2002, 124, 5457–5465. [Google Scholar] [CrossRef] [PubMed]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Göbel, C.; Schlesiger, C.; Malzer, W.; Trunschke, A.; Schomäcker, R.; Thomas, A. Solid-State Ion-Exchanged Cu/Mordenite Catalysts for the Direct Conversion of Methane to Methanol. ACS Catal. 2017, 7, 1403–1412. [Google Scholar] [CrossRef]
- Sainz-Vidal, A.; Balmaseda, J.; Lartundo-Rojas, L.; Reguera, E. Preparation of Cu-Mordenite by Ionic Exchange Reaction under Milling: A Favorable Route to Form the Mono-(μ-Oxo) Dicopper Active Species. Microporous Mesoporous Mater. 2014, 185, 113–120. [Google Scholar] [CrossRef]
- Wang, J.B.; Lin, S.C.; Huang, T.J. Selective CO Oxidation in Rich Hydrogen over CuO/Samaria-Doped Ceria. Appl. Catal. A Gen. 2002, 232, 107–120. [Google Scholar] [CrossRef]
- Contarini, S.; Kevan, L. X-Ray Photoelectron Spectroscopic Study of Copper-Exchanged X- and Y-Type Sodium Zeolites: Resolution of Two Cupric Ion Components and Dependence on Dehydration and X-Irradiation. J. Phys. Chem. 1986, 90, 1630–1632. [Google Scholar] [CrossRef]
