

Understanding the Role of Electrolyte Cations on Activity and Product Selectivity of CO2 Reduction over Cu Electrode

1
CAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology, Beijing 100190, China
2
University of Chinese Academy of Sciences, Beijing 100049, China
*
Authors to whom correspondence should be addressed.
Catalysts 2023, 13(7), 1092; https://doi.org/10.3390/catal13071092
Submission received: 13 June 2023
/
Revised: 5 July 2023
/
Accepted: 7 July 2023
/
Published: 12 July 2023
(This article belongs to the Special Issue Application of Catalysts in CO2 Capture, Production and Utilization)
Abstract
:The electrocatalytic conversion of CO2 on a Cu electrode has the potential to produce valuable chemicals such as hydrocarbons and oxygenated compounds. While the influence of electrolyte cation on the activity and selectivity of the CO2 reduction reaction (CO2RR) on Cu has been widely observed, the specific mechanism through which cation species affect the CO2RR remains unclear and subject to debate. In this work, the CO2RR in the carbonate electrolytes containing different alkali metals (Li+, Na+, K+, Rb+, and Cs+) was investigated at potentials from −0.1 to −1.1 V (vs. RHE) over a Cu electrode using electrochemical techniques. Charge transfer kinetics, adsorption of species, and mass transport were considered comprehensively during the analysis. It is found that several factors can play a role in the CO2RR, including hydrated cation adsorption, preferential hydrolysis, and interaction between the cation and adsorbed species, with the dominating factor determined by the external bias and cation species. Consequently, a coherent interpretation of the influence of electrolyte cations on the intrinsic kinetics of the CO2RR has been put forward. We envision that these insights will greatly contribute to the development of efficient catalytic systems and the optimization of catalytic conditions, thereby advancing progress toward commercial applications in this field.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The electrocatalytic CO2 reduction reaction (CO2RR) offers an important strategy to store electrical energy produced via renewable and intermittent resources like wind, hydro, and solar energy [1,2,3]. The high energy density of hydrocarbons and alcohols make these products more preferable compared to carbon monoxide and formic acid. So far, extensive efforts have been invested to seek out such catalysts that can directly produce hydrocarbons or alcohols, but only copper (Cu) is known to catalyze the C-C coupling with appreciable efficiency to generate a wide range of multicarbon products in an aqueous solution [4,5,6]. However, the application of CO2RR technology on a global scale faces significant technical challenges, including poor product selectivity, high overpotential, and slow reaction kinetics associated with Cu [7]. Consequently, gaining a comprehensive understanding of the reaction processes occurring on the surface of Cu is essential for the production of hydrocarbons and the precise tuning of target products [8]. The activity and selectivity of the CO2RR are largely influenced by factors such as catalyst structure, electrolyte composition, and pH levels during the reaction [9,10,11,12,13,14,15,16]. In order to comprehend and manipulate these influences, state-of-the-art strategies should be employed.
A number of reports have indicated that the species of electrolyte cations can play a significant role in influencing the activity and product selectivity of the CO2RR [2,17,18,19,20,21,22,23,24,25]. In the case of Cu electrodes, increasing the size of electrolyte alkali metal cations leads to higher C2 selectivity, like for ethylene and ethanol [18,19,23]. However, the origination of the effects from electrolyte cations is intricate and is still contentious today. Previous reports revealed that these cations can be adsorbed on the electrode surface at the outer Helmholtz plane (OHP), giving rise to changes in the local electric property to different extents due to hydration and/or influence on the formation/stabilization of the reduction intermediates [2,22,26,27]; while other research suggests that the surface pH can be influenced by different sized cations owing to the discrepancy in their buffer capacity, thus changing the pH-dependent reaction routes or surface CO2 concentration for the CO2RR [19,28,29]. The consistent explanation of how the electrolyte cations influence the CO2RR kinetics is challenging since the relevant factors in the electrolyte are complex (including mass transport, conductivity, and pH). Neglecting the comprehensive consideration of these factors can lead to confusion. For instance, Singh et al. have shown that cation-dependent pH at the electrode surface accounts for the different CO2RR performances under inadequate mass transfer conditions [19]. On the other hand, Resasco and coworkers reported that the interfacial dipole field dominates the cation-dependent CO2RR under the condition where influence from mass transport is avoided [2]. Therefore, robust methodologies are required to elucidate the specific role of cations in the CO2RR over Cu electrodes, taking into account various influencing factors.
Directly extracting information at the electrode/electrolyte interface during the reaction would be promising to study the influential mechanism of the electrolyte cations. According to classic catalytic theory, the properties of the electrode/electrolyte interface, such as the electrode/cation interaction, play a significant role in the reaction by regulating the local electrical field, charge, and mass transfer, as well as the adsorption/desorption process. However, the understanding of the electrode/cation interface remains limited primarily due to the lack of well-established characterization techniques for studying the electrolyte and its interaction with the electrode. Electrochemical characterization offers a mature and useful approach to comprehensively probe in an operando manner the electrode/electrolyte interface [30]. Linear scan voltammetry (LSV) and electrochemical impedance spectroscopy (EIS) are frequently used techniques to study the kinetics of electrode processes in the CO2RR [31,32,33,34,35]. These methods are valuable as they can provide direct insights into the interfacial redox reaction mechanism at a low cost. Specifically, EIS enables the qualitative analysis of the kinetics of redox processes at the electrode/electrolyte interface, the adsorption/desorption behavior of species on the electrode surface, and mass transport. Recently, LSV and EIS were successfully applied to identify the processes during the CO2RR on tin foil [36]. However, the utilization of electrochemical techniques in analyzing the effect of electrolyte cations is still lacking and requires further exploration.
Herein, we performed a systematic electrochemical study to elucidate the origination of the electrolyte cation effect in the CO2RR over the Cu electrode. The information on charge transfer processes, adsorption of intermediates, and mass transport during the CO2RR was analyzed in detail. While the cation effect on activity and selectivity of the CO2RR have been reported previously [2,19], our work offers a more comprehensive approach by investigating the mechanism across a broad range of applied potentials. The role of cations with varying sizes and their influence on the CO2RR under different potentials are thoroughly investigated.
2. Results and Discussion
It has been reported experimentally that the CO2RR dominates over the hydrogen evolution reaction (HER) in electrolytes with larger cations (K+, Rb+, Cs+) compared to the smaller ones (Li+ and Na+) [2,18,19]. Meanwhile, the driving force is an important factor regulating the CO2RR performance [37]. Thus, the following discussion is divided into different parts, large (K+, Rb+, Cs+) and small (Li+ and Na+) cations, and high (−0.6 to −1.1 V) and low (−0.1 to −0.5 V) negative applied bias voltages. In addition, it should be pointed out that here, the discussions are based on the previously reported product distribution data affected by the electrolyte cations [2,19]. As the cation size becomes larger, HER is inhibited, and selective CO2RRs (especially C2 production) are favored.
2.1. Linear Scan Voltammetry
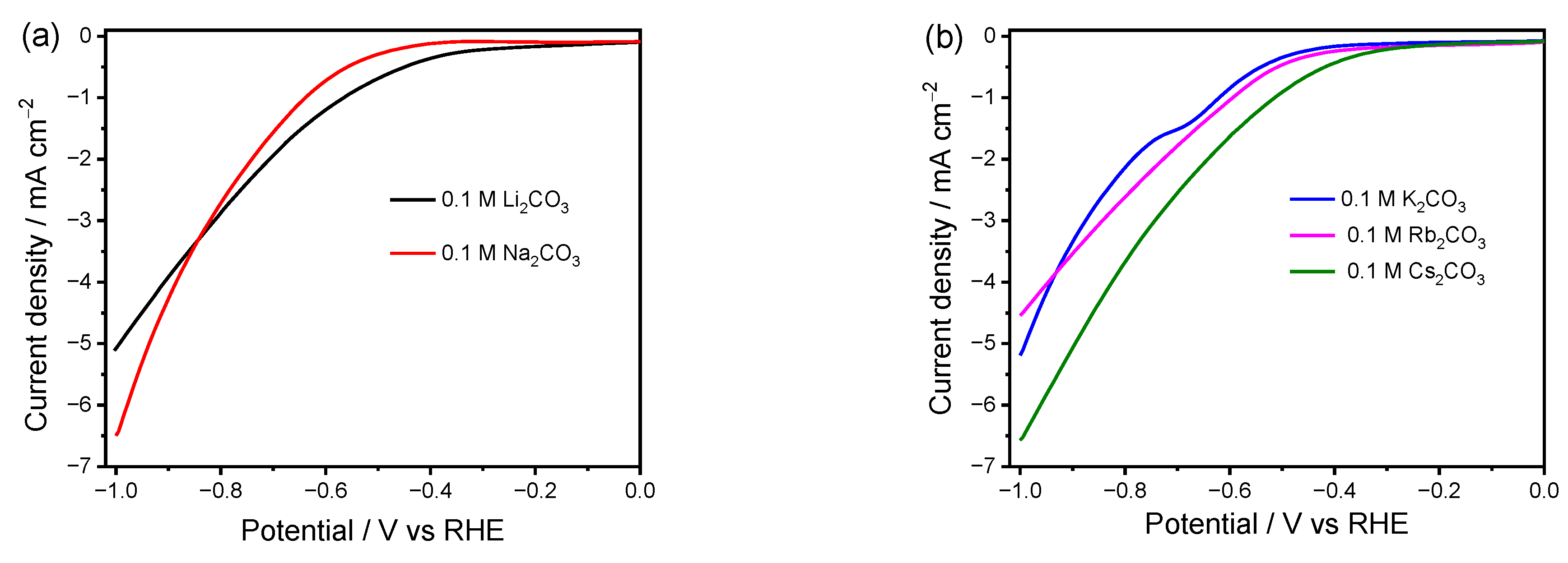
To investigate the influence of electrolyte cations on the Faradic current over the Cu electrode, LSV was carried out in CO2-saturated M2CO3 electrolytes (0.1 M, M = Li, Na, K, Rb, and Cs) (Figure 1). Figure 1a presents the LSV curves of the Cu electrode in CO2-saturated Li2CO3 and Na2CO3 solutions. A higher cathodic current density and onset potential can be observed for the reaction in the Li2CO3 electrolyte in the low negative potential region due to the higher HER and more facile charge transfer kinetics as compared to that in the Na2CO3 electrolyte. Both the onset potential and the cathodic current density increase once the cation size further increases from K+ to Cs+ (Figure 1b), demonstrating a more facile catalytic reaction in the case of a larger cation size. The level of cation hydration and extent of cation adsorption can explain this phenomenon well. The larger cations possess lower hydration power, and much more can be adsorbed on the electrode surface in the outer Helmholtz plane (OHP) [22]. Generally, the potential at OHP can be elevated by the adsorbed cations, leading to decreased concentration of positive H+ and accelerated the CO2RR by stabilizing the negatively charged intermediate CO2·-, thereby suppressing HER and enhancing the CO2RR. Therefore, for the reactions in electrolytes with large cations (K+, Rb+, Cs+) where the CO2RR dominates [18], the cathodic current density increases with the increase in cation size, as shown in Figure 1b. However, for the HER favorable cations in Figure 1a, since Na+ has a higher propensity to be adsorbed on the electrode than Li+, the repulsion of H+ from OHP is more pronounced in the Na+ electrolyte, resulting in lower current density than that in Li+.
