Abstract
A synthetic scheme was developed for the large-scale preparation of a dimethylthiophene-fused and tetrahydroquinaldine-linked dimethylcyclopentadienyl titanium complex (2), which is a high-performance homogeneous Ziegler catalyst. 2,3,4,5-Tetramethyl-4,5-dihydrocyclopenta[b]thiophen-6-one was prepared without chromatography purification on the 40-g scale in a laboratory setting, from which the ligand precursor for 2 was obtained in 65% yield on a 50-g scale in a one-pot without the need for chromatography purification. Metallation was achieved in a high yield (78%) through reaction of the dilithiated compound with TiCl4. Many derivatives were prepared by employing the same synthetic scheme as applied for 2. Among them, the titanium complex prepared from 2-methyl-4,5-dimethyl-6-(2-n-butyl-2,3,4,5-tetrahydroquinolin-8-yl)-4H-cyclopenta[b]thiophene exhibited an exceptionally high activity. Under commercially relevant high-temperature polymerization conditions (160 °C), this compound showed a higher activity than 2 (126 × 106 g/molTi∙h versus 72 × 106 g/molTi∙h), albeit with the formation of a polymer of slightly lower molecular weight (Mw, 159,000 versus 218,000) and with a slightly lower 1-octene content (9.3 mol% versus 12 mol%).
1. Introduction
In 1977, Kaminsky demonstrated that methylaluminoxane-activated metallocenes can be useful in the polymerization of olefins [1]. The industrial production of resins using metallocene catalysts began in the early 1990s, and currently, some 5 million tons of polyethylene (PE) and about 1.5 million tons of polypropylene (PP) are produced annually using the metallocene catalysts [2]. Now, there is a bright industrial future for metallocene catalysts. Kaminsky’s initial bis(cyclopentadienyl)-type metallocene catalysts were later expanded to half-metallocene catalysts, which are titanium(IV) complexes coordinated by a cyclopentadienyl-type ligand and an amido-type ligand [3,4,5,6,7,8,9]. A typical representative of the half-metallocene catalysts is the constrained-geometry catalyst (CGC) [Me2Si(η5-Me4C5)(NtBu)]TiCl2 [10,11,12]. The CGC's merits are its high activity, high α-olefin incorporation, high thermal stability, and high molecular weight in ethylene/α-olefin copolymerizations. A variety of CGC derivatives have been prepared to test their catalytic performances. We have also prepared a number of complexes through the replacement of the Me2Si bridge in the CGC with an ortho-phenylene bridge [13,14,15]. Among the prepared ortho-phenylene bridged complexes, the tetrahydroquinoline-derived one (1 in Figure 1) showed excellent catalytic performance, displaying better comonomer incorporation than the CGC as well as a high activity [16,17,18]. The resin produced with this complex had a lower density than that produced with the CGC, even for the same 1-octene content [19]. The higher number of long-chain branches was another merit observed in the resin [20]. The complex can be prepared simply and economically on a large scale, enabling its commercial use [21].
Later, the thiophene-fused analogue 2 and 3 were prepared and found to show some advantages over 1 (Figure 1). It produces a higher-molecular-weight polymer and also exhibits greater thermal stability [22]. Other metallocenes and half-metallocenes prepared using thiophene-fused cyclopentadienyls display the same trend in their catalytic performances [6,23,24,25]. These features allow higher temperature to be used, which is a merit when performing the solution process. These compounds show high activities even when activated with methylaluminoxane (MAO), whereas 1 is not active with MAO. In this work, we report a synthetic scheme for the large-scale preparation of 2 and many derivatives of 2, for which the catalytic performances were screened in ethylene/1-octene polymerization. One of these shows a better catalytic performance than 2.
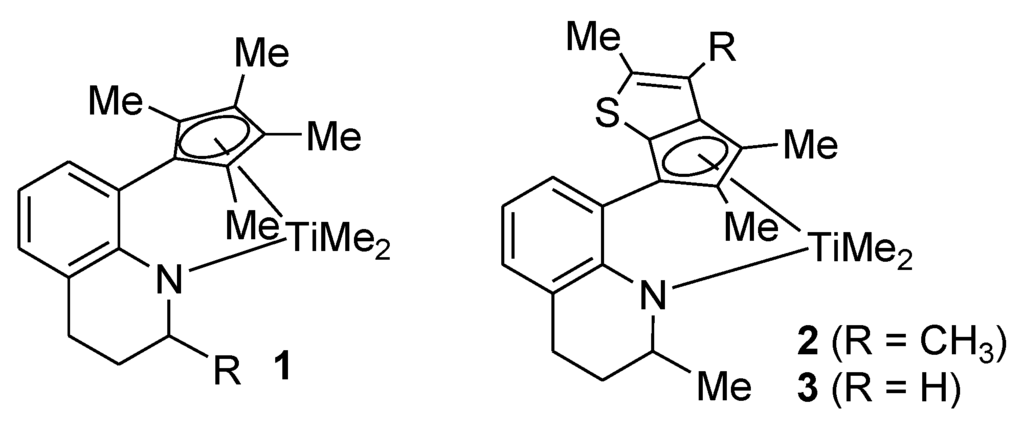
Figure 1.
Highly active catalysts for ethylene/α-olefin copolymerization.
2. Results and Discussion
2.1. Large-Scale Synthesis of 2
For use in a commercial process, the catalyst should be synthesized inexpensively and on a large scale, without the need of a chromatographic purification procedure. The thiophene-fused cyclopentanone 4 could be synthesized atom-economically by using tiglic acid and 2,3-dimethylthiophene (Scheme 1). The dissolution of the two components in polyphosphoric acid triggered, successively, the Friedel-Crafts acylation and the Nazarov cyclization, to generate 4 in a one-pot process. The yield was good, but the formation of a certain amount of side product (~10%) was problematic. The side product could not be eliminated from the main product by vacuum distillation. It could be separated by column chromatography using silica gel, but this procedure is a burden in a large-scale synthesis. The side product was identified to be a Friedel-Crafts adduct of 2,4-dimethylthiophene with tiglic acid (5). The starting material, 2,3-dimethylthiophene was prepared through the reduction of commercially available 3-formyl-2-methylthiophene. The commercial grade of 3-formyl-2-methylthiophene contains a certain amount of 4-formyl-2-methylthiophene (~10%), which was transformed to 2,4-dimethylthiophene during the reduction process. Because the boiling point of 5 is similar to that of 4, compound 5 could not be removed by vacuum distillation. Compound 5 is a good substrate for the 1,4-addition of thiol, and could be converted quantitatively to 6 when the crude mixture of 4 and 5 was treated with 1-hexanethiol (1.5 equivalents per 5) in the presence of a catalytic amount of [nBu4N]+F− (5 mol% per 5) in neat condition for 30 min at room temperature. The desired product 4 remained intact during the treatment and was distilled out in the subsequent vacuum distillation (85 °C/0.15 mmHg), whereas the converted compound 6 remained in the distillation pot owing to its higher boiling point. Through this procedure, the pure compound 4 could be prepared on a 40-g scale in a laboratory setting, without the need of column chromatography.
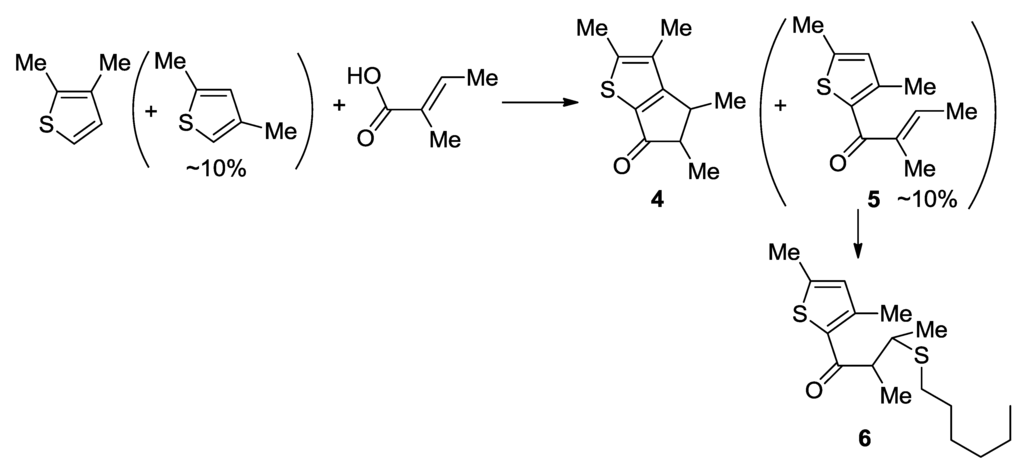
Scheme 1.
A large-scale synthesis of thiophene-fused cyclopentanone.
We have developed a simple protocol for the directed ortho-lithiation of secondary anilines. This directed ortho-lithiation is a powerful tool for the functionalization of aryl compounds, and various groups are employed as directing groups [26,27]. We found that the lithium carbamate (-N(COOLi)-) group, which was generated simply from the amino group of a secondary aniline through treatment with nBuLi and subsequent addition of CO2, was able to act as a directing group in the deprotonation of the ortho-proton. The lithium carbamate group (-N(COOLi)-) could be reconverted easily to the original amino group (-NH-) in the acidic work-up. The generated ortho-lithiated anilines were reacted directly, without isolation, with 2-cyclopentenones to prepare a selection of ligands for the ortho-phenylene-bridged half-metallocenes.
Through the same protocol, 1,2,3,4-tetrahydroquinaldine could be converted to the ligand precursor for 2, that is, compound 7 (Scheme 2). After the reaction between the ortho-lithiated compound and the thiophene derivative 4, aqueous HCl was added to trigger the elimination of the resulting tertiary alcohol. During this acid treatment, the HCl salt of the desired compound 7 precipitated as a white powder. Isolation of the precipitates by filtration and subsequent neutralization with aqueous Na2CO3 yielded the pure compound. The yield for this transformation was moderate (50%), and to make matters worse, it fluctuated from batch by batch. The yield could be improved to 65% by altering the reaction conditions and work-up procedure slightly. After charging with CO2 gas, sufficient time should be allowed for the consistent generation of the desired lithium carbamate. For the complete precipitation of the HCl salt of the desired compound 7, the ethyl acetate was replaced with the non-polar hexane solvent. The 65% yield is fairly satisfactory considering that the yields reported for other related reactions between aryllithiums and cyclopentenones are typically in the range 40%–50%. With this procedure, compound 7 could be prepared on the 50-g scale in a laboratory setting without the need for column chromatography.