- Platzman, I.; Brener, R.; Haick, H.; Tannenbaum, R. Oxidation of Polycrystalline Copper Thin Films at Ambient Conditions. J. Phys. Chem. C. 2008, 112, 1101–1108. [Google Scholar] [CrossRef]
- Shpiro, E.S.; Grünert, W.; Joyner, R.W.; Baeva, G.N. Nature, Distribution and Reactivity of Copper Species in over-Exchanged Cu-ZSM-5 Catalysts: An XPS/XAES Study. Catal. Lett. 1994, 24, 159–169. [Google Scholar] [CrossRef]
- Artiglia, L.; Sushkevich, V.L.; Palagin, D.; Knorpp, A.J.; Roy, K.; van Bokhoven, J.A. In Situ X-Ray Photoelectron Spectroscopy Detects Multiple Active Sites Involved in the Selective Anaerobic Oxidation of Methane in Copper-Exchanged Zeolites. ACS Catal. 2019, 9, 6728–6737. [Google Scholar] [CrossRef]
- Paparazzo, E. On the Curve-Fitting of XPS Ce(3d) Spectra of Cerium Oxides. Mater. Res. Bull. 2011, 46, 323–326. [Google Scholar] [CrossRef]
- Lin, T.C.; Seshadri, G.; Kelber, J.A. A Consistent Method for Quantitative XPS Peak Analysis of Thin Oxide Films on Clean Polycrystalline Iron Surfaces. Appl. Surf. Sci. 1997, 119, 83–92. [Google Scholar] [CrossRef]
- Sunding, M.F.; Hadidi, K.; Diplas, S.; Løvvik, O.M.; Norby, T.E.; Gunnæs, A.E. XPS Characterisation of in Situ Treated Lanthanum Oxide and Hydroxide Using Tailored Charge Referencing and Peak Fitting Procedures. J. Electron Spectros. Relat. Phenom. 2011, 184, 399–409. [Google Scholar] [CrossRef]
