
Nickel(BiPhePhos)-Catalyzed Hydrocyanation of Styrene—Highly Increased Catalytic Activity by Optimized Operational Procedures


Chair of Industrial Chemistry, Department of Bio- and Chemical Engineering, TU Dortmund, Emil-Figge-Straße 68, 44227 Dortmund, Germany
*
Author to whom correspondence should be addressed.
Catalysts 2024, 14(3), 210; https://doi.org/10.3390/catal14030210
Submission received: 27 February 2024
/
Revised: 13 March 2024
/
Accepted: 15 March 2024
/
Published: 21 March 2024
(This article belongs to the Section Industrial Catalysis)
Abstract
:Nitriles are versatile and highly desired chemical intermediates for a range of products. Their economic large-scale production requires highly efficient and selective synthesis. The nickel-catalyzed hydrocyanation of C=C double bonds provides such selective and 100% atom-economical access to nitriles, but the catalysts hitherto lack activity and longevity. Yet, the literature focusing on increased catalytic activity or optimized operational procedures is scarce, at the least. Here, we present a thorough investigation and optimization of operational procedures using a commercially available diphosphite ligand and styrene as a model substrate. This led us to achieve a TOF20 of more than 300,000 h−1.

1. Introduction
Nitriles can be easily converted into amines, aldehydes, esters, carboxylic acids, and other chemical functionalities (Figure 1) [1]. This high synthetic diversity can potentially enable new synthesis routes, which may be of particular interest in the development of new, sustainable processes. On a laboratory scale, nitriles can be synthesized by nucleophilic substitution reactions of an alkyl halide and an alkali cyanide in polar solvents. Nevertheless, the formation of alkali halides as byproducts excludes this method for industrial-scale synthesis of such important nitriles. These are economically, as well as ecologically, disadvantageous and difficult to handle. Hydrocyanation, the catalytic hydrogen cyanide (HCN) addition to C=C-double-bonds, provides selective and 100% atom-economical access to nitriles [1,2].
Large parts of the academic literature focus on HCN surrogates like Trimethylsilyl cyanide-MeOH [3], acetone cyanohydrin (ACH) [4,5,6,7,8,9], or zinc cyanide and water [10] for the in situ formation of HCN. More recent research focuses on transfer hydrocyanation at increased temperatures, utilizing the equilibrium established by the nickel catalyst. The remaining low boiling alkene can be evaporated from the reaction mixture (Figure 1) [11,12,13].
Synthetic routes employing HCN surrogates are usually safer to be performed on a lab scale. However, they are of little interest for larger-scale industrial application. The formation of coupled products or byproducts severely diminishes the economic value of a process and is therefore highly undesirable. Pure hydrogen cyanide, produced via the Andrussow process or the BMA process, is readily used for large-scale hydrocyanation of butadiene [1]. Unfortunately, the high toxicity of HCN requires severe safety standards, making this reasonably unattractive in purely academic research—the academic literature regarding hydrocyanation using pure HCN is limited. To fill this gap, this work explicitly focuses on the use of pure HCN and optimization of the reaction procedure in order to improve the activity of the Ni-based catalyst.
In typical lab-scale hydrocyanation experiments, the catalyst is first preformed and, after all substrates are added, hydrogen cyanide, if used in pure form, is usually added via Eppendorf pipette, either neat or in solution [14,15,16]. Early academic studies on hydrocyanation mainly used HCN surrogates like acetone cyanohydrin, which require elevated temperatures of 60 to 90 °C to liberate HCN. For reasons of comparability, these conditions were mostly kept in successive work, even when pure HCN was used. So far, there is limited research focused on optimizing the experimental methodology.
Phosphine, phosphinite, phosphonite, and phosphite ligands have been applied in Ni-catalyzed hydrocyanation [17]. The best yields and selectivities were achieved for chelating ligands with large bite angles between 105° and 120°. Rigid, large-bite-angle ligands stabilize tetragonal and trigonal bipyramidal active species and disfavor the formation of inactive square planar nickel cyanide species [17].
Electron-withdrawing π-acceptor ligands like phosphites and phosphonites improve yield and selectivity by accelerating the rate-limiting reductive elimination step [14]. Early studies with tri-o-tolylphosphite already show the positive effects of this ligand class [18,19].
Furthermore, it was shown that chelating ligands tend to form catalytically inactive Ni(bischelate) complexes. While this is reversible for diphosphines, the Ni(bischelates) formed with chelating π-acceptor ligands tend to be quite stable and may lead to complete catalyst deactivation over time, especially at elevated temperatures [14]. However, the formation of bischelates can be counteracted by sterically hindered chelating ligands [17].
Combining the mentioned characteristics, bulky and rigid diphosphites with large bite angles appear to be highly potent ligands for Ni-catalyzed hydrocyanation. Applying a diphosphonite ligand in the Ni-catalyzed hydrocyanation of styrene with pure HCN, Hofmann et al. were able to reduce the catalyst concentration to 1 mol%, still reaching a conversion of more than 90% [20]. This translates to a maximum Turnover Number (TON) of 92. These findings emphasize the need for further optimization in this particular field.
As there is a significant knowledge gap regarding the true activity of nickel complexes in hydrocyanation in terms of TOF at low conversion, in this study, we focused on the optimization of experimental procedures in order to maximize catalytic activity. For our study, we decided to apply BiPhePhos (Figure 2) as the ligand, first described in patents by Union Carbide [21]. Surprisingly, to the best of our knowledge, BiPhePhos has never been reported in the literature on Ni-catalyzed hydrocyanation.
