
Fischer-Tropsch Synthesis on Multicomponent Catalysts: What Can We Learn from Computer Simulations?



Abstract
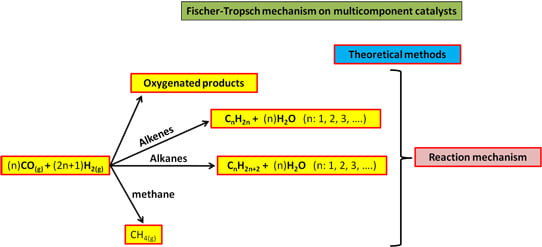
:
1. Introduction
2. Computational Studies on Catalyst Surface Models

{kind=link}
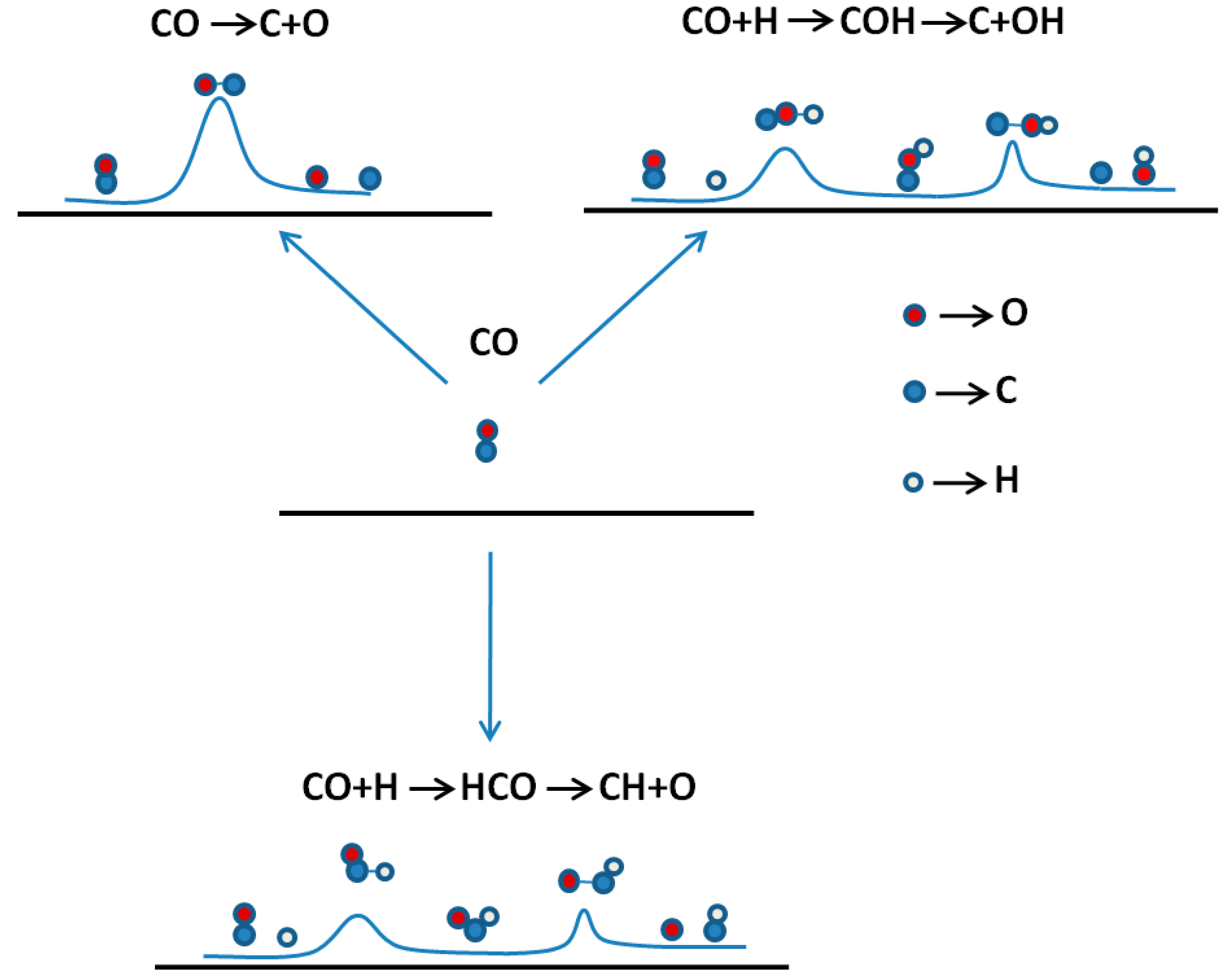{kind=link}
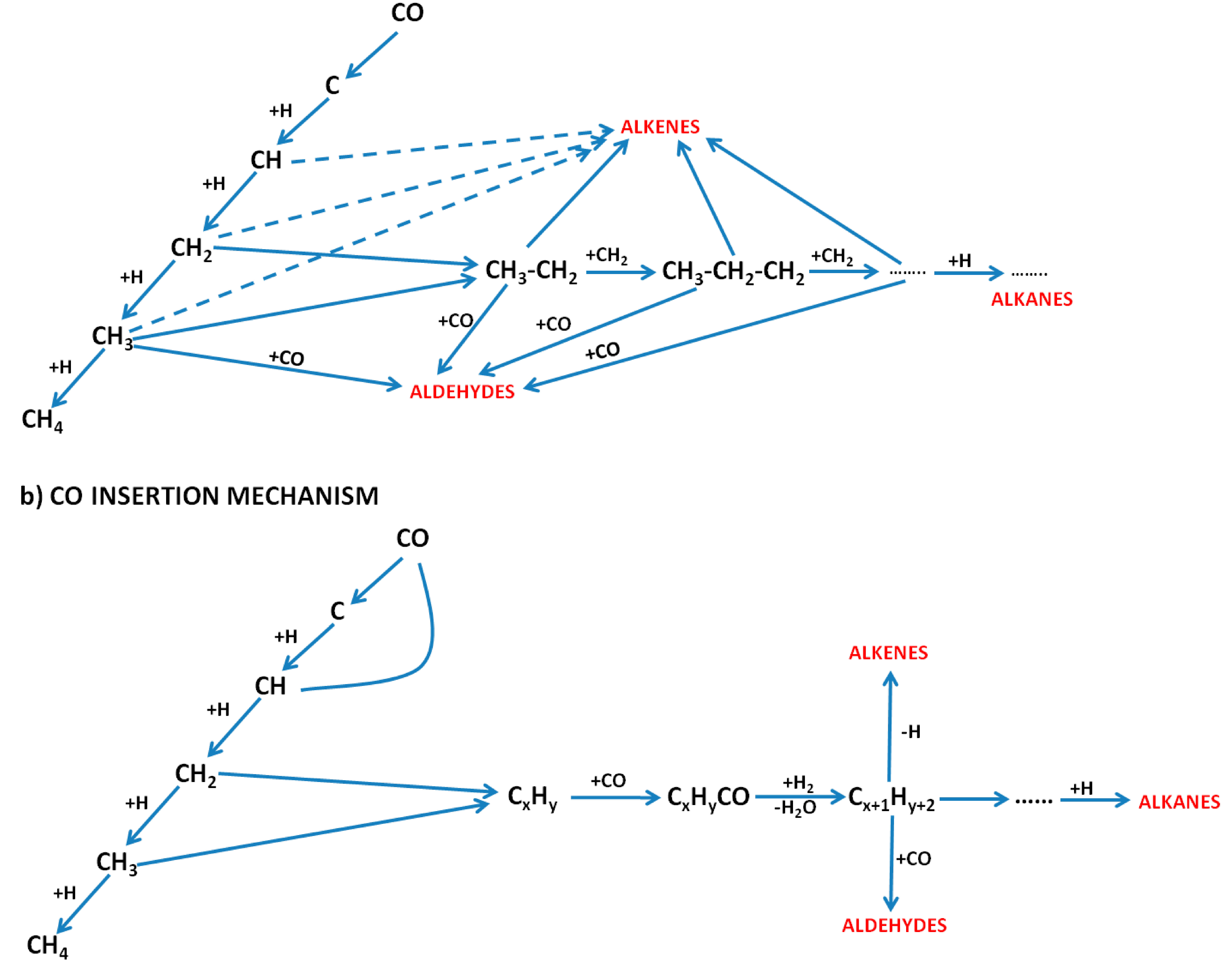{kind=link}
| Code a | DFT b | Basis set c | Transition metal surface(s) d | Ref. |
|---|---|---|---|---|
| DACAPO | PW91 | PW-USPP | Fe(110) and Co(0001) | Ojeda et al. [19] |
| DACAPO | RPBE | PW-USPP | Ru(0001) and Ru(109) | Vendelbo et al. [23] |
| VASP | PBE | PW-PAW | Co(0001), Pt@Co(0001), and Ru@Co(0001) | Balakrishnan et al. [32] |
| CASTEP | PW91 | PW-USPP | Pt(111), Pd(111), and Ru(0001) | Inderwildi et al. [49] |
| CASTEP | PW91 | PW-USPP | Co(0001) | Inderwildi et al. [50] |
| VASP | RPBE | PW-USPP | Ru(0001) | Loveless et al. [51] |
| VASP | PW91 | PW-USPP | Ru(105) | Ciobica et al. [52] |
| VASP | PBE | PW-PAW | Ru() | Shetty et al. [53] |
| VASP | PBE | PW-PAW | Ru(100)B and Co(100)B | Shetty et al. [54] |
| VASP | PBE | PW-PAW | Co(0001), Co(102), and Co(110) | Liu et al. [55] |
| VASP | PW91 | PW-PAW | Ni(111), Ni(110), Rh@Ni(111), Rh@Ni(110), Ru@Ni(111) and Ru@Ni(110) | Fajín et al. [56] |
| Siesta | PBE | DZP-TMPP | Co(0001) | Cheng et al. [57] |
| Siesta | PBE | DZP-TMPP | Ru(001) e, Fe(210), Rh(211), and Re(001)e | Cheng et al. [58] |
| VASP | PW91 | PW-PAW | Rh(111) and Rh(211) | van Grootel et al. [59] |
| SeqQuest | PBE | DZP-TMPP | Ni(111) | Mueller et al. [60] |
| - | GGA | PW-USPP | Rh(111) | Zhang et al. [61] |
| VASP | PW91 | PW-PAW | Rh(111), Ni(111), and Rh-Ni(111) | Lee et al. [62] |
| Reaction route | Fe(110) | Co(0001) | Fe(111) |
|---|---|---|---|
| CO* + * → C* + O* | 1.96 [19] | 3.80 [19]; 2.82 [50] | 1.76 [49] |
| CO* + H* → COH* + * → C* + OH* | 1.63 [19] | 3.26 [19] | |
| CO* + H* → HCO* + * → CH* + O* | 0.79 [19] | 0.95 [19]; 1.00 [50] | 1.17 [49] |
| HCO* + H* → HCOH* + * → CH* + OH* | 0.65 [19] | 1.10 [19] | |
| HCO* + H* → H2CO* + * → CH2* + O* | 3.29 [19] | 1.63 [19] |


3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fischer, F.; Tropsch, H. Synthesis of petroleum at atmospheric pressures from gasification products of coal. Brennst. Chem. 1926, 7, 97–104. [Google Scholar]
- Anderson, R.B. The Fischer Tropsh Synthesis; Academic Press: New York, NY, USA, 1984. [Google Scholar]
