Characterization of Active Sites/Entities and Redox/Catalytic Correlations in Copper-Ceria-Based Catalysts for Preferential Oxidation of CO in H2-Rich Streams


Abstract
:1. Introduction
2. Results and Discussion
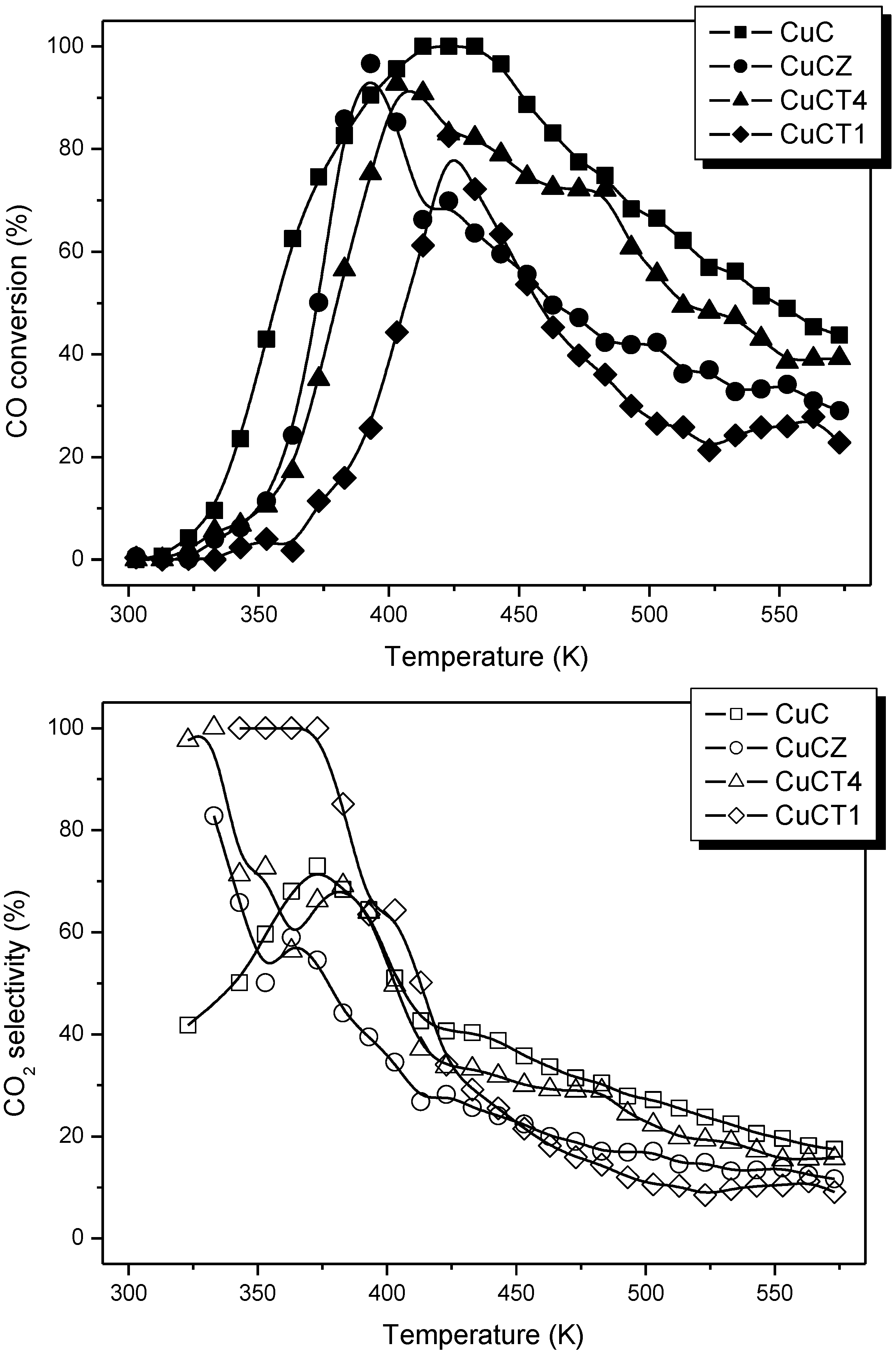
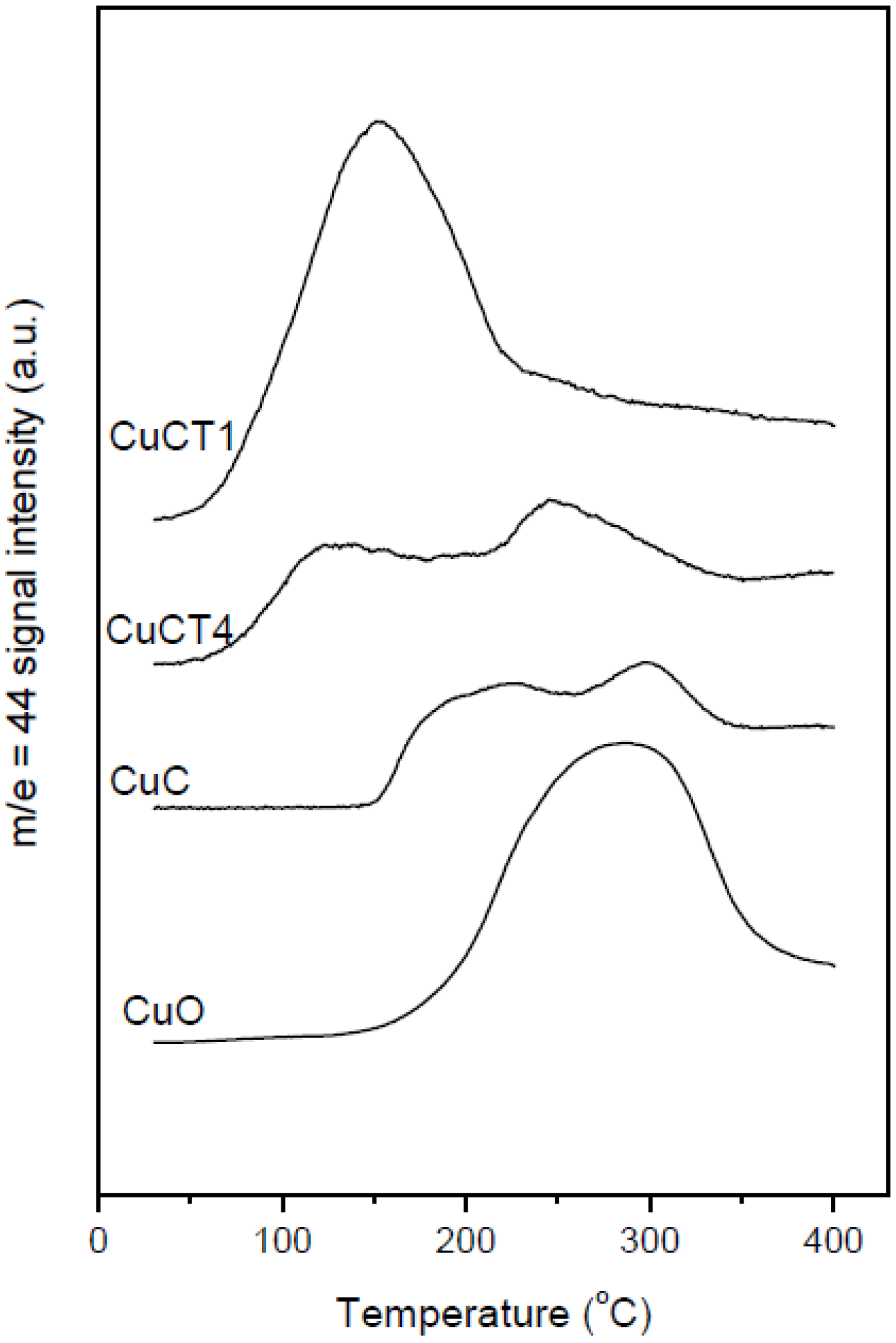
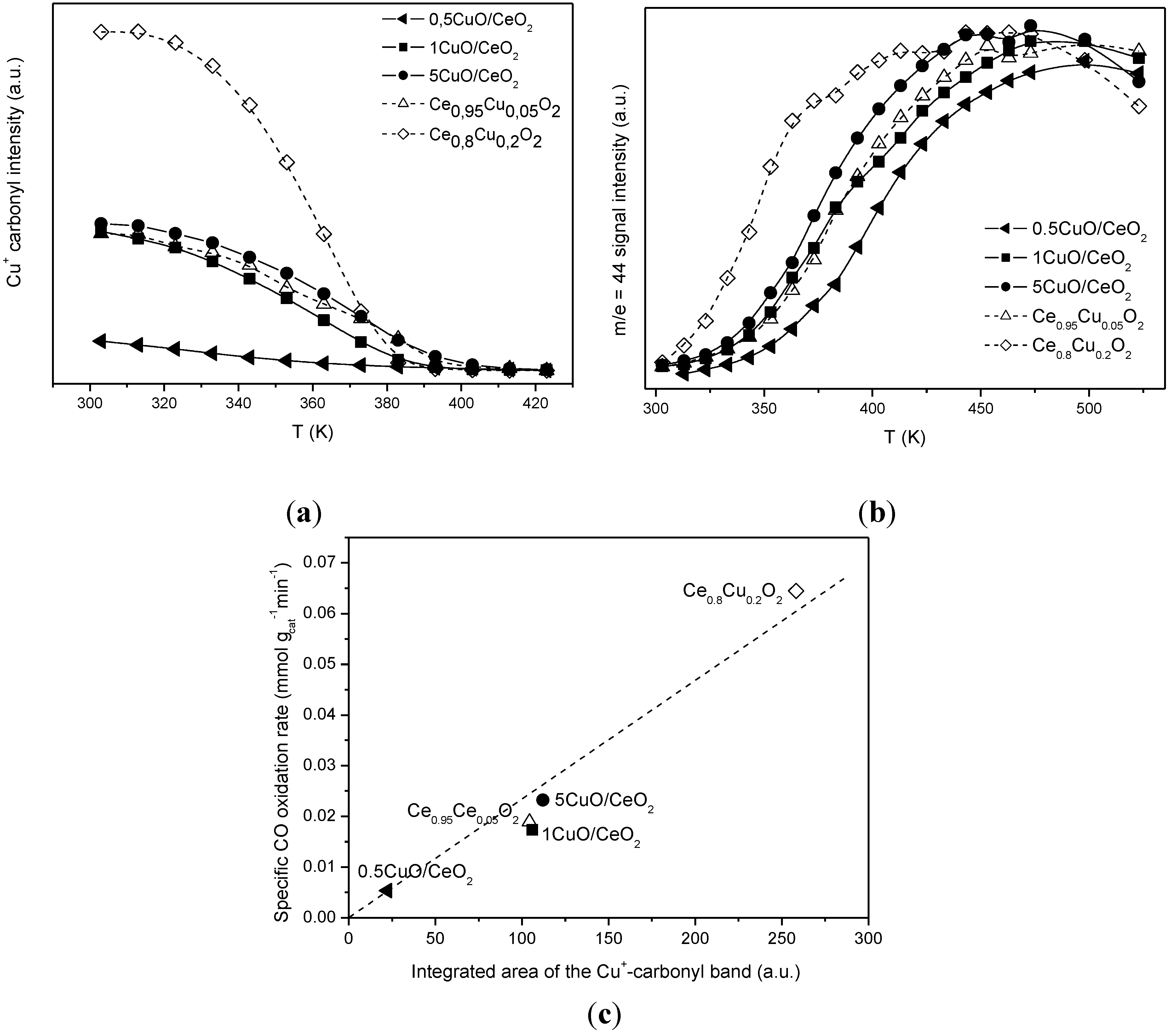
2.1. Redox and Catalytic Properties as a Function of Changes in the Ceria-Based Support Nature
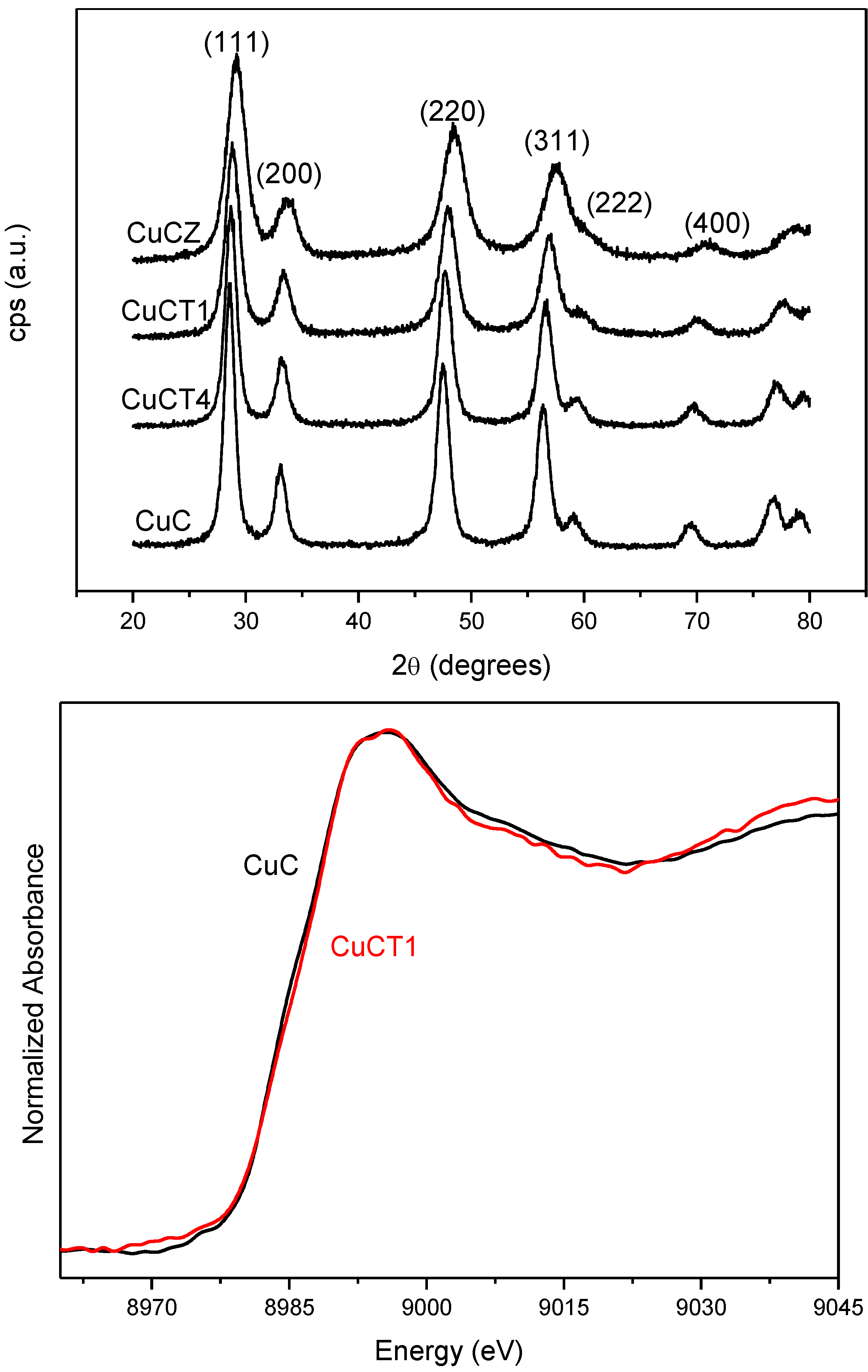
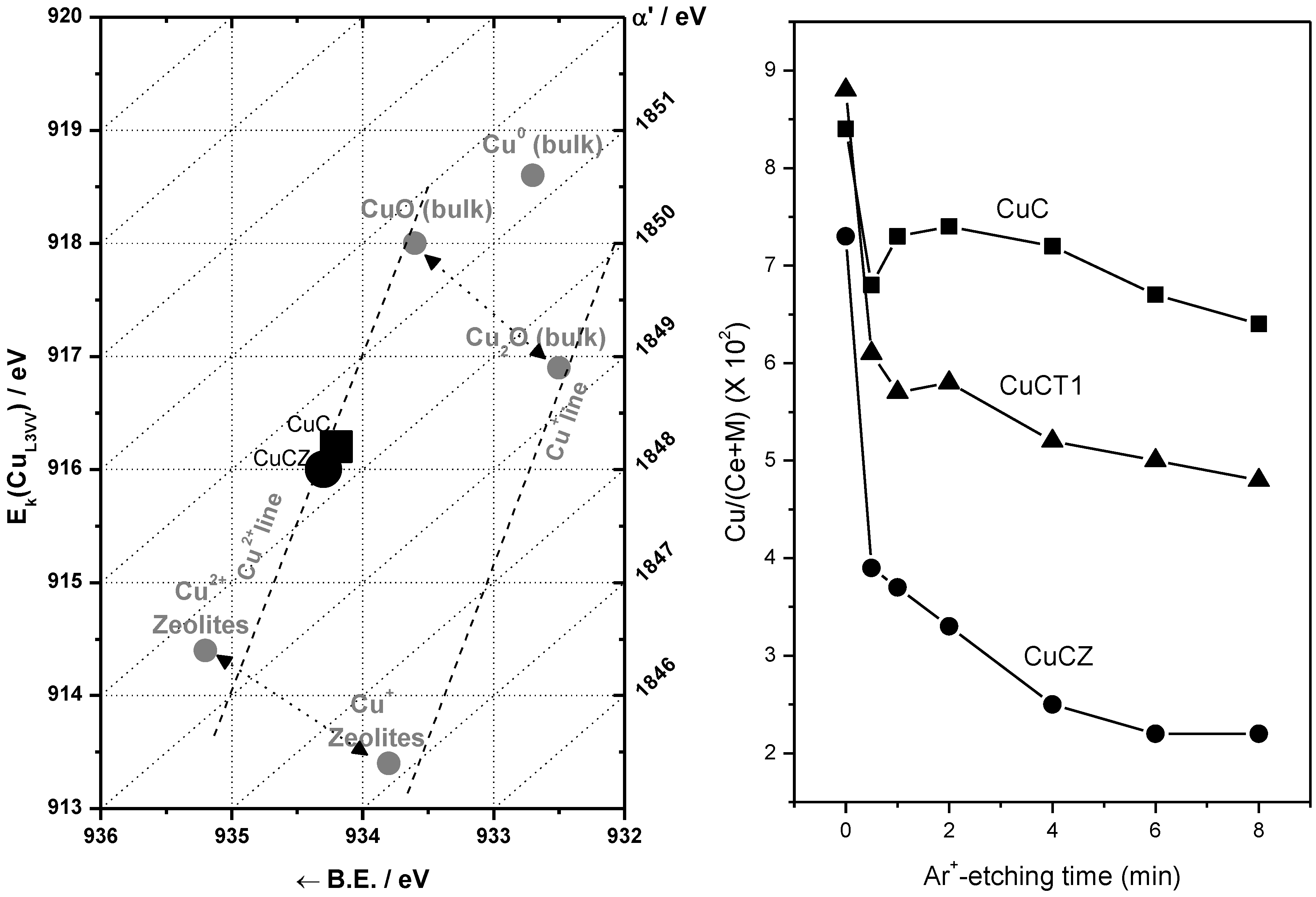
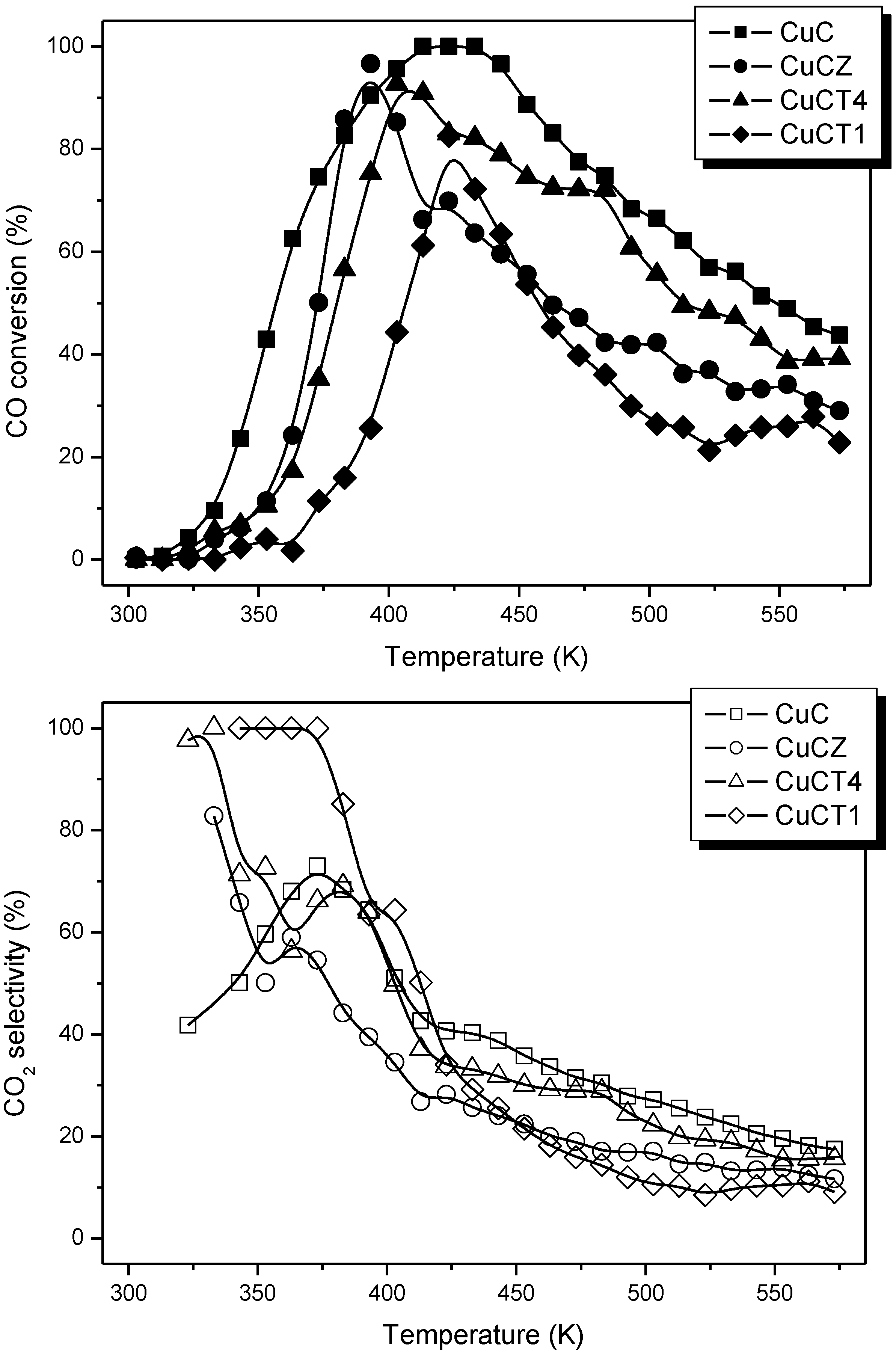
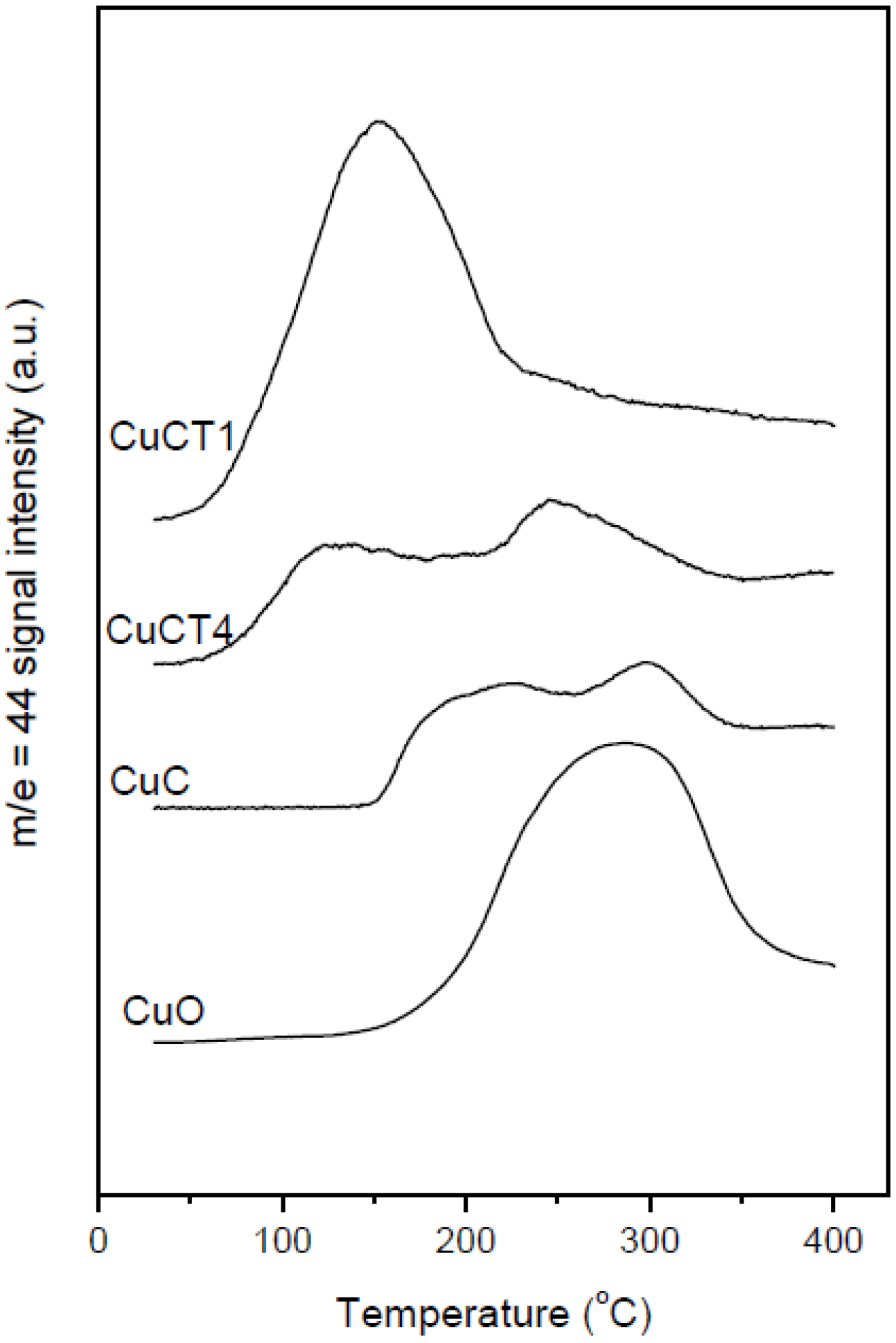
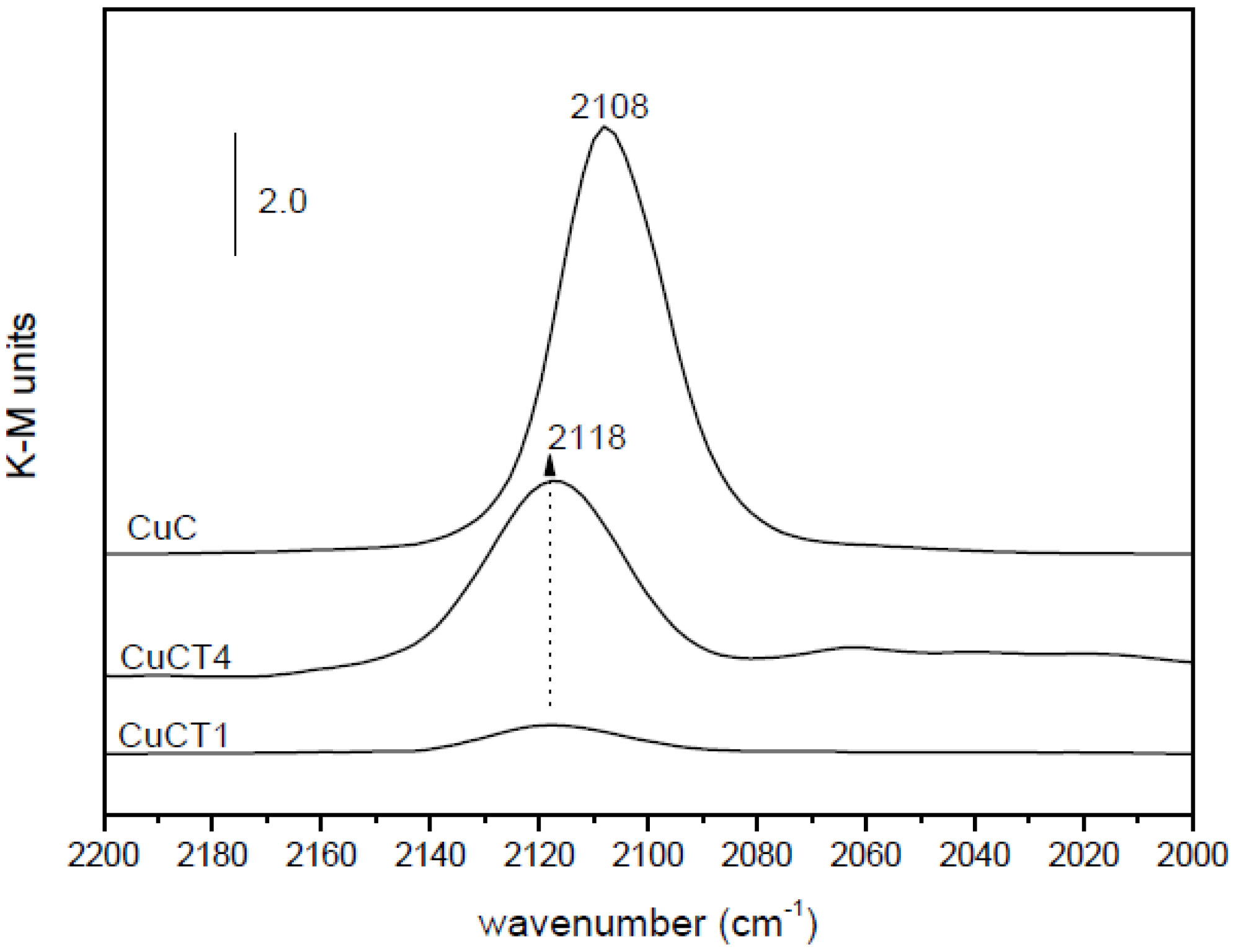
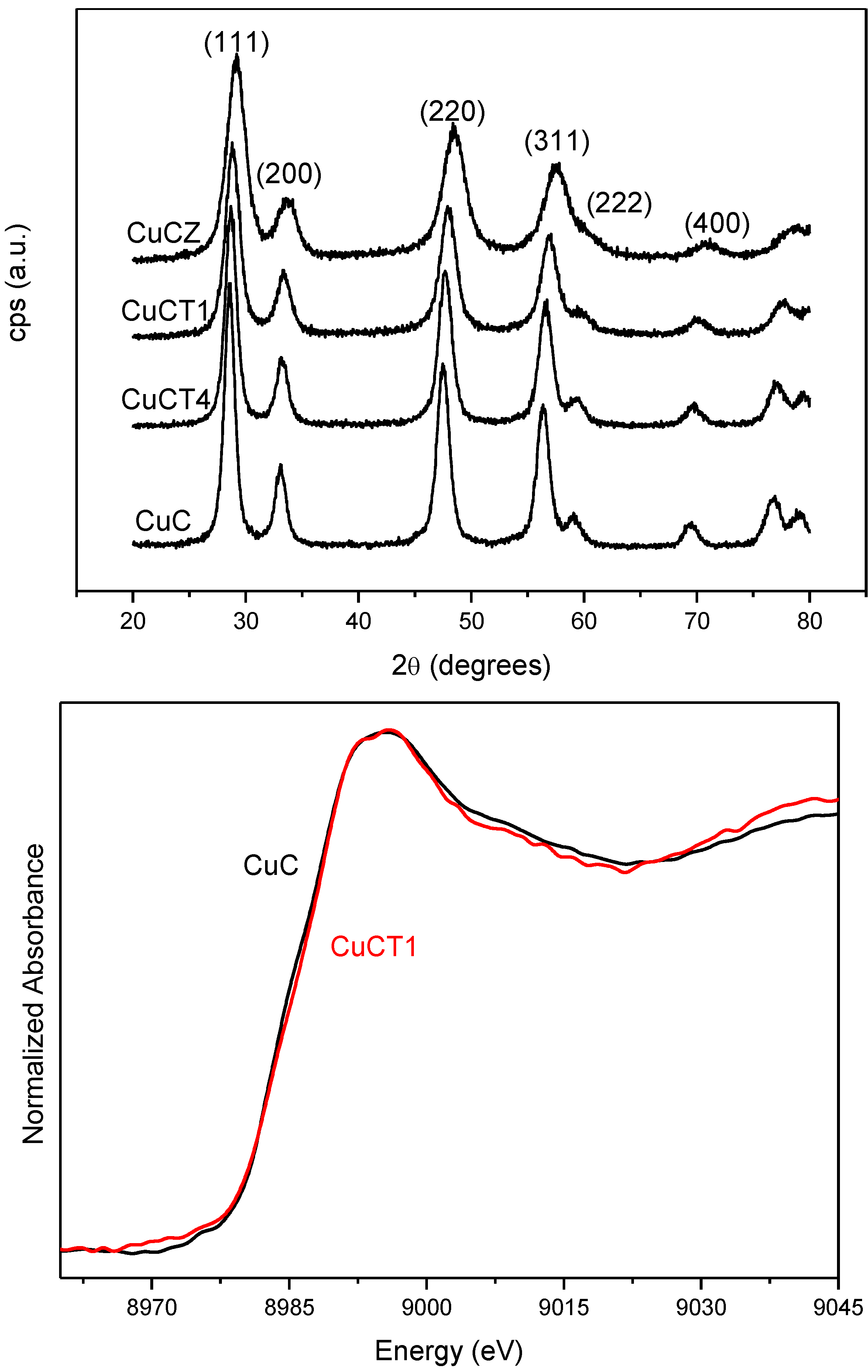
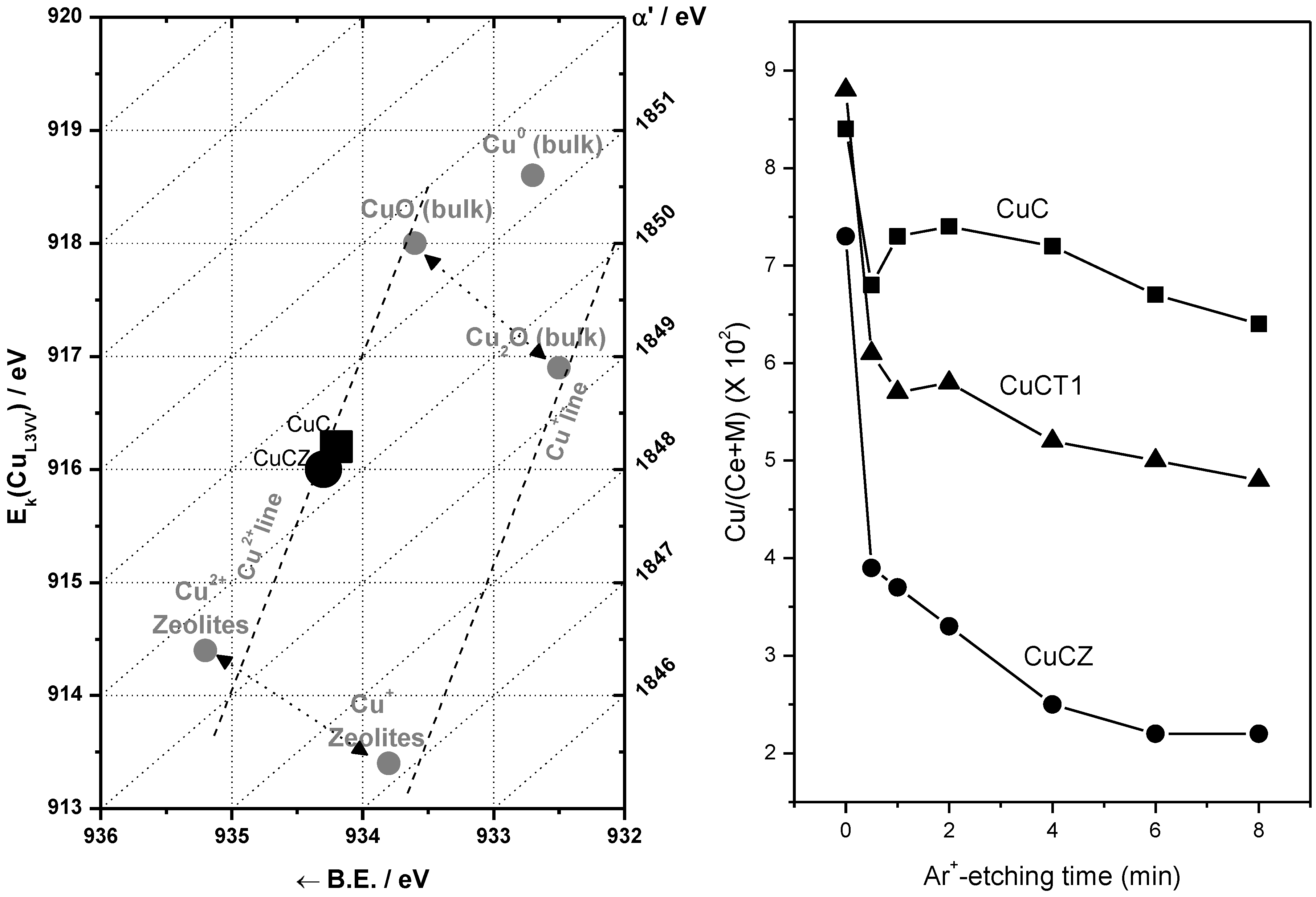
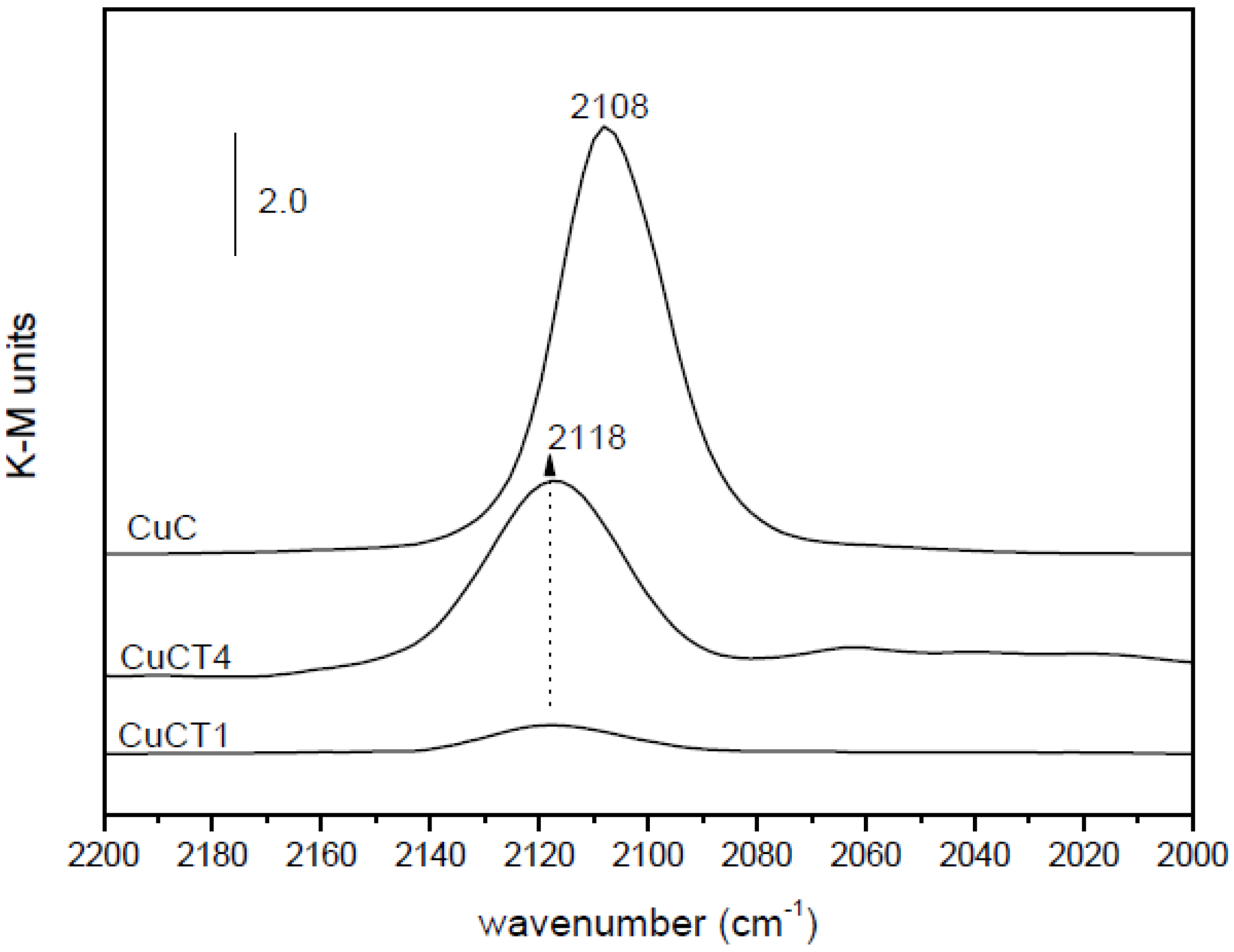
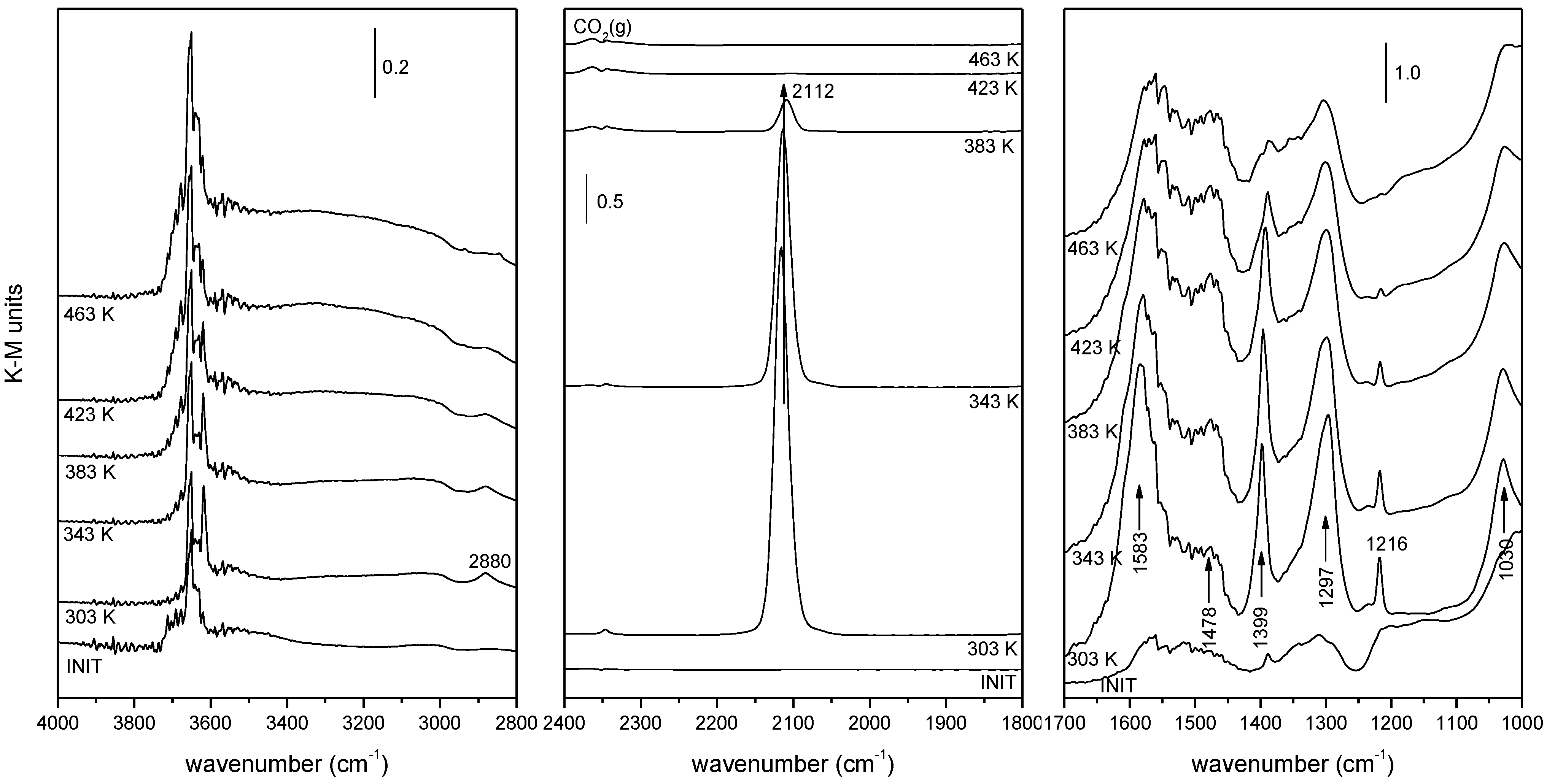
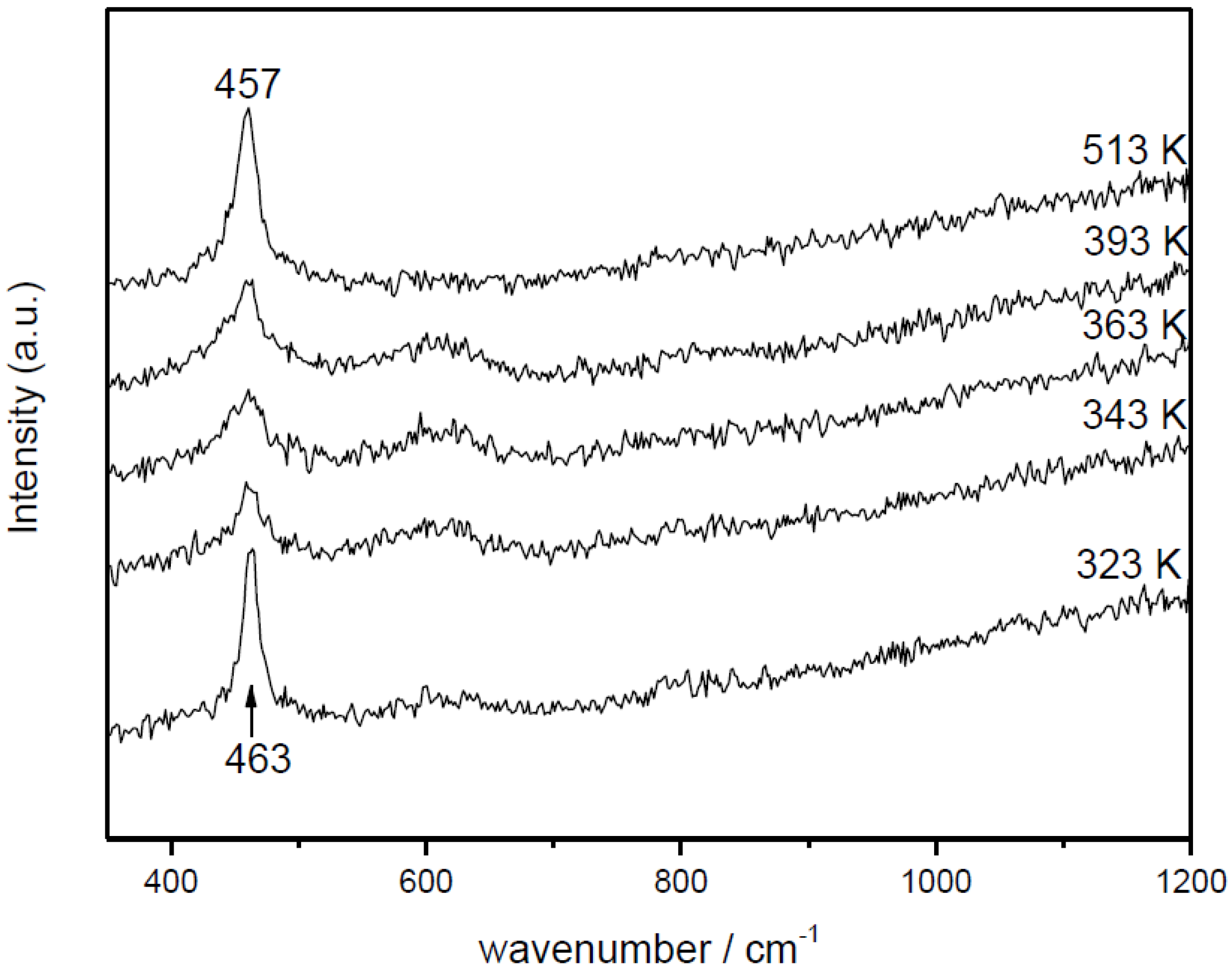
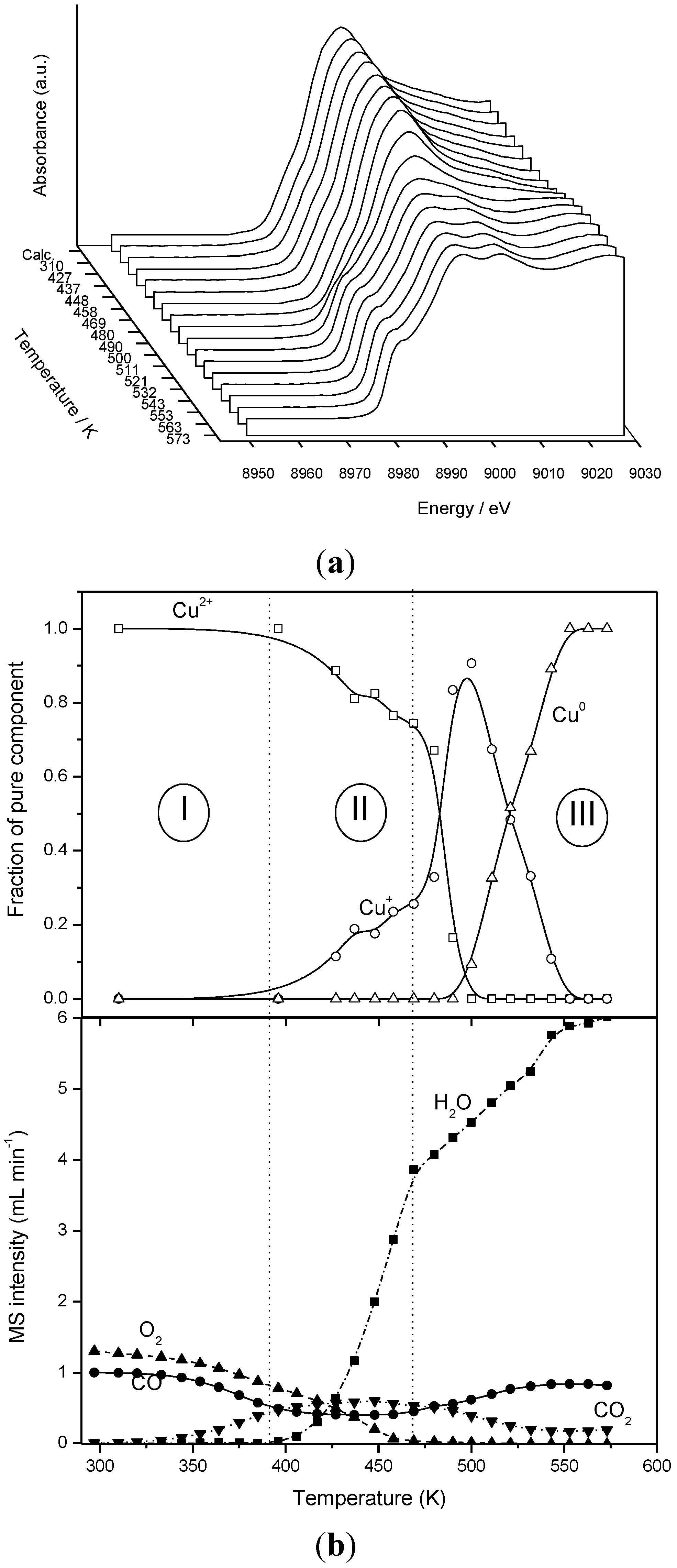
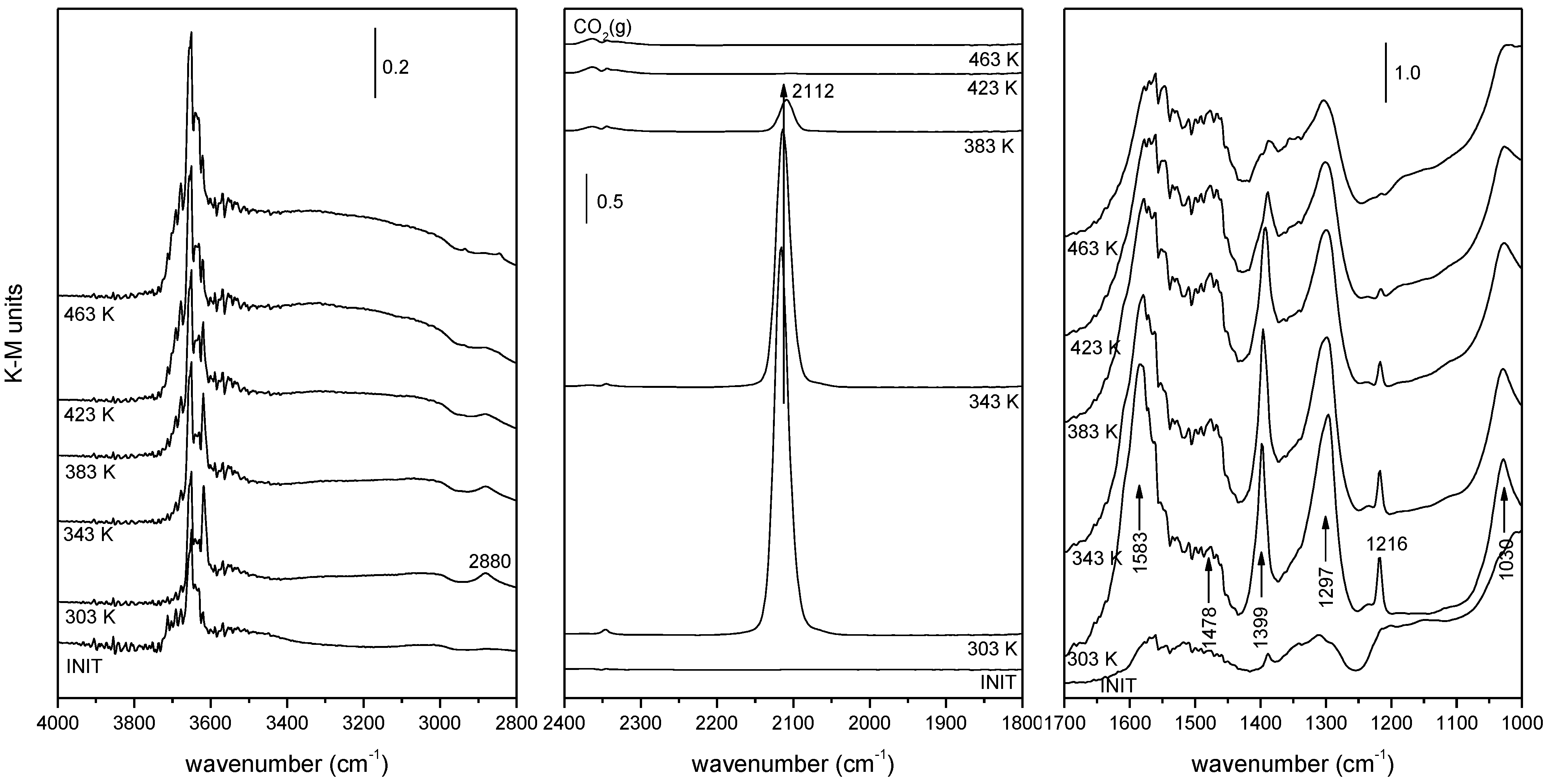
2.2. Nature of Active Sites/Entities and Further Hints on Catalytic/Redox Correlations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Synthesis method | SBET (m2/g) | Lattice parameter a (Å) | Crystal size a (nm) | Phases detected b |
|---|---|---|---|---|---|
| 0.5CuO/CeO2 | impregnation | 116 | 5.410 | 7.6 | Fluorite CeO2 |
| 1CuO/CeO2 | impregnation | 107 | 5.410 | 7.8 | Fluorite CeO2 |
| 5CuO/CeO2 | impregnation | 101 | 5.413 | 8.1 | Fluorite CeO2, tenorite CuO |
| Ce0.95Cu0.05O2 | microemulsion coprecipitation | 130 | 5.410 | 7.0 | Fluorite Ce1− xCuxO2 |
| Ce0.8Cu0.2O2 | microemulsion coprecipitation | 151 | 5.413 | 6.6 | FluoriteCe1− xCuxO2 |

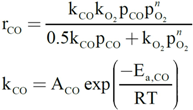
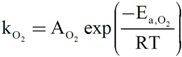
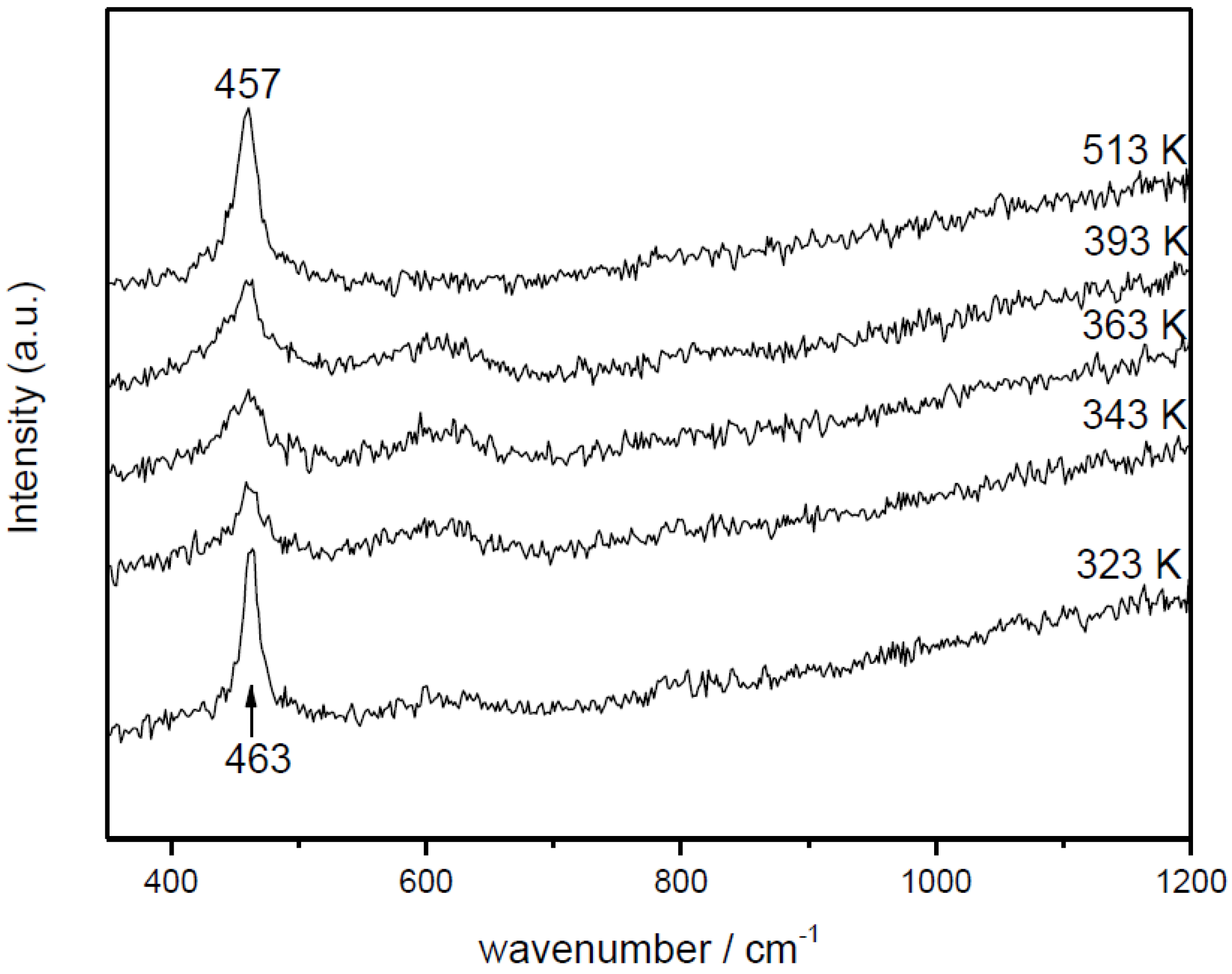
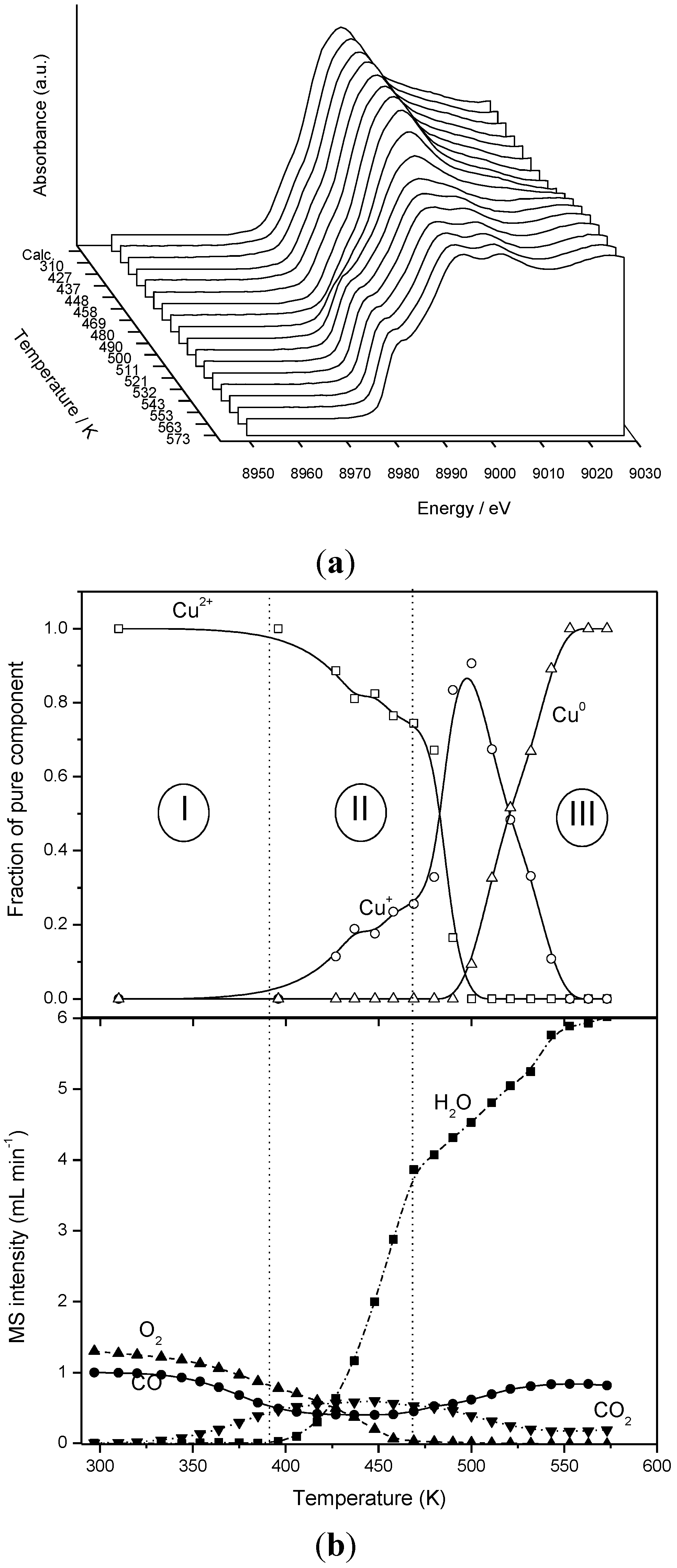
3. Summary and Main Conclusions
Acknowledgments
Conflict of Interest
References
- Rostrup-Nielsen, J.R.; Sehested, J.; Norskov, J.K. Hydrogen and synthesis gas by steam- and CO2 reforming. Adv. Catal. 2002, 47, 65–139. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Papadopoulou, C.; Batista, J.; Hocevar, S.; Matralis, H.K. A comparative study of Pt/γ-Al2O3, Au/α-Fe2O3 and CuO-CeO2 catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catal. Today 2002, 75, 157–167. [Google Scholar]
- Oh, S.H.; Sinkevitch, R.M. Carbon monoxide removal from hydrogen-rich fuel cell feedstreams by selective catalytic oxidation. J. Catal. 1993, 142, 254–262. [Google Scholar] [CrossRef]
- Wang, J.B.; Lin, S.; Huang, T. Selective CO oxidation in rich hydrogen over CuO/samaria-doped ceria. Appl. Catal. A 2002, 232, 107–120. [Google Scholar] [CrossRef]
