
Influence of Oxide Coating Layers on the Stability of Gold Catalysts for Furfural Oxidative Esterification to Methyl Furoate


Abstract
:1. Introduction
2. Results and Discussion
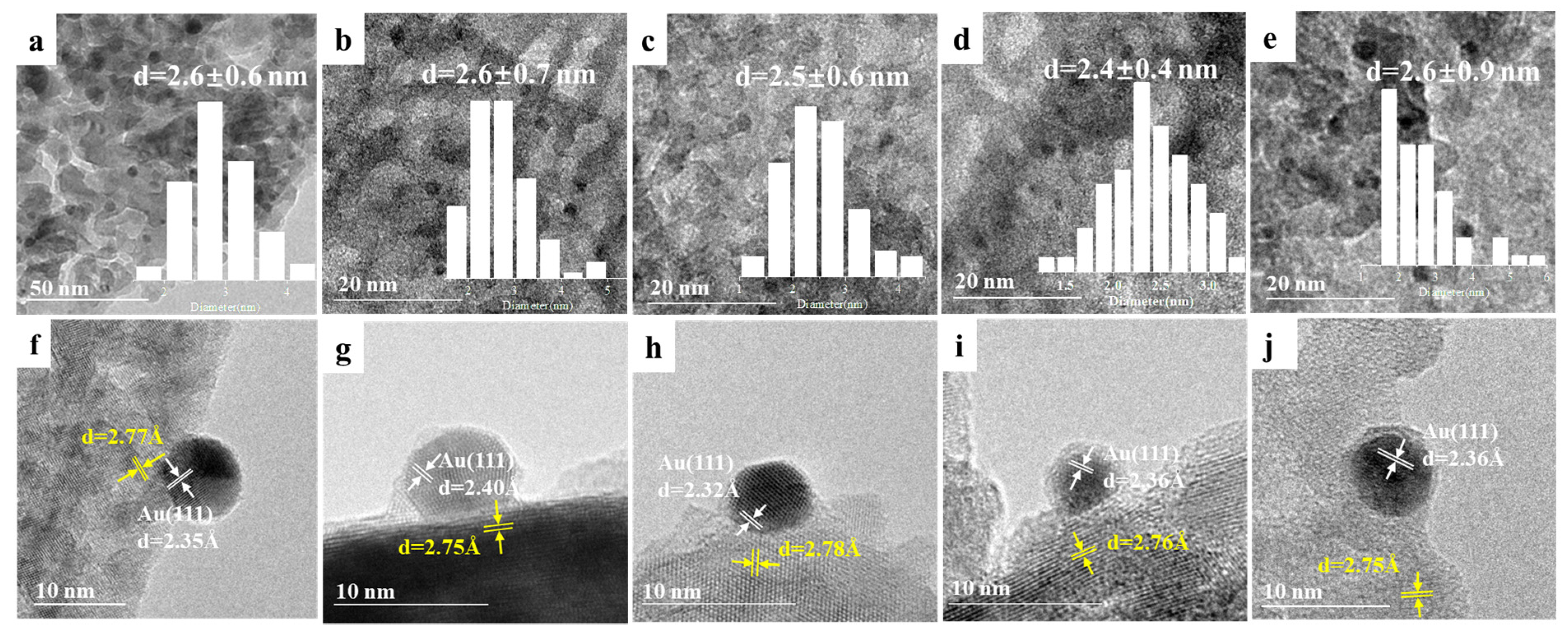
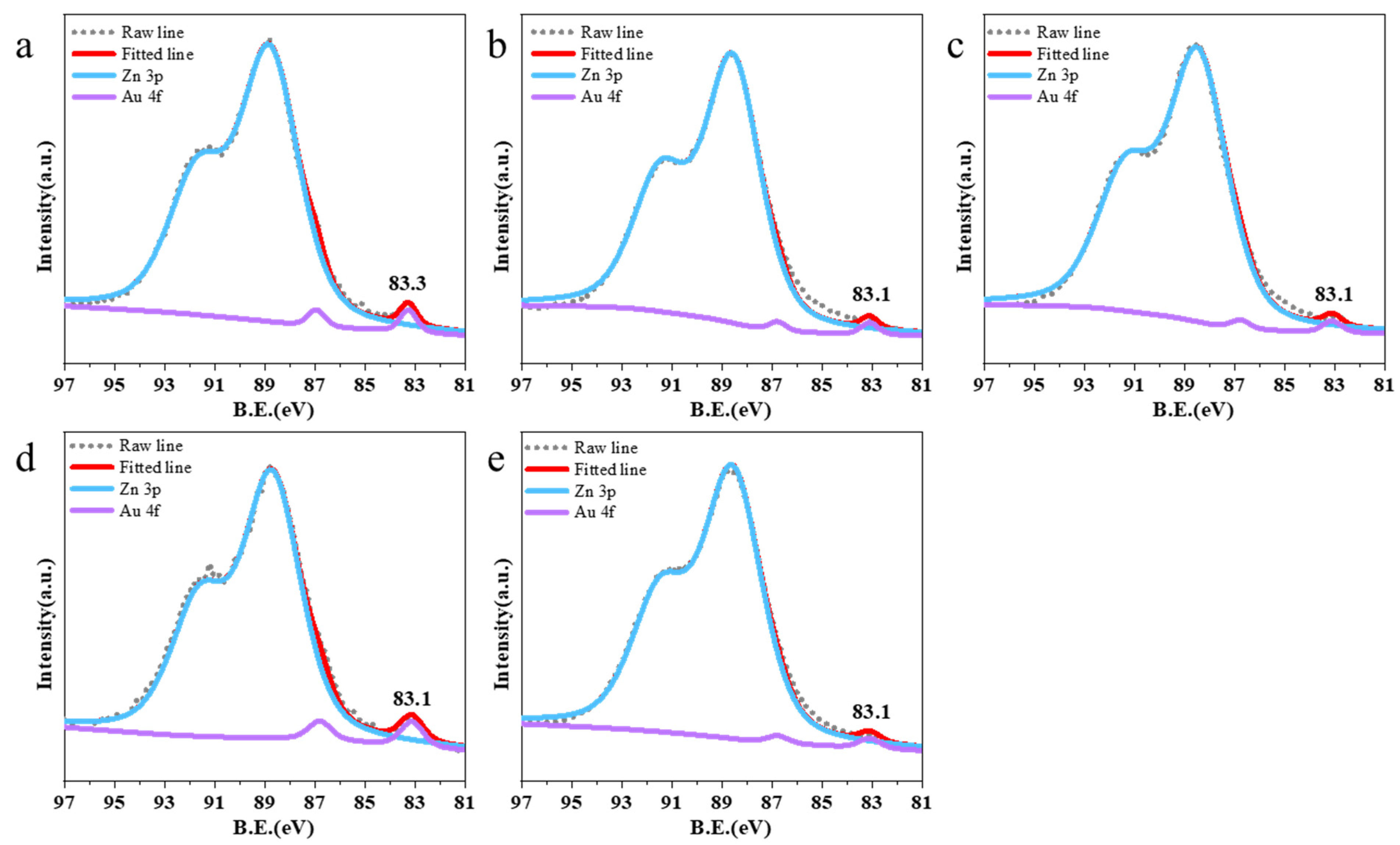
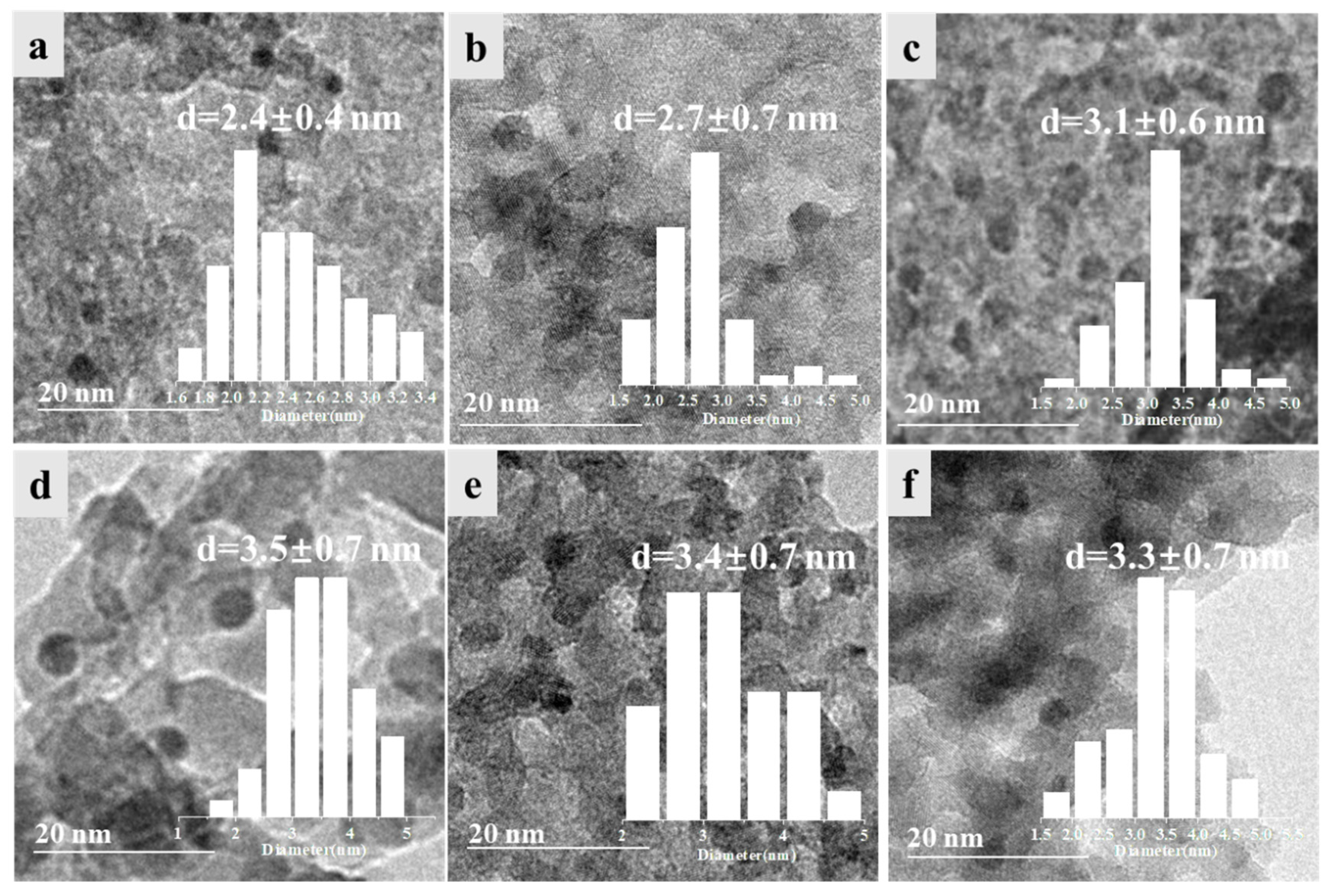
2.1. Catalyst Preparation and Structure Characterization
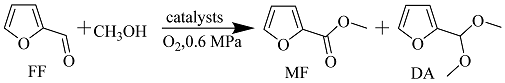
2.2. Catalytic Performances of the above Catalysts for Oxidative Esterification of FF to MF
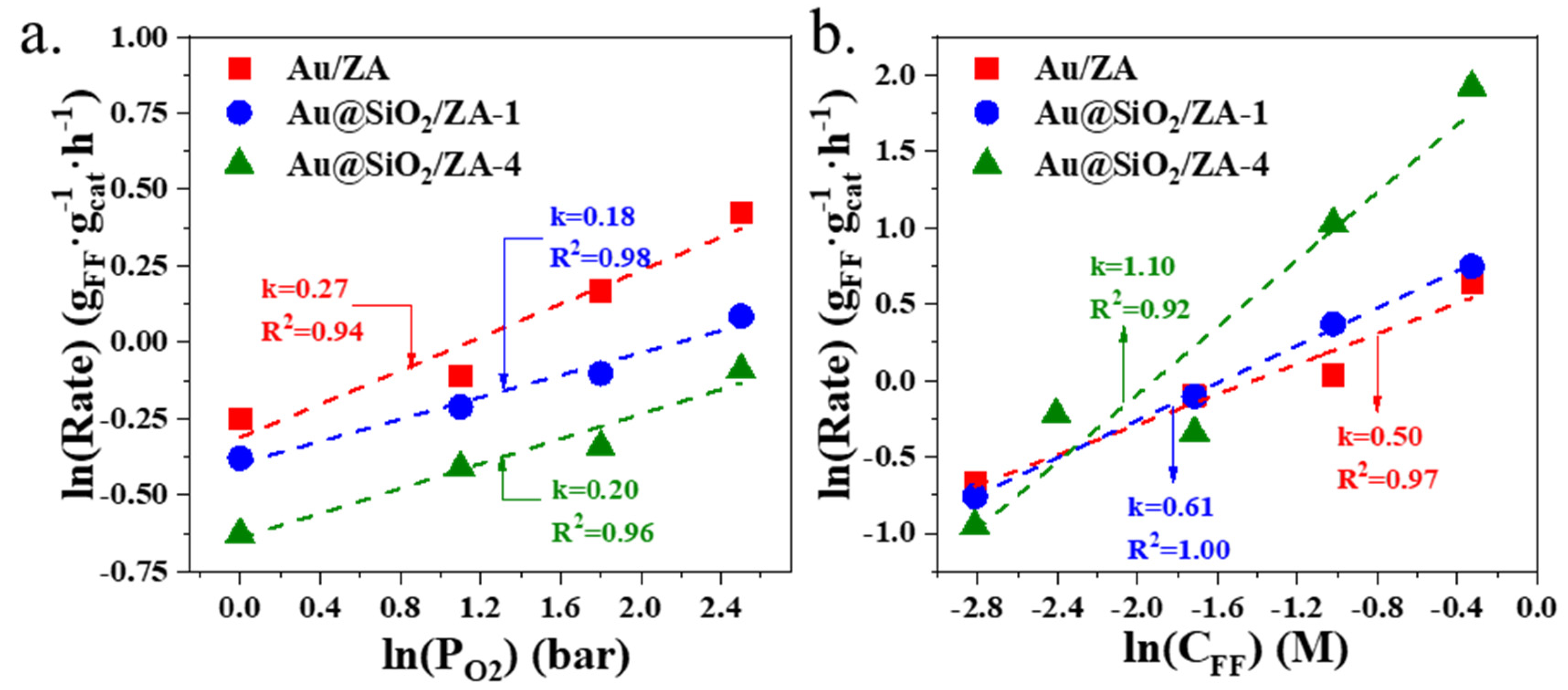
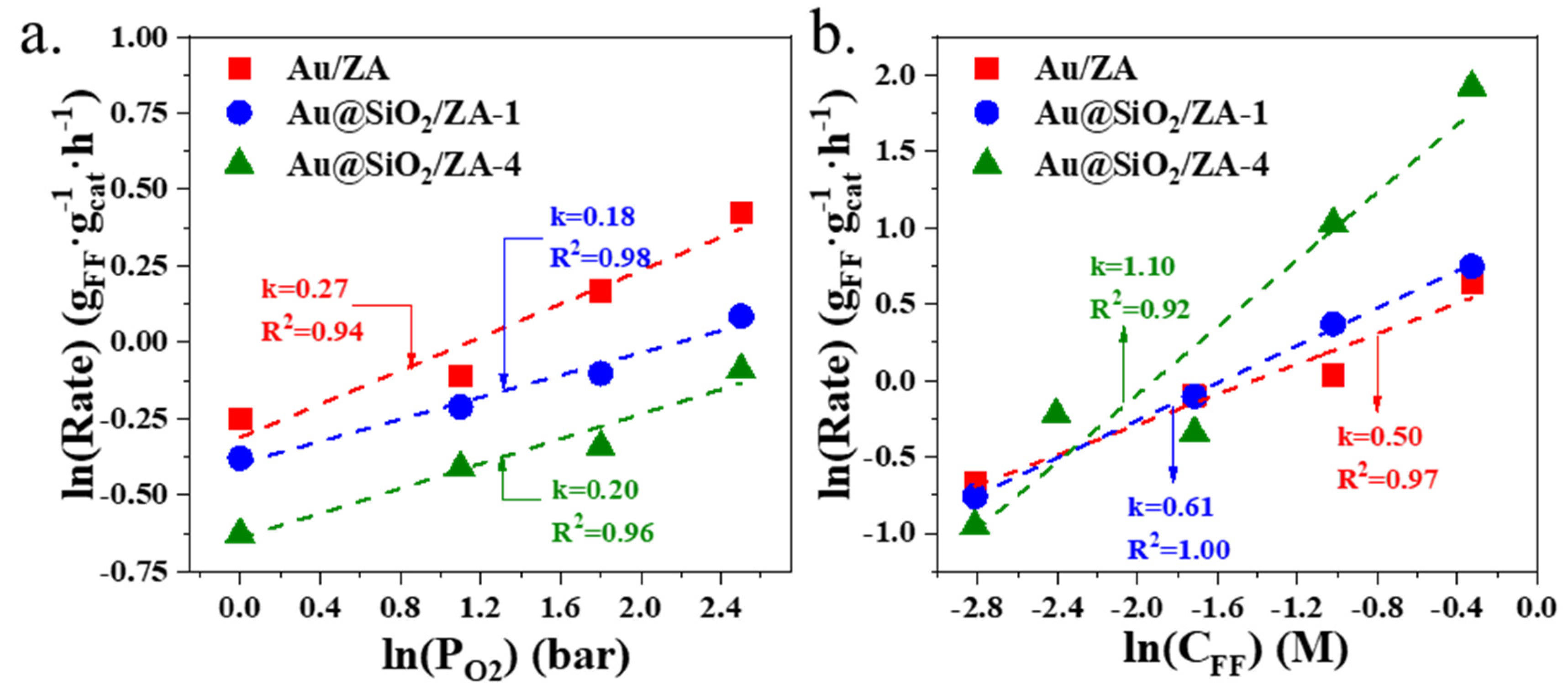
2.3. Dynamic Tests of Different Gold Catalysts for Oxidative Esterification
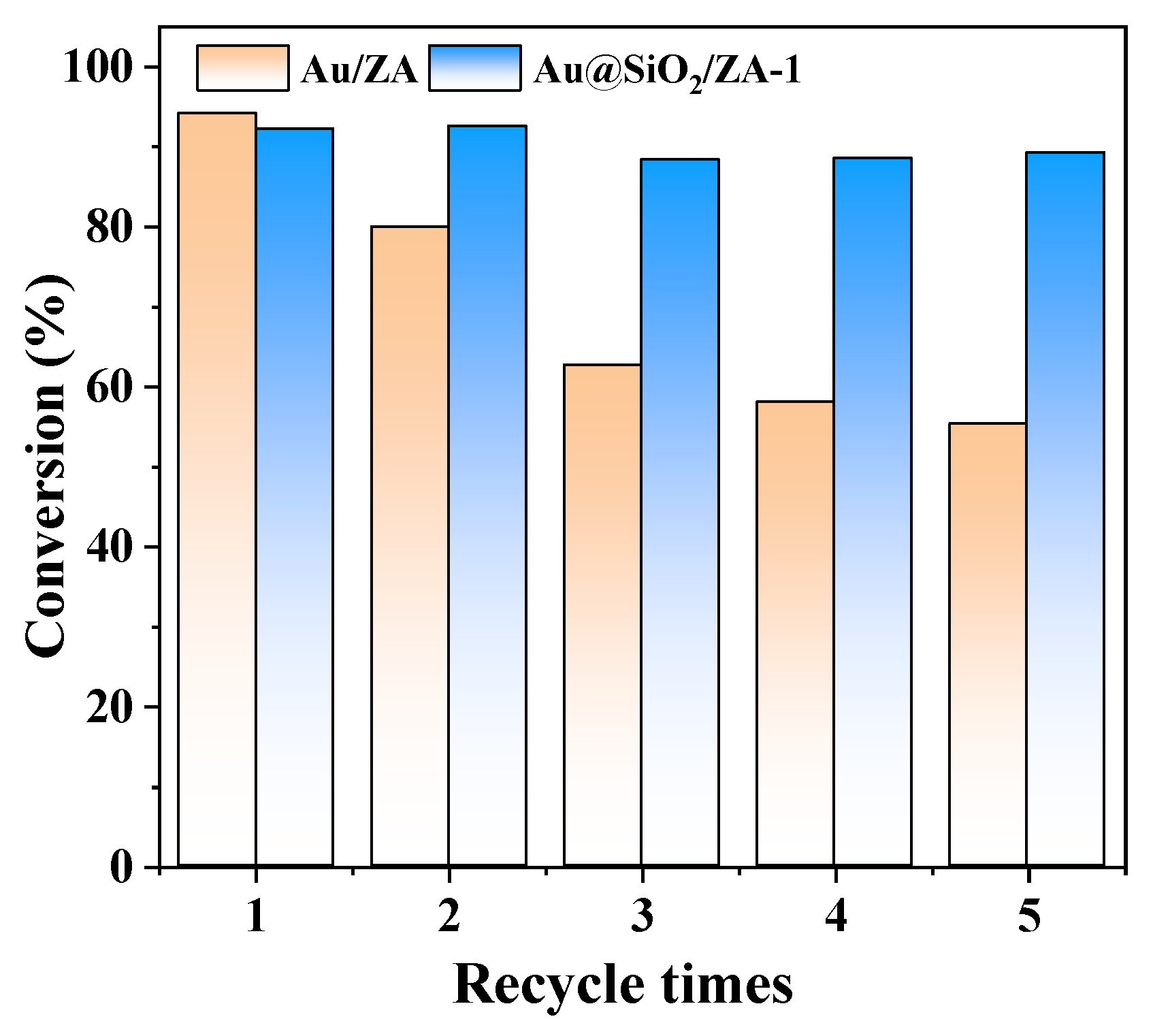
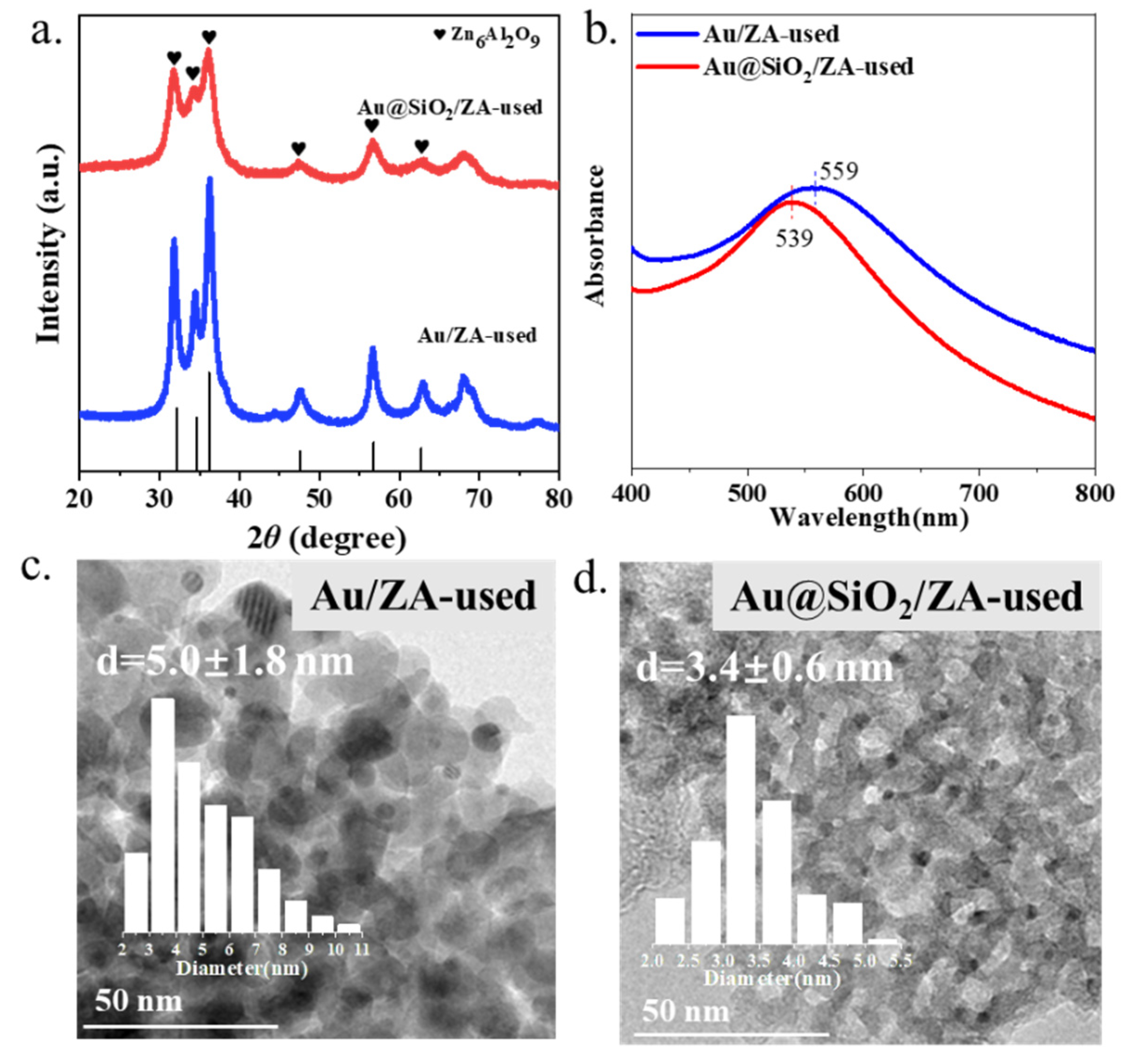
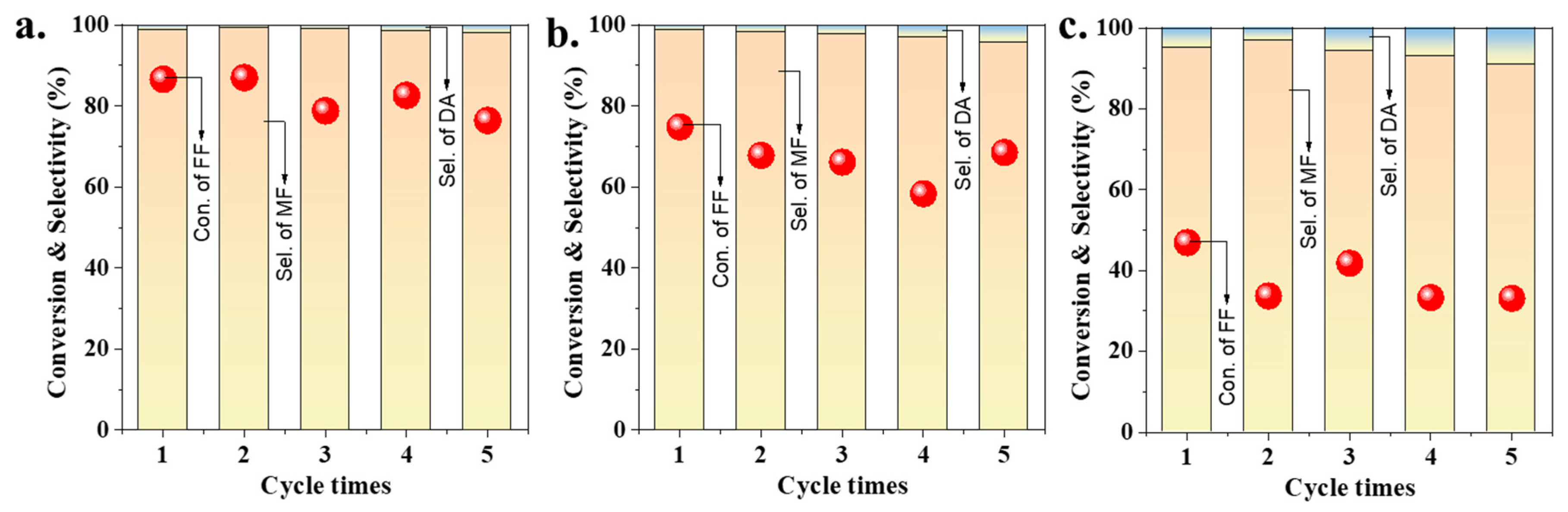
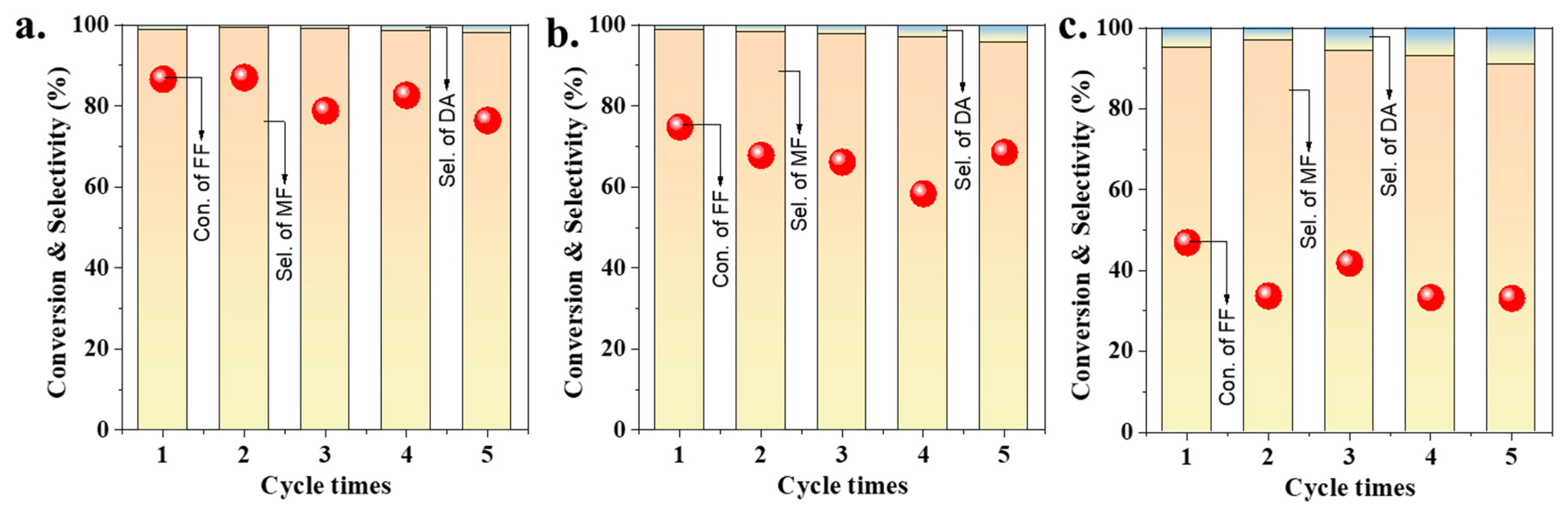
2.4. Stability Tests of Gold Catalysts with or without SiO2 Coating
2.5. Variation of Coating Layers with Other Oxides: ZrO2, TiO2, and CeO2
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of the Au/ZA Catalyst
3.3. Preparation of the Au Catalyst Coating with SiO2 (Au@SiO2/ZA-X)
3.4. Preparation of the Au Catalyst Coating with Other Metal Oxides (Au@MO2/ZA)
3.5. Catalytic Test
3.6. Characterizations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dimitrakellis, P.; Delikonstantis, E.; Stefanidis, G.D.; Vlachos, D.G. Plasma technology for lignocellulosic biomass conversion toward an electrified biorefinery. Green Chem. 2022, 24, 2680–2721. [Google Scholar] [CrossRef]
- Thanigaivel, S.; Vickram, S.; Manikandan, S.; Deena, S.R.; Subbaiya, R.; Karmegam, N.; Govarthanan, M.; Kim, W. Sustainability and carbon neutralization trends in microalgae bioenergy production from wastewater treatment: A review. Bioresour. Technol. 2022, 364, 128057. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef]