- Kusama, H.; Okabe, K.; Arakawa, H. Characterization of Rh-Co/SiO2 catalysts for CO2 hydrogenation with TEM, XPS and FT-IR. Appl. Catal. A 2001, 207, 85–94. [Google Scholar] [CrossRef]
- Schulz, H. Major and minor reactions in Fischer-Tropsch synthesis on cobalt catalysts. Top. Catal. 2003, 26, 73–85. [Google Scholar] [CrossRef]
- Lögdberg, S.; Lualdi, M.; Järås, S.; Walmsley, J.C.; Blekkan, E.A.; Rytter, E.; Holmen, A. On the selectivity of cobalt-based Fischer-Tropsch catalysts: Evidence for a common precursor for methane and long-chain hydrocarbons. J. Catal. 2010, 274, 84–98. [Google Scholar] [CrossRef]
- Guczi, L.; Stefler, G.; Koppány, Z.; Borkó, L. CO hydrogenation over Re-Co bimetallic catalyst supported over SiO2, Al2O3 and NaY zeolite. React. Kinet. Catal. Lett. 2001, 74, 259–269. [Google Scholar] [CrossRef]
- Davis, B.H. Fischer-Tropsch synthesis: Current mechanism and futuristic needs. Fuel Process. Technol. 2001, 71, 157–166. [Google Scholar] [CrossRef]
- Tupabut, P.; Jongsomjit, B.; Praserthdam, P. Impact of boron modification on MCM-41-supported cobalt catalysts for hydrogenation of carbon monoxide. Catal. Lett. 2007, 118, 195–202. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Davis, B.H.; Willauer, H.D. Influence of gas feed composition and pressure on the catalytic conversion of CO2 to hydrocarbons using a traditional cobalt-based Fischer-Tropsch catalyst. Energ. Fuel. 2009, 23, 4190–4195. [Google Scholar] [CrossRef]
- Rønning, M.; Tsakoumis, N.E.; Voronov, A.; Johnsen, R.E.; Norby, P.; van Beek, W.; Borg, Ø.; Rytter, E.; Holmen, A. Combined XRD and XANES studies of a Re-promoted Co/γ-Al2O3 catalyst at Fischer-Tropsch synthesis conditions. Catal. Today 2010, 155, 289–295. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt particle size effects in the Fischer-Tropsch reaction studied with carbon nanofiber supported catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, H.; Wang, H.; Liu, L.; Xiao, C.; Ma, D. The effects of ionic additives on the aqueous-phase Fischer-Tropsch synthesis with a ruthenium nanoparticle catalyst. Catal. Today 2012, 183, 143–153. [Google Scholar] [CrossRef]
- Inderwildi, O.R.; Jenkins, S.J. In-silico investigations in heterogeneous catalysis—Combustion and synthesis of small alkanes. Chem. Soc. Rev. 2008, 37, 2274–2309. [Google Scholar] [CrossRef] [PubMed]
