
Design of Pt/Carbon Xerogel Catalysts for PEM Fuel Cells

Abstract
:1. Introduction
2. Synthesis Techniques
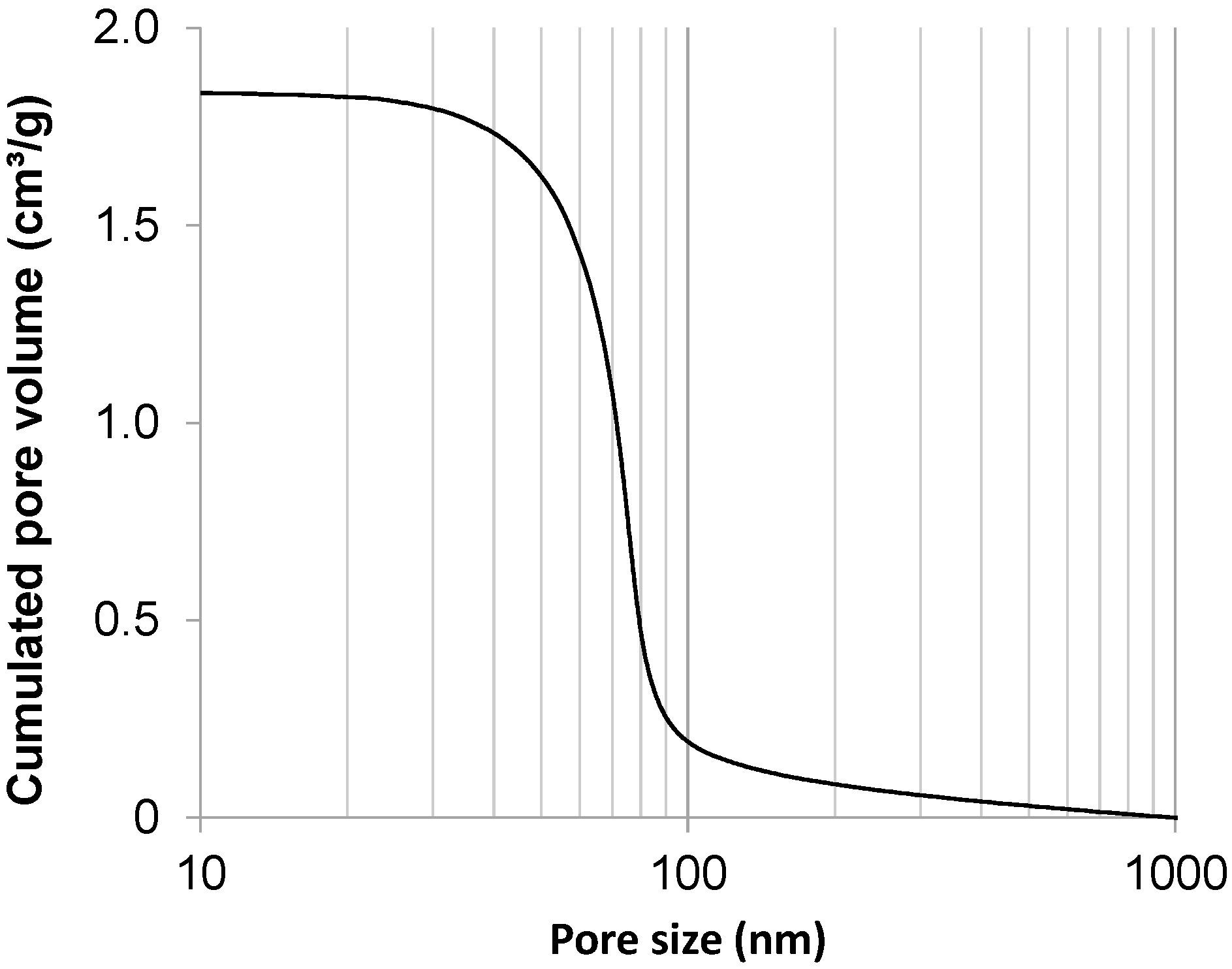
2.1. Carbon Support

2.2. Wet Impregnation
2.3. Wet Impregnation Coupled to Liquid Phase Reduction
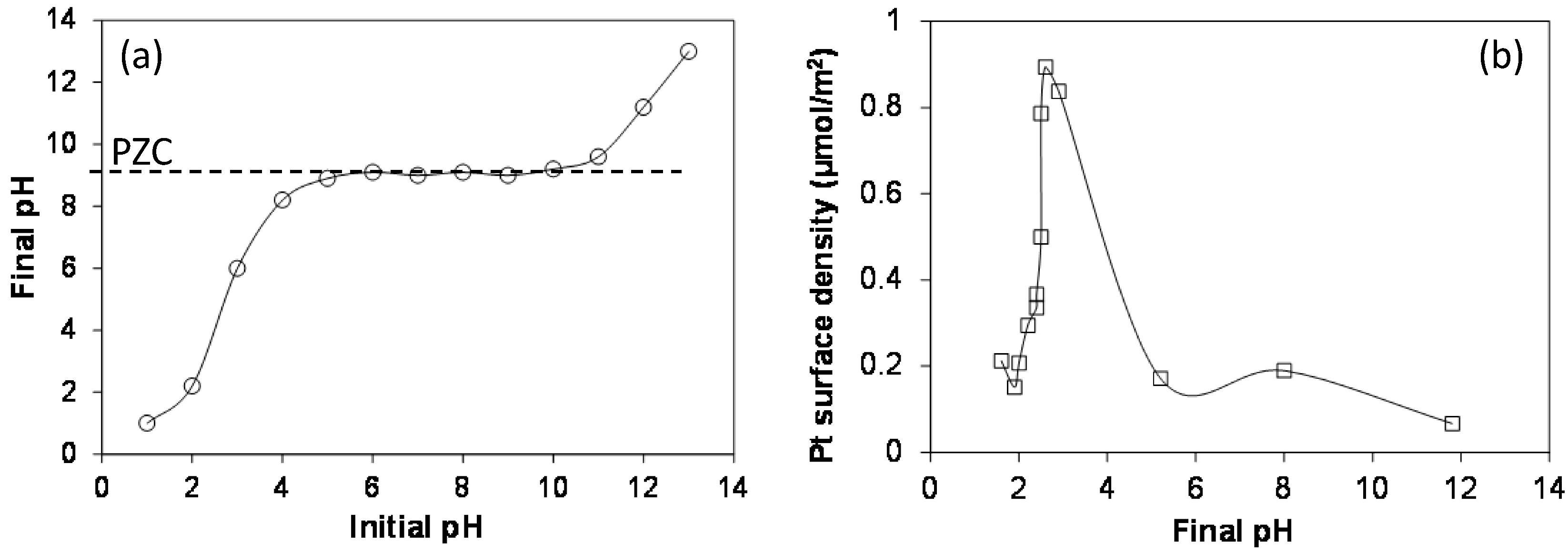
2.4. Strong Electrostatic Adsorption
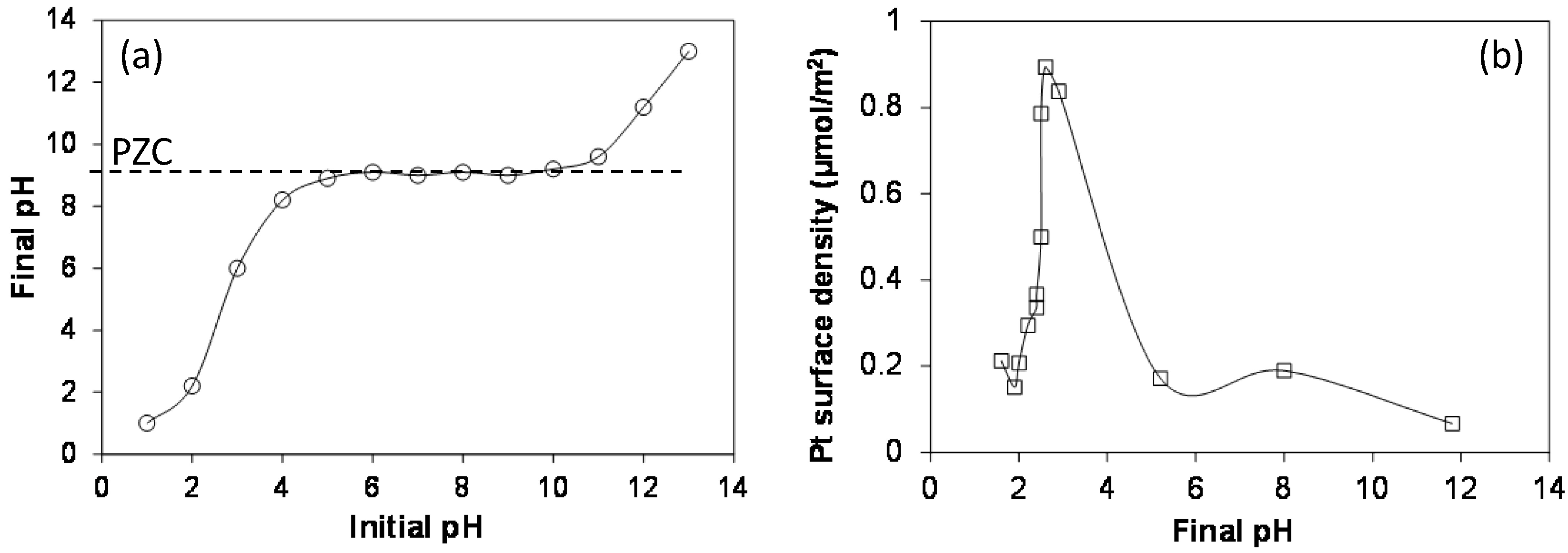
2.5. Multiple Strong Electrostatic Adsorption
2.6. Charge-Enhanced Dry Impregnation
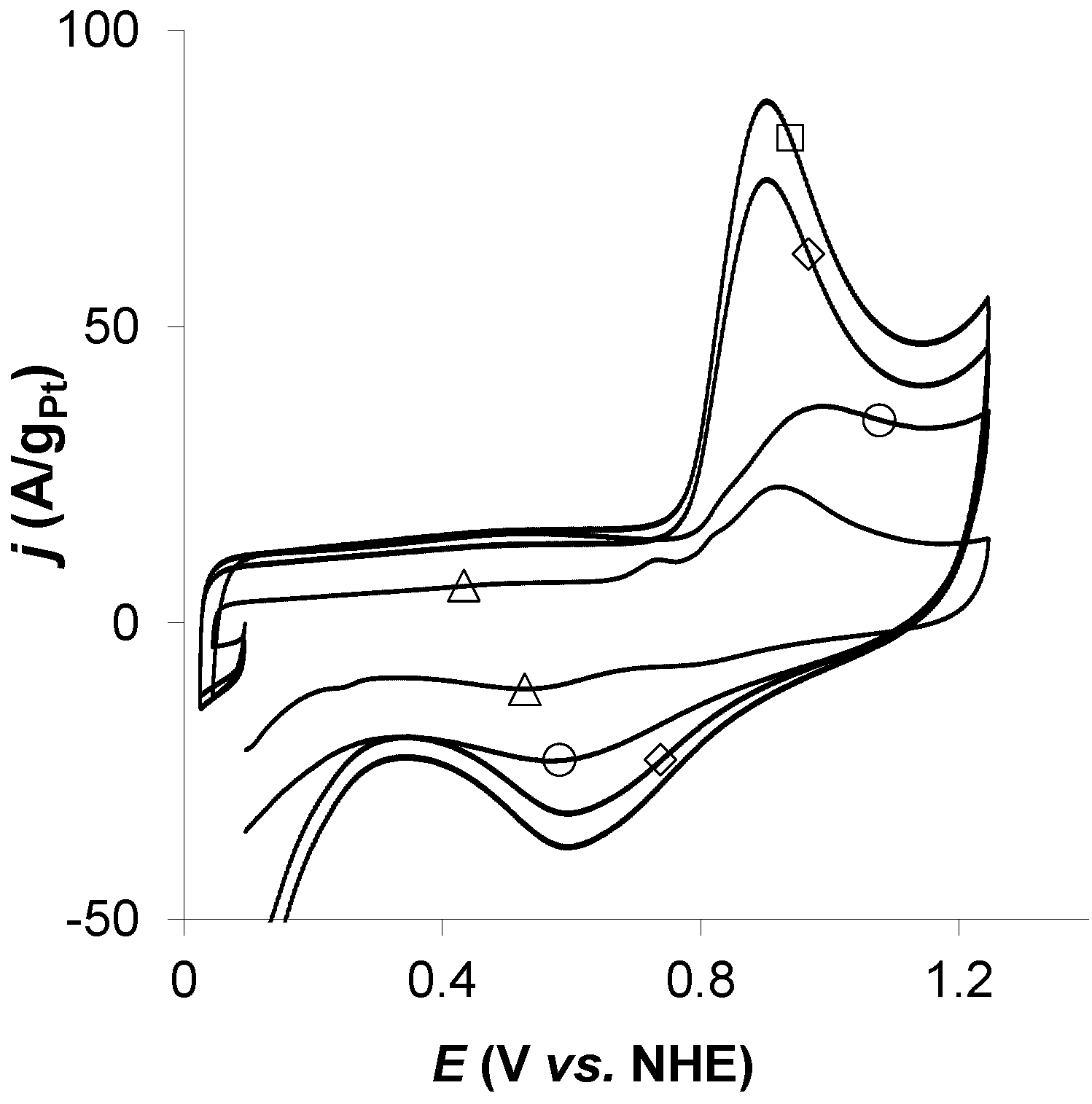
2.7. Characterization
3. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Impregnation cycles | Tr,final | tr, final | Ptth | ICP-AES | TEM | XRD | CO chemisorption | CO stripping | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PtICP | dTEM | σ | ds | dv | dXRD | ns,m | DPt | dCO | SCO-chem | SCO-strip | |||||
| (-) | (K) | (h) | (wt.%) | (wt.%) | (nm) | (nm) | (nm) | (nm) | (nm) | (mmol/gPt) | (-) | (nm) | (m2/gPt) | (m2/gPt) | |
| WI | 1 | 623 | 3 | 1.0 | 1.9 | 1.6 | 0.5 | 1.9 | 2.0 | 1.8 | 1.92 | 0.60 a | 1.9 | 153 | - b |
| WI-R | 1 | 623 | 3 | 35.0 | 31.0 | 4.1–17.7 c | - d | - d | - d | 22 e | 0.82 | 0.16 f | 6.9 | 41 | 32 |
| SEA | 1 | 473 | 1 | 8.0 | 7.5 | 2.0 | 0.7 | 2.5 | 2.7 | 2.6 | 1.15 | 0.36 a | 3.1 | 92 | 34 |
| M-SEA-473 | 2 | 473 | 1 | 16.0 | 15.0 | 1.9 | 0.8 | 2.5 | 2.8 | 2.6 | 1.10 | 0.34 a | 3.2 | 89 | 37 |