The metallation of the CGC-type ligands was a problem. In the initial report by Dow, the CGC [Me2Si(η5-Me4C5)(NtBu)]TiCl2 was obtained in just 10% yield when the dilithiated compound [Me2Si(η5-Me4C5)(NtBu)]2−Li+2 was reacted with TiCl4 [28]. The main side reaction might be electron transfer from the dilithiated compound to TiCl4. The electron transfer reaction was blocked by reacting TiCl3(THF)3 with the dilithiated compound to generate the Ti(III) complex [Me2Si(η5-Me4C5)(NtBu)]TiCl quantitatively; this was then converted quantitatively to the desired Ti(IV) complex [Me2Si(η5-Me4C5)(NtBu)]TiCl2 through the use of AgCl as an oxidant. Later, Resconi introduced a one-step metallation method: the addition of four equivalents of MeLi and then followed by the treatment with TiCl4 [29]. With this protocol, the dimethyltitanium complex 2 was generated from 7 in 58% yield. However, this procedure raises some concerns when considering large-scale synthesis. The handling of MeLi requires some caution; it is sold as a solution in diethyl ether, and should be stored and used at freezer temperature to prevent decomposition. The dimethyltitanium complex 2 is a little unstable, and decomposes slowly when stored even in a glove box. The dichlorotitanium complex 8 could be prepared on a 20-g scale in a laboratory setting through the direct reaction of the dilithiated compound of 7 with TiCl4. In this case, the electron-transfer reaction did not occur, and 8 was formed in high yield (78%). The dilithiation reaction was performed at room temperature in hexane using nBuLi. The handling of a hexane solution of nBuLi is less problematic than working with the diethyl solution of MeLi. Complex 8 was stable, and did not decompose at room temperature under an inert atmosphere. Complex 8 could be converted quantitatively to the dimethyltitanium complex 2 by using MeMgBr in diethyl ether, which also poses fewer handling burdens than MeLi.
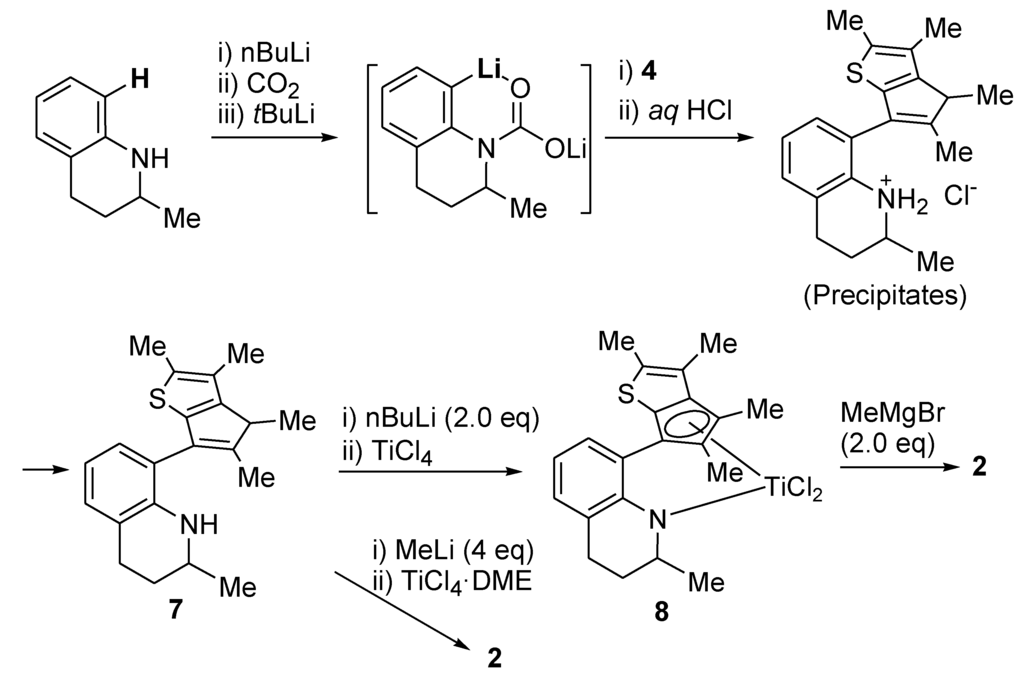
Scheme 2.
Large scale preparation of 2.
2.2. Synthesis of Derivatives
Whereas 2,3-dimethylthiophene was prepared with 2,4-dimethylthiophene (~10%) contamination from 3-formyl-2-methylthiophene, 2-methylthiophene is commercially available in 98% purity. The thiophene-fused cyclopentanone 9 (in Scheme 3) could be prepared on a large scale (50-g scale in a laboratory setting) by using 2-methylthiophene, applying the same conditions and procedure as for 4. The yield was excellent (94%) with the formation of negligible amounts of side products. The pure compound could be isolated easily by vacuum distillation (130 °C/0.25 mmHg). Thus, 9 is accessed more easily and inexpensively than 4.
Some derivatives were prepared by using 9, especially through substituent variation at the 2-position of the 1,2,3,4-tetrahydroquinoline unit (Scheme 2). Preparation methods for 1,2,3,4-tetrahydroquinoline derivatives bearing various alkyl groups at the 2-position have been reported. They can be prepared from quinoline as follows. Alkyllithium (RLi) attacks quinoline at the 2-position to give 2-R-1,2-dihydroquinoline, which is reduced with sodium metal to afford the desired 2-R-1,2,3,4-tetrahydroquinoline (R = Et, iPr, nBu). Lithium-carbamate-directed ortho-lithiation was carried out with 2-R-1,2,3,4-tetrahydroquinoline, by employing a similar procedure and conditions to those for the preparation of 7. Previously, we observed and reported that the presence of a bulky group near the nitrogen atom lowered the lithiation yield, limiting the scope of substrates for the lithium-carbamate-directed ortho-lithiation [17]. We discovered that the main problem was in the step of the CO2 reaction. A bulky group near the nitrogen atom would block or slow down the reaction between the lithium amide and CO2 in diethyl ether. This problem was solved by replacing the diethyl ether solvent with THF. Thus, ortho-lithiated compounds were generated for all three 2-R-1,2,3,4-tetrahydroquinolines (R = Et, iPr, nBu), consequently yielding the desired ligand precursors 10–12 in moderate overall yields (40%–48%) after reaction with 9. The ligand precursors 10–12 were isolated as a mixture of four isomers, respectively, owing to the presence of two chiral centers and a rotational barrier around the single bond connecting the two fused rings. The isomers could not be separated by silica gel chromatography because the four isomers were observed as a single spot in thin-layer chromatography. Consequently, the 1H and 13C NMR signals observed were somewhat complicated, but were assignable to the structures of the desired compounds 10–12, respectively. From 10–12, the dichlorotitanium complexes 13–15 were prepared by using the conditions and procedure employed for the large-scale synthesis of 8. When a hexane solution of nBuLi was added to 10–12 in hexane and the mixture was stirred overnight at room temperature, dilithiation occurred, giving rise to the precipitation of a powder or oil, which became soluble with the addition of diethyl ether. The addition of an ether solution of the dilithiated compound to a slurry of TiCl4 in diethyl ether resulted in the clean formation of the desired complexes 13–15 in high yields (75%–86%). The yields were better than those (~50%) attained with the method introduced by Resconi (4 equiv. MeLi and then TiCl4). The titanium complexes were isolated as a mixture of two isomers and two sets of signals were observed in the 1H NMR spectra. Some signals, especially that of the proton attached to the thiophene ring, were separated clearly for each isomer, enabling the calculation of the isomer ratios (1:0.8, 1:0.5, and 1:0.6 for 13, 14, and 15, respectively).
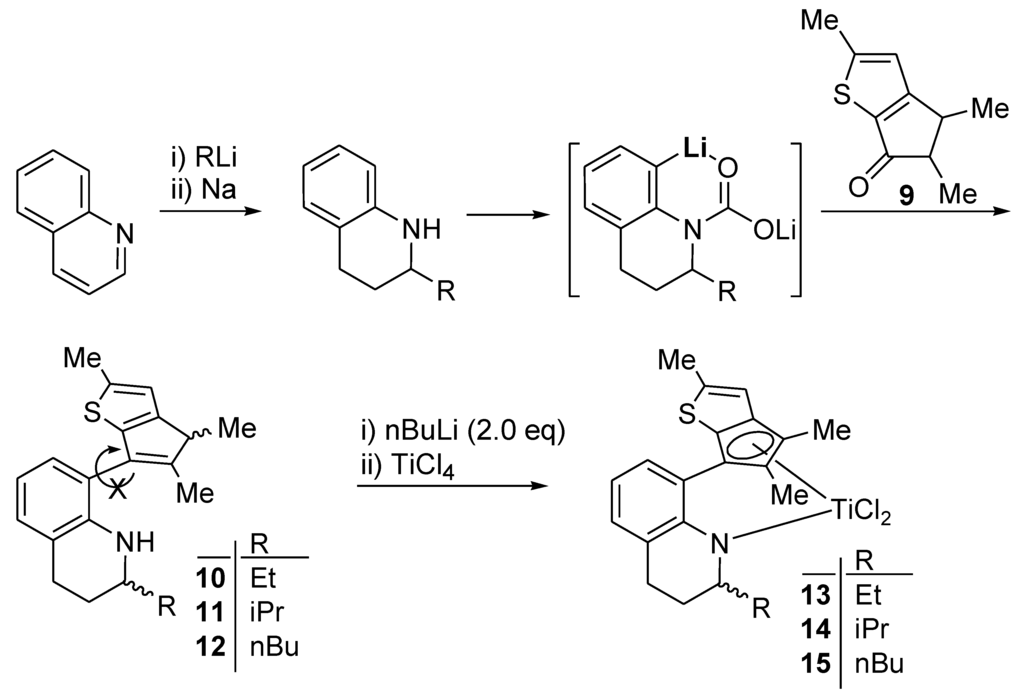
Scheme 3.
Preparation of derivatives.
The reaction of 2,3-dimethylthiophene with methacrylic acid in polyphosphoric acid was not successful, in contrast with the great success achieved with tiglic acid. When polyphosphoric acid was replaced with Eaton's reagent (7.7 wt% phosphorus pentoxide in methanesulfonic acid), the corresponding thiophene-fused cyclopentanone 16 was obtained in moderate yield (~40%). In this reaction, many side products were obtained, and product 16 was isolated by chromatography on silica gel. The reaction between 2-methylthiophene and methacrylic acid in either polyphosphoric acid or Eaton's reagent was unsuccessful.
Through the reaction of 16 with the ortho-lithiated compounds of 2-R-1,2,3,4-tetrahydroquinoline (R = Me, Et, iPr, and nBu), the desired ligand precursors 17–20 were formed in moderate yields of 42%–47% (Scheme 4). The lithium-carbamate-directed ortho-lithiation occurred under the newly set conditions even for the compounds bearing very bulky groups at the 2-position (such as 2-tert-butyl-1,2,3,4-tetrahydroquinoline); the ligand precursor 21 was also formed, albeit in rather low yield (31%). From 17–21, the corresponding dichlorotitanium complexes were obtained in high yields (73%–88%) by using the method applied for the preparation of 8 and 13–15, that is, dilithiation in hexane with two equivalents of nBuLi and subsequent treatment with TiCl4. The crude complexes were obtained as a mixture of two isomers, and two sets of signals were observed in the 1H NMR spectra. The Cp-H signals were observed clearly for each isomer separately, enabling the calculation of the isomer ratios. For 23–25, the ratios were not biased (1:0.8), but the ratios were slightly biased toward one isomer for 22 and 26 (1:0.6 and 1:0.4, respectively). The complexes were not crystalline, and were deposited as powders when the saturated hexane solution was kept in a freezer. For complex 26, a very small fraction was isolated after crystallization five times in hexane; the ratio was 1:0.1, but single crystals for X-ray crystallography could not be obtained from this highly biased mixture of the two isomers.