- Stranges, A.N. A History of the Fischer-Tropsch Synthesis in Germany 1926–45. In Fischer-Tropsch Synthesis, Catalysts, and Catalysis, 1st ed.; Davis, B.H., Occelli, M.L., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2007; pp. 1–28. [Google Scholar]
- Davis, B.H.; Occelli, M.L. Advances in Fischer-Tropsch Synthesis, Catalysts, and Catalysis; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2010. [Google Scholar]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, S.-M.; Woo, M.H.; Bae, J.W.; Jun, K.-W.; Ha, K.-S. Effects of titanium impurity on alumina surface for the activity of Co/Ti–Al2O3 Fischer-Tropsch catalyst. Appl. Catal. A 2012, 419–420, 148–155. [Google Scholar]
- Nawdali, M.; Bianchi, D. The impact of the Ru precursor on the adsorption of CO on Ru/Al2O3: Amount and reactivity of the adsorbed species. Appl. Catal. A 2002, 231, 45–54. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Iglesia, E. CO activation pathways and the mechanism of Fischer-Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Gual, A.; Godard, C.; Castillon, S.; Curulla-Ferré, D.; Claver, C. Colloidal Ru, Co and Fe-nanoparticles. Synthesis and application as nanocatalysts in the Fischer-Tropsch process. Catal. Today 2012, 183, 154–171. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Evans, J.; Agnoli, S.; Barrio, L.; Chen, T.-L.; Hrbek, J.; Rodriguez, J.A. Water-gas shift and CO methanation reactions over Ni-CeO2(111) catalysts. Top. Catal. 2011, 54, 34–41. [Google Scholar] [CrossRef]
- Bundhoo, A.; Schweicher, J.; Frennet, A.; Kruse, N. Chemical transient kinetics applied to CO hydrogenation over a pure nickel catalyst. J. Phys. Chem. C 2009, 113, 10731–10739. [Google Scholar] [CrossRef]
- Vendelbo, S.B.; Johansson, M.; Mowbray, D.J.; Andersson, M.P.; Abild-Pedersen, F.; Nielsen, J.H.; Nørskov, J.K.; Chorkendorff, I. Self blocking of CO dissociation on a stepped ruthenium surface. Top. Catal. 2010, 53, 357–364. [Google Scholar] [CrossRef]
- Williams, C.T.; Black, C.A.; Weaver, M.J.; Takoudis, C.G. Adsorption and hydrogenation of carbon monoxide on polycrystalline rhodium at high gas pressures. J. Phys. Chem. B 1997, 101, 2874–2883. [Google Scholar] [CrossRef]
- Jenewein, B.; Fuchs, M.; Hayek, K. The CO methanation on Rh/CeO2 and CeO2/Rh model catalysts: A comparative study. Surf. Sci. 2003; 532–535, 364–369. [Google Scholar]