| M-SEA-723 | 2 | 723 | 5 | 16.0 | 15.0 | 2.0 | 0.7 | 2.5 | 2.7 | 2.7 | 1.53 | 0.48 a | 2.3 | 122 | 127 |
| M-SEA-r | 2 | 723 | 5 | 16.0 | 14.7 | 2.2 | 0.7 | 2.7 | 3.0 | 2.3 | - g | - g | - g | - g | 93 |
| CEDI-523 | 1 | 523 | 1 | 10.0 | 10.0 | 2.0 | 0.4 | 1.6 | 1.8 | 1.9 | - g | - g | - g | - g | 77 |
| CEDI-723 | 1 | 723 | 1 | 10.0 | 10.0 | 2.0 | 0.4 | 1.7 | 1.8 | 2.0 | - g | - g | - g | - g | 95 |

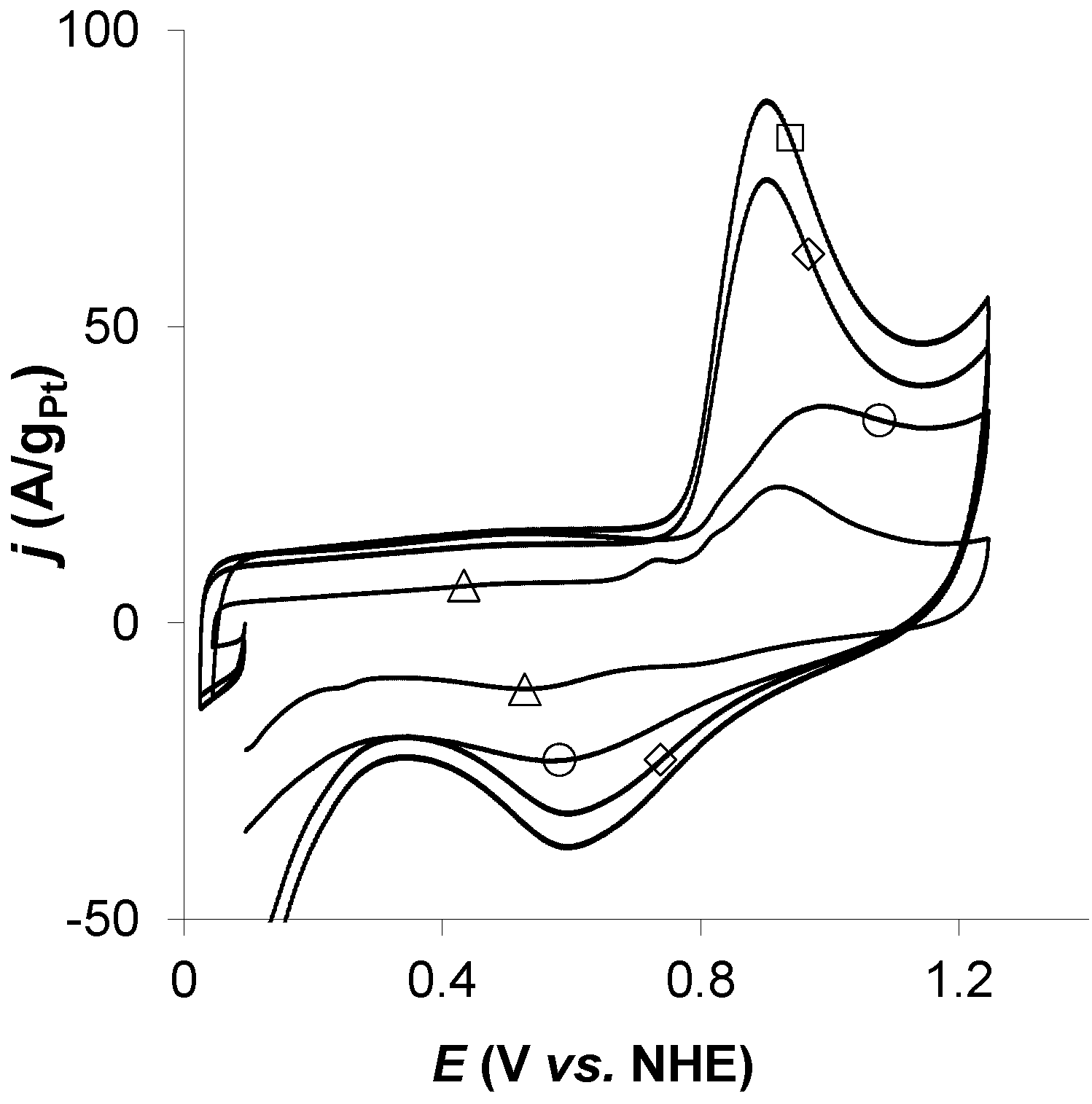
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sopian, K.; Daud, W.R.W. Challenges and future developments in proton exchange membrane fuel cells. Renew. Energy 2006, 31, 719–727. [Google Scholar] [CrossRef]
- Kinoshita, K. Particle size effects for oxygen reduction on highly dispersed platinum in acid electrolytes. J. Electrochem. Soc. 1990, 137, 845–848. [Google Scholar] [CrossRef]
- Antolini, E. Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B 2009, 88, 1–24. [Google Scholar] [CrossRef]
- Job, N.; Théry, A.; Pirard, R.; Marien, J.; Kocon, L.; Rouzaud, J.-N.; Béguin, F.; Pirard, J.-P. Carbon aerogels, xerogels and cryogels: Influence of the drying method on the textural properties of porous carbon materials. Carbon 2005, 43, 2481–2494. [Google Scholar] [CrossRef]