2. Results and Discussion
For the intended optimizations, styrene was used as a model substrate. Due to its structural properties, it is an ideal model substrate for studies on vinyl arene hydrocyanation (Figure 3).
Under the chosen conditions, styrene is almost exclusively converted to the branched product 2-phenylpropionitrile (Figure 3). This is caused by the thermodynamically favored transition state between the π-complex, which is necessary for the branched product formation, and the reductive eliminated product. This crucial step is therefore kinetically favored over the reductive elimination of the linear product to an extent where no linear product is being formed [15].
2.1. Optimizations of Operational Procedure
2.1.1. Catalyst Preformation
At first, the preforming procedure was investigated. It was found that dissolving the ligand BiPhePhos first, before the addition of Ni(cod)2, helped to achieve reproducible results. Dissolving Ni(cod)2 first, instead of BiPhePhos, often results in a cloudy solution, giving rise to non-reproducible results.
The preforming solutions were analyzed by 31P-NMR. After catalyst preformation for only 15 min at 22 °C, the characteristic signal for the (BiPhePhos)Ni(cod) complex was observed at δ = 178 ppm [22]. Due to the 1.2 equivalents of ligand applied, the signal of the free ligand also remained at δ = 145 ppm. Even stirring the catalyst solution over night at 22 °C did not show the characteristic signal of the highly undesired (BiPhePhos)2Ni bischelate complex at δ = 162 ppm. This complex was stable and catalytically inactive, as shown earlier by Fazekas (Figure 4) [22].
Our systematic study of catalyst preformation by 31P-NMR showed fast formation of the catalytically inactive bischelate complex at elevated temperatures (Figure S8). This explains why hydrocyanation at 60 or 90 °C results in a lower Turn Over Number (TON). Here, we present a new and reliable protocol for hydrocyanation reactions at an ambient temperature of 22 °C.
2.1.2. Reaction Procedure
In order to optimize the reaction protocol, operational procedures were systematically varied while maintaining identical stoichiometry (see Table 1 and the conditions given there). Initially, n-decane was used as internal standard in the reaction mixture for analysis by gas chromatography (GC) (entry 1). Hydrogen cyanide (HCN) was added neat by Eppendorf pipette at −50 °C, resulting in a mere 5% conversion of styrene, and no product was observed. Adding the GC standard (n-decane) after the reaction to mitigate the potential for oxygen contamination (entry 2), a modest enhancement in performance was achieved, with 19% conversion and 14% yield. Introducing hydrogen cyanide as a solution in toluene at 22 °C markedly elevated both conversion and yield to 96% (Table 1, entry 3). This clearly shows that lowering the temperature to −50 °C while adding pure HCN via Eppendorf pipette has a negative effect on the effectiveness of the catalyst systems tested. Utilizing a rubber septum notably streamlines the operational procedure, minimizing the risk of contamination. This results in a conversion and yield exceeding 99% (Table 1, entry 4).
To the best of our knowledge, no literature has been found that demonstrates the significant influence of the HCN addition technique and its temperature dependency. With these optimizations, the operational procedure has been established. Further investigations are carried out on the catalytic activity.
2.2. Studies on Activity
The reductive elimination, as the rate-determining step, in nickel-catalyzed hydrocyanations, is generally accelerated by electron-withdrawing ligands like the commercially available diphosphite BiPhePhos [15].
Generally, the reaction rate is strongly temperature dependent. The protocol presented above (Section 2.1.2) involves catalyst solution preformation in a glovebox, followed by attaching a rubber septum lid. In a fume hood, styrene is added via syringe through the septum. Hydrogen cyanide (HCN) is added to one-third of the solvent in a separate Schlenk tube under cold inert conditions, causing HCN to freeze out. The solution is further warmed to the HCN melting point for syringe withdrawal and then injected into the reaction vessel via the septum to start the reaction. It is to be expected that this procedure will lead to a temperature profile that can strongly influence the reaction rate. The temperature is monitored by an internal thermometer (see Figure 5).
Following the addition of the cold hydrogen cyanide (HCN) solution to the catalyst/styrene mixture, an immediate temperature drop of 1.3 °C below 22 °C (21.8 °C) is observed. Subsequently, a rapid temperature rise to 23 °C occurs within 11 min, attributed to the exothermic nature of the addition reaction. After 11 min, a noticeable temperature decline indicates the nearly complete conversion of styrene to 2-phenylpropionitrile.
Essentially, the reaction displays minimal temperature fluctuations during this phase, potentially attributed to the use of a syringe with a cannula. This configuration exposes the HCN solution in the cannula to a high surface-to-volume ratio, enabling an efficient heat exchange. However, repeating the experiment without pre-cooling the toluene eliminates the 1.5 °C temperature drop. Nevertheless, cooling of the HCN was maintained to ensure precise stoichiometry and compliance with safety parameters, thus ensuring controlled and safe execution of the reaction protocol.
The reaction progress was monitored by GC analysis of the samples withdrawn from the reaction mixture (see reaction profile Figure S9). For this study, a catalyst concentration of 0.5 mol% was selected. The investigation revealed a remarkably fast reaction, achieving total styrene conversion in just 10 min. Notably, 20% conversion was observed already after 2.5 min, resulting in a turnover frequency (TOF20) of 817 h−1. This activity can likely be further increased by reducing the solvent amount, validated by stepwise solvent reduction from 3 mL (Table 2, entry 1) to 0.5 mL (Table 2, entry 4).