- Bulushev, D.A.; Froment, G.F. A DRIFTS study of the stability and reactivity of adsorbed CO species on a Rh/γ-Al2O3 catalyst with a very low metal content. J. Mol. Catal. A 1999, 139, 63–72. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic aspects of the selective methanation of CO over Ru/TiO2 catalyst. Catal. Today 2012, 181, 138–147. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. Mechanistic study of low temperature CO2 methanation over Rh/TiO2 catalysts. J. Catal. 2013, 301, 141–153. [Google Scholar] [CrossRef]
- Izquierdo, U.; Barrio, V.L.; Bizkarra, K.; Gutierrez, A.M.; Arraibi, J.R.; Gartzia, L.; Bañuelos, J.; Lopez-Arbeloa, I.; Cambra, J.F. Ni and Rh-Ni catalysts supported on zeolites L for hydrogen and syngas production by biogas reforming processes. Chem. Eng. J. 2014, 238, 178–188. [Google Scholar] [CrossRef]
- Pirola, C.; Scavini, M.; Galli, F.; Vitali, S.; Comazzi, A.; Manenti, F.; Ghigna, P. Fischer-Tropsch synthesis: EXAFS study of Ru and Pt bimetallic Co based catalysts. Fuel 2014, 132, 62–70. [Google Scholar] [CrossRef]
- Christensen, J.M.; Medford, A.J.; Studt, F.; Jensen, A.D. High pressure CO hydrogenation over bimetallic Pt-Co catalysts. Catal. Lett. 2014, 144, 777–782. [Google Scholar] [CrossRef]
- Balakrishnan, N.; Joseph, B.; Bhethanabotla, V.R. Effect of Pt and Ru promoters on deactivation of Co catalysts by C deposition during Fischer-Tropsch synthesis: A DFT study. Appl. Catal. 2013, 462–463, 107–115. [Google Scholar]
- Weststrate, C.J.; Ciobîcă, I.M.; Saib, A.M.; Moodley, D.J.; Niemantsverdriet, J.W. Fundamental issues on practical Fischer-Tropsch catalysts: How surface science can help. Catal. Today 2014, 228, 106–112. [Google Scholar] [CrossRef]
- Bambal, A.S.; Guggilla, V.S.; Kugler, E.L.; Gardner, T.H.; Dadyburjor, D.B. Poisoning of a silica-supported cobalt catalyst due to presence of sulfur impurities in syngas during Fischer-Tropsch synthesis: Effects of chelating agent. Ind. Eng. Chem. Res. 2014, 53, 5846–5857. [Google Scholar]
- Tian, D.; Liu, Z.; Li, D.; Shi, H.; Pan, W.; Cheng, Y. Bimetallic Ni-Fe total-methanation catalyst for the production of substitute natural gas under high pressure. Fuel 2013, 104, 224–229. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R.; Takagaki, A.; Sugawara, T.; Oyama, S.T.; Satokawa, S. Effect of metal addition to Ru/TiO2 catalyst on selective CO methanation. Catal. Today 2014, 232, 16–21. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, K.; Liu, P.; Hui, H.; Tan, Y. Synthesis of light olefins from syngas over Fe-Mn-V-K catalysts in the slurry phase. J. Ind. Eng. Chem. 2013, 19, 961–965. [Google Scholar] [CrossRef]