- Martini, A.; Borfecchia, E.; Lomachenko, K.A.; Pankin, I.A.; Negri, C.; Berlier, G.; Beato, P.; Falsig, H.; Bordiga, S.; Lamberti, C. Composition-Driven Cu-Speciation and Reducibility in Cu-CHA Zeolite Catalysts: A Multivariate XAS/FTIR Approach to Complexity. Chem. Sci. 2017, 8, 6836–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipek, B.; Wulfers, M.J.; Kim, H.; Göltl, F.; Hermans, I.; Smith, J.P.; Booksh, K.S.; Brown, C.M.; Lobo, R.F. Formation of [Cu2O2]2+ and [Cu2O]2+ toward C-H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39. ACS Catal. 2017, 7, 4291–4303. [Google Scholar] [CrossRef]
- Borfecchia, E.; Lomachenko, K.A.; Giordanino, F.; Falsig, H.; Beato, P.; Soldatov, A.V.; Bordiga, S.; Lamberti, C. Revisiting the Nature of Cu Sites in the Activated Cu-SSZ-13 Catalyst for SCR Reaction. Chem. Sci. 2015, 6, 548–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure-Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Wang, Y.; Lu, J.; Zhang, J.; Zhang, Z.; Xu, S.; Liu, X.; Zhang, T.; Gocyla, M.; Heggen, M.; et al. Acid-Promoter-Free