It is noted that the driving force starts to meet the requirement for the CO2RR as the applied potential becomes more negative, like <−0.84 V (Figure 1a), which is more favorable to occur in the Na2CO3 electrolyte. A higher current density at more negative potentials in Na2CO3 than that in Li2CO3 indicates that other factors, rather than hydration discrepancy in the cations, become dominant in this region. According to previous reports, the CO2RR is mass transport limited at high cathodic current [38,39]. The hydrated cations can act as a buffer to maintain the interfacial pH value. A larger cation tends to exhibit higher power to sustain a low local pH near the electrode, and thus keeps the concentration of local CO2 at a higher level than that for a smaller cation [19,28,29]. Accordingly, the difference in buffer effect can reasonably explain the higher current density in Na2CO3 electrolyte than that in Li2CO3 under a more negative bias.
In addition, the current density in the K2CO3 solution exceeds that in Rb2CO3 at more negative potentials (Figure 1b). A hump in the LSV curve can be observed in K2CO3 at around −0.65 V, which is probably due to the reduction of CO2 to CO. However, this hump cannot be observed in any other electrolytes. Such abnormal phenomena indicate that other factors caused by the cation also play a role in the CO2RR. It is widely accepted that the catalytic performance of the CO2RR and HER can be impacted by various processes, such as mass transfer, adsorption/desorption of species, and local pH. The EIS analysis is thus carried out to further investigate the influential mechanism of cations on the catalytic performance over Cu electrodes under different applied biases.
2.2. Electrochemical Impedance Spectroscopy
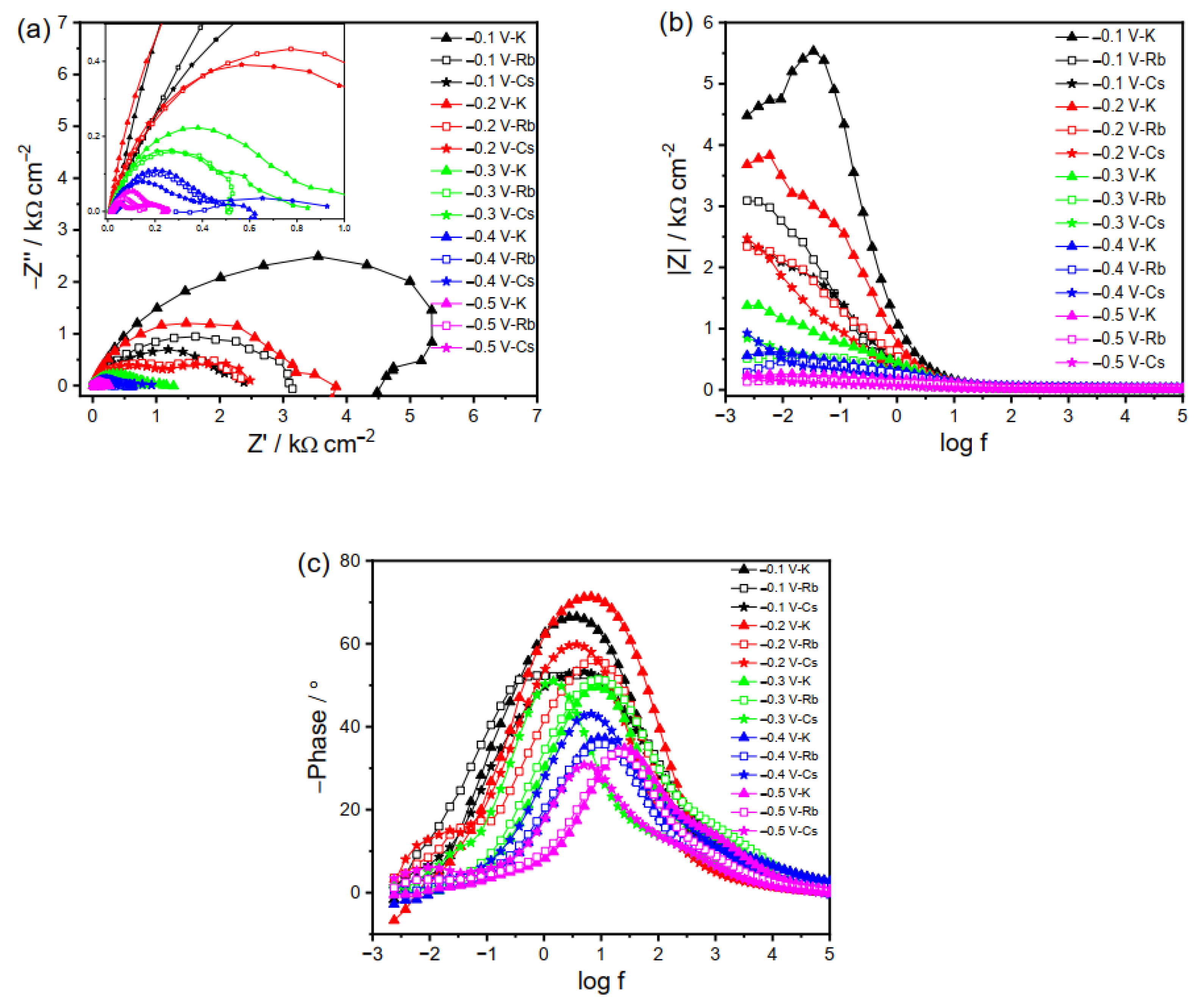
EIS is a powerful tool to probe the information at the electrode/electrolyte interface. Charge transfer kinetics and mass transport can be deduced from the EIS plot. In addition, the behavior of capacitance and inductance of an electrode process influences not only the magnitude of alternating current in the impedance spectroscopy, but also the phase, which can help analyze the adsorption/desorption phenomena. A negative value of the phase angle is usually ascribed to the adsorption of chemical species. The peak in the phase angle plot corresponds to the time constant, i.e., the charge transfer process at the electrode/electrolyte interface. Therefore, both the impedance and phase are taken into consideration in the following EIS analysis so as to study the electrochemical behavior at the electrode/electrolyte interface. The EIS was performed in a CO2-saturated 0.1 M M2CO3 electrolyte under different applied bias voltages. As analyzed in Figure 1, the Faradic reaction over the Cu electrode strongly depends on the cation size and applied potential. To clearly interpret the process, the impedance behavior of the Cu electrode in solutions with small (Li+ and Na+) and large cations (K+, Rb+, and Cs+) were studied and explained separately. For each of them, two different potential ranges, i.e., less (−0.1 to −0.5 V) and more (−0.6 to −1.1 V) negative potentials, were employed to study the process at the electrode/electrolyte interface during the electrolysis. Three different equivalent circuits are used to interpret the EIS spectra depending on the elements in the data, Rs(RctCPE), Rs((RctZW)CPE), and Rs((RctZW)CPE1)((RlL)RctCPE2)), where Rs represents series resistance; the constant phase elements CPE, CPE1, and CPE2 represent the capacitance; Rct and Rl are the charge transfer resistance; ZW is the Warburg impedance that describes diffusion process; and the inductive impedance L represents the adsorption process.