- Sedmak, G.; Hocevar, S.; Levec, J. Kinetics of selective CO oxidation in excess of H2 over the nanostructured Cu0.1Ce0.9O2−y catalyst. J. Catal. 2003, 213, 135–150. [Google Scholar] [CrossRef]
- Kim, D.H.; Cha, J.E. A CuO-CeO2 Mixed-oxide catalyst for CO clean-up by selective oxidation in hydrogen-rich mixtures. Catal. Lett. 2003, 86, 107–112. [Google Scholar]
- Korotkikh, O.; Farrauto, R. Selective catalytic oxidation of CO in H2: Fuel cell applications. Catal. Today 2000, 62, 249–254. [Google Scholar] [CrossRef]
- Ghenciu, A.F. Review of fuel processing catalysts for hydrogen production in PEM fuel cell systems. Curr. Opin. Solid State Mater. Sci. 2002, 6, 389–399. [Google Scholar] [CrossRef]
- Haruta, M.; Date, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Schubert, M.M.; Venugopal, A.; Kahlich, M.J.; Plzak, V.; Behm, R.J. Influence of H2O and CO2 on the selective CO oxidation in H2-rich gases over Au/α-Fe2O3. J. Catal. 2004, 222, 32–40. [Google Scholar]
- Luengnaruemitchai, A.; Osuwan, S.; Gulari, E. Selective catalytic oxidation of CO in the presence of H2 over gold catalyst. Int. J. Hydrog. Energy 2004, 29, 429–435. [Google Scholar] [CrossRef]
- Deng, W.; de Jesus, J.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Low-content gold-ceria catalysts for the water-gas shift and preferential CO oxidation reactions. Appl. Catal. A 2005, 291, 126–135. [Google Scholar] [CrossRef]
- Carrettin, S.; Concepcion, P.; Corma, A.; López, Nieto, J.M.; Puentes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Papavasiliou, J.; Tabakova, T.; Idakiev, V.; Ioannides, T. A comparative study of ceria-supported gold and copper oxide catalysts for preferential CO oxidation reaction. Chem. Eng. J. 2006, 124, 41–45. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Preferential oxidation of CO in H2 over CuO-CeO2 catalysts. Catal. Today 2004, 93–95, 241–246. [Google Scholar]
- Marbán, G.; Fuertes, A.B. Highly active and selective CuOx/CeO2 catalyst prepared by a single-step citrate method for preferential oxidation of carbon monoxide. Appl. Catal. B 2005, 57, 43–53. [Google Scholar]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Munuera, G. Preferential oxidation of CO in a H2-rich stream over CuO/CeO2 and CuO/(Ce,M)Ox (M = Zr, Tb) catalysts. J. Power Sour. 2005, 151, 32–42. [Google Scholar] [CrossRef]
