
Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis/Kinetic Resolution of Racemic Planar-Chiral 1,1′-Diallylferrocenes

Abstract
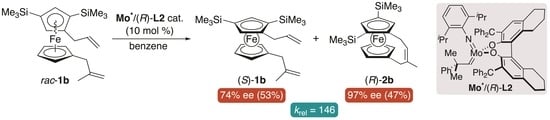
:
1. Introduction
2. Results and Discussion
2.1. Design and Preparation of Racemic Planar-Chiral 1,1′-Diallylferrocene Substrates 1a-c for the Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis/Kinetic Resolution
2.2. Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis/Kinetic Resolution of Racemic Planar-Chiral 1,1′-Diallylferroce Substrates 1a-c: Catalyst Screening Studies
3. Materials and Methods
3.1. General Information
3.2. Preparation of Racemic Diallylferrocene Substrates rac-1a-d [44]
3.3. Characterization Data of Racemic 1,1′-Diallylferrocene Substrates 1a-d [44]
3.4. General Procedure for Molybdenum-Catalyzed Enantioselective RCM/Kinetic Resolution of rac-1
3.5. Characterization Data of Ferrocenophanes 2a-d [44]
3.6. Calculation of Selectivity Factors “krel” in Table 1 and in Scheme 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Halterman, R.L. Synthesis and Applications of Chiral Cyclopentadienylmetal Complexes. Chem. Rev. 1992, 92, 965–994. [Google Scholar] [CrossRef]
- Wagner, G.; Herrmann, R. Chiral Ferrocene Derivatives. An Introduction. In Ferrocenes; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; Chapter 4; pp. 173–218. [Google Scholar]
- Togni, A. Planar-chiral Ferrocenes: Synthetic Methods and Applications. Angew. Chem. Int. Ed. 1996, 35, 1475–1477. [Google Scholar] [CrossRef]
- Richards, C.J.; Locke, A.J. Recent Advances in the Generation of Non-racemic Ferrocene Derivatives and Their Application to Asymmetric Synthesis. Tetrahedron Asymmetry 1998, 9, 2377–2407. [Google Scholar] [CrossRef]
- Štěpnička, P.; Lamač, M. Synthesis and Catalytic Use of Planar Chiral and Polydentate Ferrocene Donors. In Ferrocenes; Štěpnička, P., Ed.; Wiley: Chichester, UK, 2008; Chapter 7; pp. 237–277. [Google Scholar]
- Alba, A.-N.R.; Rios, R. Kinetic Resolution: A Powerful Tool for the Synthesis of Planar-chiral Ferrocenes. Molecules 2009, 14, 4747–4757. [Google Scholar] [CrossRef] [PubMed]
- Schaarschmidt, D.; Lang, H. Selective Syntheses of Planar-chiral Ferrocenes. Organometallics 2013, 32, 5668–5704. [Google Scholar] [CrossRef]
- Arae, S.; Ogasawara, M. Catalytic Asymmetric Synthesis of Planar-chiral Transition-metal Complexes. Tetrahedron Lett. 2015, 56, 1751–1761. [Google Scholar] [CrossRef]
- Hayashi, T. Asymmetric Catalysis with Chiral Ferrocenylphosphine Ligands. In Ferrocenes; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; Chapter 2; pp. 105–142. [Google Scholar]
- Togni, A. New Chiral Ferrocenyl Ligands for Asymmetric Catalysis. In Metallocenes; Togni, A., Halterman, R.L., Eds.; Wiley-VCH: Weinheim, Germany, 1998; Volume 2, Chapter 11; pp. 685–721. [Google Scholar]