Under all reaction conditions, the conversion and yield exceeded 90%, which excludes the possibility of catalyst deactivation due to an increased HCN concentration. Another reaction profile was recorded for the minimum amount of solvent (entry 4), which reduced the total reaction volume from the previous 8.1 mL to 1.4 mL (Figure 6).
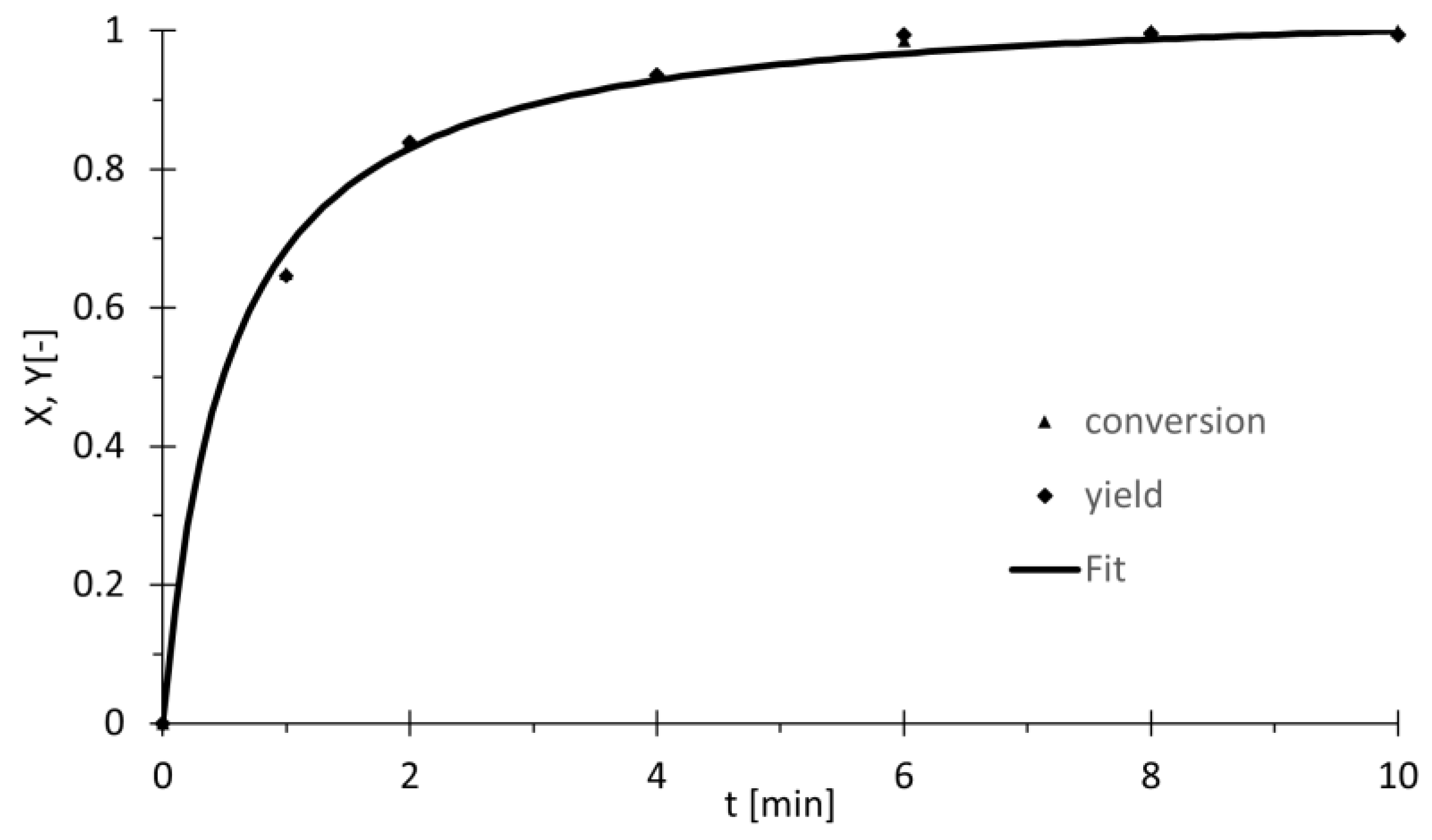
It turns out (Figure 6) that complete conversion is reached after 6 min, at which point no remaining styrene is detected. A yield of 70% is achieved after just one minute, which can be interpolated to 20% conversion within approximately 7.5 s, relating to a TOF20 of 19,000 h−1. Compared to the previous experiment, this remarkable increase in performance exceeds all values previously documented in the scientific literature. Further reducing the quantity of solvent employed in the experiment appears impractical due to the limited solubility of BiPhePhos. Alternatively, reducing the solvent amount for dilution of the highly volatile HCN risks inaccurate stoichiometry. By increasing the temperature during activity studies, the balance between deactivation and activity is investigated. The slower, gradual deactivation of the catalyst might still allow completion of the hydrocyanation reaction before full catalyst deactivation. At 60 °C, TOF20 increases to 191,000 h−1, more than ten times the results at 22 °C (Table 3, entries 1 and 2). Further raising the temperature to 90 °C boosts activity even further to 309,000 h−1 (Table 3, entry 3).