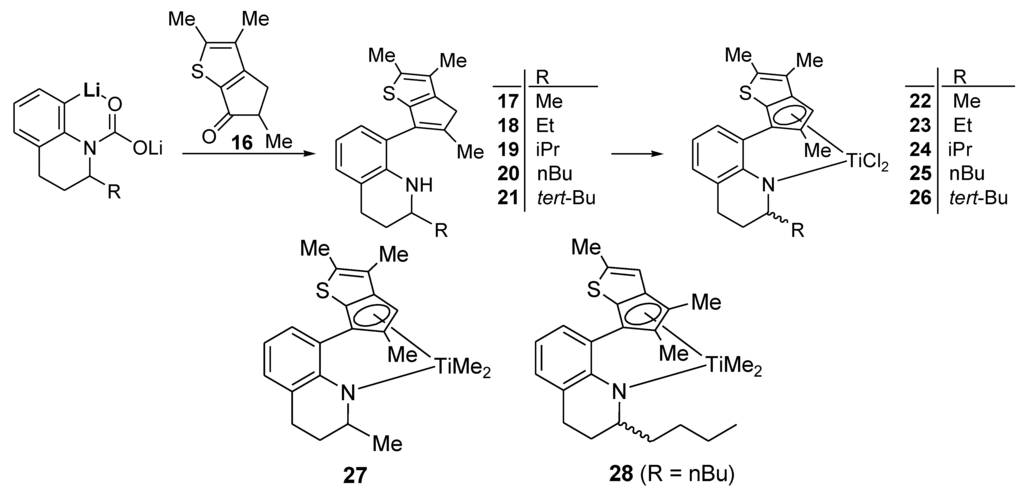
Scheme 4.
Preparation of derivatives.
2.3. X-ray Crystallographic Studies
Single crystals of the dichlorotitanium complex 15 were obtained through recrystallization in hexane at −30 °C, and the molecular structure was determined by X-ray crystallography (Figure 2). The two diastereomers are packed randomly in a single crystal. Single crystal growth was not successful for the dichlorotitanium complexes 22–26, but single crystals were obtained in hexane at −30 °C after the dichlorotitanium complex 22 was converted to the corresponding dimethyltitanium complex 27. The 1H NMR spectrum of the crystals showed a set of signals indicating the deposition of the major isomer. The molecular structure of 27 determined by X-ray crystallography is shown in Figure 3. The metrical parameters are summarized in Table 1, and compared with those of 2. The Cp(centroid)-Ti-N angles, which have been used as a qualitative measure of the “constrained geometry”, were 105.93° and 106.07° for 15 and 27, respectively. The angles were smaller than that observed for the CGC [Me2Si(η5-Me4C5)(NtBu)]TiCl2, (107.6°), indicating that 15 and 27 are more constrained than the CGC [30]. The Cl-Ti-Cl angle in 15 (103.34(3)°) and the CH3-Ti-CH3 angle in 27 (101.44(9)°) were larger than the CH3-Ti-CH3 angle observed in 2 (99.9(2)°), possibly because of the smaller number of methyl substituents in the thiophene-fused cyclopentadienyl ring. The Cp(centroid)-Ti and the Ti-N distances were also shorter in 15 and 27 than in 2, possibly owing to the smaller number of methyl substituents. The sum of the bond angles around the nitrogen atom was 360° in both complexes, indicating the π-donation of an electron pair on the nitrogen atom to the titanium through sp2-hybridization.
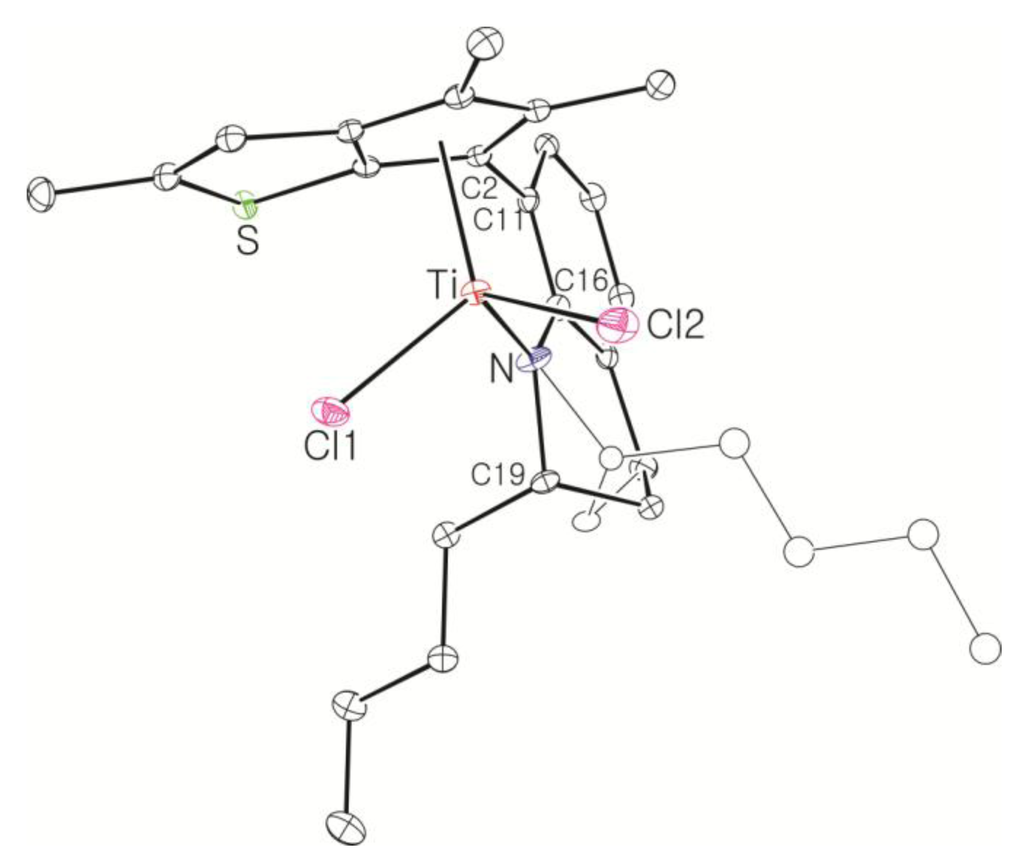
Figure 2.
Thermal ellipsoid plot (30% probability level) of 15. Hydrogen atoms are omitted for clarity.
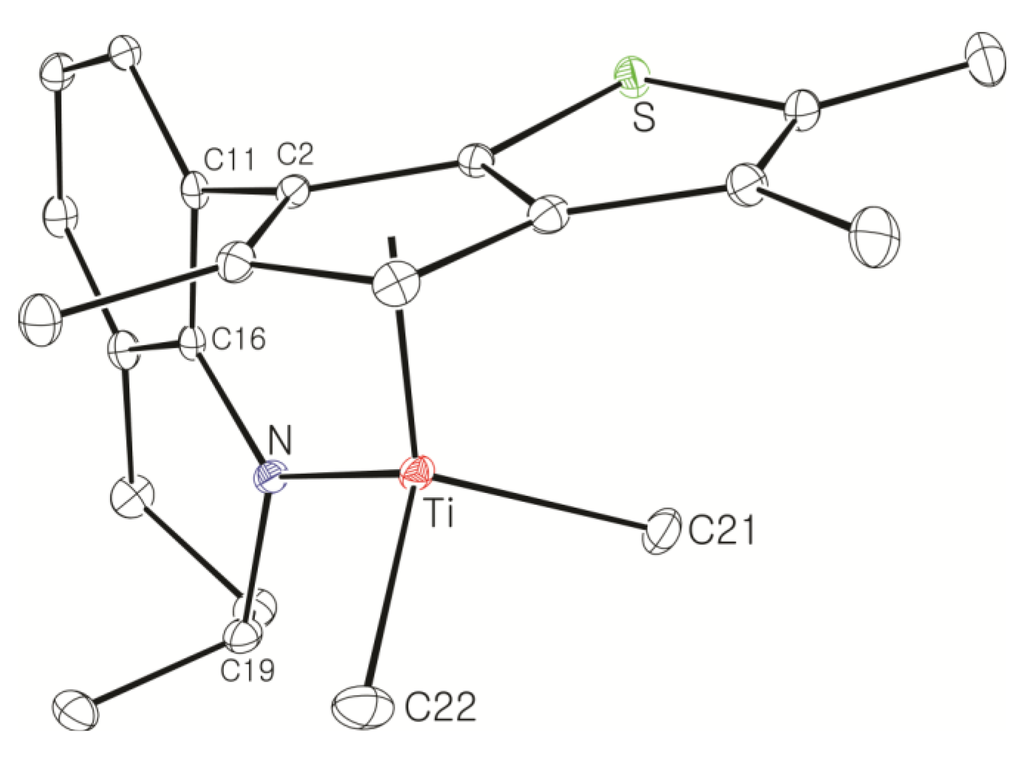
Figure 3.
Thermal ellipsoid plot (30% probability level) of 27. Hydrogen atoms are omitted for clarity.

Table 1.
Selected bond distances (Å) and angles (°) in 15, 27, and 2.
| 15 | 27 | 2 | |
|---|---|---|---|
| Ti-Cp(cent) | 2.0217(10) | 2.0393(9) | 2.041 |
| Ti-N | 1.9079(19) | 1.9270(16) | 1.934(3) |
| Ti-Cl (or Ti-CH3) | 2.2753(7) | 2.103(2) | 2.104(4) |
| 2.2754(7) | 2.104(2) | 2.100(4) | |
| S-C(bridgehead) | 1.735(2) | 1.7285(19) | 1.730(3) |
| S-C | 1.764(2) | 1.766(2) | 1.769(3) |
| Cp(cent)-Ti-N | 105.93(7) | 106.07(5) | 106.62 |
| Cl-Ti-Cl (or CH3-Ti-CH3) | 103.34(3) | 101.44(9) | 99.9(2) |
| Cp(cent)-C(2)-C(11) | 170.71 | 169.14 | 169.46 |
| C(2)-Cp(cent)-Ti | 87.78 | 88.49 | 88.00 |
| Ti-N-C(16) | 127.39(15) | 126.07(12) | 125.6(2) |
| Ti-N-C(19) | 116.96(15) | 118.21(12) | 118.8(2) |
| C(16)-N-C(19) | 115.49(19) | 115.70(15) | 115.5(3) |
| C(2)-C(11)-C(16) | 113.03(19) | 114.05(15) | 114.3(3) |
| C(11)-C(16)-N | 114.7(2) | 115.49(15) | 115.8(3) |
2.4. Polymerization Studies
The newly prepared complexes, 13–15 and 22–26, activated with [Ph3C]+[B(C6F5)4]− in the presence of iBu3Al, were screened for ethylene/1-octene copolymerization under identical conditions: a toluene solution of 1-octene (0.30 M, 30 mL), Ti (0.25 µmol), [Ph3C]+[B(C6F5)4]− (1.0 µmol), iBu3Al (0.20 mmol), ethylene (4.0 bar), initial temperature 80 °C, 3 min (Table 2). The polymerization was run for a short time, not only because of the formation of a viscous solution in such a short time, but also to minimize the drift of the 1-octene concentration. Fairly high activities (24–87 × 106 g/molTi∙h) were observed with the formation of the polymers with high 1-octene content (14~32 mol%) and high molecular weight (Mw, 200,000~350,000). Among the screened catalysts, 15 exhibited the highest activity (87 × 106 g/molTi∙h), albeit with the lowest molecular weight (Mw, 204,000). The catalytic performance of 28, which is the dimethyltitanium analogue of 15, was studied in comparison with the excellent, previously reported catalysts 2 and 3 under the polymerization conditions relevant to the high-temperature commercial solution process (hexane (1.00 L), 1-octene (250 mL), complex (7.5 µmol), [Ph3C]+[B(C6F5)4]− (45 µmol), iBu3Al (2.30 mmol), ethylene (135 g, initial pressure 41–45 bar), initial temperature 160 °C, 5 min). The activity of 28 (126 ×106 g/molTi∙h) was higher than those attained with 2 and 3 (72 × 106 and 96 × 106 g/molTi∙h, respectively) (entries 9–11). However, the 1-octene incorporation ability of 28 was slightly inferior to those of 2 and 3 ([1-Oct], 9.3 mol%versus 12–13 mol%). The molecular weight of the polymer obtained with 28 was also slightly lower than that of the polymer obtained with 2 (Mw, 159,000 versus 218,000), but comparable to that of the polymer obtained with 3 (Mw, 167,000).