- Colacot, T.J. A Concise Update on the Applications of Chiral Ferrocenyl Phosphines in Homogeneous Catalysis Leading to Organic Synthesis. Chem. Rev. 2003, 103, 3101–3118. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.X.; Tu, T.; You, S.L.; Deng, W.P.; Hou, X.L. Asymmetric Catalysis with Chiral Ferrocene Ligands. Acc. Chem. Res. 2003, 36, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, P.; Bianchini, C.; Giambastiani, G.; Parisel, S.L. Progress in Stereoselective Catalysis by Metal Complexes with Chiral Ferrocenyl Phosphines. Coord. Chem. Rev. 2004, 248, 2131–3150. [Google Scholar] [CrossRef]
- Arrayás, R.G.; Adrio, J.; Carretero, J.C. Recent Applications of Chiral Ferrocene Ligands in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef]
- Fu, G.C. Applications of Planar-chiral Heterocycles as Ligands in Asymmetric Catalysis. Acc. Chem. Res. 2006, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Ganter, C. Planar Chiral Phosphaferrocene-based Ligands. In Phosphorus Ligands in Asymmetric Catalysis; Börner, A., Ed.; Wiley-VCH: Weinheim, Germany, 2008; Chapter 4.3; pp. 393–407. [Google Scholar]
- Dai, L.-X.; Hou, X.-L. (Eds.) Chiral Ferrocenes in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Toma, Š.; Csizmadiová, J.; Mečiarová, M.; Šebesta, R. Ferrocene Phosphane-Heteroatom/Carbon Bidentate Ligands in Asymmetric Catalysis. Dalton Trans. 2014, 16557–16579. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, L.; Benson, A.; Guiry, P.J. Recent Developments in the Synthesis and Applications of Chiral Ferrocene Ligands and Organocatalysts in Asymmetric Catalysis. Org. Biomol. Chem. 2020, 18, 9329–9370. [Google Scholar] [CrossRef] [PubMed]
- Butsugan, Y.; Araki, S.; Watanabe, M. Enantioselective Addition of Dialkylzinc to Aldehydes Catalyzed by Chiral Ferrocenyl Amino Alcohols. In Ferrocenes; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; Chapter 3; pp. 143–169. [Google Scholar]
- Fu, G.C. Enantioselective Nucleophilic Catalysis with “Planar-chiral” Heterocycles. Acc. Chem. Res. 2000, 33, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.C. Asymmetric Catalysis with “Planar-chiral” Derivatives of 4-(Dimethylamino)pyridine. Acc. Chem. Res. 2004, 37, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.C. Planar-Chiral Heterocycles as Enantioselective Organocatalysts. In Asymmetric Synthesis, 2nd ed.; Christmann, M., Brase, S., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 195–199. [Google Scholar]
- Marion, N.; Fu, G.C. Applications of Aza- and Phosphaferrocenes and Related Compounds in Asymmetric Catalysis. In Chiral Ferrocenes in Asymmetric Catalysis; Dai, L.-X., Hou, X.-L., Eds.; Wiley-VCH: Weinheim, Germany, 2010; pp. 307–335. [Google Scholar]
- Zhu, J.-C.; Cui, D.-X.; Li, Y.-D.; Jiang, R.; Chen, W.-P.; Wang, P.-A. Ferrocene as a Privileged Framework for Chiral Organocatalysts. ChemCatChem 2018, 10, 907–919. [Google Scholar] [CrossRef]
