
Synthesis of New Dehydrodieugenol Derivatives via Olefin Cross Metathesis and In Vitro Evaluation of Their Trypanocidal Activity

 , , , and
, , , and
Abstract
:
1. Introduction
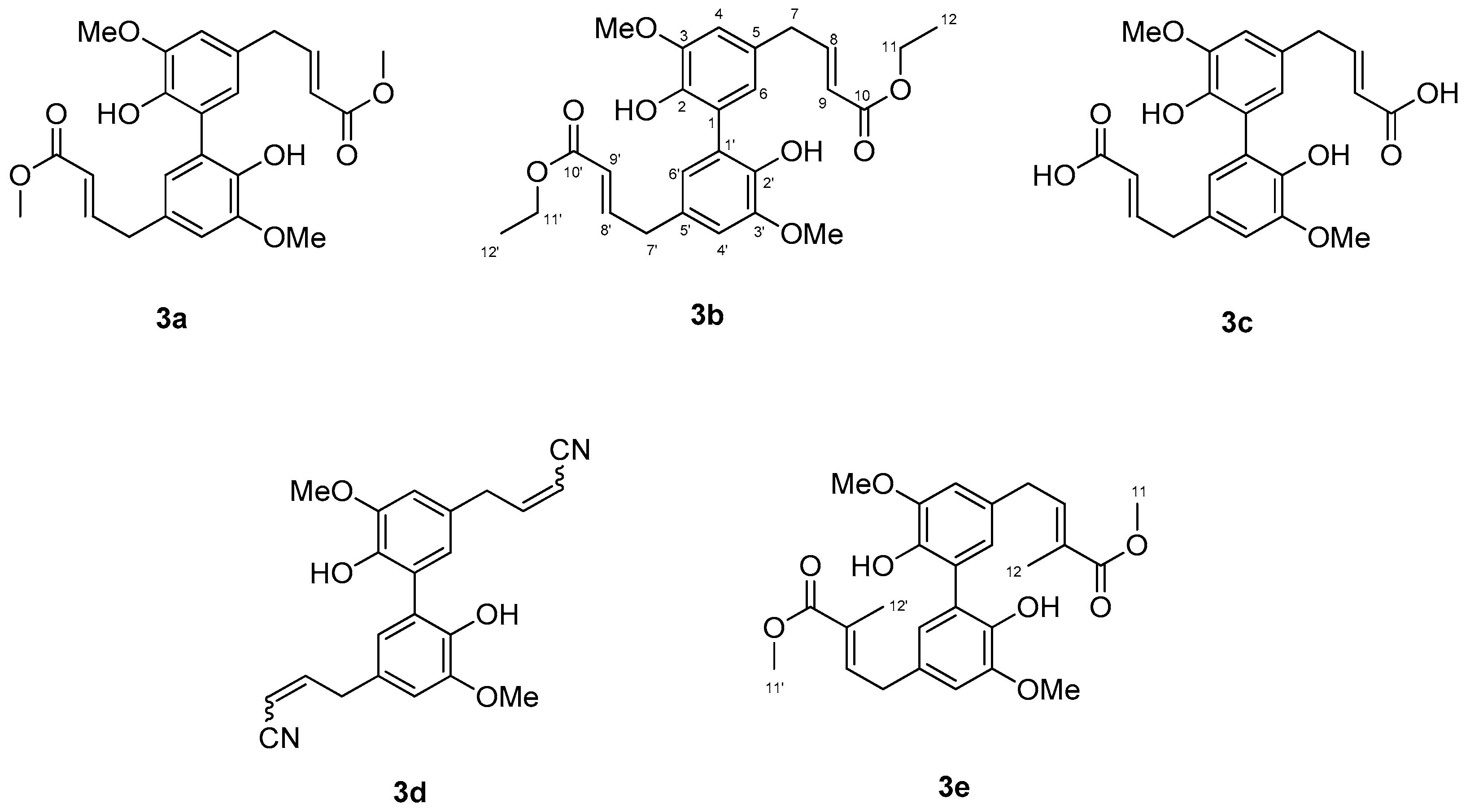
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Preparation of Dehydrodieugenol (1)
Dimethoxy-5,5′-diprop-2-enyl [1,1′-biphenyl]-2,2′-diol (biseugenol) 1
3.3. General Procedure for Cross Metathesis Reactions
- i.