- He, L.; Chen, L.; Zheng, B.; Zhou, H.; Wang, H.; Li, H.; Zhang, H.; Xu, C.C.; Yang, S. Deep eutectic solvents for catalytic biodiesel production from liquid biomass and upgrading of solid biomass into 5-hydroxymethylfurfural. Green Chem. 2023, 25, 7410–7440. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.B.T.L.; Wu, T.Y. A review on solvent systems for furfural production from lignocellulosic biomass. Renew. Sust. Energy Rev. 2021, 137, 110172. [Google Scholar] [CrossRef]
- Li, X.; Cong, L.; Lin, N.; Tang, C. Efficient electrochemical upgradation strategies for the biomass derivative furfural. J. Mater. Chem. A 2023, 11, 23133–23147. [Google Scholar] [CrossRef]
- Wen, H.; Zhang, W.; Fan, Z.; Chen, Z. Recent Advances in Furfural Reduction via Electro-And Photocatalysis: From Mechanism to Catalyst Design. ACS Catal. 2023, 13, 15263–15289. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; Granados, M.L. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Xue, Z.; Wu, S.; Fu, Y.; Luo, L.; Li, M.; Li, Z.; Shao, M.; Zheng, L.; Xu, M.; Duan, H. Efficient light-driven reductive amination of furfural to furfurylamine over ruthenium-cluster catalyst. J. Energy Chem. 2023, 76, 239–248. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, C.; Li, D.; Liu, Q.; Tan, J.; Wang, C.; Cai, C.; Ma, L. Recent advances in catalytic conversion of biomass to 5-hydroxymethylfurfural and 2, 5-dimethylfuran. Renew. Sust. Energy Rev. 2019, 103, 227–247. [Google Scholar] [CrossRef]
- Long, J.; Xu, Y.; Zhao, W.; Li, H.; Yang, S. Heterogeneous catalytic upgrading of biofuranic aldehydes to alcohols. Front. Chem. 2019, 7, 529. [Google Scholar] [CrossRef] [PubMed]
- Román, A.M.; Hasse, J.C.; Medlin, J.W.; Holewinski, A. Elucidating acidic electro-oxidation pathways of furfural on platinum. ACS Catal. 2019, 9, 10305–10316. [Google Scholar] [CrossRef]