- Bernardo, O.; González-Pelayo, S.; López, L.A. Synthesis and Applications of Ferrocene-Fused Nitrogen Heterocycles. Eur. J. Inorg. Chem. 2022, e202100911. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V. Ferrocene Derivatives with Planar Chirality and Their Enantioseparation by Liquid-Phase Techniques. Electrophoresis 2023, 44, 158–189. [Google Scholar] [CrossRef]
- Izumi, T.; Hino, T. Enzymatic Resolution of Planar Chiral Ferrocene Derivatives. J. Chem. Technol. Biotechnol. 1992, 55, 325–331. [Google Scholar] [CrossRef]
- Lambusta, D.; Nicolosi, G.; Patti, A.; Piattelli, M. Lipase-Mediated Resolution of Racemic 2-Hydroxymethyl-1-methylthioferrocene. Tetrahedron Lett. 1996, 37, 127–130. [Google Scholar] [CrossRef]
- Patti, A.; Lambusta, D.; Piattelli, M.; Nocolosi, G. Lipase-Mediated Resolution of 2-Hydroxymethyl-1-iodoferrocene: Synthesis of Ferrocenes and Biferrocenes with Planar Chirality. Tetrahedron Asymmetry 1998, 9, 3073–3080. [Google Scholar] [CrossRef]
- Marquarding, D.; Klusacek, H.; Gokel, G.; Hoffmann, P.; Ugi, I. Correlation of Central and Planar Chirality in Ferrocene Derivatives. J. Am. Chem. Soc. 1970, 92, 5389–5393. [Google Scholar] [CrossRef]
- Riant, O.; Samuel, O.; Kagan, H.B. A General Asymmetric Synthesis of Ferrocenes with Planar Chirality. J. Am. Chem. Soc. 1993, 115, 5835–5836. [Google Scholar] [CrossRef]
- Rebiere, F.; Riant, O.; Ricard, L.; Kagan, H.B. Asymmetric Synthesis and Highly Diastereoselective ortho-Lithiation of Ferrocenyl Sulfoxides. Application to the Synthesis of Ferrocenyl Derivatives with Planar Chirality. Angew. Chem. Int. Ed. Engl. 1993, 32, 568–570. [Google Scholar] [CrossRef]
- Richards, C.J.; Damalidis, T.; Hibbs, D.E.; Hursthouse, M.B. Synthesis of 2-[2-(Diphenylphosphino)ferrocenyl]oxazoline Ligands. Synlett 1995, 1995, 74–76. [Google Scholar] [CrossRef]
- Sammakia, T.; Latham, H.A.; Schaad, D.R. Highly Diastereoselective Ortho Lithiations of Chiral Oxazoline-Substituted Ferrocenes. J. Org. Chem. 1995, 60, 10–11. [Google Scholar] [CrossRef]
- Riant, O.; Samuel, O.; Flessner, T.; Tauten, S.; Kagan, H.B. An Efficient Asymmetric Synthesis of 2-Substituted Ferrocenecarboxaldehydes. J. Org. Chem. 1997, 62, 6733–6745. [Google Scholar] [CrossRef]
- Enders, D.; Peters, R.; Lochtman, R.; Raabe, G. Asymmetric Synthesis of Novel Ferrocenyl Ligands with Planar and Central Chirality. Angew. Chem. Int. Ed. 1999, 38, 2421–2423. [Google Scholar] [CrossRef]
- Geisler, F.M.; Helmchen, G. A Straightforward Synthesis of (3S)-4-Methoxybutane-1,3-diol and Its Use as Chiral Auxiliary for the Preparation of (pS)-1-(Diphenylphosphino)-2-formyl-1′,2′,3′,4′,5′-pentamethylferrocene. Synthesis 2006, 2006, 2201–2205. [Google Scholar]
- Wölfle, H.; Kopacka, H.; Wurst, K.; Ongania, K.-H.; Görtz, H.-H.; Preishuber-Pflügl, P.; Bildstein, B. Planar Chiral Ferrocene Salen-type Ligands Featuring Additional Central and Axial Chirality. J. Organomet. Chem. 2006, 691, 1197–1215. [Google Scholar] [CrossRef]