- Xu, J.-D.; Zhu, K.-T.; Weng, X.-F.; Weng, W.-Z.; Huang, C.-J.; Wan, H.-L. Carbon nanotube-supported Fe-Mn nanoparticles: A model catalyst for direct conversion of syngas to lower olefins. Catal. Today 2013, 215, 86–94. [Google Scholar] [CrossRef]
- Shi, B.; Wu, L.; Liao, Y.; Jin, C.; Montavon, A. Explanations of the formation of branched hydrocarbons during Fischer-Tropsch synthesis by alkylidene mechanism. Top. Catal. 2014, 57, 451–459. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Shafer, W.D.; Jacobs, G.; Davis, B.H. Fischer-Tropsch synthesis: Effect of reaction temperature for aqueous-phase synthesis over a platinum promoted Co/alumina catalyst. Catal. Lett. 2014, 144, 1088–1095. [Google Scholar] [CrossRef]
- Jermwongratanachai, T.; Jacobs, G.; Shafer, W.D.; Pendyala, V.R.R.; Ma, W.; Gnanamani, M.K.; Hopps, S.; Thomas, G.A.; Kitiyanan, B.; Khalid, S.; et al. Fischer-Tropsch synthesis: TPR and XANES analysis of the impact of simulated regeneration cycles on the reducibility of Co/aluminacatalysts with different promoters (Pt, Ru, Re, Ag, Au, Rh, Ir). Catal. Today 2014, 228, 15–21. [Google Scholar] [CrossRef]
- Ning, W.; Yang, S.; Chen, H.; Yamada, M. Influences of K and Cu on coprecipitated FeZn catalysts for Fischer-Tropsch reaction. Catal. Commun. 2013, 39, 74–77. [Google Scholar] [CrossRef]
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Factors influencing the activity of Co/Ca/TiO2 catalyst for Fischer-Tropsch synthesis. Catal. Today 2014, 232, 2–10. [Google Scholar] [CrossRef]
- Azzam, K.; Jacobs, G.; Ma, W.; Davis, B.H. Effect of cobalt particle size on the catalyst intrinsic activity for Fischer-Tropsch synthesis. Catal. Lett. 2014, 144, 389–394. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Jacobs, G.; Ma, W.; Klettlinger, J.L.S.; Yen, C.H.; Davis, B.H. Fischer-Tropsch synthesis: Effect of catalyst particle (sieve) size range on activity, selectivity, and aging of a Pt promoted Co/Al2O3 catalyst. Chem. Eng. J. 2014, 249, 279–284. [Google Scholar] [CrossRef]
- Ding, M.; Qiu, M.; Liu, J.; Li, Y.; Wang, T.; Ma, L.; Wu, C. Influence of manganese promoter on co-precipitated Fe-Cu based catalysts for higher alcohols synthesis. Fuel 2013, 109, 21–27. [Google Scholar] [CrossRef]