- Ayoub, N.; Toufaily, J.; Guénin, E.; Enderlin, G. Catalyst-free process for oxidation of furfural to maleic acid by high frequency ultrasonic activation. Green Chem. 2022, 24, 4164–4173. [Google Scholar] [CrossRef]
- Ning, L.; Liao, S.; Liu, X.; Guo, P.; Zhang, Z.; Zhang, H.; Tong, X. A regulatable oxidative valorization of furfural with aliphatic alcohols catalyzed by functionalized metal-organic frameworks-supported Au nanoparticles. J. Catal. 2018, 364, 1–13. [Google Scholar] [CrossRef]
- Ferraz, C.P.; Braga, A.H.; Ghazzal, M.N.; Zieliński, M.; Pietrowski, M.; Itabaiana, I., Jr.; Dumeignil, F.; Rossi, L.M.; Wojcieszak, R. Efficient oxidative esterification of furfural using Au nanoparticles supported on group 2 alkaline earth metal oxides. Catalysts 2020, 10, 430. [Google Scholar] [CrossRef]
- Tong, X.; Liu, Z.; Yu, L.; Li, Y. A tunable process: Catalytic transformation of renewable furfural with aliphatic alcohols in the presence of molecular oxygen. Chem. Commun. 2015, 51, 3674. [Google Scholar] [CrossRef] [PubMed]
- Delparish, A.; Bouter, A.W.N.d.L.D.; Yercan, A.; van der Schaaf, J.; D’Angelo, M.F.N. Bringing the promises of microreactors and gold catalysis to lignocellulosic biomass valorization: A study on oxidative transformation of furfural. Chem. Eng. J. 2023, 452, 138903. [Google Scholar] [CrossRef]
- Manzoli, M.; Menegazzo, F.; Signoretto, M.; Marchese, D. Biomass derived chemicals: Furfural oxidative esterification to methyl-2-furoate over gold catalysts. Catalysts 2016, 6, 107. [Google Scholar] [CrossRef]
- Escobar, A.; Pérez, M.; Sathicq, A.; García, M.; Paola, A.; Romanelli, G.; Blustein, G. Alkyl 2-furoates obtained by green chemistry procedures as suitable new antifoulants for marine protective coatings. J. Coat. Technol. Res. 2018, 16, 159–166. [Google Scholar] [CrossRef]