It can be concluded that the hydrocyanation reaction rate shows a higher temperature dependency compared to catalyst deactivation by bischelate formation.
2.3. Scale-Up and HCN-Dosing
A notable constraint within the present reaction setup is the required minimum catalyst concentration of 0.5 mol% to achieve full conversion and yield (Table 4, entry 1). Lowering the catalyst concentration to 0.1 mol% only yields 8% of the 2-phenyl propionitrile (Table 4, entry 2). Again, one potential reason is traces of oxygen contamination leading to catalyst deactivation considering the low amount of catalyst, 0.001 mmol, applied in the reaction. Despite the optimized reaction protocol, contamination might occur during the preparation of the HCN solution. In this case, running the reaction at a larger scale is expected to minimize that risk.
The reaction (entry 3) was scaled up by a factor of 5, maintaining the total catalyst amount (0.005 mmol Ni(cod)2) used in entry 1. The result is a conversion and yield exceeding 99% (entry 3).
Another experiment analogous to entry 2 was performed but this time HCN was added slowly over 5 min. A conversion of 59% instead of 8% was achieved at >99% selectivity (Table 4, entry 4). Both observations can be attributed to catalyst deactivation by traces of oxygen present in the hydrogen cyanide solution. If applied in a sufficient amount, i.e., exceeding the amount of the traces of oxygen, the remaining catalyst stays active and results in product formation. In the case of the fivefold preparation (entry 3), where 0.005 mmol of Ni(cod)2 are present (similar to the entry 1 conditions), this catalyst amount appears sufficient to achieve a full yield/conversion despite partial deactivation by oxygen. In entry 4, assuming the same total amount of contaminant in the HCN solution as in entry 2, the catalyst deactivation is sustained due to the slow addition. Ultimately, however, when the full amount of solution has been added, complete catalyst deactivation occurs, preventing full styrene conversion.
Excessive HCN concentrations are another potential cause of catalyst deactivation, as well. In this case, the dosage experiment (entry 4) would have been expected to show full conversion/yield due to the HCN concentration being kept low. Furthermore, the fivefold preparation (entry 3) would have been expected to show only minimal reactivity, maintaining the same Ni:HCN ratio from entry 2, but neither could be observed. This clearly suggests that traces of oxygen are still the leading cause for the low conversion/yields in entry 2.
In order to further push to the limits, an experiment at 20 mmol scale and with a reduced catalyst amount of 0.025 mol% was performed, maintaining the total catalyst amount of 0.005 mmol (entry 5). Under these conditions, 23% conversion and 94% selectivity 21% was observed. This indicates the potential for further improvement by scaling up the batch size, a possibility not explored further in this study.
These findings emphasize the potential of the BiPhePhos-nickel system for hydrocyanation applications, showcasing a notable breakthrough in catalytic activity not reported before. However, elevated temperature still leads to catalyst deactivation over time. Thus, a balanced approach is crucial for achieving optimal activity while maintaining catalyst stability.
3. Materials and Methods
All experiments were performed using standard Schlenk techniques. Working steps in the glovebox were performed in a Labmasterpro ECO from mBRAUN under an argon atmosphere. Hydrocyanations were performed in Schlenk tubes (height: 124.5 mm, inner diameter: 23.4 mm, glass thickness: 2.6 mm) with a GL-32 closure and Young stopcock connection.
3.1. Purification of Solvents and Substrates
Toluene, n-decane, and di-n-butyl ether are purified by inert distillation over sodium. Styrene is dried over P2O5 and, afterwards, carefully distilled under argon with reduced pressure. Temperatures should not exceed 50 °C to prevent polymerization. Before use, all liquids are degassed by freeze-pump thaw method. BiPhePhos, donated by Evonik AG, was applied without further purification.
3.2. Synthesis and Handling of Ni(cod)2
Ni(cod)2 is synthesized as a precipitate from dry Ni(acac)2 and n-Bu2Mg in dry and degassed tetrahydrofuran. For purification, it is washed with dry and degassed acetone based on the work of Baker et. al. [23]. The yielded yellow Ni(cod)2 is highly sensitive towards oxygen, sunlight, and increased temperatures. Therefore, it must be handled with care exclusively in an inert atmosphere. If stored in the glovebox’s freezer, it is stable for several months. Ni(cod)2 can only be handled using glass spatulas. Usage of stainless-steel spatulas results in the formation of metallic nickel black.
3.3. Handling of Hydrogen Cyanide and Safety Instructions
Hydrogen cyanide is produced from potassium cyanide and sulphuric acid in the presence of iron(III)-sulphate under a steam of argon. The yielded gaseous hydrogen cyanide is then dried by calcium chloride and condensed in a cooling trap. To remove traces of HCN from the argon stream, it is guided through two gas washing bottles containing a basic hydrogen peroxide solution.
For further purification, HCN is additionally degassed using the Freeze-Pump-Thaw method. Caution: Hydrogen cyanide is highly toxic and volatile. It must only be handled in a well-ventilated fume hood and with at least two instructed persons present. In addition to the general personal protective equipment for safe working in the laboratory (lab coat and safety goggles), a hydrogen cyanide gas detector (Honeywell BW™ Solo HCN, Poole, UK) and butyl rubber gloves (Honeywell Butoject™ 898) are required. Furthermore, HCN containers are stored at −26 °C in a lockable ATEX-certified freezer, located in a fume hood, which only instructed persons can access. The internal temperature is monitored via a battery-powered sensor connected to the Internet. For safe handling of single containers, the volume in a single storage container should not exceed 5 mL.