- Bakar, W.A.W.A.; Ali, R.; Toemen, S. Catalytic methanation reaction over supported nickel-rhodium oxide for purification of simulated natural gas. J. Nat. Gas Chem. 2011, 20, 585–594. [Google Scholar] [CrossRef]
- Carenco, S.; Tuxen, A.; Chintapalli, M.; Pach, E.; Escudero, C.; Ewers, T.D.; Jiang, P.; Borondics, F.; Thornton, G.; Alivisatos, A.P.; et al. Dealloying of cobalt from CuCo nanoparticles under Syngas exposure. J. Phys. Chem. C 2013, 117, 6259–6266. [Google Scholar]
- Inderwildi, O.R.; Jenkins, S.J.; King, D.A. Mechanistic studies of hydrocarbon combustion and synthesis on noble metals. Angew. Chem. Int. Ed. 2008, 47, 5253–5255. [Google Scholar] [CrossRef]
- Inderwildi, O.R.; Jenkins, S.J.; King, D.A. Fischer-Tropsch mechanism revisited: Alternative pathways for the production of higher hydrocarbons from synthesis gas. J. Phys. Chem. C 2008, 112, 1305–1307. [Google Scholar] [CrossRef]
- Loveless, B.T.; Buda, C.; Neurock, M.; Iglesia, E. CO chemisorption and dissociation at high coverages during CO hydrogenation on Ru catalysts. J. Am. Chem. Soc. 2013, 135, 6107–6121. [Google Scholar] [PubMed]
- Ciobica, I. M.; van Santen, R.A. Carbon monoxide dissociation on planar and stepped Ru(0001) surfaces. J. Phys. Chem. B 2003, 107, 3808–3812. [Google Scholar] [CrossRef]
- Shetty, S.; Jansen, A.P.J.; van Santen, R.A. Direct versus hydrogen-assisted CO dissociation. J. Am. Chem. Soc. 2009, 131, 12874–12875. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Jansen, A.P.J.; van Santen, R.A. Hydrogen induced CO activation on open Ru and Co surfaces. Phys. Chem. Chem. Phys. 2010, 12, 6330–6332. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-X.; Su, H.-Y.; Li, W.-X. Structure sensitivity of CO methanation on Co (0001), () and () surfaces: Density functional theory calculations. Catal. Today 2013, 215, 36–42. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S.; Gomes, J.R.B. Methanation of CO on pure and Rh or Ru doped nickel surfaces. J. Phys. Chem. 2014. submitted. [Google Scholar]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. A DFT study of the chain growth probability in Fischer-Tropsch synthesis. J. Catal. 2008, 257, 221–228. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. Chain growth mechanism in Fischer-Tropsch synthesis: A DFT study of C-C coupling over Ru, Fe, Rh, and Re surfaces. J. Phys. Chem. C 2008, 112, 6082–6086. [Google Scholar] [CrossRef]