- Huo, N.; Ma, H.; Wang, X.; Wang, T.; Wang, G.; Wang, T.; Hou, L.; Gao, J.; Xu, J. High-efficiency oxidative esterification of furfural to methylfuroate with a non-precious metal Co-NC/MgO catalyst. J. Catal. 2017, 38, 1148–1154. [Google Scholar]
- Radhakrishnan, R.; Thiripuranthagan, S.; Devarajan, A.; Kumaravel, S.; Erusappan, E.; Kannan, K. Oxidative esterification of furfural by Au nanoparticles supported CMK-3 mesoporous catalysts. Appl. Catal. A 2017, 545, 33–43. [Google Scholar] [CrossRef]
- Cho, A.; Byun, S.; Cho, J.H.; Kim, B.M. AuPd-Fe3O4 Nanoparticle-Catalyzed Synthesis of Furan-2,5-dimethylcarboxylate from 5-Hydroxymethylfurfural under Mild Conditions. ChemSusChem 2019, 12, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, H.; Liu, Z.; Akri, M.; Tan, Y.; Kang, L.; Chi, J.; Qiao, B.; Ding, Y. Synergic effect between gold and vanadate substituted hydroxyapatite support for synthesis of methyl methacrylate by one-step oxidative esterification. Chem. Eng. J. 2022, 431, 133207. [Google Scholar] [CrossRef]
- Zhao, X.; Fang, R.; Shen, Z.; Wang, F.; Li, Y. Atomic decoration of Pt on Co nanoparticles for enhanced oxidative esterification performances. AIChE J. 2023, e18340. [Google Scholar] [CrossRef]
- Li, F.; Li, X.-L.; Li, C.; Shi, J.; Fu, Y. Aerobic oxidative esterification of 5-hydroxymethylfurfural to dimethyl furan-2,5-dicarboxylate by using homogeneous and heterogeneous PdCoBi/C catalysts under atmospheric oxygen. Green Chem. 2018, 20, 3050–3058. [Google Scholar] [CrossRef]
- Wang, R.; Lu, K.; Zhang, J.; Li, X.; Zheng, Z. Regulation of the Co–Nx Active Sites of MOF-Templated Co@NC Catalysts via Au Doping for Boosting Oxidative Esterification of Alcohols. ACS Catal. 2022, 12, 14290–14303. [Google Scholar] [CrossRef]