- methyl acrylate (2a, 56 μL) to afford compound 3a
- ii.
- ethyl acrylate (2b, 65 μL) to afford compound 3b
- iii.
- acrylic acid (2c, 42 μL) to afford compound 3c
- iv.
- acrylonitrile (2d, 40 μL) to afford compound 3d
- v.
- methyl methacrylate (2e, 2 mL) to afford compound 3e
3.3.1. (9. E,9’E)-dimethyl 7,7’-(2,2’-dihydroxy-3,3’-dimethoxy-[1,1’-biphenyl]-5,5’-diyl)bis(but-9-enoate), 3a
3.3.2. (9. E,9’E)-diethyl 7,7’-(2,2’-dihydroxy-3,3’-dimethoxy-[1,1’-biphenyl]-5,5’-diyl)bis(but-2-enoate) 3b
3.3.3. (9. E,9’E)-7,7’-(2,2’-dihydroxy-3,3’-dimethoxy-[1,1’-biphenyl]-5,5’-diyl)bis(but-2-enoic acid) 3c
3.3.4. 7,7’-(2,2’-dihydroxy-3,3’-dimethoxy-[1,1’-biphenyl]-5,5’-diyl)bis(but-2-enenitrile) 3d
3.3.5. (9. E,9’E)-dimethyl 7,7’-(2,2’-dihydroxy-3,3’-dimethoxy-[1,1’-biphenyl]-5,5’-diyl)bis(9-methylbut-9-enoate) 3e
3.4. Experimental Animals
3.5. Parasites and Mammalian Cell Maintenance
3.6. Determination of Anti-Trypomastigote and Anti-Amastigote Activity
3.7. Determination of Cytotoxicity against Mammalian Cells
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crane, E.A.; Gademann, K. Capturing biological activity in natural product fragments by chemical synthesis. Angew. Chem. Int. Ed. 2016, 55, 3882–3902. [Google Scholar] [CrossRef] [Green Version]
- Szychowski, J.; Truchon, J.-F.; Bennani, Y.L. Natural Products in Medicine: Transformational Outcome of Synthetic Chemistry. J. Med. Chem. 2014, 57, 9292–9308. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [Green Version]
- Wenqiang, G.; Shufen, L.; Ruixiang, Y.; Shaokun, T.; Can, Q. Comparison of essential oils of clove buds extracted with supercritical carbon dioxide and other three traditional extraction methods. Food Chem. 2007, 101, 1558–1564. [Google Scholar]
- Juhász, L.; Kürti, L.; Antus, S. Simple Synthesis of Benzofuranoid Neolignans from Myristica fragrans. J. Nat. Prod. 2000, 63, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.; Fabbri, D.; Dettori, M.A.; Forni, A.; Casalone, G. Enantiopure 2,2′-dihydroxy-3,3′-dimethoxy-5,5′-diallyl-6,6′-dibromo-1,1′-biphenyl: A conformationally stable C2-dimer of a eugenol derivative. Tetrahedron Asymmetry 2004, 15, 275–282. [Google Scholar] [CrossRef]
- Fujisawa, S.; Kashiwagi, Y.; Atsumi, T.; Iwakura, I.; Ueha, T.; Hibino, Y.; Yokoe, I. Application of bis-eugenol to a zinc oxide eugenol cement. J. Dent. 1999, 27, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Barratt, M.D.; Basketter, D.A. Possible origin of the skin sensitization potential of isoeugenol and related compounds. Contact Dermat. 1992, 27, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Takeyoshi, M.; Noda, S.; Yamazaki, S.; Kakishima, H.; Yamasaki, K.; Kimber, I. Assessment of the skin sensitization potency of eugenol and its dimers using a non-radioisotopic modification of the local lymph node assay. J. Appl. Toxicol. 2004, 24, 77–81. [Google Scholar] [CrossRef]
- Gerosa, R.; Borin, M.; Menegazzi, G.; Puttini, M.; Cavalleri, G. In vitro evaluation of the cytotoxicity of pure eugenol. J. Endod. 1996, 22, 532–534. [Google Scholar] [CrossRef]
- De Diaz, A.M.; Gottlieb, H.E.; Gottlieb, O.R. Dehydrodieugenols from Ocotea cymbarum. Phytochemistry 1980, 19, 681–682. [Google Scholar] [CrossRef]