2.2.1. EIS Plots of Cu Electrode in Li2CO3 and Na2CO3 Electrolyte
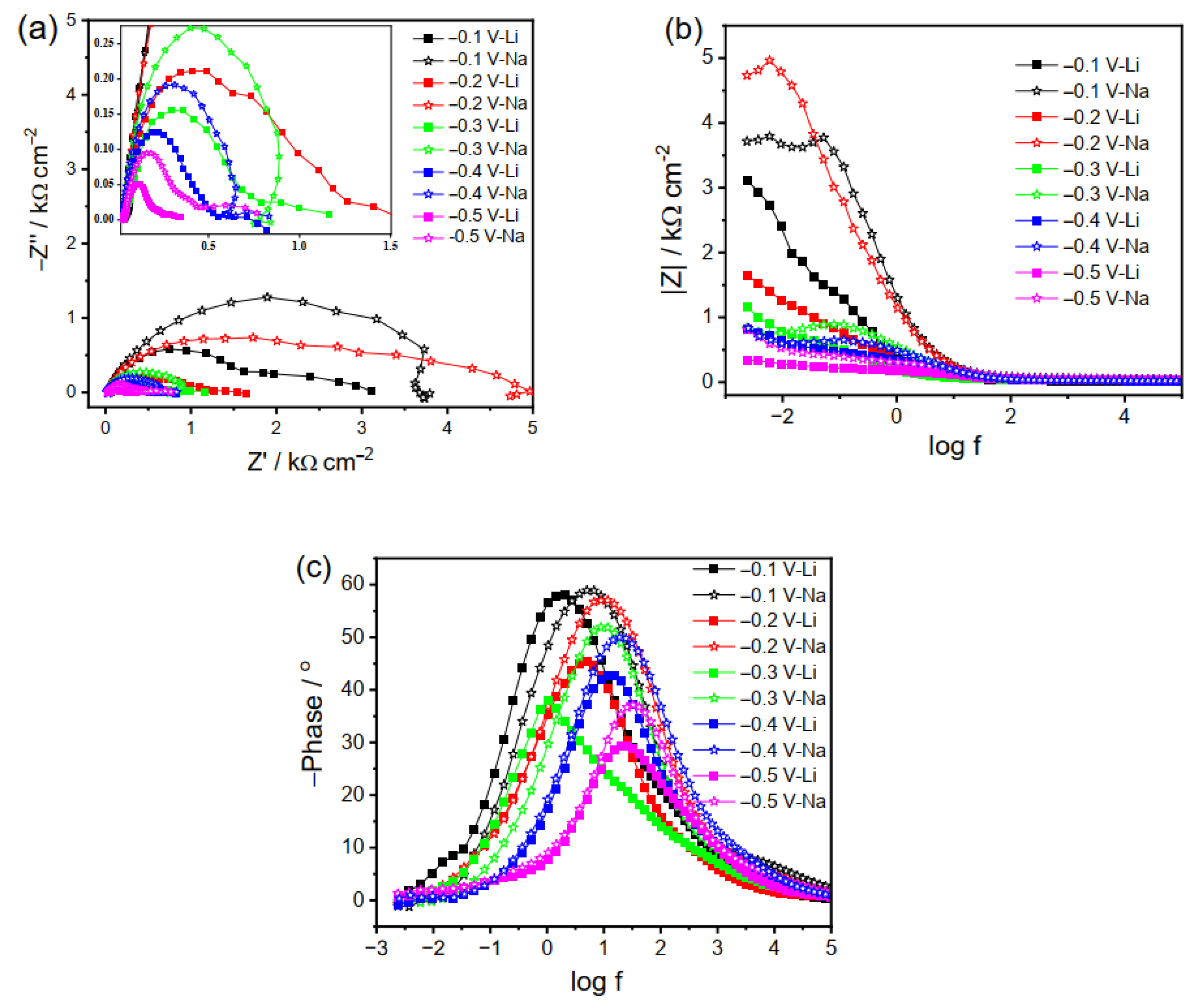
The spectra recorded at less negative potentials are presented as Nyquist (Figure 2a) and Bode plots (Figure 2b,c), respectively. The impedance modulus has almost the same value for the two systems at a high frequency that corresponds to θ = 0 in the phase angle plots, which is the solution resistance between working and reference electrodes. The difference in ionic conductivity of the electrolytes with different cations is thus not considered in this work. It is noted that in most cases, only one characteristic peak is observed in Bode plots due to the charge transfer process (Figure 2c). The charge transfer resistance (RCT) is directly proportional to the arc radius in the Nyquist plot (Figure 2a), which decreases as the applied potential becomes more negative due to faster charge kinetics upon a larger driving force. Lower RCT in Li2CO3 than in Na2CO3 is observed at the same potential, indicating a more facile charge transfer at the Cu/electrolyte interface in Li2CO3.
The spectra in Figure 2a can reflect the HER to some degree during the electrolysis in Li2CO3 under low applied bias. In comparison, a different character in the Nyquist plot is observed for the Na2CO3 electrolyte, with an inductive loop in the low-frequency region. It is speculated that this is due to the adsorption process of the CO2RR intermediate and/or CO2. Specifically, since the ability to initiate the CO2RR is stronger in Na2CO3 than in Li2CO3 and a low overpotential is not large enough to drive efficient CO2RR and consume the adsorbates, reactants are easier to be adsorbed and/or accumulated on the Cu surface in Na2CO3, manifested as an inductive loop in the Nyquist plot (Figure 2a). It is worth mentioning that the adsorbates can block the active sites on the Cu electrode surface and can thus decrease the current density [40]. Hence, in addition to the more facile HER in Li2CO3 as compared to that in Na2CO3, the greater adsorption extent in Na2CO3 can also suitably explain the lower current density in LSV at a less negative potential (Figure 1a) and higher RCT (Figure 2a) in Na2CO3 as compared to in Li2CO3.
Interestingly, a sharp decrease in RCT is observed at −0.3 V in the Na2CO3 electrolyte as compared to those at −0.1 and −0.2 V (Figure 2a,b), implying that −0.3 V is the critical potential for the beginning of the Faradic reaction. Likewise, a further decrease in the applied potential (such as −0.4 V) can result in further decrement in the RCT value. Thus, the EIS results further prove that in the less negative potential region, reactions proceed easier in Li2CO3 than in Na2CO3 due to the dominant HER and less blockage of the Cu surface by the adsorbates.
As the applied potential becomes more negative (i.e., −0.6 to −1.1 V), a reversal in the RCT occurs, i.e., faster charge transfer kinetics at the electrode/electrolyte interface in Na2CO3 as compared to Li2CO3 (Figure 3). This reversal suggests more facile charge transfer in the Na2CO3 electrolyte, as the high potential can drive charge transfer to the adsorbed species, which will otherwise passivate the electrode surface and hinder the reduction reactions at less negative potentials. Interestingly, only one arc is observed in the Nyquist plots at high frequency (Figure 3a), perhaps because of the comparable or merged time constants of different reactions. Since various products can be produced during the CO2RR via sequential reduction reactions along with HER, the apparent single time constant is indeed a combination of several time constants [36]. Similarly, the negative phase angle values at low frequency imply the intermediate’s adsorption under high applied potential.
An exception is that a charge transfer process without an inductive element is observed at a low frequency for the reaction in Li2CO3 at −0.6 V, which may be caused by the low amount of adsorbates that can be consumed instantly. As the applied potential becomes more negative, the impedance spectra in Li2CO3 electrolyte start to resemble those in Na2CO3 with diffusion and adsorption process observed, as a higher driving force can lead to faster charge kinetics and thus a high amount of adsorbed CO2RR intermediates and/or exhausted molecular CO2 at the electrode surface and, thereby, the rate-determining step of CO2 diffusion.
It is worth mentioning that the low-frequency arc representing the diffusion process can be observed at high applied potentials (Figure 3a), indicating the accelerated reactions due to rapid consumption of CO2, which is more prominent for those in Na2CO3. It is noted that the surface pH rises during the catalytic reaction, resulting in a pH gradient in the vicinity of the electrode [41,42]. The pH value adjacent to the electrode is high at large negative applied potential due to the weak buffer capacity of Li+ and Na+. As the CO2 solubility is pH dependent, the concentration of surface molecular CO2 decreases as the local pH becomes higher. Thus, dissolved molecular CO2 will transport to the electrode surface for equilibrium and supplement. Considering CO2 is consumed in a faster way in the electrolyte containing larger Na+ cations, the diffusion element can be observed in a clearer way in the Na2CO3 electrolyte than in Li2CO3.
2.2.2. EIS Plots of Cu Electrode in K2CO3, Rb2CO3 and Cs2CO3 Electrolyte
The EIS plots of Cu electrode in the electrolytes with larger cations (K+, Rb+, and Cs+) behave differently than those with smaller ones (Li+ and Na+), as the larger cations adsorbed on the electrode surface can influence the interfacial reactions in more complicated ways as compared to the smaller ones. The RCT at less negative potentials decreases in the following order: K+ < Rb+ < Cs+ (Figure 4a,b). At −0.1 V in the K2CO3 electrolyte, the inductive loop at low frequency shows adsorption behavior on the Cu electrode, as evidenced by the negative phase in Figure 4c, which is otherwise absent in the case of the Rb2CO3 and Cs2CO3 electrolytes. Such difference at −0.1 V is probably due to the neutral or positive property of the adsorbed species and greater blockage extent of the electrode surface by Rb+ and Cs+ as compared to K+. The sharp decrease in impedance at −0.3 V additionally supports the above conclusion that it is the critical CO2RR potential that is similar to that in the Na2CO3 electrolyte. Further reducing the applied potential will decrease the impedance and facilitate the CO2RR. Reactions at the potentials of −0.4 and −0.5 V give rise to a new semicircle at low frequency, probably corresponding to the reduction of the intermediates.
For the reactions in Rb2CO3, the charge transfer is more facile as compared to that in K2CO3. Although it is hard to distinguish, it seems the EIS in Rb2CO3 at −0.1 V consists of two different time constants, as manifested by the two peaks in Figure 4c. It becomes more evident at potentials < −0.2 V, with a new semicircle appearing in the Nyquist plot, representing the sequential charge transfer processes. A small inductive loop at low frequency by further reducing the potential (−0.4 and −0.5 V) suggests the intermediates’ adsorption. Likewise, the impedance plot for Cs2CO3 shows a similar phenomenon to that for Rb2CO3 at −0.1 V. However, the impedance spectra at −0.2 to −0.5 V for Cs+ cation present two semicircles related to the charge transfer processes, with no inductive loop observed. This may be due to the fact that the adsorbed species can be consumed instantly, owing to the fast reaction kinetics in the Cs2CO3 electrolyte.
Figure 5 presents the impedance spectra at high negative potentials where the CO2RR is dominant. At −0.6 V, the impedance behavior is similar in the three electrolytes, i.e., two distinct peaks corresponding to two separate time constants. The decrease in impedance magnitude with increasing negative potential is again attributed to the faster kinetics under high negative biases. At potentials from −0.7 to −1.1 V, the charge transfer kinetics follows the order of K+ < Cs+ < Rb+ (Figure 5). So far, it is still unclear why the EIS spectra exhibit an elevated charge transfer in the electrolyte with smaller-sized Rb+ than Cs+. A possible speculation is that not only the cation size but also the nature of products play a role in this potential region. It is reported that HER is more favored at −1 V in Rb2CO3 electrolytes than in the K2CO3 and Cs2CO3 electrolytes, demonstrating a lower reaction barrier in electrolytes containing Rb+ [2]. Moreover, the inductive loop in the low-frequency region becomes evident in these three electrolytes (Figure 5), indicating the intermediates’ adsorption on the electrode surface at a large driving force, which is otherwise absent under less negative bias. As manifested by the inductive loop, the greater adsorption extent for large cations shown in Figure 5 than that for the smaller ones presented in Figure 3 indicates that the large cations in the electrolyte can stabilize the intermediates on the electrode surface in a better way. These results imply that the adsorption phenomena on the electrode surface depend on the bias voltage and electrolyte cation size. Given the dynamic nature of the processes involved in the generation, stabilization, and reduction of intermediates, it is not universally valid that selective and efficient CO2RRs come from the high extent of adsorption. Figure 5 indicates that the adsorption decreases in the case of large cations and at high negative potentials like reactions in Cs2CO3 at −1 V, probably due to the rapid consumption of negatively charged intermediates and, thus, efficient generation of the final products. Moreover, mass diffusion can also be observed in Figure 5, which is more prominent than that in small cation electrolytes shown in Figure 3.