- Jiang, B.-L.; Lin, Y.; Wang, M.-L.; Liu, D.-S.; Xu, B.-H.; Zhang, S.-J. Cobalt-catalyzed direct transformation of aldehydes to esters: The crucial role of an enone as a mediator. Org. Chem. Front. 2019, 6, 801–807. [Google Scholar] [CrossRef]
- Song, F.; Cen, S.; Wan, C.; Wang, L. Nano-Au Anchored in Organic Base Group-Grafted Silica Aerogel: A Durable and Robust Catalysts for Green Oxidative Esterification of Furfural. ChemCatChem 2022, 14, e202200704. [Google Scholar] [CrossRef]
- Yang, N.; Pattisson, S.; Douthwaite, M.; Zeng, G.; Zhang, H.; Ma, J.; Hutchings, G.J. Influence of Stabilizers on the Performance of Au/TiO2 Catalysts for CO Oxidation. ACS Catal. 2021, 11, 11607–11615. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Zhang, B.; Si, R.; Han, B.; Hong, F.; Niu, Y.; Sun, L.; Li, L.; Qiao, B.; et al. Boosting the catalysis of gold by O2 activation at Au-SiO2 interface. Nat. Commun. 2020, 11, 558. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhao, G.; Sun, J.; Cao, X.; He, Y.; Feng, J.; Li, D. The effect of oxygen vacancies in ZnO at an Au/ZnO interface on its catalytic selective oxidation of glycerol. J. Catal. 2019, 377, 271–282. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Z.; Ye, B.; Huang, X.; Deng, H. Ligand effect over gold nanocatalysts towards enhanced gas-phase oxidation of alcohols. J. Catal. 2021, 400, 274–282. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Turner, M.; Golovko, V.B.; Vaughan, O.P.H.; Abdulkin, P.; Berenguer-Murcia, A.; Tikhov, M.S.; Johnson, B.F.G.; Lambert, R.M. Selective oxidation with dioxygen by gold nanoparticle catalysts derived from 55-atom clusters. Nature 2008, 454, 981–983. [Google Scholar] [CrossRef]