Table 2.
Ethylene/1-Octene Copolymerization Results a.
| Entry | Catalyst | Temp (°C) | Yield (g) | Activity b | [Oct] c (mol%) | Mw × 10−3 | Mw/Mn |
|---|---|---|---|---|---|---|---|
| 1 | 13 | 80–85 | 0.65 | 52 | 24 | 349 | 2.7 |
| 2 | 14 | 80–88 | 0.48 | 38 | 24 | 277 | 3.3 |
| 3 | 15 | 80–96 | 1.09 | 87 | 24 | 204 | 2.9 |
| 4 | 22 | 80–86 | 0.51 | 41 | 31 | 312 | 2.8 |
| 5 | 23 | 80–86 | 0.57 | 46 | 32 | 312 | 2.7 |
| 6 | 24 | 80–82 | 0.35 | 28 | 28 | 218 | 3.8 |
| 7 | 25 | 80–83 | 0.37 | 30 | 32 | 244 | 3.0 |
| 8 | 26 | 80–82 | 0.30 | 24 | 14 | 350 | 3.5 |
| 9 b | 28 | 160–183 | 79 | 126 | 9.3 | 159 | 3.5 |
| 10 b | 2 | 160–167 | 45 | 72 | 12 | 218 | 2.8 |
| 11 b | 3 | 160–175 | 60 | 96 | 13 | 167 | 3.2 |
a Polymerization conditions for entries 1–8: toluene solution of 1-octene (0.30 M, 30 mL), complex (0.25 µmol), [Ph3C]+[B(C6F5)4]−(1.0 µmol), iBu3Al (0.20 mmol), ethylene (4.0 bar), 3 min. b Polymerization conditions for entries 9–11: hexane (1.00 L), 1-octene (250 mL), complex (7.5 µmol), [Ph3C]+[B(C6F5)4]− (45 µmol), iBu3Al (2.30 mmol), ethylene (135 g, initial pressure of 41–45 bar), 5 min. b Activity in unit of 106 g/molTi∙h. c1-Octene mole fraction in the copolymer measured by the 1H NMR spectrum.
3. Experimental Section
3.1. General Remark
All manipulations were performed under an inert atmosphere using standard glove box and Schlenk techniques. Diethyl ether, THF, and C6D6 were distilled from benzophenone ketyl. Toluene (anhydrous grade) and 1-octene used for the polymerization reaction were purchased from Aldrich and purified over Na/K alloy. Ethylene was purchased from Conley Gas (99.0%) and was purified by contact with molecular sieves and copper for several days under 200 psig pressure. The 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Varian Mercury plus 400. Elemental analyses were carried out at the Analytical Center, Kyunghee University. Mass spectra were obtained on a Micromass VG Autospec. Gel permeation chromatograms (GPC) were obtained at 50 °C in toluene using a Waters Model 150-C+ GPC and the data were analyzed using a polystyrene analyzing curve.
3.2. Synthesis
Large-Scale Synthesis of Compound 4: A CH2Cl2 solution (18 mL) containing dissolved 2,3-dimethylthiophene contaminated with 2,4-dimethylthiophene (30.0 g, 267 mmol) and tiglic acid (26.7 g, 267 mmol) was added dropwise to polyphosporic acid (220 g, 85%) with a syringe pump at 50 °C for 2 h, and then the mixture was stirred at 50 °C for 2 h. Ice (350 g) was added and the product was extracted with diethyl ether (4 × 100 mL). The combined extracts were washed with saturated aqueous Na2CO3 (160 mL) and dried over anhydrous MgSO4. The solvent was removed by using a rotary evaporator to give an oily residue (56 g), which was found by analysis of the 1H NMR spectrum to be a mixture of 4 and 5 in 6:1 ratio. Hexanethiol (l7.3 g, 600 mmol) and tetrabutylammonium fluoride (3.20 g, 12.2 mmol) were added to the crude mixture of 4 (~48.0 g, 247 mmol) and 5 (~8.0 g, 41.1 mmol). The neat mixture was stirred for 30 min at room temperature, and then, the flask was connected to a vacuum distillation set. Compound 4 was distilled out at 85 °C at reduced pressure (0.15 mmHg), whereas the thiol-added compound 6 remained in the distillation pot (39.0 g, 75%). 1H NMR (CDCl3): 3.32 (quintet, J = 7.2 Hz, 0.5H), 3.00 (quintet, J = 7.2 Hz, 0.5H), 2.80 (qd, J = 7.2, 3.2 Hz, 0.5H), 2.41 (qd, J = 7.2, 3.2 Hz, 0.5H), 2.40 (s, 3H), 2.12 (s, 1.5H), 2.11 (s, 1.5H), 1.34 (d, J = 7.2 Hz, 1.5H), 1.27 (d, J = 7.2 Hz, 1.5H), 1.17 (d, J = 7.2 Hz, 1.5H), 1.15 (d, J = 7.2 Hz, 3H) ppm. 13C{1H}17R (CDCl3): 199.04, 198.73, 172.94, 171.45, 150.83, 150.43, 134.35, 134.16, 130.06, 129.83, 55.70, 50.20, 40.71, 35.31, 18.86, 15.93, 15.89, 15.16, 15.08, 11.95, 11.80, 11.54 ppm.
Large-Scale Synthesis of Compound 7: nBuLi (54.3 mL, 136 mmol, 2.5 M solution in hexane) was added dropwise to a solution of 1,2,3,4-tetrahydroquinaline (20.0 g, 136 mmol) in hexane (280 mL) at room temperature. When the solution was stirred at room temperature overnight, a white solid precipitated, which was filtered and washed with hexane. The lithium amide compound was formed in quantitative yield (21.0 g). A diethyl ether solution (200 mL) containing the lithium amide compound (22.4 g, 115 mmol) was stirred at −78 °C and CO2 gas was added. The white solid disappeared immediately. After stirring of the solution for 40 min at −78 °C, the temperature was raised slowly to room temperature while excess CO2 gas was removed through a bubbler, and the solution was stirred overnight. A white solid precipitated again. THF (9.80 g, 136 mmol) and tert-BuLi (80.0 mL, 136 mmol, 1.7 M solution in pentane) were added successively to the slurry at −20 °C, and the solution was stirred for 2 h at this temperature. A diethyl ether solution (200 mL) with dissolved 4 (22.4 g, 115 mmol) was added to the ortho-lithiated compound using a syringe at −20 °C. The solution was stirred for 1 h at −20 °C and subsequently warmed slowly to room temperature. After stirring the solution overnight, aqueous 2M HCl (250 mL) was added at 0 °C and the mixture was stirred at room temperature for 30 min. The product was extracted with diethyl ether (3 × 100 mL). The organic phases were collected, and the solvent was removed with a rotary evaporator. Aqueous HCl solution (108 mL, 6 N) was added to the solution of crude products (~60 g) in hexane (600 mL) under vigorous stirring. The precipitated white solid was collected by filtration and washed with hexane (~300 mL). The solid was suspended in ethyl acetate (500 mL), and neutralized with saturated aqueous Na2CO3 solution (300 mL). The organic phase was collected and dried over anhydrous MgSO4. Removal of the solvent with a rotary evaporator gave a yellow viscous oil (52 g, 65%), which was pure enough to be used for the metallation without further purification.
Large-Scale Synthesis of Complex 8: nBuLi (49.5 mL, 2.5 M in hexane, 120 mmol) was added dropwise to a stirred solution of 7 (20.0 g, 61.6 mmol) in hexane (100 mL), and then the solution was stirred overnight at room temperature. A white solid was deposited. Diethyl ether (54 mL) was added at −30 °C to dissolve the deposited solid. Diethyl ether (240 mL) was slowly added to another flask containing TiCl4 in toluene solution (11.7 g, 61.6 mmol) at −30 °C and the resulting solution was stirred for 1 h at room temperature to form slurry. The ether solution of the dilithiated compound was added in one portion to the flask containing the TiCl4 slurry at −30 °C. After stirring of the solution for 6 h at room temperature, the solution was filtered through Celite. The solvent was removed under vacuum to give a brown solid, which was triturated in hexane. The red powder was isolated by filtration (21.0 g, 78%). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.10 (t, J = 4.4 Hz, 1H), 6.90 (d, J = 4.4 Hz, 2H), 5.27 and 5.22 (m, 1H, NCH), 2.54–2.38 (m, 1H, CH2), 2.20–2.08 (m, 1H, CH2), 2.36 and 2.35 (s, 3H), 2.05 and 2.03 (s, 3H), 1.94 and 1.93 (s, 3H), 1.89 and 1.84 (s, 3H), 1.72–1.58 (m, 2H, CH2), 1.36–1.28 (m, 2H, CH2), 1.17 and 1.14 (d, J = 6.4, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.78, 147.91, 142.45, 142.03, 136.91, 131.12, 130.70, 130.10, 128.90, 127.17, 123.39, 121.33, 119.87, 54.18, 26.48, 21.74, 17.28, 14.46, 14.28, 13.80, 13.27 ppm. Anal. Calcd. (C21H23Cl2NSTi): C, 57.29; H, 5.27; N, 3.18%. Found: C, 57.42; H, 5.51; N, 3.46%.
Complex 2: MeMgBr (0.235 g, 0.680 mmol, 3.0 M in diethyl ether) was added dropwise to a stirred solution of 8 (0.150 g, 0.340 mmol) in diethyl ether (2 mL) at −30 °C. After stirring of the solution for 4 h at room temperature, the solvent was removed under vacuum. The resulting residue was dissolved in hexane (5 mL) and filtered through Celite. After all the volatiles were removed under vacuum, the residue was triturated in hexane (~1 mL). The red solid was isolated by decantation (0.12 g, 87%). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.13 and 7.10 (d, J = 7.2 Hz, 1H), 6.96 and 6.94 (d, J = 7.2 Hz, 1H), 6.82 and 6.81 (t, J = 7.2 Hz, 1H), 5.45 (m, 1H, NCH), 2.75–2.60 (m, 1H, CH2), 2.45–2.20 (m, 1H, CH2), 2.34 and 2.30 (s, 3H), 2.10 (s, 3H), 1.97 (s, 3H), 1.75 and 1.66 (s, 3H), 1.85–1.50 (m, 2H, CH2), 1.20 (d, J = 6.8 Hz, 3H), 0.76 and 0.72 (s, 3H, TiMe), 0.44 and 0.35 (s, 3H, TiMe) ppm. 13C{1H} NMR (C6D6): 160.13, 159.86, 141.33, 140.46, 138.39, 137.67, 136.74, 134.83, 131.48, 129.90, 129.78, 127.69, 127.65, 127.60, 127.45, 126.87, 126.81, 121.34, 121.23, 120.21, 120.15, 119.15, 118.93, 114.77, 111.60, 57.54, 55.55, 55.23, 51.73, 50.43, 50.36, 27.83, 27.67, 22.37, 22.31, 20.53, 20.26, 14.29, 13.51, 13.42, 13.06, 12.80 ppm.