2.3. Mechanism Analysis of Cations Effect on CO2RR
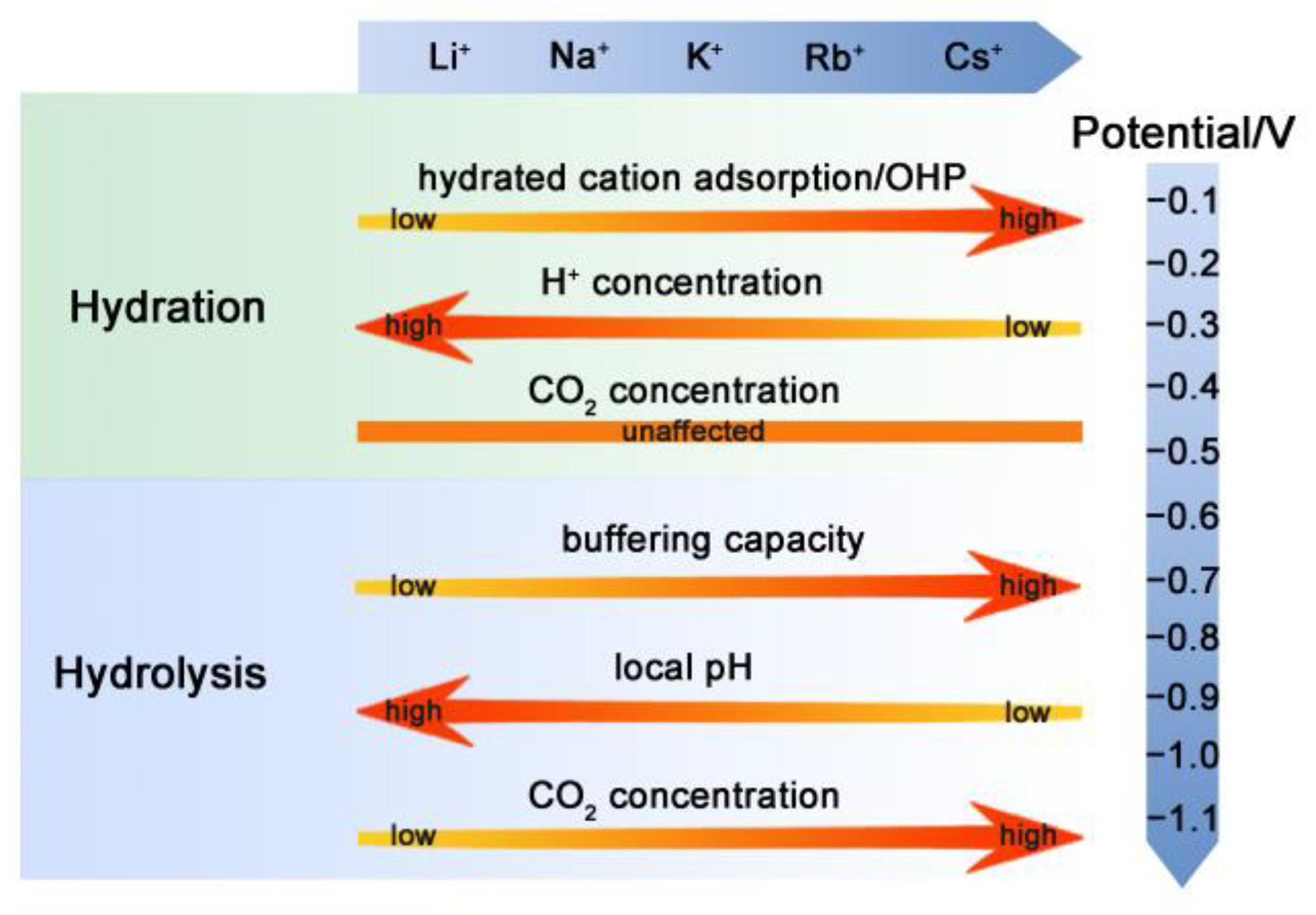
Different theories have been proposed previously for the cation effect on the CO2RR over the Cu electrode, including hydrated cation adsorption, preferential hydrolysis, and interaction between the cation and adsorbed species. However, there are still some phenomena that cannot be explained by these theories. For example, it was reported that higher local pH is favorable for the intermediates’ dimerization and hence the formation of C2 products [43]. According to this claim, it is expected that the higher local pH in the case of small cations with less buffering capacity should produce more C2 products. However, the trend is the opposite, and the C2 formation is more favorable in the electrolyte with large cations [2]. Thus, not only do the buffering capacity and modulation of local pH account for the CO2RR performance, but other factors can also play a part. On the basis of the LSV and EIS analysis here, it is found that the CO2RR performance over the Cu electrode in electrolytes with different cations is a result of the interplay among various factors, including effects from the cation hydration, hydrolysis, and adsorption processes. The dominant factor influencing the electrochemical reactions is determined by both the cations and the applied potentials, and their specific influences are summarized in Figure 6.
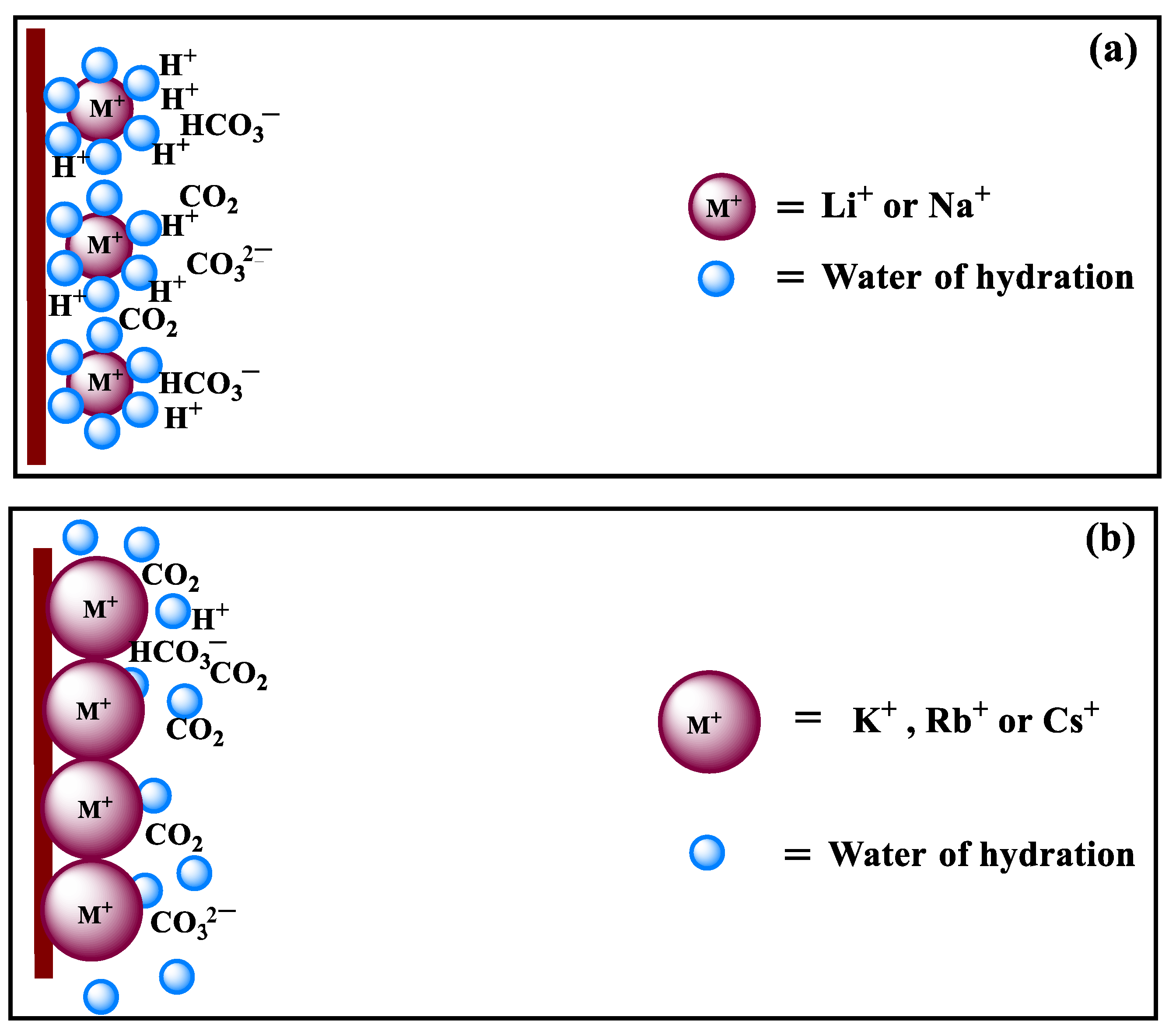
At low negative potentials, mass transport is not the rate-determining step for the CO2RR, and variation in the local pH resulting from cation hydrolysis cannot be the major factor either since it is negligible, or no CO2RR occurs under such conditions. The reaction in electrolytes with small cations (Li+ and Na+) and the dominant HER are taken as examples here to explain the influence of cation hydration. If the cations are strongly hydrated, it would be unfavorable for them to be adsorbed on the electrode. According to the literature [22], the hydration power is in the order of Li+ > Na+ > K+ > Rb+ > Cs+. Therefore, the smaller cation with a higher hydration number, like Li+, tends to adsorb less on the Cu electrode as compared to the larger ones [44], leading to a lower increase in the OHP potential with less blocked surface by the cations (Figure 6). The concentration of H+ is inversely proportional to the value of OHP potential since it is positively charged, while the concentration of CO2 is not influenced by OHP potential because it is not electrically charged (Figure 6). Thus, the cation hydration can impose a larger influence on the HER under low overpotentials. The cations adsorption and hydration propensity of small and large cations is schematically shown in Figure 7a,b, respectively. In the electrolyte with small cations and more negative OHP potential, the concentration of H+ ions at the Cu electrode surface is higher than that of the large cations, according to Equation (1) [22]:
where [H+]electrode and [H+] are the respective H+ concentration at the surface and in the bulk, F is Faraday’s constant, φ is the OHP potential, R is the general gas constant, and T is the absolute temperature. The increase in [H+]electrode leads to a higher HER and, hence, faster charge transfer kinetics at the electrode surface, as manifested in the Nyquist plots (i.e., higher HER in Li+ than in Na+ in the potential range of −0.1 V to −0.5 V). These results agree well with the previously observed phenomenon of an increase in the HER with an increase in the electrolyte concentration due to the decrement in OHP potential [45].