3.4. Gas Chromatography
GC samples were taken after removing the remaining hydrogen cyanide by a steam of argon. n-decane was added as an internal standard. About 0.7 mL of the reaction mixture is filtered through a syringe filter, placed in a GC vial, and diluted with toluene.
3.5. Calculation of Conversion (X), Yield (Y), and Selectivity (S)
First, the calibration factors for the used gas chromatography instrument (Agilent 7890A equipped with an Agilent HP-5 column equipped with an FID-detector) is determined with known masses of the analytes and n-decane or di-n-butyl ether (DBE) using the following Formula (1):
The resulting slope Rf is used for further calculation of analytes masses. The mass of styrene mstyrene within a GC sample is calculated with the weighed mass of the n-decane mdecane, the areas A of the chromatogram, and the calibration factor Rf using Formula (2):
The mass is then converted into the amount of substance n via Formula (3) to calculate the conversion X, the yield Y, and the selectivity S of the hydrocyanation according to Formulas (4)–(6):
4. Conclusions and Outlook
The experimental protocol for the Ni-catalyzed hydrocyanation of styrene as a model substrate was systematically refined. The addition of HCN as a solution at the reaction temperature was found to be critical for high catalytic activity and required stringent precautions to protect the oxygen-sensitive catalyst. The use of commercially available BiPhePhos as a ligand in the styrene hydrocyanation led to significant improvements in yield and selectivity. Together with the optimized protocol, this led to unprecedented catalytic activity with a TOF20 of up to 309,000 h−1, depending on the temperature. Surrogates require higher temperatures, resulting in more pronounced catalyst deactivation. Future improvements might be achieved by further scale-up and reducing the solvent amount—even a neat reaction can be considered. Long-term studies to balance stability, deactivation, and overall productivity are crucial for practical applications in large-scale processes.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/catal14030210/s1, Figure S1. Yellow color of the synthesized Ni(cod)2 indicates high purity. (Grey or beige color in-dicates the presence of metallic nickel, whereas green color indicates Ni(II) complexes); Figure S2. apparatus for hydrogen cyanide synthesis; Figure S3. GC-FID Calibration of Styrene with n-decane as internal standard; Figure S4. GC-FID Calibration of 2-phenylpropionitrile with n-decane as internal standard; Figure S5. GC-FID Calibration of Styrene with DBE as internal standard; Figure S6. GC-FID Calibration of 2-phenyl propionitrile with DBE as internal standard; Figure S7. 31P{1H}-NMR (162 MHz, toluene) of the preformed catalyst at room temperature. Con-ditions: Ni(cod)2 (13.8 mg, 5 mmol, 1 eq) and BiPhePhos (56.8 mg, 7 mmol, 1.2 eq) were weighed out, dissolved in toluene (3 mL) and stirred. Top: 4 h, bottom: 8 h; Figure S8. 31P{1H}-NMR (162 MHz, toluene) of the preforming solution at 90 °C. Reaction condi-tions: Ni(cod)2 (13.8 mg, 5 mmol, 1 eq) and BiPhePhos (48.7 mg, 6 mmol, 1.2 eq) were dissolved in toluene (3 mL) and stirred (90 °C, 4 h); Figure S9. Conversion/time-plot of the hydrocyanation of styrene: Reaction conditions: BiPhePhos (11.8 mg, 0.015 mmol,) is dissolved in toluene (5 mL) and Ni(cod)2 (3.4 mg, 0.013 mmol, 0.5 mol%) is added. Styrene (260.5 mg, 2.5 mmol, 1 eq) and dibutyl ether (168.6 mg) are added as internal GC standard at 22 °C. HCN (125 µL, 3.125 mmol, 1.25 eq) is dissolved in cold toluene (2.5 mL) in a separate flask and added to the reaction mixture; Figure S10. Conversion/time-plot of hydrocyanation of styrene with reduced amount of solvent: reaction conditions: BiPhePhos (11.8 mg, 0.015 mmol) is dissolved in toluene (0.5 mL) and Ni(cod)2 (3.4 mg, 0.013 mmol, 0.5 mol%) is added. Styrene (260.5 mg, 2.5 mmol, 1 eq) and dibutyl ether (174.3 mg) are added as an internal GC standard. The reaction mixture is placed inside of an oil bath at 60 °C for 5 minutes to reach 60 °C. HCN (125 µL, 3.125 mmol, 1.25 eq) is dissolved in cold toluene (0.75 mL) in a separate flask and added to the reaction mixture. X and Y based on GC analysis with DBE as internal standard; Figure S11. Conversion/time-plot of hydrocyanation of styrene with reduced amount of solvent: reaction conditions: BiPhePhos (11.8 mg, 0.015 mmol) is dissolved in toluene (0.5 mL) and Ni(cod)2 (3.4 mg, 0.013 mmol, 0.5 mol%) is added. Styrene (260.5 mg, 2.5 mmol, 1 eq) and dibutyl ether (174.3 mg) are added as an internal GC standard. The reaction mixture is placed in an oil bath at 90 °C for 5 minutes to reach 90 °C. HCN (125 µL, 3.125 mmol, 1.25 eq) is dissolved in cold toluene (0.75 mL) in a separate flask and added to the reaction mixture. X and Y based on GC analysis with DBE as internal standard; Figure S12. Temperature profile of hydrocyanation of styrene without precooling of HCN: Reaction conditions: BiPhePhos (4.7 mg, 0.006 mmol) was dissolved in toluene (2 mL) and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.5 mol%) was added. Styrene (104 mg, 1 mmol, 1 eq) was added. HCN (50 µL, 1.25 mmol, 1.25 eq) was dissolved in toluene (1 mL) in a separate flask and added to the reaction mix-ture. After the reaction time of 3 h at room temperature, n-decane was added as an internal standard after the reaction. The temperature was recorded with an EnviroPad-TC thermometer; Table S1. Gas flow profile of the GC method; Table S2. Temperature profile of the GC method.