- Mariño, F.; Descorme, C.; Duprez, D. Supported base metal catalysts for the preferential oxidation of carbon monoxide in the presence of excess hydrogen (PROX). Appl. Catal. B 2005, 58, 175–183. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Munuera, G.; Gamarra, D. Preferential oxidation of CO in rich H2 over CuO/CeO2: Details of selectivity and deactivation under the reactant stream. Appl. Catal. B 2006, 65, 207–216. [Google Scholar] [CrossRef]
- Park, J.-W.; Jeong, J.-H.; Yoon, W.-L.; Jung, H.; Lee, H.-T.; Lee, D.-K.; Park, Y.-K.; Rhee, Y.W. Activity and characterization of the Co-promoted CuO-CeO2/γ-Al2O3 catalyst for the selective oxidation of CO in excess hydrogen. Appl. Catal. A 2004, 274, 25–32. [Google Scholar] [CrossRef]
- Jobbagy, M.; Mariño, F.; Schönbrod, B.; Baronetti, G.; Laborde, M. Synthesis of copper-promoted CeO2 catalysts. Chem. Mater. 2006, 18, 1945–1950. [Google Scholar] [CrossRef]
- Moretti, E.; Lenarda, M.; Storaro, L.; Talon, A.; Montanari, T.; Busca, G.; Rodríguez-Castellón, E.; Jiménez-López, A.; Turco, M.; Bagnasco, G.; et al. One-step synthesis of a structurally organized mesoporous CuO-CeO2-Al2O3 system for the preferential CO oxidation. Appl. Catal. A 2008, 335, 46–55. [Google Scholar] [CrossRef]
- Gurbani, A.; Ayastuy, J.L.; González-Marcos, M.P.; Herrero, J.E.; Guil, J.M.; Gutiérrez-Ortiz, M.A. Comparative study of CuO-CeO2 catalysts prepared by wet impregnation and deposition-precipitation. Int. J. Hydrog. Energy 2009, 34, 547–553. [Google Scholar]
- Martínez-Arias, A.; Soria, J.; Cataluña, R.; Conesa, J.C.; Cortés Corberán, V. Influence of ceria dispersión on the catalytic performance of Cu/CeO2/Al2O3 catalysts for the CO oxidation reaction. Stud. Surf. Sci. Catal. 1998, 116, 591–600. [Google Scholar] [CrossRef]
- Liu, W.; Sarofim, A.F.; Flytzani-Stephanopoulos, M. Complete oxidation of carbon monoxide and methane over metal-promoted fluorite oxide catalysts. Chem. Eng. Sci. 1995, 49, 4871–4888. [Google Scholar]
- Martínez-Arias, A.; Fernández-García, M.; Gálvez, O.; Coronado, J.M.; Anderson, J.A.; Conesa, J.C.; Soria, J.; Munuera, G. Comparative study on redox properties and catalytic behavior for CO Oxidation of CuO/CeO2 and CuO/ZrCeO4 catalysts. J. Catal. 2000, 195, 207–216. [Google Scholar]
- Skårman, B.; Grandjean, D.; Benfield, R.E.; Hinz, A.; Andersson, A.; Wallenberg, L.R. Carbon monoxide oxidation on nanostructured CuOx/CeO2 composite particles characterized by HREM, XPS, XAS, and high-energy diffraction. J. Catal. 2002, 211, 119–133. [Google Scholar]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Munuera, G. Interfacial redox processes under CO/O2 in a nanoceria-supported copper oxide catalyst. J. Phys. Chem. B 2004, 108, 17983–17991. [Google Scholar]