Compound 10: nBuLi (17.0 mL, 27.3 mmol, 1.6 M solution in hexane) was added dropwise to a solution of 2-ethyl-1,2,3,4-tetrahydroquinoline (4.00 g, 24.8 mmol) in hexane (34 mL) at room temperature. When the solution was stirred at room temperature overnight, a white solid precipitated, which was filtered and washed with hexane. The lithium amide compound was formed in quantitative yield (4.02 g). THF (6 mL) was added dropwise to a flask containing the lithium amide compound (0.440 g, 2.63 mmol) at −78 °C. A white suspension was formed, to which CO2 gas was added at −78 °C. The white solid was dissolved immediately by the addition of CO2 gas. After stirring for 1 h at −78 °C, the temperature was slowly raised to 0 °C while excess CO2 gas was removed through a bubbler. The solvent was removed by vacuum, and then diethyl ether (6 mL) was added. After being cooled to −20 °C, THF (0.40 mL) and tert-BuLi (1.70 mL, 2.89 mmol, 1.7 M solution in pentane) were added successively to the slurry. The solid was dissolved through treatment with tert-BuLi, and the resulting solution was stirred for 2 h at −20 °C. Compound 9 (0.403 g, 2.24 mmol) dissolved in diethyl ether (6 mL) was added dropwise, and the resulting solution was stirred for 1 h at −20 °C. After stirring of the solution overnight, aqueous HCl (2 N, 13 mL) was added to the solution at 0 °C. The two-phase solution was stirred for 30 min at room temperature. The organic phase was collected and the water phase was extracted further with ethyl acetate (3 × 15 mL).The collected organic phases were washed with aqueous saturated NaHCO3 (20 mL) and dried over anhydrous MgSO4. The solvent was removed with a rotary evaporator to give a residue which was purified by column chromatography on silica gel, eluting with hexane and ethyl acetate (v/v, 50:1). The product was obtained as a pale yellow viscous oil (0.41 g, 48%). 1H NMR (CDCl3): δ 7.05 and 7.01 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.75 (s, 1H), 6.68–6.58 (m, 1H), 4.20–3.90 (m, 1H, NH), 3.35–3.10 (m, 2H, NCHMe, CHMe), 3.00–2.75 (m, 2H, CH2), 2.52 (s, 3H, CH3), 1.97 and 1.96 (s, 3H, CH3), 1.75–1.55 (m, 2H, CH2), 1.55–1.40 (m, 2H, CH2), 1.37, 1.36, 1.35 and 1.33 (s, 3H, CH3), 0.91 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 150.80, 150.70, 145.83, 145.71, 145.30, 143.57, 143.31, 143.01, 141.95, 139.22, 139.08, 131.40, 131.18, 130.96, 130.69, 128.58, 127.83, 127.57, 127.46, 127.21, 121.30, 120.02, 119.99, 119.80, 119.71, 119.34, 115.91, 53.45, 53.39, 53.17, 53.08, 45.88, 45.74, 45.66, 45.56, 29.99, 29.86, 29.59, 28.10, 27.71, 27.34, 27.19, 27.13, 27.07, 26.94, 16.38, 16.35, 16.00, 14.34, 14.18, 13.87, 10.54, 10.39, 10.37 ppm. HRMS(EI): m/z Calcd. ([M+] C21H25NS) 323.1708. Found: 323.1709.
Compound 11: The compound was synthesized using the same conditions and procedure as those for 10 with 2-isopropyl-1,2,3,4-tetrahydroquinaline (0.480 g, 2.68 mmol) and 9 (0.410 g, 2.28 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 53% yield (0.41 g). 1H NMR (CDCl3): δ 7.07 and 7.03 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.75 (s, 1H), 6.68–6.58 (m, 1H), 4.20–3.83 (m, 1H, NH), 3.34–3.16 (m, 1H, NCHMe), 3.12–2.96 (m, 1H, CHMe), 2.96–2.76 (m, 2H, CH2), 2.53 (s, 3H, CH3), 1.99, 1.98, 1.97 and 1.96 (s, 3H, CH3), 2.04–1.92 (m, 1H, CH), 1.80–1.54 (m, 2H, CH2), 1.37, 1.36, 1.35 and 1.34 (s, 3H, CH3), 1.00–0.80 (m, 6H, CH3) ppm. 13C{1H} NMR (CDCl3): 150.80, 150.66, 150.60, 145.96, 145.76, 145.21, 143.75, 143.51, 143.48, 143.21, 142.35, 142.22, 139.19, 139.06, 139.02, 131.23, 130.90, 130.66, 128.52, 128.43, 127.72, 127.48, 127.28, 127.10, 121.32, 121.09, 120.02, 120.00, 119.76, 119.66, 119.49, 119.12, 115.88, 115.63, 57.82, 57.43, 45.87, 45.74, 45.67, 45.53, 33.23, 33.07, 33.05, 32.96, 27.59, 27.50, 27.44, 25.32, 24.73, 24.70, 19.21, 19.11, 18.90, 18.83, 18.74, 18.56, 16.54, 16.48, 16.40, 16.02, 14.47, 14.31, 13.90, 13.85 ppm. HRMS(EI): m/z Calcd. ([M+] C22H27NS) 337.1864. Found: 337.1863.
Compound 12: The compound was synthesized using the same conditions and procedure as those for compound 10 with 2-n-butyl-1,2,3,4-tetrahydroquinaline (1.50 g, 7.68 mmol) and 9 (1.18 g, 6.53 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 40% yield (1.1 g). 1H NMR (CDCl3): δ 7.06 and 7.02 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.80–6.70 (m, 1H), 6.70–6.58 (m, 1H, CH), 4.20–3.94 (m, 1H, NH), 3.38–3.16 (m, 2H, NCHMe, CHMe), 3.00–2.74 (m, 2H, CH2), 2.53 (s, 3H, CH3), 1.98 and 1.97 (s, 3H, CH3), 1.76–1.56 (m, 2H, CH2), 1.56–1.18 (m, 6H, CH2), 1.38, 1.37, 1.36 and 1.34 (s, 3H, CH3), 0.89 (t, J = 5.6 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 150.80, 150.71, 145.80, 145.75, 145.33, 145.26, 143.54, 143.30, 143.23, 142.97, 141.95, 139.21, 139.08, 131.38, 131.17, 130.98, 130.71, 128.57, 127.82, 127.58, 127.38, 127.18, 121.28, 120.00, 119.80, 119.70, 119.30, 115.89, 52.00, 51.82, 51.72, 51.56, 45.87, 45.72, 45.67, 45.58, 36.90, 36.80, 36.48, 28.66, 28.41, 28.29, 28.23, 27.88, 27.24, 27.19, 26.96, 23.13, 23.04, 16.37, 15.99, 14.47, 14.34, 14.18, 13.87 ppm. HRMS(EI): m/z Calcd. ([M+] C23H29NS) 351.2021. Found: 351.2021.
Complex 13: The complex was synthesized using the same conditions and procedure as those for 8 with 10 (1.00 g, 3.09 mmol). It was obtained as a red solid in 70% yield (1.12 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.5 ratio. 1H NMR (C6D6): δ 7.08 (t, J = 4.0 Hz, 1H), 6.94–6.84 (m, 2H), 6.41 and 6.29 (s, 1H), 5.07–4.83 (m, 1H, NCH), 2.54–2.36 (m, 1H, CH2), 2.28 and 2.27 (s, 3H, CH3), 2.25–2.15 (m, 1H, CH2), 2.04 and 2.02 (s, 3H, CH3), 1.76 (s, 3H, CH3), 1.74–1.65 (m, 2H, CH2), 1.10–0.85 (m, 2H, CH2), 0.78 and 0.75 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 163.17, 151.31, 149.74, 147.16, 146.21, 142.20, 140.41, 137.21, 129.90, 129.70, 129.43, 126.70, 126.54, 123.00, 122.89, 121.49, 120.33, 119.93, 118.94, 118.64, 118.10, 63.62, 63.19, 32.09, 23.04, 22.55, 21.06, 20.84, 20.44, 19.98, 16.72, 16.63, 14.65, 13.84 ppm. Anal. Calcd. (C21H23Cl2NSTi): C, 57.29; H, 5.27; N, 3.18%. Found: C, 57.56; H, 5.49; N, 3.32%.
Complex 14: The complex was synthesized using the same conditions and procedure as those for 8 with 11 (0.185 g, 0.550 mmol). It was obtained as a red solid in 86% yield (0.22 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.5 ratio. 1H NMR (C6D6): δ 7.08 (t, J = 4.4 Hz, 1H), 6.92–6.82 (m, 2H), 6.39 and 6.27 (s, 1H), 5.10–5.34 (m, 1H, NCH), 2.56–2.34 (m, 2H, CH2), 2.28 and 2.27 (s, 3H, CH3), 2.30–2.18 (m, 1H, NCH) 2.12–2.04 (m, 1H, CH), 2.02 and 2.01 (s, 3H, CH3), 1.96 and 1.76 (s, 3H, CH3), 1.74–1.58 (m, 2H, CH2), 0.99 and 0.92 (d, J = 6.8 Hz, 3H, CH3), 0.76 and 0.75 (d, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.68, 162.63, 148.09, 146.90, 144.40, 143.25, 142.41, 142.15, 140.73, 138.82, 132.45, 132.32, 129.93, 129.91, 129.80, 127.13, 126.70, 126.60, 126.49, 123.31, 123.23, 120.26, 119.90, 106.32, 104.54, 60.42, 60.07, 32.20, 23.32, 23.11, 22.55, 21.65, 21.57, 21.12, 16.70, 16.19, 14.66, 14.26, 12.34, 12.22, 11.21 ppm. Anal. Calcd. (C22H25Cl2NSTi): C, 58.17; H, 5.55; N, 3.08%. Found: C, 58.34; H, 5.72; N, 3.29%.