- Job, N.; Heinrichs, B.; Lambert, S.; Pirard, J.-P.; Colomer, J.-F.; Vertruyen, B.; Marien, J. Carbon xerogels as catalyst supports: Study of mass transfer. AICHE J. 2006, 52, 2663–2676. [Google Scholar] [CrossRef]
- Job, N.; Marie, J.; Lambert, S.; Berthon-Fabry, S.; Achard, P. Carbon xerogels as catalyst supports for PEM fuel cell cathode. Energy Convers. Manag. 2008, 49, 2461–2470. [Google Scholar]
- Liu, B.; Creager, S. Carbon xerogels as Pt catalyst supports for polymer electrolyte membrane fuel-cell applications. J. Power Source 2010, 195, 1812–1820. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. Synthesis and functionalization of carbon xerogels to be used as supports for fuel cell catalysts. J. Energy Chem. 2013, 22, 195–201. [Google Scholar] [CrossRef]
- Job, N.; Pereira, M.F.R.; Lambert, S.; Cabiac, A.; Delahay, G.; Colomer, J.-F.; Marien, J.; Figueiredo, J.L.; Pirard, J.-P. Highly dispersed platinum catalysts prepared by impregnation of texture-tailored carbon xerogels. J. Catal. 2006, 240, 160–171. [Google Scholar] [CrossRef]
- Brunelle, J.P. Preparation of catalysts by metallic complex adsorption on mineral oxides. Pure Appl. Chem. 1978, 50, 1211–1229. [Google Scholar] [CrossRef]
- Regalbuto, J.R. Strong Electrostatic adsorption of metals onto catalyst support. In Catalyst Preparation: Science and Engineering; Regalbuto, J.R., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2007; pp. 297–318. [Google Scholar]