- Ilichev, A.N.; Firsova, A.A.; Korchak, V.N. Mechanism of CO oxidation in excess H2 over CuO/CeO2 catalysts: ESR and TPD studies. Kinet. Catal. 2006, 47, 585–592. [Google Scholar] [CrossRef]
- Gamarra, D.; Hornés, A.; Koppány, Z.; Schay, Z.; Munuera, G.; Soria, J.; Martínez-Arias, A. Catalytic processes during preferential oxidation of CO in H2-rich streams over catalysts based on copper-ceria. J. Power Sour. 2007, 169, 110–116. [Google Scholar] [CrossRef]
- Luo, M.-F.; Song, Y.-P.; Lu, J.Q.; Wang, X.-Y.; Pu, Z.-Y. Identification of CuO species in high surface area CuO-CeO2 catalysts and their catalytic activities for CO oxidation. J. Phys. Chem. C 2007, 111, 12686–12692. [Google Scholar]
- Luo, M.-F.; Ma, J.-M.; Lu, J.-Q.; Song, Y.-P.; Wang, Y.-J. High-surface area CuO-CeO2 catalysts prepared by a surfactant-templated method for low-temperature CO oxidation. J. Catal. 2007, 246, 52–59. [Google Scholar]
- Pintar, A.; Batista, J.; Hocevar, S. TPR, TPO, and TPD examinations of Cu0.15Ce0.85O2−y mixed oxides prepared by co-precipitation, by the sol-gel peroxide route, and by citric acid-assisted synthesis. J. Coll. Interf. Sci. 2005, 285, 218–231. [Google Scholar] [CrossRef]
- Manzoli, M.; di Monte, R.; Boccuzzi, F.; Coluccia, S.; Kaspar, J. CO oxidation over CuOx-CeO2-ZrO2 catalysts: Transient behaviour and role of copper clusters in contact with ceria. Appl. Catal. B 2005, 61, 192–205. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Matralis, H. Influence of the preparation method on the performance of CuO-CeO2 catalysts for the selective oxidation of CO. Appl. Catal. B 2005, 56, 87–93. [Google Scholar] [CrossRef]
- Wang, X.Q.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martínez-Arias, A.; Fernández-García, M. Unusual physical and chemical properties of Cu in Ce1−xCuxO2 oxides. J. Phys. Chem. B 2005, 109, 19595–19603. [Google Scholar]
- Wang, J.B.; Tsai, D.-H.; Huang, T.-J. Synergistic Catalysis of carbon monoxide oxidation over copper oxide supported on samaria-doped ceria. J. Catal. 2002, 208, 370–380. [Google Scholar] [CrossRef]
- Lee, H.C.; Kim, D.H. Kinetics of CO and H2 oxidation over CuO-CeO2 catalyst in H2 mixtures with CO2 and H2O. Catal. Today 2008, 132, 109–116. [Google Scholar] [CrossRef]