- Ogata, M.; Hoshi, M.; Urano, S.; Endo, T. Antioxidant Activity of Eugenol and Related Monomeric and Dimeric Compounds. Chem. Pharm. Bull. 2000, 48, 1467–1469. [Google Scholar] [CrossRef] [Green Version]
- Bortolomeazzi, R.; Verardo, G.; Liessi, A.; Callea, A. Formation of dehydrodiisoeugenol and dehydrodieugenol from the reaction of isoeugenol and eugenol with DPPH radical and their role in the radical scavenging activity. Food Chem. 2010, 118, 256–265. [Google Scholar] [CrossRef]
- Murakami, Y.; Shoji, M.; Hanazawa, S.; Tanaka, S.; Fujisawa, S. Preventive effect of bis-eugenol, a eugenol ortho dimer, on lipopolysaccharide-stimulated nuclear factor kappa B activation and inflammatory cytokine expression in macrophages. Biochem. Pharmacol. 2003, 66, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Shoji, M.; Hirata, A.; Tanaka, S.; Yokoe, I.; Fujisawa, S. Dehydrodiisoeugenol, an isoeugenol dimer, inhibits lipopolysaccharide-stimulated nuclear factor kappa B activation and cyclooxygenase-2 expression in macrophages. Arch. Biochem. Biophys. 2005, 434, 326–332. [Google Scholar] [CrossRef]
- Fujisawa, S.; Ishihara, M.; Yokoe, I. Computer-Aided Synthesis of Dimerized Eugenol. Int. Elec. J. Mol. Des. 2004, 3, 241–246. [Google Scholar]
- Shen, J.-L.; Man, K.-M.; Huang, P.-H.; Chen, W.-C.; Chen, D.-C.; Cheng, Y.-W.; Liu, P.-L.; Chou, M.-C.; Chen, Y.-H. Honokiol and Magnolol as Multifunctional Antioxidative Molecules for Dermatologic Disorders. Molecules 2010, 15, 6452–6465. [Google Scholar] [CrossRef] [Green Version]
- Mascia, M.P.; Fabbri, D.; Dettori, M.A.; Ledda, G.; Delogu, G.; Biggio, G. Hydroxylated biphenyl derivatives are positive modulators of human GABAA receptors. Eur. J. Pharmacol. 2012, 693, 45–50. [Google Scholar] [CrossRef]
- Wu, L.; Zou, H.; Xia, W.; Wang, L. Role of magnolol in the proliferation of vascular smooth muscle cells. Herz 2015, 40, 542–548. [Google Scholar] [CrossRef]
- Kleiner, H.E.; Vulimiri, S.V.; Miller, L.; Johnson, W.H., Jr.; Whitman, C.P.; DiGiovanni, J. Oral administration of naturally occurring coumarins leads to altered phase I and II enzyme activities and reduced DNA adduct formation by polycyclic aromatic hydrocarbons in various tissues of SENCAR mice. Carcinogenesis 2001, 22, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.-Y.; Tsai, C.-C.; Chen, C.-P.; Huang, J.-S.; Lin, C.-C. Antimicrobial activity of honokiol and magnolol isolated from Magnolia officinalis. Phytother. Res. 2001, 15, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Geng, Y.; Li, F.; Zheng, C. Isolation and purification of honokiol and magnolol from cortex Magnoliae officinalis by high-speed counter-current chromatography. J. Chromatogr. A 2004, 1036, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-R.; Chen, H.-H.; Ko, C.-H.; Chan, M.-H. Effects of honokiol and magnolol on acute and inflammatory pain models in mice. Life Sci. 2007, 81, 1071–1078. [Google Scholar] [CrossRef]
- Wang, J.-H.; Shih, K.-S.; Liou, J.-P.; Wu, Y.-W.; Chang, A.S.-Y.; Wang, K.-L.; Tsai, C.-L.; Yang, C.-R. Anti-Arthritic Effects of Magnolol in Human Interleukin 1β-Stimulated Fibroblast-Like Synoviocytes and in a Rat Arthritis Model. PLoS ONE 2012, 7, e31368. [Google Scholar] [CrossRef] [PubMed]