Complex 15: The complex was synthesized using the same conditions and procedure as those for 8 with 12 (0.681 g, 1.94 mmol). It was obtained as a brown solid in 75% yield (0.68 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.6 ratio. 1H NMR (C6D6): δ 7.15–7.06 (m, 1H), 6.96–6.84 (m, 2H), 6.39 and 6.32 (s, 1H), 5.26–5.00 (m, 1H, NH), 2.53–2.36 (m, 1H, CH2), 2.32–2.24 (m, 1H, CH2) 2.28 and 2.27 (s, 3H, CH3), 2.18–2.06 (m, 1H, NCHMe), 2.04 and 2.02 (s, 3H, CH3), 1.88 and 1.82 (s, 3H, CH3), 1.72–1.54 (m, 2H, CH2), 1.44–1.00 (m, 6H, CH2), 0.85 and 0.79 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.77, 162.58, 150.94, 150.11, 147.23, 146.42, 145.33, 143.20, 139.52, 137.21, 130.87, 130.33, 129.92, 129.80, 126.81, 126.67, 123.15, 123.07, 120.48, 119.95, 119.75, 118.88, 118.64, 118.13, 58.48, 58.39, 29.45, 29.26, 28.95, 28.92, 23.01, 22.96, 22.28, 22.08, 21.36, 16.77, 16.68, 14.64, 14.34, 14.29, 13.88, 13.72 ppm. Anal. Calcd. (C23H27Cl2NSTi): C, 58.99; H, 5.81; N, 2.99%. Found: C, 58.78; H, 5.67; N, 2.88%.
Compound 16: Methacrylic acid (22.7 g, 264 mmol) and 2,3-dimethylthiophene (24.7 g, 220 mmol) were mixed and the mixture was added dropwise to Eaton’s reagent (220 mL) for 3 h at 80 °C. The mixture was then stirred for 30 min at 80 °C. The resulting solution was poured slowly into a flask containing a two-phase mixture of water (440 mL) and diethyl ether (75 mL) under vigorous stirring. The organic phase was collected, and the water phase was further extracted with additional diethyl ether (3 × 80 mL). The collected organic phases were combined and washed with saturated aqueous NaHCO3 (200 mL). The combined organic phases were dried over anhydrous MgSO4 and the solvent was removed by using a rotary evaporator. The oily residue was purified by column chromatography on silica gel, eluting with hexane and ethyl acetate (v/v, 20:1). The product was obtained as a mixture of two diastereomers in a 1:1 ratio (14.3 g, 36%). 1H NMR (C6D6): 2.58 (qd, J = 7.6, 3.2 Hz, 1H), 2.48 and 2.44 (d, J = 7.2 Hz, 1H), 1.90 (s, 3H, CH3), 1.62 (s, 3H, CH3), 1.16 (d, J = 7.6 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 199.52, 168.13, 150.54, 134.95, 130.04, 46.23, 46.14, 32.54, 32.31, 17.35, 17.13, 15.24, 15.05, 11.50, 11.29 ppm. HRMS(EI): m/z Calcd. ([M+] C10H12OS) 181.0609. Found: 181.0608.
Compound 17: The compound was synthesized using the same conditions and procedure as those for 7 with 1,2,3,4-tetrahydroquinaldine (20.0 g, 139 mmol) and 16 (1.50 g, 8.33 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 48% yield (1.50 g). 1H NMR (CDCl3): δ 7.14 (d, J = 7.2 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H), 6.75 (t, J = 7.2 Hz, 1H), 4.14–3.92 (m, 1H, NH), 3.62–3.44 (m, 1H, NCHMe), 3.31 (d, J = 5.2 Hz, 2H, CH2), 3.10–2.86 (m, 2H, CH2), 2.50 (s, 3H, CH3), 2.26 (s, 3H, CH3), 2.19 (s, 3H, CH3), 2.10–2.00 (m, 1H, CH2), 1.82–1.68 (m, 1H, CH2), 1.29, 1.28 and 1.26 (s, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 145.65, 142.30, 142.11, 141.70, 141.35, 139.96, 139.56, 133.17, 132.65, 132.30, 132.12, 128.59, 128.29, 127.97, 127.75, 127.39, 120.87, 120.50, 119.15, 118.96, 115.84, 115.50, 47.34, 47.20, 40.07, 39.97, 30.39, 29.89, 27.25, 26.93, 23.06, 22.78, 16.26, 15.90, 14.09, 12.44 ppm. HRMS(EI): m/z Calcd. ([M+] C20H23NS) 309.1551. Found: 309.1551.
Compound 18: The compound was synthesized using the same conditions and procedure as those for 10 with 2-ethyl-1,2,3,4-tetrahydroquinoline (0.500 g, 2.99 mmol) and 16 (0.458 g, 2.54 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 44% yield (0.43 g). 1H NMR (CDCl3): δ 7.03 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 6.8 Hz, 1H), 6.68–6.58 (m, 1H, CH), 4.12–3.90 (m, 1H, NH), 3.30–3.10 (m, 3H, NCH, CH2), 2.97–2.75 (m, 2H, CH2), 2.39 (s, 3H, CH3), 2.15 (s, 3H, CH3), 2.08 (s, 3H, CH3), 2.05–1.94 (m, 1H, CH2), 1.72–1.57 (m, 1H, CH2), 1.57–1.41 (m, 2H, CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 145.61, 142.11, 141.95, 141.71, 141.44, 139.96, 139.47, 132.98, 132.50, 132.25, 132.08, 128.48, 128.27, 127.99, 127.58, 127.34, 121.17, 120.87, 119.17, 115.68, 53.28, 52.98, 40.06, 39.94, 29.83, 29.48, 27.61, 27.21, 27.07, 26.88, 16.30, 15.91, 14.07, 12.42, 10.49, 10.31 ppm. HRMS(EI): m/z Calcd. ([M+] C21H25NS) 323.1708. Found: 323.1709.
Compound 19: The compound was synthesized using the same conditions and procedure as those for 10 with 2-isopropyl-1,2,3,4-tetrahydroquinaline (0.500 g, 2.76 mmol) and 16 (0.423 g, 2.35 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 40% yield (0.34 g). 1H NMR (C6D6): δ 7.33 and 7.25 (d, J = 6.8 Hz, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.88–6.70 (m, 1H), 4.22–4.00 (m, 1H, NH), 3.05–2.50 (m, 5H, NCH, CH2), 2.15 (s, 3H, CH3), 2.01 (d, J = 7.2 Hz, 3H, CH3), 1.93 (s, 3H, CH3), 1.70–1.45 (m, 2H, CH2), 1.44–1.30 (m, 1H, CH), 0.73 (d, J = 6.8 Hz, 6H, CH3) ppm. 13C{1H} NMR (C6D6): 146.07, 145.97, 143.03, 142.89, 142.35, 142.11, 139.64, 139.17, 134.30, 133.76, 132.51, 132.29, 129.10, 128.15, 127.94, 121.70, 121.43, 119.98, 119.66, 116.71, 116.43, 57.98, 57.49, 40.07, 39.93, 33.26, 33.14, 27.85, 25.06, 24.98, 19.11, 18.96, 18.72, 18.44, 16.32, 15.83, 13.98, 12.35 ppm. HRMS(EI): m/z Calcd. ([M+] C22H27NS) 337.1864. Found: 337.1865.
Compound 20: The compound was synthesized using the same conditions and procedure as those for 10 with 2-n-butyl-1,2,3,4-tetrahydroquinaline (1.00 g, 5.12 mmol) and 16 (0.785 g, 4.35 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 42% yield (0.76 g). 1H NMR (C6D6): δ 7.32 and 7.26 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 7.2 Hz, 1H), 6.86–6.74 (m, 1H), 4.24–4.06 (m, 1H, NH), 3.14–2.98 (m, 1H, NCH), 2.56–2.96 (m, 4H, CH2), 2.15 (s, 3H, CH3), 2.01 (s, 3H, CH3), 1.93 (s, 3H, CH3), 1.74–1.62 (m, 1H, CH2), 1.56–1.42 (m, 1H, CH2), 1.34–1.00 (m, 6H, CH2), 0.78 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 145.61, 142.13, 141.87, 141.65, 141.43, 139.94, 139.46, 132.99, 132.53, 132.23, 132.09, 128.49, 128.25, 127.96, 127.58, 127.35, 121.17, 120.87, 119.19, 118.92, 115.68, 115.41, 51.83, 51.56, 40.07, 39.96, 36.81, 36.45, 28.25, 28.20, 28.13, 27.78, 27.12, 26.91, 23.05, 16.29, 15.91, 14.37, 14.05, 12.42 ppm. HRMS(EI): m/z Calcd. ([M+] C23H29NS) 351.2021. Found: 351.2021.
Compound 21: The compound was synthesized using the same conditions and procedure as those for 10 with 2-tert-butyl-1,2,3,4-tetrahydroquinaline (1.00 g, 5.12 mmol) and 16 (0.785 g, 4.35 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). A light yellow viscous oil was obtained in 31% yield (0.56 g). 1H NMR (C6D6): δ 7.35 and 7.28 (d, J = 7.6 Hz, 1H), 7.00 (d, J = 7.2 Hz, 1H), 6.85–6.74 (m, 1H) 4.40–4.16 (m, 1H, NH), 3.06–2.54 (m, 5H, NCH, CH2), 2.14 and 2.12 (s, 3H, CH3), 2.04 and 1.99 (s, 3H, CH3), 1.91 and 1.90 (s, 3H, CH3), 1.70–1.60 (m, 1H, CH2), 1.58–1.40 (m, 1H, CH2), 0.76 (s, 9H, CH3) ppm. 13C{1H} NMR (C6D6): 146.07, 145.95, 143.32, 143.22, 142.37, 142.16, 139.65, 138.97, 134.29, 133.64, 132.52, 132.23, 128.97, 128.89, 121.79, 121.51, 120.27, 119.77, 116.86, 116.52, 61.47, 60.95, 40.08, 39.89, 33.79, 33.58, 28.46, 28.41, 26.32, 23.69, 23.53, 16.49, 15.81, 13.98, 12.35 ppm. HRMS(EI): m/z Calcd ([M+] C23H29NS) 351.2021. Found: 351.2020.
Complex 22: The complex was synthesized using the same conditions and procedure as those for 8 with 17 (0.500 g, 1.62 mmol). It was obtained as a brown solid in 73% yield (0.50 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.4 ratio. 1H NMR (C6D6): δ 7.25–6.98 (m, 1H), 6.94–6.78 (m, 2H), 6.15 and 6.09 (s, 1H), 5.44–5.26 (m, 1H, NCH), 2.52–2.32 (m, 1H, CH2), 2.16–1.98 (m, 1H, CH2), 1.95 (s, 3H, CH3), 1.90 (s, 3H, CH3), 1.88 (s, 3H, CH3), 1.70–1.50 (m, 2H, CH2), 1.14 and 1.12 (d, J = 6.8, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 161.99, 161.92, 148.63, 147.61, 144.25, 143.22, 142.63, 142.32, 140.58, 138.83, 132.54, 132.40, 129.91, 129.88, 129.57, 129.49, 126.93, 126.61, 126.52, 123.26, 123.24, 119.78, 119.54, 106.07, 106.57, 54.30, 59.96, 26.25, 21.33, 16.78, 16.75, 16.46, 16.41, 14.78, 14.75, 12.64, 12.58 ppm. Anal. Calcd. (C20H21Cl2NSTi): C, 56.36; H, 4.97; N, 3.29%. Found: C, 56.23; H, 4.79; N, 3.02%.