- Gamarra, D.; Munuera, G.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Midgley, P.A.; Wang, X.Q.; Hanson, J.C.; Rodriguez, J.A.; Martínez-Arias, A. Structure-activity relationship in nanostructured copper-ceria-based preferential CO oxidation catalysts. J. Phys. Chem. C 2007, 111, 11026–11038. [Google Scholar] [CrossRef]
- Gamarra, D.; Belver, C.; Fernández-García, M.; Martínez-Arias, A. Selective CO oxidation in excess H2 over copper-ceria catalysts: Identification of active entities/species. J. Am. Chem. Soc. 2007, 129, 12064–12065. [Google Scholar] [CrossRef]
- Gamarra, D.; Lopez Camara, A.; Monte, M.; Rasmussen, S.B.; Chinchilla, L.E.; Hungria, A.B.; Munuera, G.; Gyorffy, N.; Schay, Z.; Corberan, V.C.; et al. Preferential oxidation of CO in excess H2 over CuO/CeO2 catalysts: Characterization and performance as a function of the exposed face present in the CeO2 support. Appl. Catal. B 2013, 130–131, 224–238. [Google Scholar]
- Bion, N.; Epron, F.; Moreno, M.; Mariño, F.; Duprez, D. Preferential oxidation of carbon monoxide in the presence of hydrogen (PROX) over noble metals and transition metal oxides: Advantages and drawbacks. Top. Catal. 2008, 51, 76–88. [Google Scholar] [CrossRef]
- López, I.; Valdés-Solís, T.; Marbán, G. An attempt to rank copper-based catalysts used in the CO-PROX reaction. Int. J. Hydrog. Energy 2008, 33, 197–205. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalysis by Ceria and Related Materials; Imperial College Press: London, UK, 2002. [Google Scholar]
- Hungría, A.B.; Martínez-Arias, A.; Fernández-García, M.; Iglesias-Juez, A.; Guerrero-Ruiz, A.; Calvino, J.J.; Conesa, J.C.; Soria, J. Structural, morphological, and oxygen handling properties of nanosized cerium-terbium mixed oxides prepared by microemulsion. Chem. Mater. 2003, 15, 4309–4316. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Hungría, A.B.; Conesa, J.C.; Munuera, G. Spectroscopic characterization of heterogeneity and redox effects in zirconium-cerium (1:1) mixed oxides prepared by microemulsion methods. J. Phys. Chem. B 2003, 107, 2667–2677. [Google Scholar]
- Hornés, A.; Bera, P.; López Cámara, A.; Gamarra, D.; Munuera, G.; Martínez-Arias, A. CO-TPR-DRIFTS-MS in situ study of CuO/Ce1−xTb2O2−y (x = 0, 0.2 axnd 0.5) catalysts: Support effects on redox properties and CO oxidation catalysis. J. Catal. 2009, 268, 367–375. [Google Scholar] [CrossRef]
- Bera, P.; López Cámara, A.; Hornés, A.; Martínez-Arias, A. Comparative in situ DRIFTS-MS study of 12CO- and 13CO-TPR on CuO/CeO2 catalyst. J. Phys. Chem. C 2009, 113, 10689–10695. [Google Scholar]