- Jada, S.; Doma, M.R.; Singh, P.P.; Kumar, S.; Malik, F.; Sharma, A.; Khan, I.A.; Qazi, G.N.; Kumar, H.M.S. Design and synthesis of novel magnolol derivatives as potential antimicrobial and antiproliferative compounds. Eur. J. Med. Chem. 2012, 51, 35–41. [Google Scholar] [CrossRef]
- Ikeda, K.; Nagase, H. Magnolol Has the Ability to Induce Apoptosis in Tumor Cells. Biol. Pharm. Bull. 2002, 25, 1546–1549. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Tang, W.; Du, G.; Kokudo, N. Targeting apoptosis pathways in cancer with magnolol and honokiol, bioactive constituents of the bark of Magnolia officinalis. Drug Discov. Ther. 2011, 5, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, M.; Hisama, M. Antimutagenic Activity of Phenylpropanoids from Clove (Syzygium aromaticum). J. Agric. Food Chem. 2003, 51, 6413–6422. [Google Scholar] [CrossRef]
- Okada, N.; Hirata, A.; Murakami, Y.; Shoji, M.; Sakagami, H.; Fujisawa, S. Induction of cytotoxicity and apoptosis and inhibition of cyclooxygenase-2 gene expression by eugenol-related compounds. Anticancer Res. 2005, 25, 3263–3269. [Google Scholar]
- Cho, W.C.-S. Nasopharyngeal carcinoma: Molecular biomarker discovery and progress. Mol. Cancer 2007, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Grecco, S.S.; Costa-Silva, T.A.; Jerz, G.; de Sousa, F.S.; Conserva, G.A.A.; Mesquita, J.T.; Galuppo, M.K.; Tempone, A.G.; Neves, B.J.; Andrade, C.H.; et al. Antitrypanosomal activity and evaluation of the mechanism of action of dehydrodieugenol isolated from Nectandra leucantha (Lauraceae) and its methylated derivative against Trypanosoma cruzi. Phytomedicine 2017, 24, 62–67. [Google Scholar] [CrossRef]
- Higman, C.S.; Lummiss, J.A.M.; Fogg, D.E. Olefin metathesis at the dawn of implementation in pharmaceutical and specialty-chemicals manufacturing. Angew. Chem. Int. Ed. 2016, 55, 3552–3565. [Google Scholar] [CrossRef]
- Van Lierop, B.J.; Lummiss, J.A.M.; Fogg, D.E. Olefin Metathesis, Theory and Practice; Grela, K., Ed.; Wiley: Hoboken, NJ, USA, 2014; pp. 85–152. [Google Scholar]
- Cossy, J. Applications of olefin metathesis reactions: Applications in the synthesis of natural products and biologically active molecules. In Olefin Metathesis, Theory and Practice; Grela, K., Ed.; Wiley & Sons Inc.: Hoboken, NJ, USA, 2014; pp. 287–309. [Google Scholar]
- Cossy, J.; Willis, C.; Bellosta, V. Enantioselective allyltitanation and cross metathesis. synthesis of (−)-prosophylline. Synlett 2001, 10, 1578–1580. [Google Scholar] [CrossRef]
- Van Zijl, A.W.; Szymanski, W.; López, F.; Minnaard, A.J.; Feringa, B.L. Catalytic Enantioselective Synthesis of Vicinal Dialkyl Arrays. J. Org. Chem. 2008, 73, 6994–7002. [Google Scholar] [CrossRef] [PubMed]
- Bressin, R.K.; Osman, S.; Pohorilets, I.; Basu, U.; Koide, K. Total Synthesis of Meayamycin B. J. Org. Chem. 2020, 85, 4637–4647. [Google Scholar] [CrossRef]
- Li, J.; Ahmed, T.S.; Xu, C.; Stoltz, B.M.; Grubbs, R.H. Concise Syntheses of Δ12-Prostaglandin J Natural Products via Stereoretentive Metathesis. J. Am. Chem. Soc. 2019, 141, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Brummer, O.; Ruckert, A.; Blechert, S. Olefin cross metathesis with monosubstituted olefins. Chem. Eur. J. 1997, 3, 441–446. [Google Scholar] [CrossRef]