- Regalbuto, J.R. Electrostatic adsorption. In Synthesis of Solid Catalysts; de Jong, K.P., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 33–58. [Google Scholar]
- Schreier, M.; Regalbuto, J.R. A fundamental study of Pt tetraammine impregnation of silica: 1. The electrostatic nature of platinum adsorption. J. Catal. 2004, 225, 190–202. [Google Scholar] [CrossRef]
- Miller, J.T.; Schreier, M.; Kropf, A.J.; Regalbuto, J.R. A fundamental study of platinum tetraammine impregnation of silica: 2. The effect of method of preparation, loading, and calcination temperature on (reduced) particle size. J. Catal. 2004, 225, 203–212. [Google Scholar] [CrossRef]
- Spieker, W.A.; Liu, J.; Hao, X.; Miller, J.T.; Kropf, A.J.; Regalbuto, J.R. An EXAFS study of the coordination chemistry of hydrogen hexachloroplatinate (IV): 2. Speciation of complexes adsorbed onto alumina. Appl. Catal. A 2003, 243, 53–66. [Google Scholar] [CrossRef]
- Hao, X.; Quach, L.; Korah, J.; Spieker, W.A.; Regalbuto, J.R. The control of platinum impregnation by PZC alteration of oxides and carbon. J. Mol. Catal. A 2004, 219, 97–107. [Google Scholar] [CrossRef]
- Hao, X.; Barnes, S.; Regalbuto, J.R. A fundamental study of Pt impregnation of carbon: Adsorption equilibrium and particle synthesis. J. Catal. 2011, 279, 48–65. [Google Scholar] [CrossRef]
