
Oxygen Reduction Reaction Activity and Durability of Pt Catalysts Supported on Titanium Carbide


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Characterization of Pt/TiC Catalysts



2.2. Electrochemical Characterization of Pt/TiC Catalysts




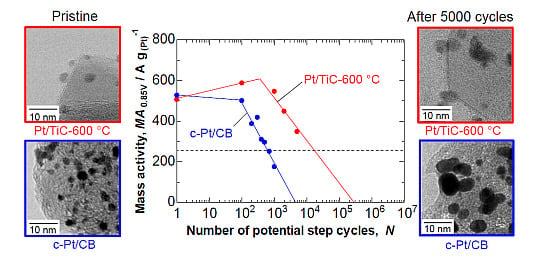
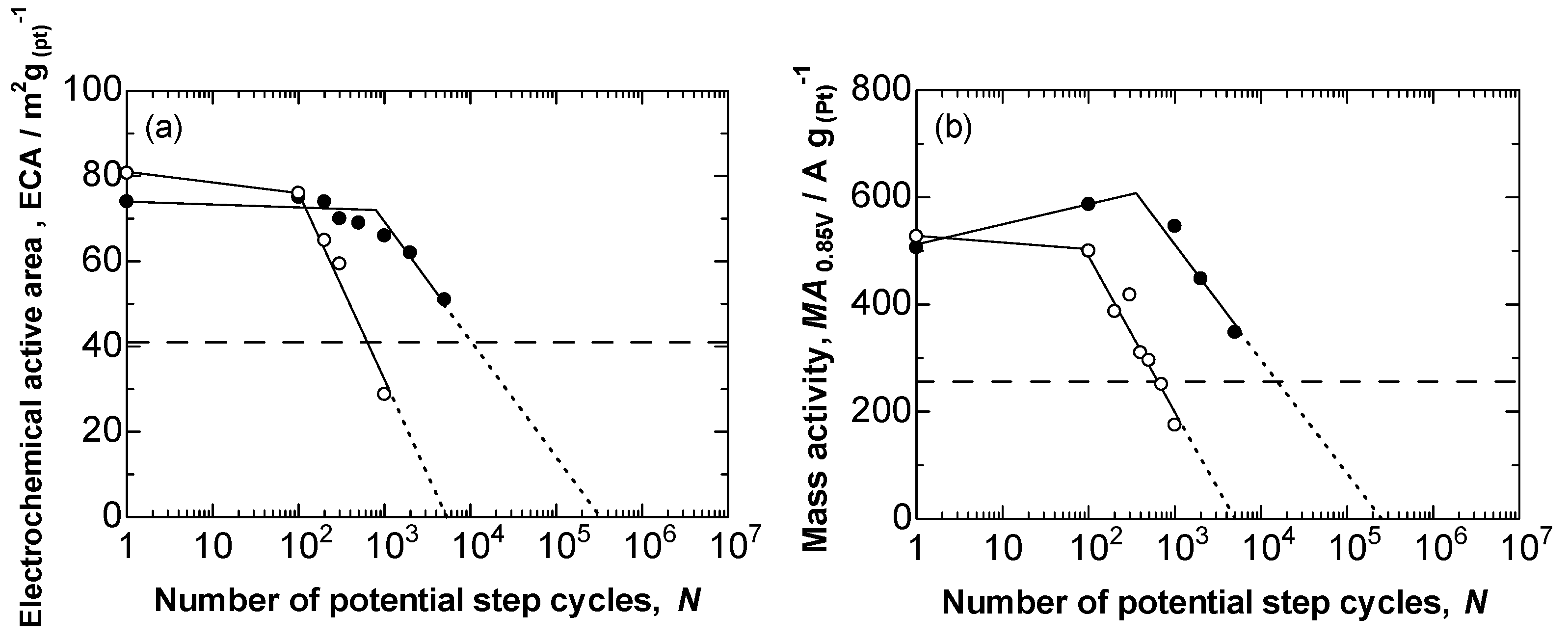
2.3. Durability of Pt/TiC-600 °C in the Potential Step Cycle Test