- Bilel, H.; Hamdi, N.; Fischmeister, C.; Bruneau, C. Transformations of bio-sourced 4-hydroxyphenylpropanoids based on olefin metathesis. ChemCatChem 2020, 12, 5000–5021. [Google Scholar] [CrossRef]
- Zaja, M.; Blechert, S. Concise enantioselective synthesis of (−)-lasubine II. Tetrahedron 2004, 60, 9629–9634. [Google Scholar] [CrossRef]
- Bilel, H.; Hamdi, N.; Zagrouba, F.; Fischmeister, C.; Bruneau, C. Eugenol as a renewable feedstock for the production of polyfunctional alkenes via olefin cross metathesis. RSC Adv. 2012, 2, 9584–9589. [Google Scholar] [CrossRef]
- Vieira, G.M.; Granato, A.V.; Gusevskaya, E.V.; dos Santos, E.N.; Dixneuf, P.H.; Fischmeister, C.; Bruneau, C. Tandem hydroformylation/isomerization/hydrogenation of bio-derived 1-arylbutadienes for the regioselective synthesis of branched aldehydes. Appl. Catal. A Gen. 2020, 598, 117583. [Google Scholar] [CrossRef]
- Dixneuf, P.H.; Bruneau, C.; Fischmeister, C. Alkene Metathesis Catalysis: A Key for Transformations of Unsaturated Plant Oils and Renewable Derivatives. Oil Gas Sci. Technol. 2016, 71, 19. [Google Scholar] [CrossRef] [Green Version]
- Galhardo, T.S.; Ueno, A.K.; Costa-Silva, T.A.; Tempone, A.G.; Carvalho, W.A.; Fischmeister, C.; Bruneau, C.; Mandelli, D.; Lago, J.H.G. New derivatives from dehydrodieugenol B and its methyl ether displayed high anti-Trypanosoma cruzi activity and cause depolarization of the plasma membrane and collapse the mitochondrial membrane potential. Chem. Biol. Interact. 2022, 366, 110129. [Google Scholar] [CrossRef]
- Rodrigues, L.C.; Barbosa-Filho, J.M.; de Oliveira, M.R.; Néris, P.L.N.; Borges, F.V.P.; Mioso, R. Synthesis and Antileishmanial Activity of Natural Dehydrodieugenol and Its Mono- and Dimethyl Ethers. Chem. Biodivers. 2016, 13, 870–874. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Hassam, M.; Taher, A.; Arnott, G.E.; Green, I.R.; van Otterlo, W.A.L. Isomerization of allylbenzenes. Chem. Rev. 2015, 115, 5462–5569. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Dimethyl Carbonate: An Eco-Friendly Solvent in Ruthenium-Catalyzed Olefin Metathesis Transformations. Chemsuschem 2008, 1, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Dixneuf, P.H.; Fischmeister, C.; Bruneau, C. A green route to nitrogen-containing groups: The acrylonitrile cross metathesis and applications to plant oil derivatives. Green Chem. 2011, 13, 2258–2271. [Google Scholar] [CrossRef]
- Miao, X.; Malacea, R.; Fischmeister, C.; Bruneau, C.; Dixneuf, P.H. Ruthenium–alkylidene catalysed cross metathesis of fatty acid derivatives with acrylonitrile and methyl acrylate: A key step toward long-chain bifunctional and amino acid compounds. Green Chem. 2011, 13, 2911–2919. [Google Scholar] [CrossRef]
- Bidange, J.; Fischmeister, C.; Bruneau, C.; Dubois, J.-L.; Couturier, J.-L. Cross metathesis of bio-sourced fatty nitriles with acrylonitrile. Monatsh. Chem. 2015, 146, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Bruneau, C.; Fischmeister, C.; Miao, X.; Malacea, R.; Dixneuf, P.H. Cross metathesis with acrylonitrile and applications to fatty acid derivatives. Eur. J. Lipid Sci. Technol. 2010, 112, 3–9. [Google Scholar] [CrossRef]