- Wang, J.B.; Shih, W.-H.; Huang, T.-J. Study of Sm2O3-doped CeO2/Al2O3-supported copper catalyst for CO oxidation. Appl. Catal. A 2000, 203, 191–199. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Soria, J.; Conesa, J.C. Spectroscopic study of a Cu/CeO2 catalyst subjected to redox treatments in carbon monoxide and oxygen. J. Catal. 1999, 182, 367–377. [Google Scholar]
- Caputo, T.; Lisi, L.; Pirone, R.; Russo, G. On the role of redox properties of CuO/CeO2 catalysts in the preferential oxidation of CO in H2-rich gases. Appl. Catal. A 2008, 348, 42–53. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Iglesias-Juez, A.; Soria, J.; Conesa, J.C.; Anderson, J.A.; Munuera, G. Operando DRIFTS study of the redox and catalytic properties of CuO/Ce1−xTbxO2−d (x = 0–0.5) catalysts: Evidence of an induction step during CO oxidation. Phys. Chem. Chem. Phys. 2012, 14, 2144–2151. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.-C. An FTIR study of surface ceria hydroxy groups during a redox process with H2. J. Chem. Soc. Faraday Trans. 1996, 92, 4669–4673. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.-I.; Onishi, T. Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by fourier-transform infrared spectroscopy. Part 1.—Formation of carbonate species on dehydroxylated CeO2, at room temperature. J. Chem. Soc. Faraday Trans. 1 1989, 85, 929–943. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.-I.; Onishi, T. Adsorption of carbon monoxide and carbon dioxide on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 2.—Formation of formate species on partially reduced CeO2 at room temperature. J. Chem. Soc. Faraday Trans. 1 1989, 85, 1451–1461. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Teschner, D.; Wootsch, A.; Kröhnert, J.; Steinhauer, B.; Sauer, H.; Toth, L.; Jentoft, F.C.; Knop-Gericke, A.; Paál, Z.; et al. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2 under reaction conditions. J. Catal. 2006, 237, 1–16. [Google Scholar] [CrossRef]
- Vayssilov, N.; Mihaylov, M.; Petkov, P.S.; Hadjiivanov, K.I.; Neyman, K.M. reassignment of the vibrational spectra of carbonates, formates, and related surface species on ceria: A combined density functional and infrared spectroscopy investigation. J. Phys. Chem. C 2011, 115, 23435–23454. [Google Scholar] [CrossRef]