Author Contributions
Conceptualization, T.K.; Data curation, T.K. and B.R.; Formal analysis, T.K. and B.R.; Investigation, T.K. and B.R.; Methodology, T.K.; Project administration, T.K. and D.V.; Supervision, D.V.; Validation, T.K.; Visualization, T.K.; Writing—original draft, T.K. and B.R.; Writing—review and editing, D.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in the Supplementary Materials.
Acknowledgments
We would like to acknowledge Evonik Industries AG for providing the BiPhePhos ligand. We would also like to acknowledge the TU Dortmund University and the chair of industrial chemistry for providing laboratory support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bohnet, M.; Ullmann, F. (Eds.) Ullmann’s Encyclopedia of Industrial Chemistry, 6th ed.; Wiley VCH: Hoboken, NJ, USA, 2003. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice, 1st ed. paperback; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Falk, A.; Göderz, A.-L.; Schmalz, H.-G. Enantioselective nickel-catalyzed hydrocyanation of vinylarenes using chiral phosphine-phosphite ligands and TMS-CN as a source of HCN. Angew. Chem. Int. Ed. 2013, 52, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, K.; Nagafuchi, T.; Tominaga, K.; Sato, K. Efficient nickel-catalyzed hydrocyanation of alkenes using acetone cyanohydrin as a safer cyano source. Tetrahedron Lett. 2016, 57, 3199–3203. [Google Scholar] [CrossRef]
- Baker, M.J.; Harrison, K.N.; Orpen, A.G.; Pringle, P.G.; Shaw, G. Chelating diphosphite complexes of nickel(0) and platinum(0): Their remarkable stability and hydrocyanation activity. J. Chem. Soc., Chem. Commun. 1991, 12, 803. [Google Scholar] [CrossRef]
- Yu, R.; Rajasekar, S.; Fang, X. Enantioselective Nickel-Catalyzed Migratory Hydrocyanation of Nonconjugated Dienes. Angew. Chem. Int. Ed. 2020, 59, 21436–21441. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Xing, Y.; Fang, X. Regio-, Chemo-, and Enantioselective Ni-Catalyzed Hydrocyanation of 1,3-Dienes. Organ. Lett. 2021, 23, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ni, J.; Yu, R.; Cheng, G.-J.; Fang, X. Ni-Catalyzed Isomerization-Hydrocyanation Tandem Reactions: Access to Linear Nitriles from Aliphatic Internal Olefins. Org. Lett. 2021, 23, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Jiao, M.; Ni, J.; Yu, R.; Cheng, G.-J.; Fang, X. Nickel-Catalyzed Migratory Hydrocyanation of Internal Alkenes: Unexpected Diastereomeric-Ligand-Controlled Regiodivergence. Angew. Chem. Int. Ed. 2021, 60, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xie, X.; Xu, W.; Liu, Y. Nickel-catalyzed highly regioselective hydrocyanation of alkenes with Zn(CN)2. Org. Chem. Front. 2019, 6, 2037–2042. [Google Scholar] [CrossRef]
- Reisenbauer, J.C.; Finkelstein, P.; Ebert, M.-O.; Morandi, B. Mechanistic Investigation of the Nickel-Catalyzed Transfer Hydrocyanation of Alkynes. ACS Catal. 2023, 13, 11548–11555. [Google Scholar] [CrossRef]
- Fang, X.; Yu, P.; Morandi, B. Catalytic reversible alkene-nitrile interconversion through controllable transfer hydrocyanation. Science 2016, 351, 832–836. [Google Scholar] [CrossRef]
- Reisenbauer, J.C.; Bhawal, B.N.; Jelmini, N.; Morandi, B. Development of an Operationally Simple, Scalable, and HCN-Free Transfer Hydrocyanation Protocol Using an Air-Stable Nickel Precatalyst. Org. Process Res. Dev. 2022, 26, 1165–1173. [Google Scholar] [CrossRef]
- Goertz, W.; Keim, W.; Vogt, D.; Englert, U.; Boele, M.D.K.; van der Veen, L.A.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Electronic effects in the nickel-catalysed hydrocyanation of styrene applying chelating phosphorus ligands with large bite angles. J. Chem. Soc. Dalton Trans. 1998, 2981–2988. [Google Scholar] [CrossRef]
- Bini, L.; Müller, C.; Vogt, D. Mechanistic Studies on Hydrocyanation Reactions. ChemCatChem 2010, 2, 590–608. [Google Scholar] [CrossRef]
- Hewat, A. Zerovalent Nickel Complexes as Catalysts in the Hydrocyanation and Isomerization of functionalized Olefins. Ph.D. Thesis, RWTH Aachen University, Aachen, Germany, 2000. [Google Scholar]
- Bini, L.; Müller, C.; Vogt, D. Ligand development in the Ni-catalyzed hydrocyanation of alkenes. Chem. Commun. 2010, 46, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.W.; Swift, H.E. The addition of hydrogen cyanide to α-olefins catalyzed by nickel(0) complexes. J. Catal. 1972, 26, 254–260. [Google Scholar] [CrossRef]
- Tolman, C.A.; Seidel, W.C.; Druliner, J.D.; Domaille, P.J. Catalytic hydrocyanation of olefins by nickel(0) phosphite complexes—Effects of Lewis acids. Organometallics 1984, 3, 33–38. [Google Scholar] [CrossRef]
- Göthlich, A.P.V.; Tensfeldt, M.; Rothfuss, H.; Tauchert, M.E.; Haap, D.; Rominger, F.; Hofmann, P. Novel Chelating Phosphonite Ligands: Syntheses, Structures, and Nickel-Catalyzed Hydrocyanation of Olefins. Organometallics 2008, 27, 2189–2200. [Google Scholar] [CrossRef]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R. Transition Metal Complex Catalyzed Processes. U.S. Patent 12,329, 6 September 1988. [Google Scholar]
- Fazekas, E. Iron and Nickel Complexes as Catalysts in CO2/Epoxide Coupling, Radical Polymerisation and Hydrocyanation Reactions. Ph.D. Thesis, The University of Edinburgh, Edinburgh, UK, 2018. Available online: https://era.ed.ac.uk/handle/1842/35688 (accessed on 1 February 2024).