- Torker, S.; Koh, M.J.; Khan, R.K.M.; Hoveyda, A.H. Regarding a persisting puzzle in olefin metathesis with ru complexes: Why are transformations of alkenes with a small substituent Z-selective? Organometallics 2016, 35, 543–562. [Google Scholar] [CrossRef]
- Don, R.; Ioset, J.-R. Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology 2014, 141, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Rea, A.; Tempone, A.G.; Pinto, E.G.; Mesquita, J.T.; Rodrigues, E.; Silva, L.G.M.; Sartorelli, P.; Lago, J.H.G. Soulamarin Isolated from Calophyllum brasiliense (Clusiaceae) Induces Plasma Membrane Permeabilization of Trypanosoma cruzi and Mytochondrial Dysfunction. PLoS Neglected Trop. Dis. 2013, 7, e2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, H.; Shiho, O.; Kuroshima, K.; Koyama, M.; Tsukamoto, K. An improved colorimetric assay for interleukin 2. J. Immunol. Methods 1986, 93, 157–165. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 2a (Eq.) | DMC (mL) | Ru1 (mol%) | Time (h) | Conversion (%) |
|---|---|---|---|---|---|
| 1 | 4 | 2 | 2 | 16 | 91 |
| 2 | 4 | 2 | 2 | (2 + 3) b | 96 |
| 3 | 4 | 2 | 4 | (2 + 3) b | 97 |
| 4 | 12 | 2 | 2 | (2 + 3) b | 96 |
| 5 | 4 | 10 | 2 | (2 + 3) b | 87 |
| Entry | Substrate | Olefin | Product | Conversion (%) | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1 | Methyl acrylate (2a) | 3a | 96 | 52 |
| 2 | 1 | Ethyl acrylate (2b) | 3b | 94 | 25 |
| 3 | 1 | Acrylic acid (2c) | 3c | 71 | 32 |
| 4 b | 1 | Acrylonitrile (2d) | 3d | 88 | 51 |
| 5 c | 1 | Methyl methacrylate (2e) | 3e | 81 | 29 |
| Compound | IC50/µM | CC50/µM | SI | ||
|---|---|---|---|---|---|
| Trypomastigote | Amastigote | NCTC | Trypomastigote | Amastigote | |
| 1 | 11.5 ± 2.5 | 15.1 ± 3.4 | 58.2 | 3.9 | 3.8 |
| 3a | 19.9 ± 0.1 | NA | >200 | >10.0 | - |
| 3b | 11.9 ± 0.7 | 17.7 ± 8.0 | >200 | >16.8 | >11.2 |
| 3c | 79.5 ± 9.3 | NA | >200 | >2.5 | - |
| 3d | 23.5 ± 2.0 | 15.5 ± 1.8 | >200 | >8.5 | >12.9 |
| 3e | 11.6 ± 2.1 | NA | >200 | >17.2 | - |
| benznidazole | 5.5 ± 0.9 | 18.7 ± 4.1 | >200 | >36.4 | >10.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galhardo, T.S.; Ueno, A.K.; Carvalho, W.A.; Costa-Silva, T.A.; Gonçalves, M.M.; Abiuzi, M.B.; Tempone, A.G.; Lago, J.H.G.; Mandelli, D.; Fischmeister, C.; et al. Synthesis of New Dehydrodieugenol Derivatives via Olefin Cross Metathesis and In Vitro Evaluation of Their Trypanocidal Activity. Catalysts 2023, 13, 1097. https://doi.org/10.3390/catal13071097
Galhardo TS, Ueno AK, Carvalho WA, Costa-Silva TA, Gonçalves MM, Abiuzi MB, Tempone AG, Lago JHG, Mandelli D, Fischmeister C, et al. Synthesis of New Dehydrodieugenol Derivatives via Olefin Cross Metathesis and In Vitro Evaluation of Their Trypanocidal Activity. Catalysts. 2023; 13(7):1097. https://doi.org/10.3390/catal13071097
Chicago/Turabian StyleGalhardo, Thalita S., Anderson K. Ueno, Wagner A. Carvalho, Thais A. Costa-Silva, Marina M. Gonçalves, Mariana B. Abiuzi, Andre G. Tempone, João Henrique G. Lago, Dalmo Mandelli, Cedric Fischmeister, and et al. 2023. "Synthesis of New Dehydrodieugenol Derivatives via Olefin Cross Metathesis and In Vitro Evaluation of Their Trypanocidal Activity" Catalysts 13, no. 7: 1097. https://doi.org/10.3390/catal13071097