- Van Grootel, P.W.; Hensen, E.J.M.; van Santen, R.A. The CO formation reaction pathway in steam methane reforming by rhodium. Langmuir 2010, 26, 16339–16348. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.E.; van Duin, A.C.T.; Goddard III, W.A. Structures, energetics, and reaction barriers for CHx bound to the nickel (111) surface. J. Phys. Chem. C 2009, 113, 20290–20306. [Google Scholar] [CrossRef]
- Zhang, C.J.; Hu, P.; Lee, M.-H. A density functional theory study on the interaction between chemisorbed CO and S on Rh(111). Surf. Sci. 1999, 432, 305–315. [Google Scholar] [CrossRef]
- Lee, K.; Song, C.; Janik, M.J. Density functional theory study of sulfur tolerance of CO adsorption and dissociation on Rh-Ni binary metals. Appl. Catal. A 2010, 389, 122–130. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 67, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Shustorovich, E.; Sellers, H. The UBI-QEP method: A practical theoretical approach to understanding chemistry on transition metal surfaces. Surf. Sci. Rep. 1998, 31, 1–119. [Google Scholar] [CrossRef]
- Storsæter, S.; Chen, D.; Holmen, A. Microkinetic modelling of the formation of C1 and C2 products in the Fischer-Tropsch synthesis over cobalt catalysts. Surf. Sci. 2006, 600, 2051–2063. [Google Scholar] [CrossRef]
- Luo, W.; Asthagiri, A. Density functional theory study of methanol steam reforming on Co(0001) and Co(111) surfaces. J. Phys. Chem. C 2014, 118, 15274–15285. [Google Scholar]
- Eckle, S.; Anfang, H.-G.; Behm, R.J. Reaction intermediates and side products in the methanation of CO and CO2 over supported Ru catalysts in H2-rich reformate gases. J. Phys. Chem. C 2011, 115, 1361–1367. [Google Scholar] [CrossRef]
- Li, T.; Wang, H.; Yang, Y.; Xiang, H.; Li, Y. Study on an iron-nickel bimetallic Fischer-Tropsch synthesis catalyst. Fuel Proc. Technol. 2014, 118, 117–124. [Google Scholar] [CrossRef]
- Enger, B.C.; Holmen, A. Nickel and Fischer-Tropsch synthesis. Catal. Rev.: Sci. Eng. 2012, 54, 437–488. [Google Scholar] [CrossRef]
- Van Santen, R.A.; Markvoort, A.J.; Filot, I.A.W.; Ghouri, M.M.; Hensen, E.J.M. Mechanism and microkinetics of the Fischer-Tropsch reaction. Phys. Chem. Chem. Phys. 2013, 15, 17038–17063. [Google Scholar] [CrossRef] [PubMed]