- Mamane, V. The Diastereoselective Ortho-lithiation of Kagan’s Ferrocenyl Acetal. Generation and Reactivity of Chiral 2-Substituted Ferrocenecarboxaldehydes. Tetrahedron Asymmetry 2010, 21, 1019–1029. [Google Scholar] [CrossRef]
- Siegel, S.; Schmalz, H.-G. Insertion of Carbenoids into Cp-H Bonds of Ferrocenes: An Enantioselective-Catalytic Entry to Planar-Chiral Ferrocenes. Angew. Chem. Int. Ed. Engl. 1997, 36, 2456–2458. [Google Scholar] [CrossRef]
- Genet, C.; Canipa, S.J.; O’Brien, P.; Taylor, S.J. Catalytic Asymmetric Synthesis of Ferrocenes and P-Stereogenic Bisphosphines. J. Am. Chem. Soc. 2006, 128, 9336–9337. [Google Scholar] [CrossRef] [PubMed]
- Bueno, A.; Rosol, M.; García, J.; Moyano, A. Asymmetric Dihydroxylation of 2-Substituted 1-Vinylferrocenes: The First Non-Enzymatic Kinetic Resolution of Planar-Chiral Ferrocenes. Adv. Synth. Catal. 2006, 348, 2590–2596. [Google Scholar] [CrossRef]
- Ogasawara, M.; Watanabe, S.; Fan, L.; Nakajima, K.; Takahashi, T. Kinetic Resolution of Planar-Chiral Ferrocenes by Mo-Catalyzed Enantioselective Metathesis. Organometallics 2006, 25, 5201–5203. [Google Scholar] [CrossRef]
- Zhu, D.-Y.; Chen, P.; Xia, J.-B. Synthesis of Planar Chiral Ferrocenes by Transition-Metal-Catalyzed Enantioselective C-H Activation. ChemCatChem 2016, 8, 68–73. [Google Scholar] [CrossRef]
- Gao, D.-W.; Gu, Q.; Zheng, C.; You, S.-L. Synthesis of Planar Chiral Ferrocenes via Transition-Metal-Catalyzed Direct C−H Bond Functionalization. Acc. Chem. Res. 2017, 50, 351–365. [Google Scholar] [CrossRef]
- Liu, C.-X.; Gu, Q.; You, S.-L. Asymmetric C-H Bond Functionalization of Ferrocenes: New Opportunities and Challenges. Trends Chem. 2020, 2, 737–749. [Google Scholar] [CrossRef]
- Ogasawara, M. Enantioselective Preparation of Planar-Chiral Transition Metal Complexes by Asymmetric Olefin-Metathesis Reactions in Metal Coordination Spheres. Chem. Rec. 2021, 21, 3509–3519. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.B.; Gladysz, J.A. Metal-Catalyzed Olefin Metathesis in Metal Coordination Spheres. In Handbook of Metathesis; Grubbs, R.H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Volume 2, Chapter 2.11; pp. 403–431. [Google Scholar]
- Fiedler, T.; Gladysz, J.A. Multifold Ring-Closing Olefin Metatheses in Syntheses of Organometallic Molecules with Unusual Connectivities. In Olefin Metathesis; Grela, K., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp. 311–328. [Google Scholar]
- Ogasawara, M.; Nagano, T.; Hayashi, T. Metathesis Route to Bridged Metallocenes. J. Am. Chem. Soc. 2002, 124, 9068–9069, Erratum in J. Am. Chem. Soc. 2002, 124, 12626. [Google Scholar] [CrossRef]
- Ogasawara, M.; Wu, W.-Y.; Arae, S.; Nakajima, K.; Takahashi, T. Inter- versus Intraannular Ring-Closing Metathesis on Polyallylferrocenes: Five-Fold RCM within a Single Molecule. Organometallics 2013, 32, 6593–6598. [Google Scholar] [CrossRef]