Ethylene Methoxycarbonylation over Ru-Clusters/Ceria: The Catalysis of Interfacial Lewis Acid-Base Pair. J. Am. Chem. Soc. 2018, 140, 4172–4181. [Google Scholar] [CrossRef]
- Xue, H.; Huang, X.; Zhan, E.; Ma, M.; Shen, W. Selective Dealumination of Mordenite for Enhancing Its Stability in Dimethyl Ether Carbonylation. Catal. Commun. 2013, 37, 75–79. [Google Scholar] [CrossRef]
- Maurin, G.; Senet, P.; Devautour, S.; Gaveau, P.; Henn, F.; Van Doren, V.E.; Giuntini, J.C. Combining the Monte Carlo Technique with 29SI NMR Spectroscopy: Simulations of Cation Locations in Zeolites with Various Si/Al Ratios. J. Phys. Chem. B. 2001, 105, 9157–9161. [Google Scholar] [CrossRef]
- Dimitrijevic, R.; Lutz, W.; Ritzmann, A. Hydrothermal Stability of Zeolites: Determination of Extra-Framework Species of H-Y Faujasite-Type Steamed Zeolite. J. Phys. Chem. Solids 2006, 67, 1741–1748. [Google Scholar] [CrossRef]
Figure 1.
Cu2p3/2 photoelectron spectra of: (a) Cu-MOR1, (b) Cu-MOR1-water, (c) Cu-MOR1 after calcination at 450 °C for 2 h, and (d) Cu-MOR1-water after calcination at 450 °C for 2 h.
Figure 1.