- Sicard, A.J.; Baker, R.T. Safe and Expeditious Preparation of Ni(cod)2 for Same-Day High-Throughput Screening. Org. Process Res. Dev. 2020, 24, 2950–2952. [Google Scholar] [CrossRef]

Figure 2.
BiPhePhos.

Figure 3.
Hydrocyanation of styrene to 2-phenylpropionitrile and 3-phenylpropionitrile.

Figure 4.
Nickel chelates and their corresponding 31P-NMR shifts.

Figure 5.
Temperature profile of hydrocyanation of styrene with precooling of HCN. Reaction conditions: BiPhePhos (4.7 mg, 0.006 mmol) was dissolved in toluene (2 mL), and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.5 mol%) was added. Styrene (104 mg, 1 mmol, 1 eq) was then added. HCN (50 µL, 1.25 mmol, 1.25 eq) was dissolved in cold toluene (1 mL) in a separate flask and added to the reaction mixture. After the reaction time of 3 h at 22 °C, n-decane was added as an internal standard.
Figure 5.
Temperature profile of hydrocyanation of styrene with precooling of HCN. Reaction conditions: BiPhePhos (4.7 mg, 0.006 mmol) was dissolved in toluene (2 mL), and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.5 mol%) was added. Styrene (104 mg, 1 mmol, 1 eq) was then added. HCN (50 µL, 1.25 mmol, 1.25 eq) was dissolved in cold toluene (1 mL) in a separate flask and added to the reaction mixture. After the reaction time of 3 h at 22 °C, n-decane was added as an internal standard.

Figure 6.
Reaction profile for styrene hydrocyanation with reduced amount of solvent. Reaction conditions: BiPhePhos (11.8 mg, 0.015 mmol) is dissolved in toluene (0.5 mL), and Ni(cod)2 (3.4 mg, 0.0125 mmol, 0.5 mol%) is added. Styrene (260.5 mg, 2.5 mmol, 1 eq) and di-n-butyl ether (174.3 mg) as an internal GC standard are added. HCN (125 µL, 3.125 mmol, 1.25 eq) is dissolved in cold toluene (0.75 mL) in a separate flask and added to the reaction mixture. Conversion and yield based on GC analysis with DBE as internal standard. Fit: X(t) = (1.05375t)/(0.5381627 + t).
Figure 6.
Reaction profile for styrene hydrocyanation with reduced amount of solvent. Reaction conditions: BiPhePhos (11.8 mg, 0.015 mmol) is dissolved in toluene (0.5 mL), and Ni(cod)2 (3.4 mg, 0.0125 mmol, 0.5 mol%) is added. Styrene (260.5 mg, 2.5 mmol, 1 eq) and di-n-butyl ether (174.3 mg) as an internal GC standard are added. HCN (125 µL, 3.125 mmol, 1.25 eq) is dissolved in cold toluene (0.75 mL) in a separate flask and added to the reaction mixture. Conversion and yield based on GC analysis with DBE as internal standard. Fit: X(t) = (1.05375t)/(0.5381627 + t).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Different operational procedures for styrene hydrocyanation.