- Du, X.; Huang, Y.; Pan, X.; Han, B.; Su, Y.; Jiang, Q.; Li, M.; Tang, H.; Li, G.; Qiao, B. Size-dependent strong metal-support interaction in TiO2 supported Au nanocatalysts. Nat. Commun. 2020, 11, 5811. [Google Scholar] [CrossRef] [PubMed]
- Higaki, T.; Li, Y.; Zhao, S.; Li, Q.; Li, S.; Du, X.; Yang, S.; Chai, J.; Jin, R. Atomically Tailored Gold Nanoclusters for Catalytic Application. Angew. Chem. 2019, 131, 8377–8388. [Google Scholar] [CrossRef]
- Pei, Y.; Wang, P.; Ma, Z.; Xiong, L. Growth-Rule-Guided Structural Exploration of Thiolate-Protected Gold Nanoclusters. Acc. Chem. Res. 2019, 52, 23–33. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Yang, W.; Mao, G.; Meng, Z.; Wu, Z.; Jiang, H.L. Surface-Clean Au25 Nanoclusters in Modulated Microenvironment Enabled by Metal–Organic Frameworks for Enhanced Catalysis. J. Am. Chem. Soc. 2022, 144, 22008–22017. [Google Scholar] [CrossRef]
- Zhong, Y.; Liao, P.; Kang, J.; Liu, Q.; Wang, S.; Li, S.; Liu, X.; Li, G. Locking Effect in Metal@MOF with Superior Stability for Highly Chemoselective Catalysis. J. Am. Chem. Soc. 2023, 145, 4659–4666. [Google Scholar] [CrossRef]
- Wu, K.; Wang, X.Y.; Guo, L.L.; Xu, Y.J.; Zhou, L.; Lyu, Z.Y.; Liu, K.Y.; Si, R.; Zhang, Y.W.; Sun, L.D.; et al. Facile synthesis of Au embedded CuOx-CeO2 core/shell nanospheres as highly reactive and sinter-resistant catalysts for catalytic hydrogenation of p-nitrophenol. Nano Res. 2020, 13, 2044–2055. [Google Scholar] [CrossRef]
- Liu, S.; Xu, W.; Niu, Y.; Zhang, B.; Zheng, L.; Liu, W.; Li, L.; Wang, J. Ultrastable Au nanoparticles on titania through an encapsulation strategy under oxidative atmosphere. Nat. Commun. 2019, 10, 5790. [Google Scholar] [CrossRef]