At high negative potentials, CO2 is consumed quickly, and a local pH gradient is built [46]. Thus, the hydrolysis of cations starts to take action, which can provide a buffering effect on the local pH (Figure 6). The reaction in electrolytes with large cations (K+, Rb+, and Cs+) and dominant CO2RRs are taken as an example. In general, the cations undergo hydrolysis by following Equation (2):
The distribution of dissolved CO2 among molecular CO2, HCO3−, and CO32− is highly pH dependent, and a high pH value leads to reduced concentration of molecular CO2 due to its rapid consumption with hydroxyl anions to form HCO3− and CO32− at high negative potentials. A reduction in the pH near the cathode surface induced by the buffering action of large alkali metal cations can cause an increase in the concentration of dissolved molecular CO2 at the electrode surface and, thereby, increase selective CO2RR. Usually, a larger cation possesses a stronger buffering capacity (Figure 6). Among the three large cations, the Cs+ ions are expected to give rise to minimal local pH, followed by Rb+ and K+. The low local pH can help to maintain the local CO2 at a high level in the electrolyte with large cations than with small ones, favoring the selective CO2RR and suppressing the HER from the aspect of CO2 supply like Cs+ (Figure 6). Another important factor worth mentioning is the ability to adsorb/stabilize the intermediates, as manifested by the EIS results. Cations play a significant role in the adsorption of CO2 and other intermediates by changing their local concentration and stabilities. The interaction between cations and the species in solution can work through both the medium-range and short-range effects. For the medium-range effects (i.e., field-dipole interactions), the higher concentration of larger cations at OHP can result in stronger electrostatic interaction between cations and the adsorbed intermediates having large dipole moments, and thereby enhance the CO2RR compared to the smaller cations [2]. Moreover, a large cation can interact with more than one intermediate simultaneously, favoring the formation of C2 products [47]. For the short-range (i.e., electrostatic interactions), the stabilization of negatively charged CO2RR intermediates can be strengthened by coordinating with partially dehydrated cations [48]. Thus, various effects played by the electrolyte cations balance in the case of different conditions, eventually leading to various CO2RR performances in diverse electrolytes.
3. Materials and Methods
3.1. Materials
Various metal carbonates (99.999% metal basis) were purchased from Sigma-Aldrich (Shanghai, China). Copper (Cu) foil (99.9999% metal basis) was bought from Alfa Aesar (Shanghai, China). CO2 (≥99.999%) and argon (Ar, ≥99.9992%) were used to purge the electrolyte. Ultra high-purity water (18.2 MΩ cm) obtained from a Millipore system (Molsheim, France) was used through the experiments.
3.2. Electrode and Electrolyte Preparation
Cu foil (Figure S1) was cut into 1 × 1 cm2 pieces, which were cleaned by sonication consecutively in acetone, isopropyl alcohol, and water for 30 min. Then, the Cu electrode was electro-polished at 2 V for 30 min in 85% phosphoric acid, followed by being rinsed with water and dried by Ar stream (Figure S2). Aqueous electrolyte of 0.1 M metal carbonate (M2CO3, M = Li, Na, K, Rb, and Cs) was prepared and purged with Ar for 30 min and CO2 for another 60 min before the measurements.
3.3. Electrochemical Measurements
Electrochemical characterizations were carried out using an IM6 electrochemical workstation (Zahner, Kronach, Germany). Linear scan voltammetry (LSV) and electrochemical impedance spectroscopy (EIS) were performed with a homemade electrochemical cell, using Pt foil as the counter electrode and Ag/AgCl as the reference electrode (Figure S2). The potentials in this work were all converted to RHE scale by using the equation E(vs. RHE) = E(vs. Ag/AgCl) + 0.059pH +0.197. A slight negative potential was first applied to the electrode before all the electrochemical measurements so as to avoid the formation of oxides on the Cu surface. LSV was carried out from 0 to −1 V at a scanning rate of 50 mV s−1. EIS spectra were collected in the frequency range of 1.5 mHz to 100 kHz in a potentiostatic mode with an AC amplitude of 100 mV. The applied bias in EIS was varied from −0.1 to −1.1 V in order to analyze the potential dependent electrocatalytic performance. The above experiments were repeated at least three times to ensure the results were reasonable and reproducible.
4. Conclusions
In summary, LSV and EIS were performed to probe the influence mechanism of different alkali metal cations in electrolytes on the CO2RR over the Cu electrode. The results indicate that the influence of cations is not solely on the basis of their function from one aspect. Several factors, including hydrated cation adsorption, preferential hydrolysis, and interaction between the cation and adsorbed species, should be taken into account comprehensively. The dominant factor in the reaction varies with the external bias and cations. Specifically, at low negative potentials, the alteration of potential at OHP and interaction between the cations and adsorbed species in OHP have an impact on the performance, especially in the case of small cations like Li+ and Na+. When at high negative potentials, the influence of the difference in cation hydrolysis and surface adsorption dominates in the reaction, especially for the large cations of K+, Rb+, and Cs+. Rational control of the delicate balance between these factors should lead to better regulation of the CO2RR over the Cu electrode. We envision that this work will take a step forward in understanding the role of cations in tuning the electrode/electrolyte interface and, thus, the activity and selectivity of the CO2RR.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/catal13071092/s1. Figure S1: SEM images of the Cu electrode (a) before and (b) after electropolishing; Figure S2: Setup for electrochemical measurements.
Author Contributions
Conceptualization, Y.W. and T.H.; methodology, Y.W., T.H. and A.H.S.; validation, Y.W., T.H. and A.H.S.; formal analysis, Y.W. and A.H.S.; investigation, A.H.S., Y.G. and A.R.W.; resources, T.H.; data curation, A.H.S. and Y.G.; writing—original draft preparation, A.H.S.; writing—review and editing, Y.W. and T.H.; supervision, T.H. and Y.W.; project administration, T.H.; funding acquisition, T.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36000000), the BRICS STI Framework Programme (52261145703), and the National Natural Science Foundation of China (21972029). A.H.S. and A.R.W. thank the CAS-TWAS President’s Fellowship for International Ph.D. Students.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Resasco, J.; Chen, L.D.; Clark, E.; Tsai, C.; Hahn, C.; Jaramillo, T.F.; Chan, K.; Bell, A.T. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J. Am. Chem. Soc. 2017, 139, 11277–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Kim, T.; Jung, K.Y.; Park, K.T. Recent progress in electrocatalytic CO2 reduction to pure formic acid using a solid-state electrolyte device. Catalysts 2023, 13, 955. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Zarandi, R.F.; Rezaei, B.; Ghaziaskar, H.S.; Ensafi, A.A. Electrochemical reduction of CO2 to ethanol using copper nanofoam electrode and 1-butyl-3-methyl-imidazolium bromide as the homogeneous co-catalyst. J. Environ. Chem. Eng. 2019, 7, 103141. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Sa, Y.J.; Lee, C.W.; Lee, S.Y.; Na, J.; Lee, U.; Hwang, Y.J. Catalyst-electrolyte interface chemistry for electrochemical CO2 reduction. Chem. Soc. Rev. 2020, 49, 6632–6665. [Google Scholar] [CrossRef]
- Hahn, C.; Hatsukade, T.; Kim, Y.G.; Vailionis, A.; Baricuatro, J.H.; Higgins, D.C.; Nitopi, S.A.; Soriaga, M.P.; Jaramillo, T.F. Engineering Cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons. Proc. Natl. Acad. Sci. USA 2017, 114, 5918–5923. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. A direct grain-boundary-activity Correlation for CO electroreduction on Cu nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Raciti, D.; Livi, K.J.; Wang, C. Highly dense Cu nanowires for low-overpotential CO2 reduction. Nano Lett. 2015, 15, 6829–6835. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Raciti, D.; Li, C.; Livi, K.J.T.; Rottmann, P.F.; Hemker, K.J.; Mueller, T.; Wang, C. Mechanistic insights for low-overpotential electroreduction of CO2 to CO on copper nanowires. ACS Catal. 2017, 7, 8578–8587. [Google Scholar] [CrossRef]