- Feltes, T.E.; Smit, E.D.; D’Souza, L.; Meyer, R.J.; Weckhuysen, B.M.; Regalbuto, J.R. Selective adsorption of manganese onto cobalt for optimized Mn/Co/TiO2 Fischer-Tropsch catalysts. J. Catal. 2010, 270, 95–102. [Google Scholar] [CrossRef]
- D’Souza, L.; Regalbuto, J.R. Strong electrostatic adsorption for the preparation of Pt/Co/C and Pd/Co/C bimetallic electrocatalysts. Stud. Surf. Sci. Catal. 2010, 175, 715–718. [Google Scholar]
- Lambert, S.; Job, N.; D’Souza, L.; Pereira, M.F.R.; Pirard, R.; Figueiredo, J.L.; Heinrichs, B.; Pirard, J.-P.; Regalbuto, J.R. Synthesis of very highly dispersed platinum catalysts supported on carbon xerogels by the strong electrostatic adsorption method. J. Catal. 2009, 261, 23–33. [Google Scholar] [CrossRef]
- Job, N.; Lambert, S.; Chatenet, M.; Gommes, C.J.; Maillard, F.; Berthon-Fabry, S.; Regalbuto, J.R.; Pirard, J.-P. Preparation of highly loaded Pt/carbon xerogel catalysts for PEM fuel cells by the Strong Electrostatic Adsorption method. Catal. Today 2010, 150, 119–127. [Google Scholar] [CrossRef]
- Zubiaur, A.; Chatenet, M.; Maillard, F.; Lambert, S.D.; Pirard, J.-P.; Job, N. Using the Multiple SEA method to synthesize Pt/Carbon xerogel electrocatalysts for PEMFC applications. Fuel Cells 2014, 14, 343–349. [Google Scholar] [CrossRef]