Complex 23: The complex was synthesized using the same conditions and procedure as those for 8 with 18 (0.271 g, 0.838 mmol). It was obtained as a brown solid in 80% yield (0.30 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.13 and 7.09 (t, J = 3.2 Hz, 1H), 6.95–6.87 (m, 2H), 6.40 and 6.33 (s, 1H), 5.20–5.00 (m, 1H, NCH), 2.50–2.38 (m, 1H, CH2), 2.29 and 2.27 (s, 3H, CH3), 2.18–2.05 (m, 1H, CH2), 2.05 and 2.03 (s, 3H, CH3), 1.88 and 1.82 (s, 3H, CH3), 1.68–1.54 (m, 2H, CH2), 1.36–1.16 (m, 2H, CH2), 0.85 and 0.79 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.81, 162.61, 150.94, 150.11, 147.24, 146.42, 145.32, 143.21, 139.51, 137.19, 130.85, 130.37, 130.34, 129.92, 129.79, 126.80, 126.67, 123.12, 123.04, 120.48, 119.99, 119.78, 118.84, 118.62, 118.12, 58.47, 58.37, 29.46, 29.27, 28.94, 23.01, 22.96, 22.25, 22.05, 21.34, 16.73, 16.65, 14.64, 14.32, 13.85, 13.70 ppm. Anal. Calcd. (C21H23Cl2NSTi): C, 57.29; H, 5.27; N, 3.18%. Found: C, 57.42; H, 5.49; N, 3.33%.
Complex 24: The complex was synthesized using the same conditions and procedure as those for 8 with 19 (0.050 g, 0.15 mmol). It was obtained as a brown solid in 81% yield (0.06 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.05 (t, J = 4.0 Hz, 1H), 6.94–6.80 (m, 2H), 6.17 and 6.14 (s, 1H), 5.20–4.90 (m, 1H, NCH), 2.60–2.34 (m, 1H, CH2), 2.26–2.10 (m, 1H, CH2), 2.07 and 1.90 (s, 3H, CH3), 1.93 and 1.90 (s, 3H, CH3), 1.85 (s, 3H, CH3), 1.80–1.68 (m, 2H, CH2), 1.70–1.56 (m, 1H, CH), 0.97 and 0.90 (d, J = 6.8 Hz, 3H), 0.75 and 0.73 (d, J = 7.2 Hz, 3H) ppm. 13C{1H} NMR (C6D6): 163.37, 163.16, 148.24, 146.68, 144.82, 143.61, 142.15, 141.43, 141.15, 138.96, 131.53, 131.32, 129.90, 129.71, 129.15, 129.00, 127.01, 126.73, 126.54, 126.41, 123.24, 123.12, 120.59, 120.05, 107.22, 104.82, 64.12, 63.27, 32.12, 31.64, 22.99, 22.47, 22.38, 22.31, 21.23, 20.76, 20.45, 19.96, 17.05, 16.25, 14.26, 14.20, 12.41, 12.18 ppm. Anal. Calcd. (C22H25Cl2NSTi): C, 58.17; H, 5.55; N, 3.08%. Found: C, 58.37; H, 5.75; N, 3.29%.
Complex 25: The complex was synthesized using the same conditions and procedure as those for 8 with 20 (1.50 g, 9.79 mmol). The product was obtained as a brown solid in 88% yield (1.50 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.9 ratio. 1H NMR (C6D6): δ 7.11 (t, J = 4.4 Hz, 1H), 6.90 (d, J = 5.6 Hz, 2H), 6.19 and 6.12 (s, 1H), 5.26–5.10 (m, 1H, NCH), 2.52–2.36 (m, 1H, CH2), 2.18–2.02 (m, 1H, CH2), 1.97 (s, 3H, CH3), 1.92 (s, 3H, CH3), 1.91 (s, 3H, CH3), 1.70–1.54 (m, 2H, CH2), 1.36–1.00 (m, 6H, CH2), 0.84 and 0.77 (t, J = 7.6 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.74, 162.68, 148.11, 146.92, 144.38, 143.21, 142.46, 142.17, 140.71, 138.84, 132.44, 132.29, 129.94, 129.87, 129.82, 127.08, 126.71, 126.65, 126.54, 123.37, 123.29, 120.20, 119.89, 106.30, 104.55, 58.96, 58.64, 29.41, 28.92, 23.00, 22.93, 22.21, 22.15, 21.32, 16.70, 16.24, 14.62, 14.30, 14.25, 12.35, 12.26 ppm. Anal. Calcd. (C23H27Cl2NSTi): C, 58.99; H, 5.81; N, 2.99%. Found: C, 58.73; H, 5.57; N, 2.69%.
Complex 26: The complex was synthesized using the same conditions and procedure as those for 8 with 21 (0.050 g, 0.142 mmol). It was obtained as a brown solid in 82% yield (0.050 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.4 ratio. 1H NMR (C6D6): δ 7.13 (t, J = 6.8 Hz, 1H), 6.90–6.70 (m, 2H), 6.18 and 6.16 (s, 1H), 5.30–4.86 (m, 1H, NCH), 2.58–2.42 (m, 1H, CH2), 2.16 (s, 3H, CH3), 2.08–1.94 (m, 1H, CH2), 1.89 (s, 6H, CH3), 1.90–1.74 (m, 2H, CH2), 0.92 (s, 9H, CH3) ppm. 13C{1H} NMR (C6D6): 163.57, 144.82, 144.69, 144.45, 142.69, 129.66, 129.43, 126.32, 126.19, 122.77, 120.35, 108.65, 105.06, 66.99, 41.29, 30.73, 30.63 23.29, 23.19, 16.12, 14.32, 12.00 ppm. Anal. Calcd. (C23H27Cl2NSTi): C, 58.99; H, 5.81; N, 2.99%. Found: C, 58.69; H, 5.51; N, 2.65%.
Complex 27: The complex was synthesized using the same conditions and procedure as those for 2 with 22 (0.500 g, 1.17 mmol). It was obtained as red viscous oil in 86% yield (0.40 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.7 ratio. 1H NMR (C6D6): δ 7.12 and 7.08 (d, J = 6.8 Hz, 1H), 6.94 and 6.92 (d, J = 7.6 Hz, 1H), 6.79 (t, J = 7.6 Hz, 1H), 6.31 and 6.12 (s, 1H), 5.60–5.44 (m, 1H, NCH), 2.75–2.60 (m, 1H, NCHMe), 2.00 and 1.99 (s, 3H, CH3), 1.95 (s, 3H, CH3), 1.84 and 1.73 (s, 3H, CH3), 1.72–1.64 (m, 1H, CH2), 1.60–1.50 (m, 1H, CH2), 1.18 and 1.17 (s, 3H, CH3), 0.83 and 0.80 (s, 3H, TiMe), 0.35 and 0.27 (s, 3H, TiMe) ppm. 13C{1H} NMR (C6D6): 159.47, 141.38, 138.38, 138.10, 135.95, 129.64, 129.51, 127.11, 127.02, 125.75, 122.82, 120.11, 119.98, 119.01, 102.05, 99.27, 58.55, 55.61, 52.30, 50.42, 50.12, 27.53, 27.28, 21.96, 20.15, 19.71, 15.31, 15.12, 14.01, 12.50, 12.37 ppm. Anal. Calcd. (C22H27Cl2NSTi): C, 68.56; H, 7.06; N, 3.63%. Found: C, 68.77; H, 7.24; N, 3.79%.
Complex 28: The complex was synthesized using the same conditions and procedure as those for 2 with 15 (0.500 g, 1.07 mmol). It was obtained as an red viscous oil in 85% yield (0.39 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.7 ratio. 1H NMR (C6D6): δ 7.11 and 7.08 (d, J = 7.2 Hz, 1H), 6.95 (t, J = 7.6 Hz, 1H), 6.81 and 6.79 (d, J = 7.6 Hz, 1H), 6.48 and 6.47 (s, 1H), 5.50–5.38 (m, 1H, NCH), 2.78–2.60 (m, 1H, CH2), 2.44–2.26 (m, 1H, CH2), 2.28 and 2.22 (s, 3H, CH3), 2.09 (s, 3H, CH3), 1.84–1.52 (m, 2H, CH2), 1.73 and 1.63 (s, 3H, CH3), 1.40–1.15 (m, 6H, CH2), 1.18 (t, J = 5.6 Hz, 3H, CH3), 0.74 and 0.68 (s, 3H, TiMe), 0.46 and 0.37 (s, 3H, TiMe) ppm. 13C{1H} NMR (C6D6): 159.73, 159.42, 145.84, 144.82, 140.65, 139.82, 139.13, 138.75, 135.18, 131.50, 129.60, 129.49, 127.41, 127.36, 127.28, 127.14, 121.22, 121.10, 119.96, 119.88, 118.80, 118.65, 117.91, 117.66, 113.77, 110.31, 57.83, 55.23, 54.84, 51.63, 50.20, 50.06, 27.54, 27.24, 22.01, 21.99, 20.24, 19.83, 16.61, 16.58, 13.04, 13.00, 12.91, 12.60 ppm. Anal. Calcd. (C25H33Cl2NSTi): C, 70.24; H, 7.78; N, 3.28%. Found: C, 70.46; H, 7.98; N, 3.53%.
3.3. Ethylene/1-Octene Copolymerization
In a glove box, 30 mL of toluene solution of 1-octene (1.0 g, 0.30 M) was added to a dried 60 mL glass reactor. The reactor was assembled and brought out from the glove box. The reactor was then heated to 80 °C using a mantle. After an activated catalyst, which was prepared by mixing the complex (0.25 mol), (iBu)3Al (0.20 mmol, Al/Ti = 800), and [C(C6H5)3]+[B(C6F5)4]− (1.0 µmol), was added via a syringe, the ethylene gas (60 psig) was fed immediately. In the case of highly active catalysts, the mantle was removed immediately after the injection of the activated catalyst to remove the generated heat. After polymerization was conducted for 3 min, the ethylene gas was vented and methanol (10 mL) was added immediately. The solution was stirred for 10 min. Then, solvent was removed using a rotary evaporator. The residue was taken after wetting with methanol and dried under vacuum at 150 °C for several hours. The 1-octene contents were calculated by the analysis of the 1H NMR spectra of the copolymers. In the 1H NMR spectra, the methyl (CH3) signals (0.93–1.02 ppm) are well isolated from the methine (CH) and methylene (CH2) signals (1.30–1.50 ppm), and the 1-octene contents can be calculated from the integration values of the two regions. The copolymer (5 mg) was dissolved in C6D6, and the 1H NMR spectra were recorded at 80 °C.
3.4. X-ray Crystallography
The crystallographic measurements were performed at 100 K using a Bruker APEX II CCD–based diffractometer with graphite–monochromated Mo K α radiation (λ = 0.7107 Å). The reflection data were collected as multi–scan frames with 0.5°/frame and an exposure time of 10 s/frame. Cell parameters were determined and refined by SMART program. Data reduction was performed using SAINT software. The data were corrected for Lorentz and polarization effects. An empirical absorption correction was applied using the SADABS program. The structures of the compounds were solved by direct methods and refined by full matrix least–squares methods using the SHELXTL program package with anisotropic thermal parameters for all non–hydrogen atoms. Hydrogen atoms were calculated at idealized positions and refined riding on the corresponding carbon atoms with isotropic thermal parameters. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre. Crystallographic data for 15: C23H27Cl2NSTi∙C6H6, M = 546.42, Triclinic, a = 9.8285(3), b = 9.8426(3), c = 15.0264(4) Å, α = 77.802(2)°, β = 73.294(2)°, γ = 78.3100(10)°, V = 1345.12(7) Å3, T = 100(2), space group P-1, Z = 2, 19992 reflections measured, 4722 unique (R(int) = 0.0196) which were used in all calculations. The final wR2 was 0.0919 (I > 2σ(I)). Crystallographic data for 27: C42H39Cl2CoN2O5, M = 781.58, triclinic, a = 9.8572(2), b = 12.4423(9), c = 17.1598(12) Å, α = 111.160(2)°, β = 102.001(3)°, γ = 91.009(3)°, V = 1909.9(2) Å3, T = 100 (2) K, space group P-1, Z = 2, 27710 reflections measured, 6689 unique (R(int) = 0.0156) which were used in all calculations. The final wR2 was 0.0915 (I > 2σ(I)). Crystallographic data for 26: C22H27NSTi, M = 385.41, orthorhombic, a = 6.99320(10), b = 14.9918(2), c = 37.2626(6) Å, V = 3906.64(10) Å3, T = 296(2) K, space group Pbca, Z = 8, 63784 reflections measured, 4657 unique (R(int) = 0.0382) which were used in all calculations. The final wR2 was 0.0952 (I > 2σ(I)).