Cu2p3/2 photoelectron spectra of: (a) Cu-MOR1, (b) Cu-MOR1-water, (c) Cu-MOR1 after calcination at 450 °C for 2 h, and (d) Cu-MOR1-water after calcination at 450 °C for 2 h.
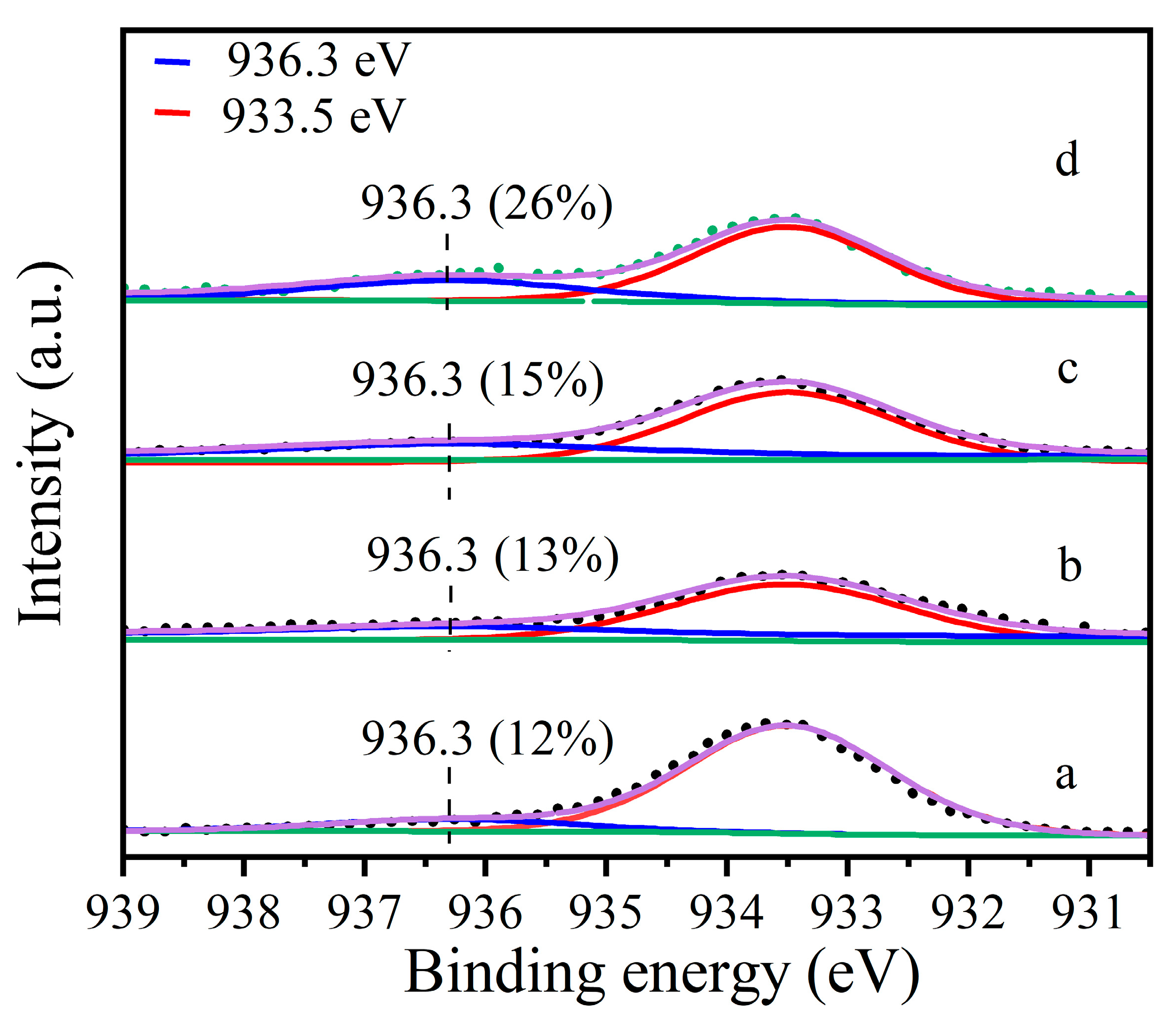
Figure 2.
In situ diffuse reflectance FTIR spectra: (a) Cu-MOR1 after calcination at 450 °C; (b) Cu-MOR1-water after calcination at 450 °C.
Figure 2.
In situ diffuse reflectance FTIR spectra: (a) Cu-MOR1 after calcination at 450 °C; (b) Cu-MOR1-water after calcination at 450 °C.
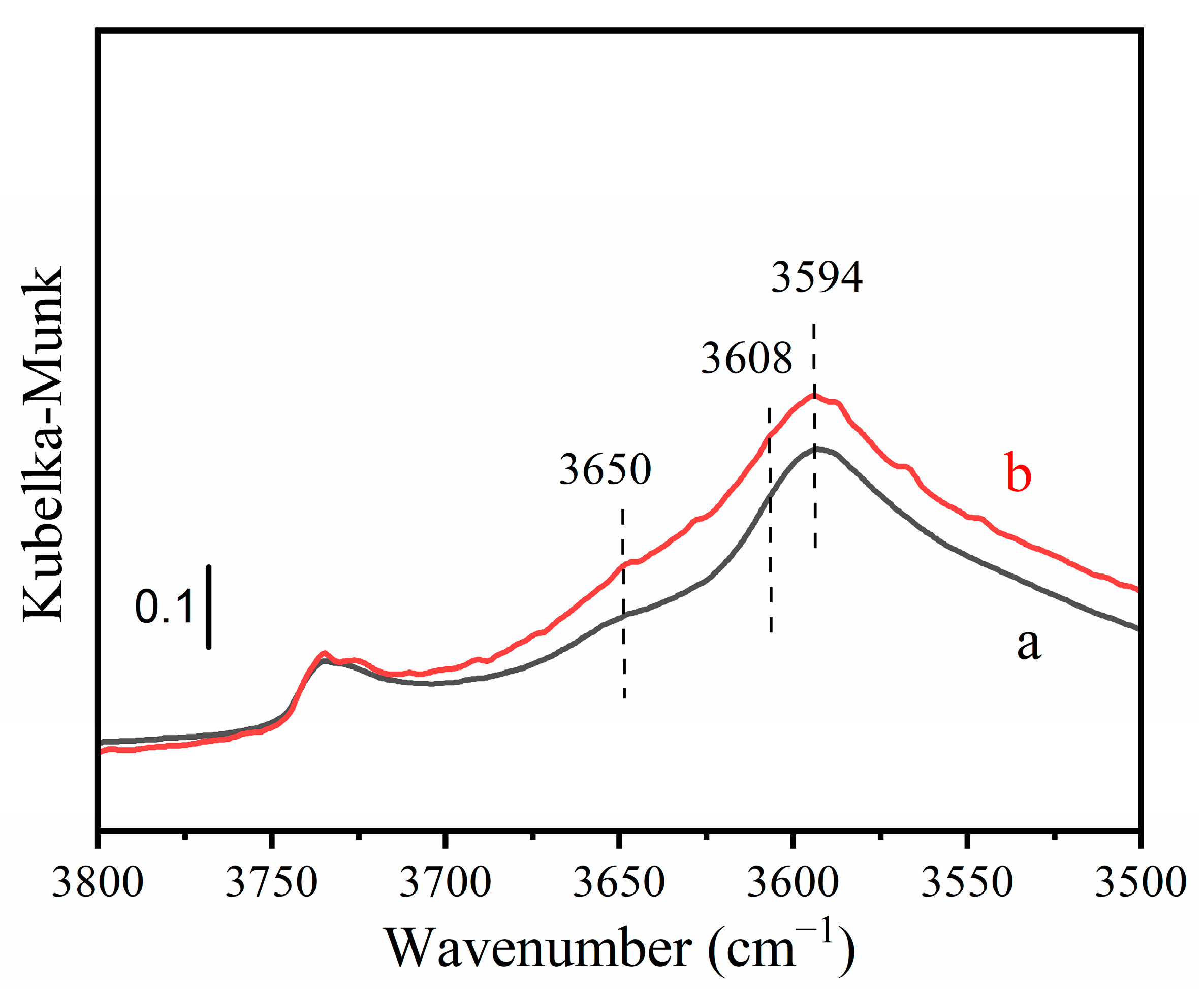
Figure 3.
27Al NMR spectra: (a) Cu-MOR1; (b) Cu-MOR1 after calcination at 450 °C for 2 h. (c) Cu-MOR1-water after calcination at 450 °C for 2 h.
Figure 3.
27Al NMR spectra: (a) Cu-MOR1; (b) Cu-MOR1 after calcination at 450 °C for 2 h. (c) Cu-MOR1-water after calcination at 450 °C for 2 h.
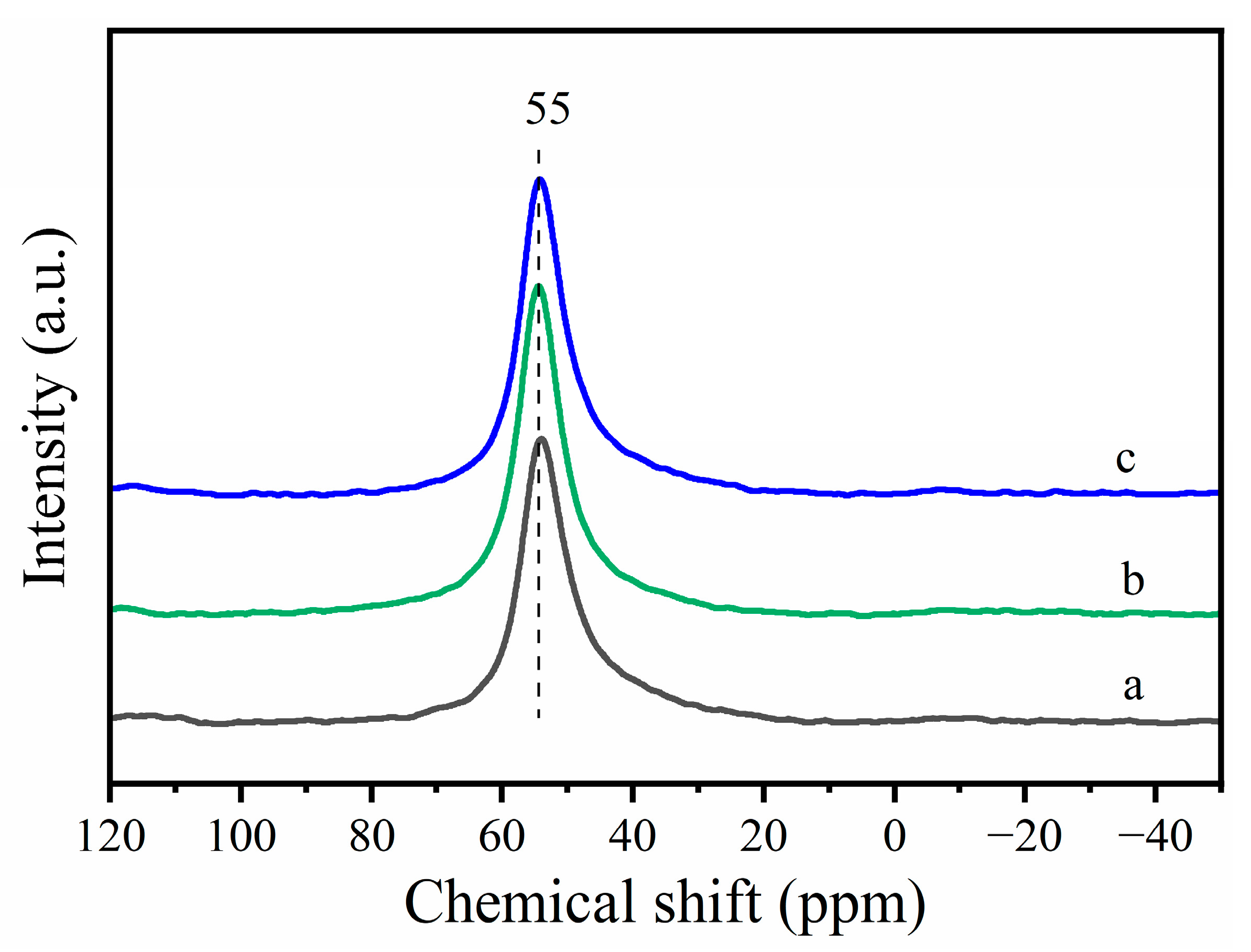
Figure 4.
XRD patterns: (a) Cu-MOR1; (b) Cu-MOR1-water; (c) Cu-MOR1 after calcination at 450 °C for 2 h; (d) Cu-MOR1-water after calcination at 450 °C for 2 h.
Figure 4.
XRD patterns: (a) Cu-MOR1; (b) Cu-MOR1-water; (c) Cu-MOR1 after calcination at 450 °C for 2 h; (d) Cu-MOR1-water after calcination at 450 °C for 2 h.
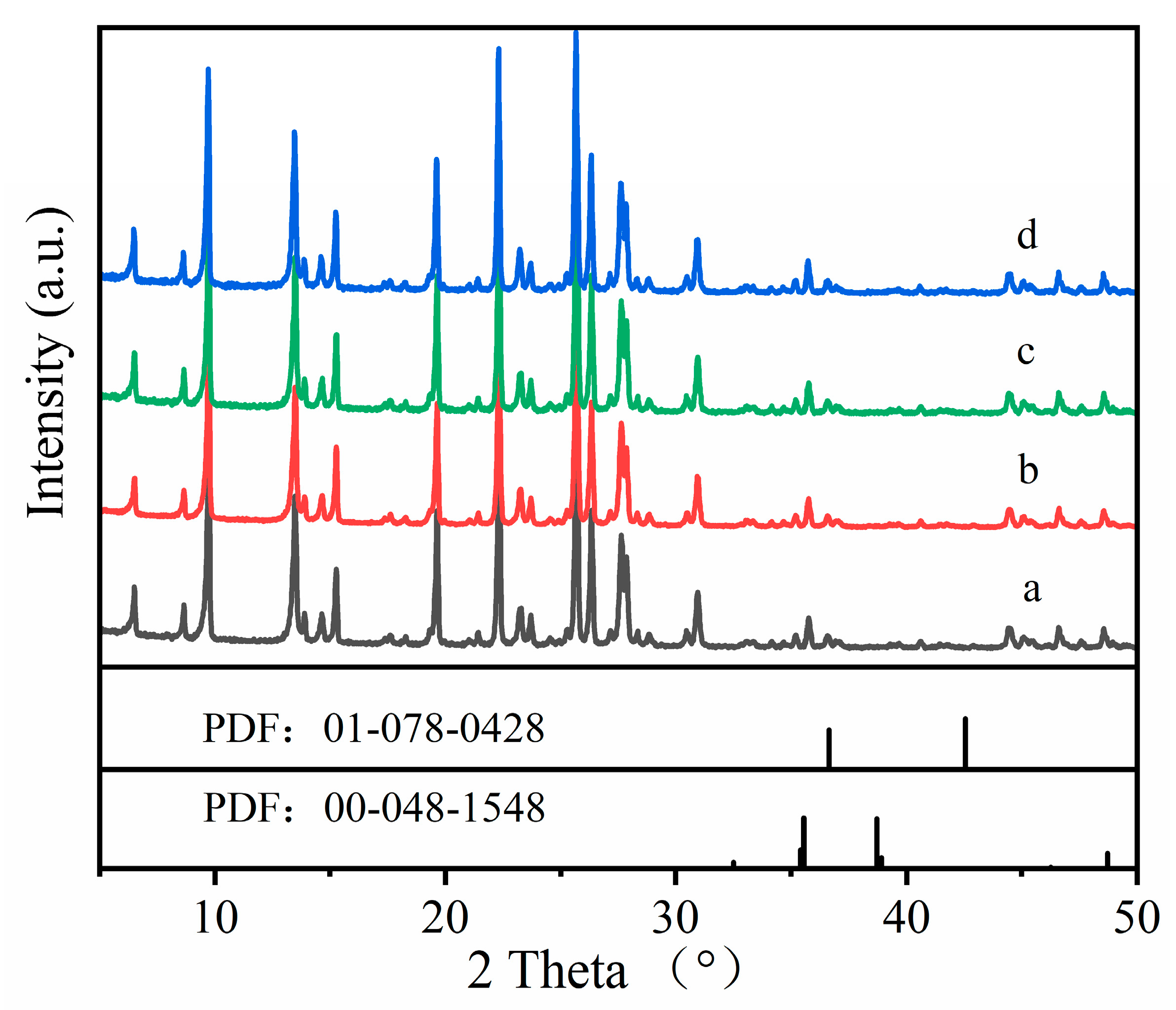
Figure 5.