- Scarano, D.; Bordiga, S.; Lamberti, C.; Spoto, G.; Ricchiardi, G.; Zecchina, A.; Otero Areán, C. FTIR study of the interaction of CO with pure and silica-supported copper(I) oxide. Surf. Sci. 1998, 411, 272–285. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Hungría, A.B.; Iglesias-Juez, A.; Gálvez, O.; Anderson, J.A.; Conesa, J.C.; Soria, J.; Munuera, G. Redox interplay at copper oxide-(Ce,Zr)Ox interfaces: Influence of the presence of NO on the catalytic activity for CO oxidation over CuO/CeZrO4. J. Catal. 2003, 214, 261–272. [Google Scholar]
- Martínez-Arias, A.; Gamarra, D.; Fernández-García, M.; Wang, X.Q.; Hanson, J.C.; Rodriguez, J.A. Comparative study on redox properties of nanosized CeO2 and CuO/CeO2 under CO/O2. J. Catal. 2006, 240, 1–7. [Google Scholar] [CrossRef]
- Polster, C.S.; Nair, H.; Baertsch, C.D. Study of active sites and mechanism responsible for highly selective CO oxidation in H2 rich atmospheres on a mixed Cu and Ce oxide catalysts. J. Catal. 2009, 266, 308–319. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Vannice, M.A. An analysis of the Mars-van Krevelen rate expression. Catal. Today 2007, 123, 18–22. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Gamarra, D.; Fernández-García, M.; Hornés, A.; Belver, C. Spectroscopic study on the nature of active entities in copper-ceria CO-PROX catalysts. Top. Catal. 2009, 52, 1425–1432. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Cataluña, R.; Conesa, J.C.; Soria, J. Effect of copper-ceria interactions on copper reduction in a Cu/CeO2/Al2O3 catalyst subjected to thermal treatments in CO. J. Phys. Chem. B 1998, 102, 809–817. [Google Scholar] [CrossRef]
- Guzman, J.; Carrettin, S.; Corma, A. spectroscopic evidence for the supply of reactive oxygen during CO oxidation catalyzed by gold supported on nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef]
- Pushkarev, V.V.; Kovalchuk, V.I.; d’Itri, J.L. probing defect sites on the ceo2 surface with dioxygen. J. Phys. Chem. B 2004, 108, 5341–5348. [Google Scholar] [CrossRef]
- Spanier, J.E.; Robinson, R.D.; Zhang, F.; Chan, S.-W.; Herman, I.P. Size-dependent properties of CeO2−y nanoparticles as studied by Raman scattering. Phys. Rev. B 2001, 64, 245407. [Google Scholar] [CrossRef]
- Kim, J.Y.; Rodriguez, J.A.; Hanson, J.C.; Frenkel, A.I.; Lee, P.L. Reduction of CuO and Cu2O with H2: H embedding and kinetic effects in the formation of suboxides. J. Am. Chem. Soc. 2003, 125, 10684–10692. [Google Scholar]
- Fernández-García, M.; Martínez-Arias, A.; Hanson, J.C.; Rodriguez, J.A. Nanostructured oxides in chemistry: Characterization and properties. Chem. Rev. 2004, 104, 4063–4104. [Google Scholar] [CrossRef]