4. Conclusions
One of the main obstacles to the commercial use of homogeneous Ziegler catalysts is the cost of constructing the elaborate ligands and their metallation. For commercial viability, the complex should be synthesized inexpensively on a large scale without any need of the chromatographic purification. A synthetic scheme was developed for large-scale preparation of the dimethylthiophene-fused and tetrahydroquinaldine-linked dimethylcyclopentadienyl titanium complex 2, which is a high-performance homogeneous Ziegler catalyst. For 2, metallation could be carried out in a high yield (78%) by reacting the dilithiated compound directly with TiCl4; this was not successful under the same conditions for the CGC and tetramethylcyclopentadienyl analogue 1 owing to the severe electron-transfer side reaction. Many derivatives of 2 were prepared through the variation of either the number of methyl groups in the thiophene-fused cyclopentadienyl unit or of the substituent at the 2-position of the tetrahydroquinoline unit. Among the newly prepared complexes, complex 15, with an n-butyl group at the 2-position in the tetrahydroquinoline unit and three methyl groups in the thiophene-fused cyclopentadienyl unit, showed exceptionally high activity. Under commercially relevant high-temperature conditions (160 °C), 28 (the dimethyltitanium analogue of the dichlorotitanium complex 15) showed a higher activity than 2 (126 × 106 g/molTi∙h versus 72 × 106 g/molTi∙h), albeit with the formation of a polymer of lower molecular weight (Mw, 159000 versus 218000) and with a slightly lower 1-octene content (9.3 mol%versus 12 mol%).
Acknowledgments
This work was supported by Mid-carrier Researcher Program (No. 2012-003303) and Priority Research Centers Program (2012-0006687) through NRF grant funded by the MEST.
Conflict of Interest
The authors declare no conflict of interest.
References
- Kaminsky, W. Discovery of Methylaluminoxane as Cocatalyst for Olefin Polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Tullo, A.H. Metallocene rise again. Chemical & Engineering News 2010, 88, 10–16. [Google Scholar]
- Nomura, K. Half-titanocenes containing anionic ancillary donor ligands as promising new catalysts for precise olefin polymerisation. Dalton Trans. 2009, 41, 8811–8823. [Google Scholar] [CrossRef]
- Stephan, D.W. The Road to Early-Transition-Metal Phosphinimide Olefin Polymerization Catalysts. Organometallics 2005, 24, 2548–2560. [Google Scholar]
- Kim, T.-J.; Kim, S.-K.; Kim, B.-J.; Hahn, J.S.; Ok, M.-A.; Song, J.H.; Shin, D.-H.; Ko, J.; Cheong, M.; Kim, J.; et al. Half-Metallocene Titanium(IV) Phenyl Phenoxide for High Temperature Olefin Polymerization: Ortho-Substituent Effect at Ancillary o-Phenoxy Ligand for Enhanced Catalytic Performance. Macromolecules 2009, 42, 6932–6943. [Google Scholar]
- Senda, T.; Hanaoka, H.; Okado, Y.; Oda, Y.; Tsurugi, H.; Mashima, K. Titanium Complexes of Silicon-Bridged Cyclopentadienyl−Phenoxy Ligands Modified with Fused-Thiophene: Synthesis, Characterization, and Their Catalytic Performance in Copolymerization of Ethylene and 1-Hexene. Organometallics 2009, 28, 6915–6926. [Google Scholar]
- Liu, K.; Wu, Q.; Luo, X.; Gao, W.; Mu, Y. Synthesis, characterization, and catalytic properties of new half-sandwich zirconium(iv) complexes. Dalton Trans. 2012, 41, 3461–3467. [Google Scholar]
- Shi, X.C.; Jin, G.X. Syntheses, reactions, and ethylene polymerization of half-sandwich titanium complexes containing salicylbenzoxazole and salicylbenzothiazole ligands. Dalton Trans. 2011, 40, 11914–11919. [Google Scholar]
- Manz, T.A.; Caruthers, J.M.; Sharma, S.; Phomphrai, K.; Thomson, K.T.; Nicholas Delgass, W.; Abu-Omar, M.M. Structureactivity correlation for relative chain initiation to propagation rates in single-site olefin polymerization catalysis. Organometallics 2012, 31, 602–618. [Google Scholar]
- Cano, J.; Kunz, K. How to synthesize a constrained geometry catalyst (CGC)—A survey. J. Organomet. Chem. 2007, 692, 4411–4423. [Google Scholar]
- McKnight, A.L.; Waymouth, R.M. Group 4 ansa-Cyclopentadienyl-Amido Catalysts for Olefin Polymerization. Chem. Rev. 1998, 98, 2587–2598. [Google Scholar]
- Arriola, D.J.; Bokota, M.; Campbell, R.E.; Klosin, J.; LaPointe, R.E.; Redwine, O.D.; Shankar, R.B.; Timmers, F.J.; Abboud, K.A. Penultimate Effect in Ethylene−Styrene Copolymerization and the Discovery of Highly Active Ethylene−Styrene Catalysts with Increased Styrene Reactivity. J. Am. Chem. Soc. 2007, 129, 7065–7076. [Google Scholar]
- Cho, D.J.; Wu, C.J.; Sujith, S.; Han, W.S.; Kang, S.O.; Lee, B.Y. O-phenylene-bridged Cp/amido titanium complexes for ethylene/1-hexene copolymerizations. Organometallics 2006, 25, 2133–2134. [Google Scholar]
- Joung, U.G.; Wu, C.J.; Lee, S.H.; Lee, C.H.; Lee, E.J.; Han, W.S.; Kang, S.O.; Lee, B.Y. Phenylene-bridged Cp/carboxamide ligands for titanium complexes of various binding modes and their ethylene/1-octene copolymerization. Organometallics 2006, 25, 5122–5130. [Google Scholar]
- Lee, S.H.; Wu, C.J.; Joung, U.G.; Lee, B.Y.; Park, J. Bimetallic phenylene-bridged Cp/amide titanium complexes and their olefin polymerization. Dalton Trans. 2007, 40, 4608–4614. [Google Scholar]
- Wu, C.J.; Lee, S.H.; Yun, H.; Lee, B.Y. Ortho lithiation of tetrahydroquinoline derivatives and its use for the facile construction of polymerization catalysts. Organometallics 2007, 26, 6685–6687. [Google Scholar]
- Wu, C.J.; Lee, S.H.; Yu, S.T.; Na, S.J.; Yun, H.; Lee, B.Y. CO2-mediated ortho-lithiatium of N-alkylanilines and its use for the construction of polymerization catalysts. Organometallics 2008, 27, 3907–3917. [Google Scholar]
- Yu, S.T.; Na, S.J.; Lim, T.S.; Lee, B.Y. Preparation of a bulky cycloolefin/ethylene copolymer and its tensile properties. Macromolecules 2010, 43, 725–730. [Google Scholar]
- Park, D.K.; Lim, B.K.; Ha, J.J.; Kum, H.H.; Kim, Y.K.; Lee, C.H.; Jung, S.W. Ethylene alpha-olefin copolymer. WO Patent 2008/140205, 12 November 2008. [Google Scholar]
- Lee, C.H.; Lim, B.K.; Lee, E.J.; Ha, J.J.; Jung, S.W.; Lee, J.A.; Ro, K.S.; Kum, D.H.; Park, D.K. Long chain-branched ethylene-alpha olefin copolymer. WO Patent 2008/140280, 20 November 2008. [Google Scholar]
- Tremblay, J.-F. Dow Chemical Loses Elastomers Patent Lawsuit Against LG Chem. Chemical & Engineering News 2012, 90, 7. [Google Scholar]
- Park, J.H.; Do, S.H.; Cyriac, A.; Yun, H.; Lee, B.Y. Preparation of half-metallocenes of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl ligands for ethylene/α-olefin copolymerization. Dalton Trans. 2010, 39, 9994–10002. [Google Scholar]
- Ewen, J.A.; Elder, M.J.; Jones, R.L.; Rheingold, A.L.; Liable-Sands, L.M.; Sommer, R.D. Chiral Ansa Metallocenes with Cp Ring-Fused to Thiophenes and Pyrroles: Syntheses, Crystal Structures, and Isotactic Polypropylene Catalysts. J. Am. Chem. Soc. 2001, 123, 4763–4773. [Google Scholar]
- De Rosa, C.; Auriemma, F.; Resconi, L. Metalloorganic polymerization catalysis as a tool to probe crystallization properties of polymers: The case of isotactic poly(1-butene). Angew. Chem. Int. Ed. 2009, 48, 9871–9874. [Google Scholar]
- Ryabov, A.N.; Voskoboynikov, A.Z. Constrained geometry complexes of titanium (IV) and zirconium (IV) involving cyclopentadienyl fused to thiophene ring. J. Organomet. Chem. 2005, 690, 4213–4221. [Google Scholar]
- Whisler, M.C.; MacNeil, S.; Snieckus, V.; Beak, P. Beyond thermodynamic acidity: A perspective on the complex-induced proximity effect (CIPE) in deprotonation reactions. Angew. Chem. Int. Ed. 2004, 43, 2206–2225. [Google Scholar]
- Mugesh, G.; Singh, H.B. Heteroatom-directed aromatic lithiation: A versatile route to the synthesis of organochalcogen (Se, Te) compounds. Acc. Chem. Res. 2002, 35, 226–236. [Google Scholar]
- Timmers, F.J. Olefin polymers formed by use of constrained geometry addition polymerization catalysts. US Patent 6670432, 30 December 2003. [Google Scholar]
- Grandini, C.; Camurati, I.; Guidotti, S.; Mascellani, N.; Resconi, L.; Nifant'ev, I.E.; Kashulin, I.A.; Ivchenko, P.V.; Mercandelli, P.; Sironi, A. Heterocycle-Fused Indenyl Silyl Amido Dimethyl Titanium Complexes as Catalysts for High Molecular Weight Syndiotactic Amorphous Polypropylene. Organometallics 2004, 23, 344–360. [Google Scholar]
- Carpenetti, D.W.; Kloppenburg, L.; Kupec, J.T.; Petersen, J.L. Application of Amine Elimination for the Efficient Preparation of Electrophilic ansa-Monocyclopentadienyl Group 4 Complexes Containing an Appended Amido Functionality. Structural Characterization of [(C5H4)SiMe2(N-t-Bu)]ZrCl2(NMe2H). Organometallics 1996, 15, 1572–1581. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).