- Birot, A.; Epron, F.; Descorme, C.; Duprez, D. Ethanol steam reforming over Rh/CexZr1−xO2 catalysts: Impact of the CO–CO2–CH4 interconversion reactions on the H2 production. Appl. Catal. B 2008, 79, 17–25. [Google Scholar] [CrossRef]
- Lv, X.; Chen, J.-F.; Tan, Y.; Zhang, Y. A highly dispersed nickel supported catalyst for dry reforming of methane. Catal. Commun. 2012, 20, 6–11. [Google Scholar] [CrossRef]
- Freni, S.; Cavallaro, S.; Mondello, N.; Spadaro, L.; Frusteri, F. Production of hydrogen for MC fuel cell by steam reforming of ethanol over MgO supported Ni and Co catalysts. Catal. Commun. 2003, 4, 259–268. [Google Scholar] [CrossRef]
- Karim, A.M.; Su, Y.; Sun, J.; Yang, C.; Strohm, J.J.; King, D.L.; Wang, Y. A comparative study between Co and Rh for steam reforming of ethanol. Appl. Catal. B 2010, 96, 441–448. [Google Scholar] [CrossRef]
- Lin, S.S.-Y.; Kim, D.H.; Ha, S.Y. Hydrogen production from ethanol steam reforming over supported cobalt catalysts. Catal. Lett. 2008, 122, 295–301. [Google Scholar] [CrossRef]
- Batista, M.S.; Santos, R.K.S.; Assaf, E.M.; Assaf, J.M.; Ticianelli, E.A. Characterization of the activity and stability of supported cobalt catalysts for the steam reforming of ethanol. J. Power Sources 2003, 124, 99–103. [Google Scholar] [CrossRef]
- Batista, M.S.; Santos, R.K.S.; Assaf, E.M.; Assaf, J.M.; Ticianelli, E.A. High efficiency steam reforming of ethanol by cobalt-based catalysts. J. Power Sources 2004, 134, 27–32. [Google Scholar] [CrossRef]
- Czekaj, I.; Struis, R.; Wambach, J.; Biollaz, S. Sulphur poisoning of Ni catalysts used in the SNG production from biomass: Computational studies. Catal. Today 2011, 176, 429–432. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajín, J.L.C.; Cordeiro, M.N.D.S.; Gomes, J.R.B. Fischer-Tropsch Synthesis on Multicomponent Catalysts: What Can We Learn from Computer Simulations? Catalysts 2015, 5, 3-17. https://doi.org/10.3390/catal5010003
Fajín JLC, Cordeiro MNDS, Gomes JRB. Fischer-Tropsch Synthesis on Multicomponent Catalysts: What Can We Learn from Computer Simulations? Catalysts. 2015; 5(1):3-17. https://doi.org/10.3390/catal5010003
Chicago/Turabian StyleFajín, José L. C., M. Natália D. S. Cordeiro, and José R. B. Gomes. 2015. "Fischer-Tropsch Synthesis on Multicomponent Catalysts: What Can We Learn from Computer Simulations?" Catalysts 5, no. 1: 3-17. https://doi.org/10.3390/catal5010003