- Locke, A.J.; Jones, C.; Richards, C.J. A Rapid Approach to Ferrocenophanes via Ring-Closing Metathesis. J. Organomet. Chem. 2001, 637-639, 669–676. [Google Scholar] [CrossRef]
- Hüerländere, D.; Kleigrewe, N.; Kehr, G.; Erker, G.; Fröhlich, R. Synthesis, Structural and Chemical Characterization of Unsaturated C4- and C10-Bridged Group-4 ansa-Metallocenes Obtained Through a Ring-Closing Olefin Metathesis Reaction. Eur. J. Inorg. Chem. 2002, 2002, 2633–2642. [Google Scholar] [CrossRef]
- Buchowicz, W.; Jerzykiewicz, L.B.; Krasińska, A.; Losi, S.; Pietzykowski, A.; Zanello, P. ansa-Nickelocenes by the Ring-Closing Metathesis Route: Syntheses, X-ray Crystal Structures, and Physical Properties. Organometallics 2006, 25, 5076–5082. [Google Scholar] [CrossRef]
- Schrock, R.R.; Murdzek, J.S.; Bazan, G.C.; Robbins, J.; DiMare, M.; O’Regan, M. Synthesis of Molybdenum Imido Alkylidene Complexes and Some Reactions Involving Acyclic Olefins. J. Am. Chem. Soc. 1990, 112, 3875–3886. [Google Scholar] [CrossRef]
- Schrock, R.R. The Discovery and Development of High Oxidation State Mo and W Imido Alkylidene Complexes for Alkene Metathesis. In Handbook of Metathesis; Grubbs, R.H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Volume 1, Chapter 1.3; pp. 8–32. [Google Scholar]
- Nguyen, S.T.; Johnson, L.K.; Grubbs, R.H.; Ziller, J.W. Ring-Opening Metathesis Polymerization (ROMP) of Norbornene by a Group VIII Carbene Complex in Protic Media. J. Am. Chem. Soc. 1992, 114, 3974–3975. [Google Scholar] [CrossRef]
- Schwab, P.; Grubbs, R.H.; Ziller, J.W. Synthesis and Applications of RuCl2(=CHR’)(PR3)2: The Influence of the Alkylidene Moiety on Metathesis Activity. J. Am. Chem. Soc. 1996, 118, 100–110. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.T.; Trnka, T.M. The Discovery and Development of Well-Defined, Ruthenium-Based Olefin Metathesis Catalysts. In Handbook of Metathesis; Grubbs, R.H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Volume 1, Chapter 1.6; pp. 61–85. [Google Scholar]
- Ogasawara, M.; Wu, W.-Y.; Arae, S.; Watanabe, S.; Morita, T.; Takahashi, T.; Kamikawa, K. Kinetic Resolution of Planar-Chiral (h6-Arene)chromium Complexes by Molybdenum-Catalyzed Asymmetric Ring-Closing Metathesis. Angew. Chem. Int. Ed. 2012, 51, 2951–2955. [Google Scholar] [CrossRef]
- Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Kinetic Resolution of Planar-Chiral 1,2-Disubstituted Ferrocenes by Molybdenum-Catalyzed Asymmetric Intraannular Ring-Closing Metathesis. Chem. Eur. J. 2013, 19, 4151–4154. [Google Scholar] [CrossRef]
- Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Kinetic Resolution of Planar-Chiral Ferrocenylphosphine Derivatives by Molybdenum-Catalyzed Asymmetric Ring-Closing Metathesis and Their Application in Asymmetric Catalysis. ACS Catal. 2016, 6, 1308–1315. [Google Scholar] [CrossRef]