- Li, J.; Chang, K.; Zhang, H.; He, M.; Goddard, W.A.; Chen, J.G.; Cheng, M.-J.; Lu, Q. Effectively increased efficiency for electroreduction of carbon monoxide using supported polycrystalline copper powder electrocatalysts. ACS Catal. 2019, 9, 4709–4718. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Takahashi, I.; Koga, O.; Hoshi, N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A Chem. 2003, 199, 39–47. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, I.; Koga, O.; Hoshi, N. Selective formation of C2 compounds from electrochemical reduction of CO2 at a series of copper single crystal electrodes. J. Phys. Chem. B 2001, 106, 15–17. [Google Scholar] [CrossRef]
- Perez-Gallent, E.; Marcandalli, G.; Figueiredo, M.C.; Calle-Vallejo, F.; Koper, M.T.M. Structure- and potential-dependent cation effects on CO reduction at copper single-crystal electrodes. J. Am. Chem. Soc. 2017, 139, 16412–16419. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Gauthier, J.A.; Dickens, C.F.; Brown, K.S.; Ringe, S.; Chan, K.; Nørskov, J.K. Atomistic insight into cation effects on binding energies in Cu-catalyzed carbon dioxide reduction. J. Phys. Chem. C 2020, 124, 24765–24775. [Google Scholar] [CrossRef]
- Murata, A.; Hori, Y. Product selectivity affected by cationic species in electrochemical reduction of CO2 and CO at a Cu electrode. Bull. Chem. Soc. Jpn. 1991, 64, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.R.; Kwon, Y.; Lum, Y.; Ager, J.W., 3rd; Bell, A.T. Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 2016, 138, 13006–13012. [Google Scholar] [CrossRef] [Green Version]
- Paik, W.; Andersen, T.N.; Eyring, H. Kinetic studies of the electrolytic reduction of carbon dioxide on the mercury electrode. Electrochim. Acta 1969, 14, 1217–1232. [Google Scholar] [CrossRef]
- Frumkin, A.N. Influence of cation adsorption on the kinetics of electrode processes. Trans. Faraday Soc. 1959, 55, 156–167. [Google Scholar] [CrossRef]
- Thorson, M.R.; Siil, K.I.; Kenis, P.J.A. Effect of cations on the electrochemical conversion of CO2 to CO. J. Electrochem. Soc. 2012, 160, F69–F74. [Google Scholar] [CrossRef] [Green Version]
- Kaneco, S.; Iiba, K.; Katsumata, H.; Suzuki, T.; Ohta, K. Effect of sodium cation on the electrochemical reduction of CO2 at a copper electrode in methanol. J. Solid State Electrochem. 2006, 11, 490–495. [Google Scholar] [CrossRef]
- Ringe, S.; Clark, E.L.; Resasco, J.; Walton, A.; Seger, B.; Bell, A.T.; Chan, K. Understanding cation effects in electrochemical CO2 reduction. Energy Environ. Sci. 2019, 12, 3001–3014. [Google Scholar] [CrossRef]
- Wu, J.; Risalvato, F.G.; Ke, F.-S.; Pellechia, P.J.; Zhou, X.-D. Electrochemical reduction of carbon dioxide I. effects of the electrolyte on the selectivity and activity with Sn electrode. J. Electrochem. Soc. 2012, 159, F353–F359. [Google Scholar] [CrossRef]
- Gao, D.; McCrum, I.T.; Deo, S.; Choi, Y.-W.; Scholten, F.; Wan, W.; Chen, J.G.; Janik, M.J.; Roldan Cuenya, B. Activity and selectivity control in CO2 electroreduction to multicarbon products over CuOx catalysts via electrolyte design. ACS Catal. 2018, 8, 10012–10020. [Google Scholar] [CrossRef]
- Xi, C.; Zheng, F.; Gao, G.; Ye, M.; Dong, C.; Du, X.-W.; Wang, L.-W. Distribution of alkali cations near the Cu (111) surface in aqueous solution. J. Mater. Chem. A 2020, 8, 24428–24437. [Google Scholar] [CrossRef]
- Ayemoba, O.; Cuesta, A. Spectroscopic evidence of size-dependent buffering of interfacial pH by cation hydrolysis during CO2 electroreduction. ACS Appl. Mater. Interfaces 2017, 9, 27377–27382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Co, A.C. Direct evidence of local pH change and the role of alkali cation during CO2 electroreduction in aqueous media. Angew Chem. Int. Ed. Engl. 2020, 59, 1674–1681. [Google Scholar] [CrossRef]
- Karthik, P.E.; Jothi, V.R.; Pitchaimuthu, S.; Yi, S.; Anantharaj, S. Alternating current techniques for a better understanding of photoelectrocatalysts. ACS Catal. 2021, 11, 12763–12776. [Google Scholar] [CrossRef]
- Sacco, A. Electrochemical impedance spectroscopy as a tool to investigate the electroreduction of carbon dioxide: A short review. J. CO2 Util. 2018, 27, 22–31. [Google Scholar] [CrossRef]
- Köleli, F.; Röpke, T.; Hamann, C.H. Electrochemical impedance spectroscopic investigation of CO2 reduction on polyaniline in methanol. Electrochim. Acta 2003, 48, 1595–1601. [Google Scholar] [CrossRef]
- Yang, D.-w.; Li, Q.-y.; Shen, F.-x.; Wang, Q.; Li, L.; Song, N.; Dai, Y.-n.; Shi, J. Electrochemical impedance studies of CO2 reduction in ionic liquid/organic solvent electrolyte on Au electrode. Electrochim. Acta 2016, 189, 32–37. [Google Scholar] [CrossRef]
- Bienen, F.; Kopljar, D.; Löwe, A.; Geiger, S.; Wagner, N.; Klemm, E.; Friedrich, K.A. Revealing mechanistic processes in gas-diffusion electrodes during CO2 reduction via impedance spectroscopy. ACS Sustain. Chem. Eng. 2020, 8, 13759–13768. [Google Scholar] [CrossRef]
- Sartori, A.; Orlandi, M.; Berardi, S.; Mazzi, A.; Bazzanella, N.; Caramori, S.; Boaretto, R.; Natali, M.; Fernandes, R.; Patel, N.; et al. Functionalized p-silicon photocathodes for solar fuels applications: Insights from electrochemical impedance spectroscopy. Electrochim. Acta 2018, 271, 472–480. [Google Scholar] [CrossRef]
- Bienen, F.; Kopljar, D.; Geiger, S.; Wagner, N.; Friedrich, K.A. Investigation of CO2 electrolysis on tin foil by electrochemical impedance spectroscopy. ACS Sustain. Chem. Eng. 2020, 8, 5192–5199. [Google Scholar] [CrossRef]
- Marcandalli, G.; Goyal, A.; Koper, M.T.M. Electrolyte effects on the faradaic efficiency of CO2 reduction to CO on a gold electrode. ACS Catal. 2021, 11, 4936–4945. [Google Scholar] [CrossRef]
- Zhang, B.A.; Ozel, T.; Elias, J.S.; Costentin, C.; Nocera, D.G. Interplay of homogeneous reactions, mass transport, and kinetics in determining selectivity of the reduction of CO2 on gold electrodes. ACS Cent. Sci. 2019, 5, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.F.C.; Harrington, D.A.; Marshall, A.T. Effects of mass transfer on the electrocatalytic CO2 reduction on Cu. Electrochim. Acta 2017, 238, 56–63. [Google Scholar] [CrossRef]
- Goyal, A.; Koper, M.T.M. The interrelated effect of cations and electrolyte pH on the hydrogen evolution reaction on gold electrodes in alkaline media. Angew Chem. Int. Ed. Engl. 2021, 60, 13452–13462. [Google Scholar] [CrossRef]
- Gupta, N.; Gattrell, M.; MacDougall, B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2005, 36, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.R.; Clark, E.L.; Bell, A.T. Effects of electrolyte, catalyst, and membrane composition and operating conditions on the performance of solar-driven electrochemical reduction of carbon dioxide. Phys. Chem. Chem. Phys. 2015, 17, 18924–18936. [Google Scholar] [CrossRef] [Green Version]
- Kas, R.; Kortlever, R.; Yılmaz, H.; Koper, M.T.M.; Mul, G. Manipulating the hydrocarbon selectivity of copper nanoparticles in CO2 electroreduction by process conditions. ChemElectroChem 2015, 2, 354–358. [Google Scholar] [CrossRef]
- Bohra, D.; Chaudhry, J.H.; Burdyny, T.; Pidko, E.A.; Smith, W.A. Modeling the electrical double layer to understand the reaction environment in a CO2 electrocatalytic system. Energy Environ. Sci. 2019, 12, 3380–3389. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Murata, A.; Takahashi, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 1 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Dunwell, M.; Yang, X.; Setzler, B.P.; Anibal, J.; Yan, Y.; Xu, B. Examination of near-electrode concentration gradients and kinetic impacts on the electrochemical reduction of CO2 using surface-enhanced infrared spectroscopy. ACS Catal. 2018, 8, 3999–4008. [Google Scholar] [CrossRef]
- Liu, H.; Liu, J.; Yang, B. Promotional role of a cation intermediate complex in C2 formation from electrochemical reduction of CO2 over Cu. ACS Catal. 2021, 11, 12336–12343. [Google Scholar] [CrossRef]
- Monteiro, M.C.O.; Dattila, F.; Hagedoorn, B.; García-Muelas, R.; López, N.; Koper, M.T.M. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 2021, 4, 654–662. [Google Scholar] [CrossRef]
Figure 1.
LSV curves of Cu electrode in 0.1 M CO2-saturated electrolytes. (a) Li2CO3 and Na2CO3 and (b) K2CO3, Rb2CO3, and Cs2CO3. Scanning rate of 50 mV s−1.
Figure 1.
LSV curves of Cu electrode in 0.1 M CO2-saturated electrolytes. (a) Li2CO3 and Na2CO3 and (b) K2CO3, Rb2CO3, and Cs2CO3. Scanning rate of 50 mV s−1.