- Cao, C.; Yuang, G.; Dubau, L.; Maillard, F.; Lambert, S.D.; Pirard, J.-P.; Job, N. Highly dispersed Pt/C catalysts prepared by the Charge Enhanced Dry Impregnation method. Appl. Catal. B 2014, 150–151, 101–106. [Google Scholar]
- Zhu, X.; Cho, H.-R.; Pasupong, M.; Regalbuto, J.R. Charge-enhanced dry impregnation: A simple way to improve the preparation of supported metal catalysts. ACS Catal. 2013, 3, 625–630. [Google Scholar] [CrossRef]
- Job, N.; Pirard, R.; Alié, C.; Pirard, J.-P. Non intrusive mercury porosimetry: Pyrolysis of resorcinol-formaldehyde xerogels. Part. Part. Syst. Charact. 2006, 23, 72–81. [Google Scholar] [CrossRef]
- Bergeret, G.; Gallezot, P. Particle size and dispersion measurements. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 1997; pp. 439–464. [Google Scholar]
- Trasatti, S. Real surface area measurements in electrochemistry. J. Electroanal. Chem. 1992, 327, 353–376. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Rodríguez-Ramos, I.; Moreno-Castilla, C.; Guerrero-Ruiz, A.; López-González, J.D. Platinum catalysts supported on activated carbons: I. Preparation and characterization. J. Catal. 1986, 99, 171–183. [Google Scholar] [CrossRef]