- Ogasawara, M.; Watanabe, S.; Nakajima, K.; Takahashi, T. Enantioselective Synthesis of Planar-Chiral Phosphaferrocenes by Molybdenum-Catalyzed Asymmetric Interannular Ring-Closing Metathesis. J. Am. Chem. Soc. 2010, 132, 2136–2137. [Google Scholar] [CrossRef]
- Kamikawa, K.; Arae, S.; Wu, W.-Y.; Nakamura, C.; Takahashi, T.; Ogasawara, M. Simultaneous Induction of Axial and Planar Chirality in Arene-Chromium Complexes by Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis. Chem. Eur. J. 2015, 21, 4954–4957. [Google Scholar] [CrossRef]
- Ogasawara, M.; Tseng, Y.-Y.; Uryu, M.; Ohya, N.; Chang, N.; Ishimoto, H.; Arae, S.; Takahashi, T.; Kamikawa, K. Molybdenum-Catalyzed Enantioselective Synthesis of Planar-Chiral (h5-Phosphacyclopentadienyl)manganese(I) Complexes and Application in Asymmetric Catalysis. Organometallics 2017, 36, 4061–4069. [Google Scholar] [CrossRef]
- Alexander, J.B.; La, D.S.; Cefalo, D.R.; Hoveyda, A.H.; Schrock, R.R. Catalytic Enantioselective Ring-Closing Metathesis by a Chiral Biphen-Mo Complex. J. Am. Chem. Soc. 1998, 120, 4041–4042. [Google Scholar] [CrossRef]
- Aeilts, S.L.; Cefalo, D.R.; Bonitatebus, P.J., Jr.; Houser, J.H.; Hoveyda, A.H.; Schrock, R.R. A Readily Available and User-Friendly Chiral Catalyst for Efficient Enantioselective Olefin Metathesis. Angew. Chem. Int. Ed. 2001, 40, 1452–1456. [Google Scholar] [CrossRef]
- Schrock, R.R.; Jamieson, J.Y.; Dolman, S.J.; Miller, S.A.; Bonitatebus, P.J., Jr.; Hoveyda, A.H. Synthesis of Enantiomerically Pure Molybdenum Imido Alkylidene Catalysts for Asymmetric Olefin Metathesis that Contain Diolate Ligands Based on 3,3′-Disubstituted Octahydrobinaphtholate and 2,6-Dichlorophenylimido Combinations. Organometallics 2002, 21, 409–417. [Google Scholar] [CrossRef]
- Singh, R.; Czekelius, C.; Schrock, R.R.; Müller, P.; Hoveyda, A.H. Molybdenum Imido Alkylidene Metathesis Catalysts that Contain Electron Withdrawing Biphenoxides or Biphenolates. Organometallics 2007, 26, 2528–2539. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.S.; Schrock, R.R.; Hoveyda, A.H. Dipyrrolyl Precursors to Bisalkoxide Molybdenum Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2006, 128, 16373–16375. [Google Scholar] [CrossRef] [PubMed]
- Bunel, E.; Valle, L.; Manriquez, J.M. Pentamethylcyclopentadienyl Acetylacetonate Complexes of Iron(II), Cobalt(II), and Nickel(II). Convenient Synthetic Entries to Mono-h5-C5Me5 Derivatives. Organometallics 1985, 4, 1680–1682. [Google Scholar] [CrossRef]
- Kagan, H.B.; Fiaud, J.C. Kinetic Resolution. In Topics in Stereochemistry; Eliel, E.L., Wilen, S.H., Eds.; John Wiley & Sons: New York, NY, USA, 1988; Volume 18, pp. 249–330. [Google Scholar]
- Vedejs, E.; Jure, M. Efficiency in Nonenzymatic Kinetic Resolution. Angew. Chem. Int. Ed. 2005, 44, 3974–4001. [Google Scholar] [CrossRef] [PubMed]
- Venier, C.G.; Casserly, E.W. Di-tert-butylcyclopentadiene and Tri-tert-butylcyclopentadiene. J. Am. Chem. Soc. 1990, 112, 2808–2809. [Google Scholar] [CrossRef]