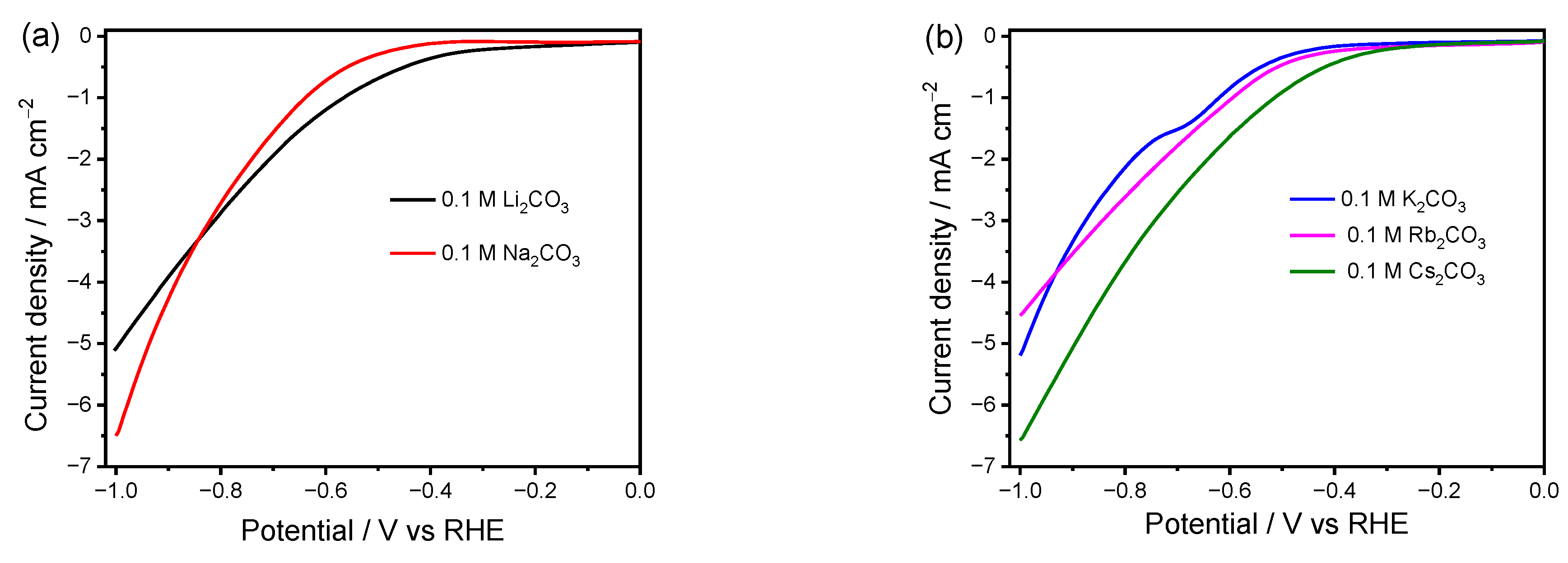
Figure 2.
EIS spectra of Cu electrode in CO2-saturated 0.1 M Li2CO3 and Na2CO3 under less negative bias voltages (−0.1 to −0.5 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
Figure 2.
EIS spectra of Cu electrode in CO2-saturated 0.1 M Li2CO3 and Na2CO3 under less negative bias voltages (−0.1 to −0.5 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.

Figure 3.
EIS spectra of Cu electrode in CO2-saturated 0.1 M Li2CO3 and Na2CO3 under more negative bias voltage (−0.6 to −1.1 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
Figure 3.
EIS spectra of Cu electrode in CO2-saturated 0.1 M Li2CO3 and Na2CO3 under more negative bias voltage (−0.6 to −1.1 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
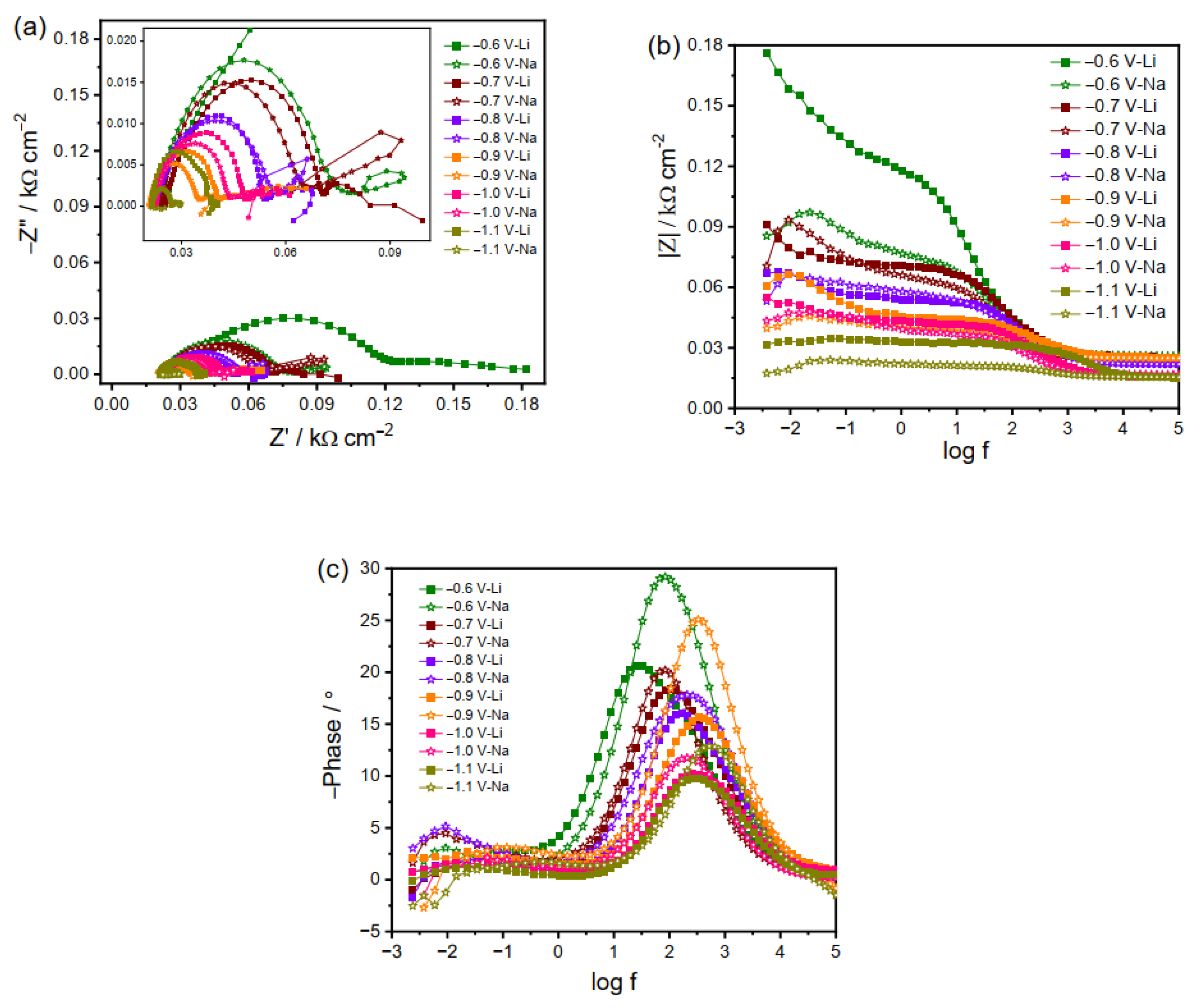
Figure 4.
EIS spectra of Cu electrode in CO2-saturated 0.1 M K2CO3, Rb2CO3, and Cs2CO3 at less negative bias voltage (−0.1 to −0.5 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
Figure 4.
EIS spectra of Cu electrode in CO2-saturated 0.1 M K2CO3, Rb2CO3, and Cs2CO3 at less negative bias voltage (−0.1 to −0.5 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
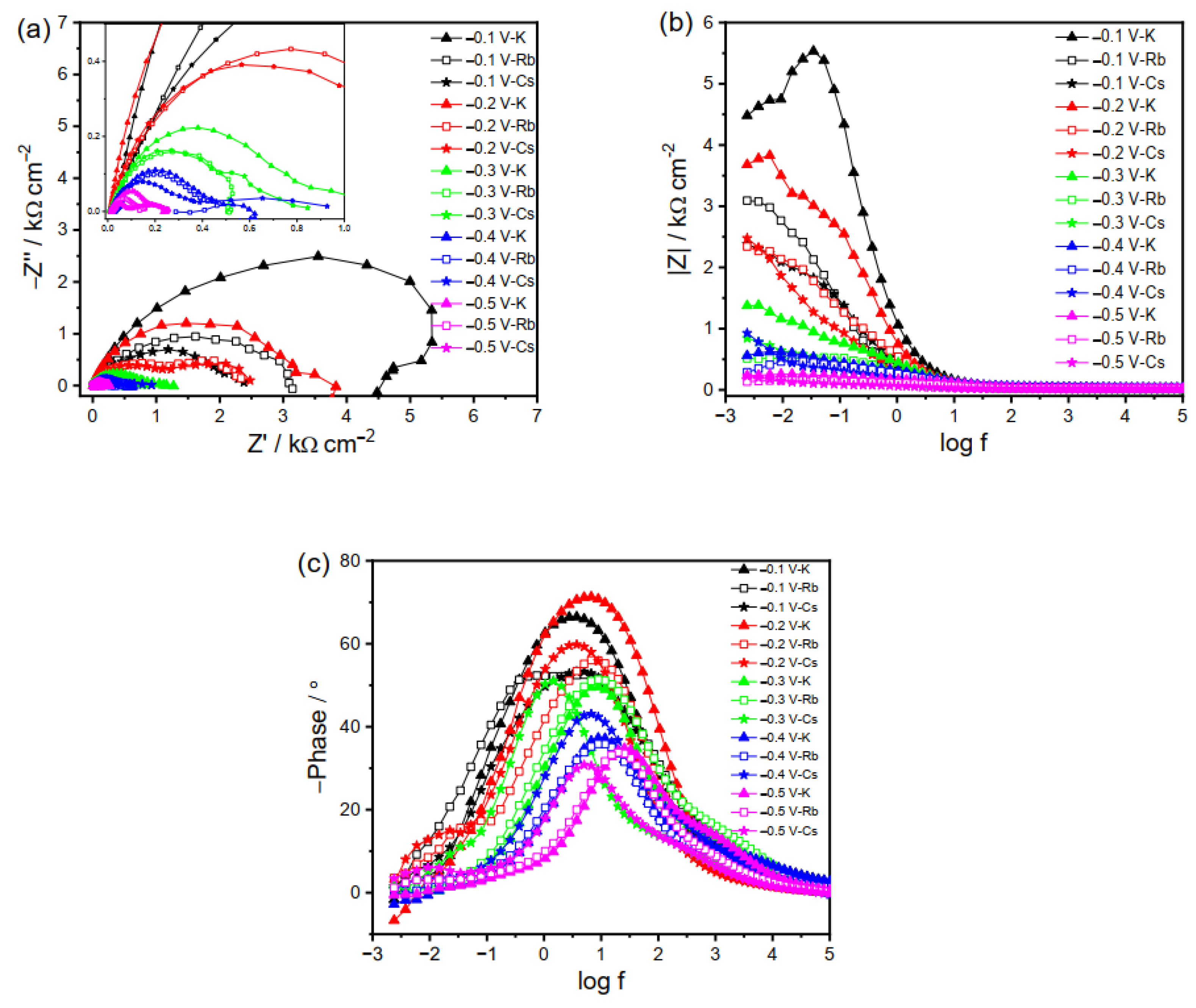
Figure 5.