- Alegre, C.; Gálvez, M.E.; Moliner, R.; Baglio, V.; Aricò, A.S.; Lázaro, M.J. Towards an optimal synthesis route for the preparation of highly mesoporous carbon xerogel-supported Pt catalysts for the oxygen reduction reaction. Appl. Catal. B 2014, 147, 947–957. [Google Scholar] [CrossRef]
- Job, N.; Chatenet, M.; Berthon-Fabry, S.; Hermans, S.; Maillard, F. Efficient Pt/carbon electrocatalysts for Proton Exchange Membrane fuel cells: Avoid chloride-based Pt salts! J. Power Sources 2013, 240, 294–305. [Google Scholar] [CrossRef]
- Holscher, A.A.; Sachtler, W.M.H. Chemisorption and surface corrosion in the tungsten + carbon monoxide system, as studied by field emission and field ion microscopy. Discuss. Faraday Soc. 1966, 41, 29–42. [Google Scholar] [CrossRef]
- Maillard, F.; Eikerling, M.; Cherstiouk, O.V.; Schreier, S.; Savinova, E.; Stimming, U. Size effects on reactivity of Pt nanoparticles in CO monolayer oxidation: The role of surface mobility. Faraday Discuss. 2004, 125, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Maillard, F.; Schreier, S.; Hanzlik, M.; Savinova, E.R.; Weinkauf, S.; Stimming, U. Influence of particle agglomeration on the catalytic activity of carbon-supported Pt nanoparticles in CO monolayer oxidation. Phys. Chem. Chem. Phys. 2005, 7, 385–393. [Google Scholar] [CrossRef]
- Maillard, F.; Savinova, E.R.; Stimming, U. CO monolayer oxidation on Pt nanoparticles: Further insights into the particle size effects. J. Electroanal. Chem. 2007, 599, 221–232. [Google Scholar] [CrossRef]