- Taarning, E.; Nielsen, I.S.; Egeblad, K.; Madsen, R.; Christensen, C.H. Chemicals from renewables: Aerobic oxidation of furfural and hydroxymethylfurfural over gold catalysts. ChemSusChem 2008, 1, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tan, Y.; Chen, X.; Yang, W.; Huang, C.; Li, J.; Ding, Y. Efficient synthesis of methyl methacrylate by one step oxidative esterification over Zn-Al-mixed oxides supported gold nanocatalysts. Catalysts 2021, 11, 162. [Google Scholar] [CrossRef]
- Ortiz, G.R.; Lartundo-Rojas, L.; Samaniego-Benítez, J.E.; Jiménez-Flores, Y.; Calderón, H.; Mantilla, A. Photocatalytic behavior for the phenol degradation of ZnAl layered double hydroxide functionalized with SDS. J. Environ. Manag. 2021, 277, 111399. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shi, J.; Shen, H. High-dispersed catalysts of core–shell structured Au@SiO2 for formaldehyde catalytic oxidation. Chem. Eng. J. 2020, 385, 123887. [Google Scholar] [CrossRef]
- Hernández-Ramíreza, E.; Wanga, J.A.; Chena, L.F.; Valenzuelaa, M.A.; Dalaib, A.K. Development of CeO2-and TiO2-Based Au Nanocatalysts for Catalytic Applications. Appl. Surf. Sci. 2016, 399, 77–85. [Google Scholar]
- Tan, Y.; Liu, X.Y.; Zhang, L.; Wang, A.; Li, L.; Pan, X.; Miao, S.; Haruta, M.; Wei, H.; Wang, H.; et al. Angew. Chem. Int. Ed. 2017, 56, 2709–2713. [Google Scholar] [CrossRef]
- Liu, Z.; Tan, Y.; Li, J.; Li, X.; Xiao, Y.; Su, J.; Chen, X.; Qiao, B.; Ding, Y. Ag substituted Au clusters supported on Mg-Al-hydrotalcite for highly efficient base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid. Green Chem. 2022, 24, 8840–8852. [Google Scholar] [CrossRef]
- Alfayate, A.; Sepúlveda, R.; Sánchez-Sánchez, M.; Pérez-Pariente, J. Influence of Si Incorporation into the Novel Ti(III)APO-5 Catalysts on the Oxidation of Cyclohexene in Liquid Phase. Top. Catal. 2016, 59, 326–336. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalysts | Au Loadings (%) a | Si Loadings (%) a | SBET (m2/g) b | Total Volume (cm3/g) b | Pore Size (nm) b |