- Kydd, R.; Ferri, D.; Hug, P.; Scott, J.; Teoh, W.Y.; Amal, R. Temperature-induced evolution of reaction sites and mechanisms during preferential oxidation of CO. J. Catal. 2011, 277, 64–71. [Google Scholar] [CrossRef]
- Yen, H.; Seo, Y.; Kaliaguine, S.; Kleitz, F. Tailored mesostructured copper/ceria catalysts with enhanced performance for preferential oxidation of CO at low temperature. Angew. Chem. Int. Ed. 2012, 51, 12032–12035. [Google Scholar] [CrossRef]
- Hornés, A.; Hungría, A.B.; Bera, P.; López Cámara, A.; Fernández-García, M.; Martínez-Arias, A.; Barrio, L.; Estrella, M.; Zhou, G.; Fonseca, J.J.; et al. Inverse CeO2/CuO catalyst as an alternative to classical direct configurations for preferential oxidation of CO in hydrogen-rich stream. J. Am. Chem. Soc. 2010, 132, 34–35. [Google Scholar] [CrossRef]
- Gamarra, D.; Martínez-Arias, A. Preferential oxidation of CO in rich H2 over CuO/CeO2: Operando-DRIFTS analysis of deactivating effect of CO2 and H2O. J. Catal. 2009, 263, 189–195. [Google Scholar] [CrossRef]
- Gamarra, D.; Fernández-García, M.; Belver, C.; Martínez-Arias, A. Operando DRIFTS and XANES study of deactivating effect of CO2 on a Ce0.8Cu0.2O2 CO-PROX catalyst. J. Phys. Chem. C 2010, 114, 18576–18582. [Google Scholar]
- Zeng, S.H.; Liu, Y.; Wang, Y.Q. CuO-CeO2/Al2O3 /FeCrAl monolithic catalysts prepared by sol-pyrolysis method for preferential oxidation of carbon monoxide. Catal. Lett. 2007, 117, 119–125. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martínez-Arias, A.; Gamarra, D.; Hungría, A.B.; Fernández-García, M.; Munuera, G.; Hornés, A.; Bera, P.; Conesa, J.C.; Cámara, A.L. Characterization of Active Sites/Entities and Redox/Catalytic Correlations in Copper-Ceria-Based Catalysts for Preferential Oxidation of CO in H2-Rich Streams. Catalysts 2013, 3, 378-400. https://doi.org/10.3390/catal3020378
Martínez-Arias A, Gamarra D, Hungría AB, Fernández-García M, Munuera G, Hornés A, Bera P, Conesa JC, Cámara AL. Characterization of Active Sites/Entities and Redox/Catalytic Correlations in Copper-Ceria-Based Catalysts for Preferential Oxidation of CO in H2-Rich Streams. Catalysts. 2013; 3(2):378-400. https://doi.org/10.3390/catal3020378
Chicago/Turabian StyleMartínez-Arias, Arturo, Daniel Gamarra, Ana B. Hungría, Marcos Fernández-García, Guillermo Munuera, Aitor Hornés, Parthasarathi Bera, José C. Conesa, and Antonio López Cámara. 2013. "Characterization of Active Sites/Entities and Redox/Catalytic Correlations in Copper-Ceria-Based Catalysts for Preferential Oxidation of CO in H2-Rich Streams" Catalysts 3, no. 2: 378-400. https://doi.org/10.3390/catal3020378