- Maillard, F.; Pronkin, S.; Savinova, E.R. Influence of size on the electrocatalytic activities of supported metal nanoparticles in fuel cells related reactions. In Handbook of Fuel Cells—Electrocatalysis, Materials, Diagnostics and Durability; Vielstich, W., Gasteiger, H.A., Yokokawa, H., Eds.; John Wiley & Sons: New York, NY, USA, 2009; Volume 5, pp. 91–111. [Google Scholar]
- Andreaus, B.; Maillard, F.; Kocylo, J.; Savinova, E.R.; Eikerling, M. Kinetic modeling of COad monolayer oxidation on carbon-supported platinum nanoparticles. J. Phys. Chem. B 2006, 110, 21028–21040. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Job, N.; Lambert, S.D.; Zubiaur, A.; Cao, C.; Pirard, J.-P. Design of Pt/Carbon Xerogel Catalysts for PEM Fuel Cells. Catalysts 2015, 5, 40-57. https://doi.org/10.3390/catal5010040
Job N, Lambert SD, Zubiaur A, Cao C, Pirard J-P. Design of Pt/Carbon Xerogel Catalysts for PEM Fuel Cells. Catalysts. 2015; 5(1):40-57. https://doi.org/10.3390/catal5010040
Chicago/Turabian StyleJob, Nathalie, Stéphanie D. Lambert, Anthony Zubiaur, Chongjiang Cao, and Jean-Paul Pirard. 2015. "Design of Pt/Carbon Xerogel Catalysts for PEM Fuel Cells" Catalysts 5, no. 1: 40-57. https://doi.org/10.3390/catal5010040