|---|---|---|---|---|---|---|
| 1 | Au/ZA | 1.2 | - | 89.3 | 0.23 | 3.8 |
| 2 | Au@SiO2/ZA-1 | 1.1 | 2.7 | 113.2 | 0.43 | 3.3 |
| 3 | Au@SiO2/ZA-2 | 1.1 | 3.1 | 118.2 | 0.48 | 3.3 |
| 4 | Au@SiO2/ZA-3 | 1.0 | 3.7 | 122.6 | 0.48 | 3.4 |
| 5 | Au@SiO2/ZA-4 | 1.1 | 4.2 | 117.3 | 0.36 | 3.8 |
 | |||||
|---|---|---|---|---|---|
| Entry | Catalysts | Conversion (%) | Selectivity (%) | TOF (h−1) b | |
| MF | DA | ||||
| 1 | ZA | 0 | 0 | 0 | |
| 2 | Au/ZA | 94.2 | 94.9 | 5.1 | 232.2 |
| 3 | Au/ZA a | 99.9 | 98.9 | 1.1 | |
| 4 | Au@SiO2/ZA-1 | 92.3 | 95.0 | 5.0 | 231.4 |
| 5 | Au@SiO2/ZA-1 a | 99.7 | 98.4 | 1.6 | |
| 6 | Au@SiO2/ZA-2 | 65.0 | 90.1 | 9.9 | 219.4 |
| 7 | Au@SiO2/ZA-2 a | 94.1 | 92.2 | 7.8 | |
| 8 | Au@SiO2/ZA-3 | 62.2 | 75.4 | 24.6 | 218.0 |
| 9 | Au@SiO2/ZA-3 a | 93.2 | 81.3 | 18.7 | |
| 10 | Au@SiO2/ZA-4 | 60.5 | 80.2 | 19.8 | 132.6 |
| 11 | Au@SiO2/ZA-4 a | 90.3 | 79.7 | 20.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J.; Zhan, N.; Tan, Y.; Min, X.; Xiao, Y.; Qiao, B. Influence of Oxide Coating Layers on the Stability of Gold Catalysts for Furfural Oxidative Esterification to Methyl Furoate. Catalysts 2024, 14, 192. https://doi.org/10.3390/catal14030192
Su J, Zhan N, Tan Y, Min X, Xiao Y, Qiao B. Influence of Oxide Coating Layers on the Stability of Gold Catalysts for Furfural Oxidative Esterification to Methyl Furoate. Catalysts. 2024; 14(3):192. https://doi.org/10.3390/catal14030192
Chicago/Turabian StyleSu, Juan, Nannan Zhan, Yuan Tan, Xiangting Min, Yan Xiao, and Botao Qiao. 2024. "Influence of Oxide Coating Layers on the Stability of Gold Catalysts for Furfural Oxidative Esterification to Methyl Furoate" Catalysts 14, no. 3: 192. https://doi.org/10.3390/catal14030192