- Ustynyuk, Y.A.; Kisin, A.V.; Pribytkova, I.M.; Zenkin, A.A.; Antonova, N.D. Nuclear Magnetic Resonance Spectroscopy of Metal Cyclopentadienyls X. Proton Magnetic Resonance Spectra of, and Dynamic Behaviour in, Bis(trimethylsilyl)cyclopentadiene. J. Organomet. Chem. 1972, 42, 47–63. [Google Scholar] [CrossRef]
- Clark, T.J.; Killian, C.M.; Luthra, S.; Nile, T.A. Synthesis and Properties of Sterically Congested Cyclopentadienes and Their Transition Metal Complexes. J. Organomet. Chem. 1993, 462, 247–257. [Google Scholar] [CrossRef]
- Trnka, T.M.; Morgan, J.P.; Sanford, M.S.; Wilhelm, T.E.; Scholl, M.; Choi, T.-L.; Ding, S.; Day, M.D.; Grubbs, R.H. Synthesis and Activity of Ruthenium Alkylidene Complexes Coordinated with Phosphine and N-Heterocyclic Carbene Ligands. J. Am. Chem. Soc. 2003, 125, 2546–2558. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate b | Ligand | Conditions | Yields (%) of 1/2/3 c | % ee of (S)-1/(R)-2 d,e | krel f |
| 1 | 1a (0.005) | (R)-L1 | 50 °C, 24 h | 46/52/2 | 99/91 | 109 |
| 2 | 1a (0.005) | (R)-L2 | 50 °C, 24 h | 45/55/0 | 97/88 | 65 |
| 3 | 1a (0.005) | (R)-L2 | 25 °C, 24 h | 53/47/0 | 67/96 | 95 |
| 4 g | 1a (0.05) | (R)-L2 | 25 °C, 1 h | 55/45/0 | 75/96 | 110 |
| 5 | 1a (0.005) | (R)-L3 | 50 °C, 24 h | 35/45/20 | 94/91 | 75 |
| 6 | 1a (0.005) | (R)-L4 | 50 °C, 24 h | 0/100/0 | --- h/--- h | --- h |
| 7 | 1a (0.005) | (R)-L4 | 25 °C, 24 h | 12/88/0 | 99/18 | 5.5 |
| 8 | 1b (0.005) | (R)-L1 | 50 °C, 24 h | 51/38/11 | 89/95 | 117 |
| 9 | 1b (0.005) | (R)-L2 | 50 °C, 24 h | 45/55/0 | 92/88 | 51 |
| 10 | 1b (0.005) | (R)-L2 | 25 °C, 24 h | 45/55/0 | 97/91 | 89 |
| 11 g | 1b (0.05) | (R)-L2 | 25 °C, 2 h | 53/47/0 | 74/97 | 146 |
| 12 | 1b (0.005) | (R)-L3 | 50 °C, 24 h | 44/52/4 | 91/92 | 76 |
| 13 | 1b (0.005) | (R)-L4 | 50 °C, 24 h | 0/100/0 | --- h/--- h | --- h |
| 14 | 1b (0.005) | (R)-L4 | 25 °C, 24 h | 20/80/0 | 90/26 | 4.4 |
| 15 | 1c (0.005) | (R)-L1 | 50 °C, 24 h | 28/52/20 | 98/54 | 15 |
| 16 | 1c (0.005) | (R)-L2 | 50 °C, 24 h | 1/99/0 | --- h/5 | --- h |
| 17 g | 1c (0.05) | (R)-L2 | 25 °C, 1 h | 45/55/0 | 84/59 | 9.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imazu, H.; Masaoka, K.; Uike, S.; Ogasawara, M. Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis/Kinetic Resolution of Racemic Planar-Chiral 1,1′-Diallylferrocenes. Catalysts 2024, 14, 123. https://doi.org/10.3390/catal14020123
Imazu H, Masaoka K, Uike S, Ogasawara M. Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis/Kinetic Resolution of Racemic Planar-Chiral 1,1′-Diallylferrocenes. Catalysts. 2024; 14(2):123. https://doi.org/10.3390/catal14020123
Chicago/Turabian StyleImazu, Haruna, Kakeru Masaoka, Saki Uike, and Masamichi Ogasawara. 2024. "Molybdenum-Catalyzed Enantioselective Ring-Closing Metathesis/Kinetic Resolution of Racemic Planar-Chiral 1,1′-Diallylferrocenes" Catalysts 14, no. 2: 123. https://doi.org/10.3390/catal14020123