The proposed mechanism of water increased Cu-MOR activity.

Figure 6.
Reusability studies of Cu-MOR1-water. Reaction conditions: 0.2 g catalyst, 200 °C, 12 h, 1 Mpa CH4.
Figure 6.
Reusability studies of Cu-MOR1-water. Reaction conditions: 0.2 g catalyst, 200 °C, 12 h, 1 Mpa CH4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Methanol yields of Cu-MOR and Cu-MOR-water.
| Entry | Sample | Methanol Yield (μmol/g) 1 | Efficiency (mol CH3OH/mol Cu) | CH3OH/(CH3OH + DME) |
|---|---|---|---|---|
| 1 | Cu-MOR1 2 | 36 | 0.07 | 100% |
| 2 | Cu-MOR1-water 2 | 92 | 0.17 | 86% |
| 3 | Cu-MOR1-water 3 | 92 | 0.17 | 83% |
| 4 | Cu-MOR1-water 4 | 90 | 0.17 | 83% |
| 5 | Cu-MOR1-water 5 | 26 | 0.05 | 100% |
| 6 | Cu-MOR1-water 6 | 46 | 0.09 | 100% |
| 7 | Cu-MOR2 7 | 50 | 0.06 | 100% |
| 8 | Cu-MOR2-water 7 | 128 | 0.15 | 93% |
1 Reaction conditions: 0.2 g catalyst, 200 °C, 12 h, 1 MPa CH4. The yield of methanol was calculated based on the total amount of methanol and dimethyl ether, where each dimethyl ether was calculated as two methanol. 2 Cu-MOR1 (SiO2/Al2O3 = 15.5, Cu/Al = 0.37). 3 Reaction conditions: 0.2 g catalyst, 200 °C, 8 h, 1 MPa CH4. 4 Reaction conditions: 0.2 g catalyst, 200 °C, 4 h, 1 MPa CH4. 5 Reaction conditions: 0.2 g catalyst, 150 °C, 12 h, 1 MPa CH4. 6 Reaction conditions: 0.2 g catalyst, 175 °C, 12 h, 1 MPa CH4. 7 Cu-MOR2 (SiO2/Al2O3= 8.73, Cu/Al = 0.38).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guan, X.; Wang, Y.; Liu, X.; Du, H.; Guo, X.; Zhang, Z. Enhancing the Activity of Cu-MOR by Water for Oxidation of Methane to Methanol. Catalysts 2023, 13, 1066. https://doi.org/10.3390/catal13071066
AMA Style
Guan X, Wang Y, Liu X, Du H, Guo X, Zhang Z. Enhancing the Activity of Cu-MOR by Water for Oxidation of Methane to Methanol. Catalysts. 2023; 13(7):1066. https://doi.org/10.3390/catal13071066
Chicago/Turabian StyleGuan, Xi’an, Yehong Wang, Xiumei Liu, Hong Du, Xinwen Guo, and Zongchao Zhang. 2023. "Enhancing the Activity of Cu-MOR by Water for Oxidation of Methane to Methanol" Catalysts 13, no. 7: 1066. https://doi.org/10.3390/catal13071066
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.