EIS spectra of Cu electrode in CO2-saturated 0.1 M K2CO3, Rb2CO3, and Cs2CO3 under more negative bias voltages (−0.6 to −1.1 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
Figure 5.
EIS spectra of Cu electrode in CO2-saturated 0.1 M K2CO3, Rb2CO3, and Cs2CO3 under more negative bias voltages (−0.6 to −1.1 V). (a) Nyquist plots and corresponding Bode plots of (b) impedance vs. frequency and (c) phase vs. frequency. Inset is zoom-in of the figure.
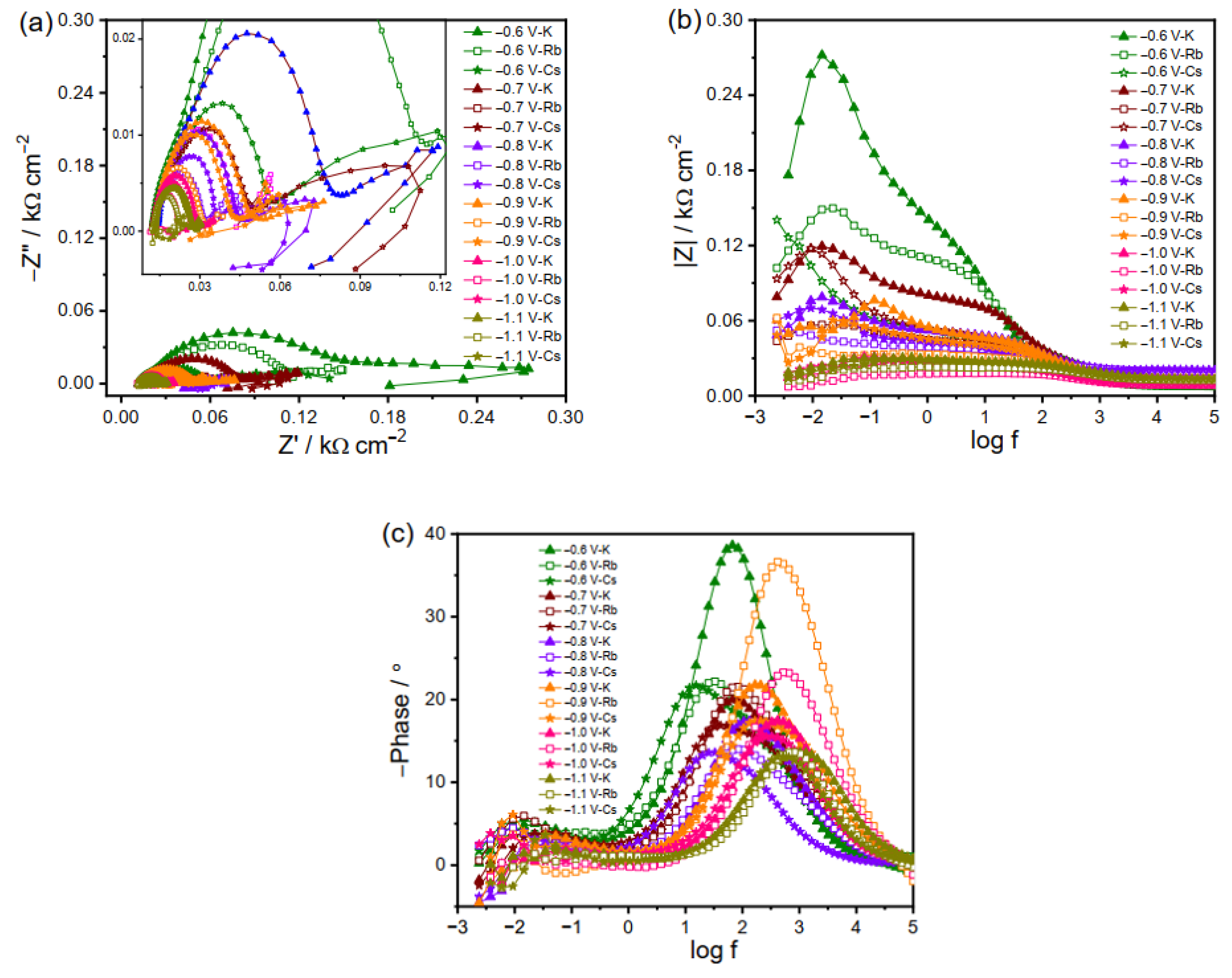
Figure 6.
Effects of electrolyte cations on CO2RR.
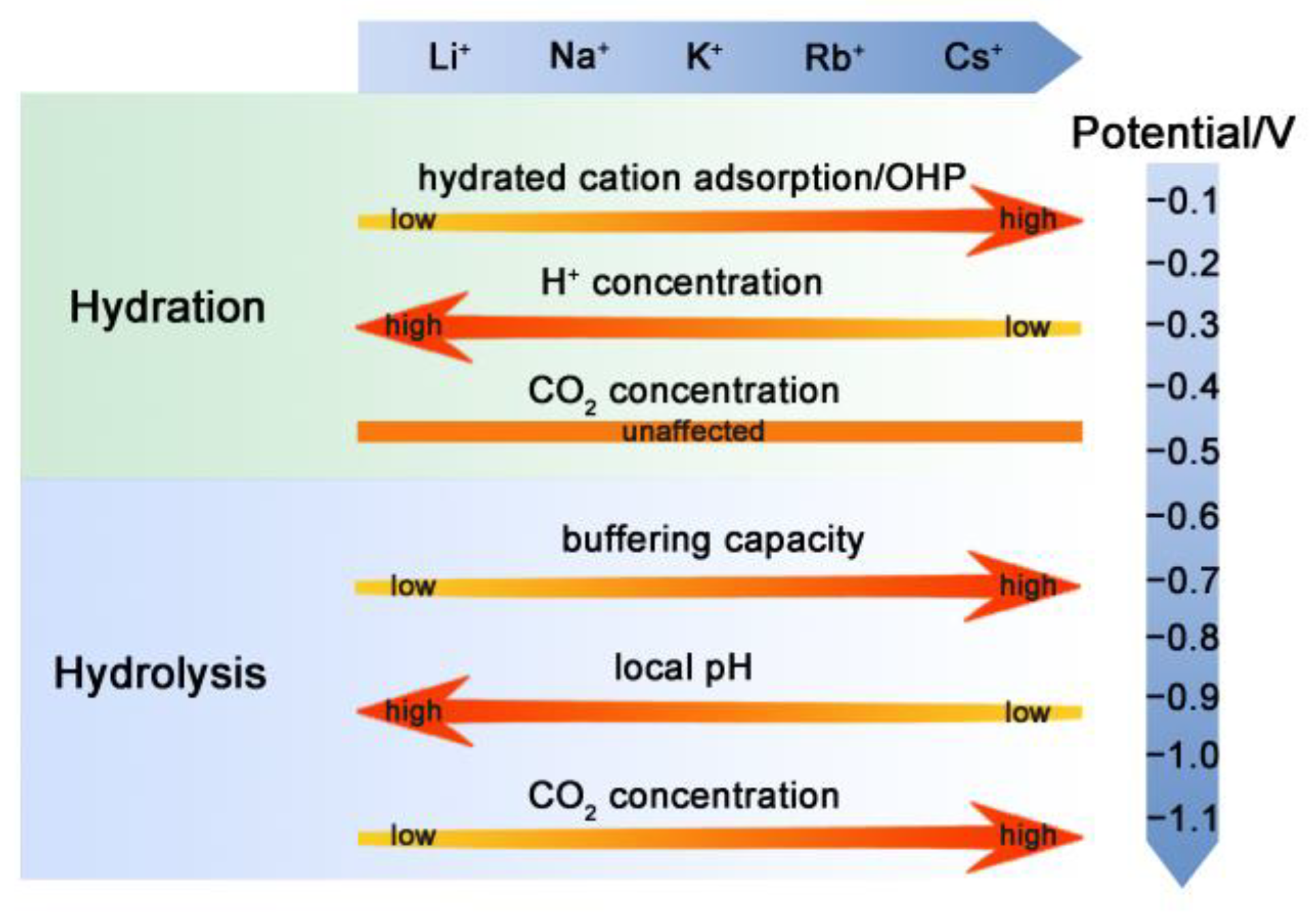
Figure 7.
Illustration of cation hydration on the surface of Cu electrode during the CO2RR in aqueous carbonate media: (a) small and (b) large alkali metallic cations.
Figure 7.
Illustration of cation hydration on the surface of Cu electrode during the CO2RR in aqueous carbonate media: (a) small and (b) large alkali metallic cations.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shah, A.H.; Gong, Y.; Wang, Y.; Woldu, A.R.; He, T. Understanding the Role of Electrolyte Cations on Activity and Product Selectivity of CO2 Reduction over Cu Electrode. Catalysts 2023, 13, 1092. https://doi.org/10.3390/catal13071092
AMA Style
Shah AH, Gong Y, Wang Y, Woldu AR, He T. Understanding the Role of Electrolyte Cations on Activity and Product Selectivity of CO2 Reduction over Cu Electrode. Catalysts. 2023; 13(7):1092. https://doi.org/10.3390/catal13071092
Chicago/Turabian StyleShah, Aamir Hassan, Yue Gong, Yanjie Wang, Abebe Reda Woldu, and Tao He. 2023. "Understanding the Role of Electrolyte Cations on Activity and Product Selectivity of CO2 Reduction over Cu Electrode" Catalysts 13, no. 7: 1092. https://doi.org/10.3390/catal13071092
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.