| Entry | n-Addition | T @ HCN Addition [°C] | Septum Use | X (a) [%] | Y (a),(b) [%] | S (c) [%] |
|---|---|---|---|---|---|---|
| 1 | before reaction (d) | −50 | No | 5 | 0 | 0 |
| 2 | after reaction (e) | −50 | No | 19 | 14 | 75 |
| 3 | after reaction (f) | RT | No | 96 | 96 | >99 |
| 4 | after reaction (f) | RT | Yes | >99 | >99 | >99 |
(a): Based on GC analysis with n-decane as an internal standard. (b): Yield of the branched product. (c): Selectivity to product nitriles. (d): Reaction conditions: BiPhePhos (4.7 mg, 0.006 mmol) was dissolved in toluene (3 mL), and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.005 eq) was added. n-decane was added as an internal standard before the addition of styrene (104 mg, 1 mmol, 1 eq). The reaction mixture was cooled down (−50 °C), and HCN (50 µL, 1.25 mmol, 1.25 eq) was added. The reaction mixture was then warmed to 22 °C for 3 h. (e): BiPhePhos (4.7 mg, 0.006 mmol) was dissolved in toluene (3 mL), and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.5 mol%) was added. Styrene (104 mg, 1 mmol, 1 eq) was then added. The reaction mixture was cooled down (−50 °C), and HCN (50 µL, 1.25 mmol, 1.25 eq) was added. The reaction mixture was warmed to 22 °C. After the reaction time of 3 h at 22 °C, n-decane was added as an internal standard after the reaction. (f): BiPhePhos (4.7 mg, 0.006 mmol) was dissolved in toluene (2 mL) and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.5 mol%) was added. Styrene (104 mg, 1 mmol, 1 eq) was then added. HCN (50 µL, 1.25 mmol, 1.25 eq) was dissolved in cold toluene (1 mL) in a separate flask and added to the reaction mixture. After the reaction time of 3 h at 22 °C, n-decane was added as an internal standard.
Table 2.
Reduction of solvent amount.
| Entry | c(styrene) [mol/L] | Total Solvent Amount [mL] | X (a) [%] | Y (a),(b) [%] | S (c) [%] |
|---|---|---|---|---|---|
| 1 | 0.32 | 3 (d) | >99 | >99 | >99 |
| 2 | 0.54 | 1.7 | 94 | 99 | >99 |
| 3 | 0.79 | 1.1 | 93 | 98 | >99 |
| 4 | 1.51 | 0.5 (e) | >99 | >99 | >99 |
Reaction conditions: BiPhePhos (4.7 mg, 0.006 mmol) is dissolved in toluene (0.2 mL), and Ni(cod)2 (1.4 mg, 0.005 mmol, 0.5 mol%) is added. Additional toluene is added if necessary, and styrene (104 mg, 1 mmol, 1 eq) is added. HCN (50 µL, 1.25 mmol, 1.25 eq) is dissolved in cold toluene (0.5 mL) in a separate flask and added to the reaction mixture. After the reaction time of 3 h, n-decane is added as an internal standard. (a): Based on GC analysis with n-decane as an internal standard. (b): Yield of the branched product. (c): Selectivity to product nitriles. (d): HCN dissolved in 1 mL toluene. (e) Addition of HCN dissolved in 0.3 mL toluene.
Table 3.
Activity of nickel/BiPhePhos catalyst in styrene hydrocyanation at 22, 60 and 90 °C.
| Entry | T [°C] | TOF20 [×103 h−1] |
|---|---|---|
| 1 | 22 | 19 |
| 2 | 60 | 191 |
| 3 | 90 | 309 |
Reaction conditions: BiPhePhos (11.8 mg, 0.015 mmol) is dissolved in toluene (0.5 mL), and Ni(cod)2 (3.4 mg, 0.0125 mmol, 0.5 mol%) is added. Styrene (260.5 mg, 2.5 mmol, 1 eq) and di-n-butyl ether (174.3 mg) as an internal GC standard are then added. HCN (125 µL, 3.125 mmol, 1.25 eq) is dissolved in cold toluene (0.75 mL) in a separate flask and added to the reaction mixture.
Table 4.
Scale-up and dosing of HCN.
| Entry | Batch Size (Styrene) [mmol] | eq.(Ni(cod)2) [mol%] | n(Ni(cod)2) [mmol] | Time of Dosiation [min] | X (a) [%] | Y (a),(b) [%] | S (c) [%] |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 0.5 | 0.005 | 0 | >99 | >99 | >99 |
| 2 | 1 | 0.1 | 0.001 | 0 | 8 | <1 | 4 |
| 3 | 5 | 0.1 | 0.005 | 0 | >99 | >99 | >99 |
| 4 | 1 | 0.1 | 0.001 | 5 | 59 | 59 | >99 |
| 5 | 20 (d) | 0.025 | 0.005 | 0 | 23 | 21 | 94 |
Reaction conditions: BiPhePhos is dissolved in toluene, and Ni(cod)2 is added. HCN is dissolved in cold toluene in a separate flask and added to the reaction mixture. After the reaction time of 3 h at 22 °C, n-decane is added as an internal standard. (a): Based on GC analysis with n-decane as an in-ternal standard. (b): Yield of the branched product. (c): Selectivity to product nitriles. (d): Half solvent amount (0.5 mL toluene per 1 mmol Styrene).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Köhler, T.; Rienhoff, B.; Vogt, D. Nickel(BiPhePhos)-Catalyzed Hydrocyanation of Styrene—Highly Increased Catalytic Activity by Optimized Operational Procedures. Catalysts 2024, 14, 210. https://doi.org/10.3390/catal14030210
AMA Style
Köhler T, Rienhoff B, Vogt D. Nickel(BiPhePhos)-Catalyzed Hydrocyanation of Styrene—Highly Increased Catalytic Activity by Optimized Operational Procedures. Catalysts. 2024; 14(3):210. https://doi.org/10.3390/catal14030210
Chicago/Turabian StyleKöhler, Till, Bernd Rienhoff, and Dieter Vogt. 2024. "Nickel(BiPhePhos)-Catalyzed Hydrocyanation of Styrene—Highly Increased Catalytic Activity by Optimized Operational Procedures" Catalysts 14, no. 3: 210. https://doi.org/10.3390/catal14030210
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.