3. Experimental Section
3.1. Preparation of Pt/TiC Catalyst
3.2. Characterization of Pt/TiC
3.3. Electrochemical Measurements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilson, M.S.; Garzon, F.H.; Sickafus, K.E.; Gottesfeld, S. Surface Area Loss of Supported Platinum in Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 1993, 140, 2872–2877. [Google Scholar] [CrossRef]
- Willsaw, J.; Heitbaum, J. The Influence of Pt-activation on the Corrosion of Carbon in Gas Diffusion Electrodes—A DEMS Study. J. Electroanal. Chem. 1984, 161, 93–101. [Google Scholar] [CrossRef]
- Yu, X.; Ye, S. Recent Advances in Activity and Durability Enhancement of Pt/C Catalytic Cathode in PEMFC: Part II: Degradation Mechanism and Durability Enhancement of Carbon Supported Platinum Catalyst. J. Power Sources 2007, 172, 145–154. [Google Scholar] [CrossRef]
- Tang, H.; Qi, Z.G.; Ramani, M.; Elter, J.F. PEM Fuel Cell Cathode Carbon Corrosion Due to the Formation of Air/Fuel Boundary at the Anode. J. Power Sources 2006, 158, 1306–1312. [Google Scholar] [CrossRef]
- Meyers, J.P.; Darling, R.M. Model of Carbon Corrosion in PEM Fuel Cells. J. Electrochem. Soc. 2006, 153, A1432–A1442. [Google Scholar] [CrossRef]
- Reiser, C.A.; Bregoli, L.; Patterson, T.W.; Yi, J.S.; Yang, D.; Perry, M.L.; Jarvi, T.D. A Reverse-Current Decay Mechanism for Fuel Cells. Electrochem. Solid-State Lett. 2005, 8, A273–A276. [Google Scholar] [CrossRef]
- Borup, R.L.; Davey, J.R.; Garzon, H.F.; Wood, D.J.; Inbody, M.A. PEM Fuel Cell Electrocatalyst Durability Measurements. J. Power Sources 2006, 163, 76–81. [Google Scholar] [CrossRef]
- Roen, L.M.; Paik, C.H.; Jarvi, T.D. Electrocatalytic Corrosion of Carbon Support in PEMFC Cathodes. Electrochem. Solid-State Lett. 2004, 7, A19–A22. [Google Scholar] [CrossRef]
- Passalacqua, E.; Antonucci, P.L.; Vivadi, M.; Patti, A.; Antonucci, V.; Giordano, N.; Kinoshita, K. The Influence of Pt on the Electrooxidation Behaviour of Carbon in Phosphoric Acid. Electrochim. Acta 1992, 37, 2725–2730. [Google Scholar] [CrossRef]
- Ferreira, P.J.; Lao, G.J.; Shao-Horn, Y.; Morgan, D.; Makharia, R.; Kocha, S.; Gasteiger, H.A. Instability of Pt/C Electrocatalysts in Proton Exchange Membrane Fuel Cells A Mechanistic Investigation. J. Electrochem. Soc. 2005, 152, A2256–A2271. [Google Scholar] [CrossRef]
- Darling, R.M.; Meyers, J.P. Kinetic Model of Platinum Dissolution in PEMFCs. J. Electrochem. Soc. 2003, 150, A1523–A1527. [Google Scholar] [CrossRef]
- Xie, J.; Wood, D.L.; Wayne, D.M.; Zawodinski, T.A.; Atanassov, P.; Borup, R.L. Durability of PEFCs at High Humidity Conditions. J. Electrochem. Soc. 2005, 152, A104–A113. [Google Scholar] [CrossRef]
- Xie, J.; Wood, D.L.; More, K.L.; Atanassov, P.; Borup, R.L. Microstructural Changes of Membrane Electrode Assemblies during PEFC Durability Testing at High Humidity Conditions. J. Electrochem. Soc. 2005, 152, A1011–A1020. [Google Scholar] [CrossRef]
- Stevens, D.A.; Hicks, M.T.; Haugen, G.M.; Dahn, J.R. Ex situ and in situ Stability Studies of PEMFC Catalysts Effect of Carbon Type and Humidification on Degradation of the Carbon. J. Electrochem. Soc. 2005, 152, A2309–A2315. [Google Scholar] [CrossRef]
- Patterson, T.W.; Darling, R.M. Damage to the Cathode Catalyst of a PEM Fuel Cell Caused by Localized Fuel Starvation. Electrochem. Solid-State Lett. 2006, 9, A183–A185. [Google Scholar] [CrossRef]
- Yoda, T.; Uchida, H.; Watanabe, M. Effects of Operating Potential and Temperature on Degradation of Electrocatalyst Layer for PEFCs. Electrochim. Acta 2007, 52, 5997–6006. [Google Scholar] [CrossRef]
- Hara, M.; Lee, M.; Liu, C.-Y.; Chen, B.-H.; Yamashita, Y.; Uchida, M.; Uchida, H.; Watanabe, M. Electrochemical and Raman Spectroscopic Evaluation of Pt/Graphitized Carbon Black Catalyst Durability for the Start/Stop Operating Condition of Polymer Electrolyte Fuel Cells. Electrochim. Acta 2012, 70, 171–181. [Google Scholar] [CrossRef]
- Masao, A.; Noda, S.; Takasaki, F.; Ito, K.; Sasaki, K. Carbon-Free Pt Electrocatalysts Supported on SnO2 for Polymer Electrolyte Fuel Cells. Electrochem. Solid-State Lett. 2009, 12, B119–B122. [Google Scholar] [CrossRef]
- Takasaki, F.; Matsuie, S.; Takabatake, Y.; Noda, Z.; Hayashi, A.; Shiratori, Y.; Ito, K.; Sasaki, K. Carbon-Free Pt Electrocatalysts Supported on SnO2 for Polymer Electrolyte Fuel Cells: Electrocatalytic Activity and Durability. J. Electrochem. Soc. 2011, 158, B1270–B1275. [Google Scholar] [CrossRef]
- Mentus, S.V. Oxygen Reduction on Anodically Formed Titanium Dioxide. Electrochim. Acta 2007, 50, 27–32. [Google Scholar] [CrossRef]
- Ioroi, T.; Akita, T.; Yamazaki, S.; Siroma, Z.; Fujiwara, N.; Yasuda, K. Corrosion-Resistant PEMFC Cathode Catalysts Based on a Magnéli-Phase Titanium Oxide Support Synthesized by Pulsed UV Laser Irradiation. J. Electrochem. Soc. 2011, 158, C329–C334. [Google Scholar] [CrossRef]
- Huang, S.Y.; Ganesan, P.; Park, S.; Popov, B.N. Development of a Titanium Dioxide-Supported Platinum Catalyst with Ultrahigh Stability for Polymer Electrolyte Membrane Fuel Cell Applications. J. Am. Chem. Soc. 2009, 131, 13898–13899. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Ganesan, P.; Park, S.; Popov, B.N. Titania Supported Platinum Catalyst with High Electrocatalytic Activity and Stability for Polymer Electrolyte Membrane Fuel Cell. Appl. Catal. B 2011, 102, 71–77. [Google Scholar] [CrossRef]
- Ioroi, T.; Senoh, H.; Yamazaki, S.; Siroma, Z.; Fujiwara, N.; Yasuda, K. Stability of Corrosion-Resistant Magnéli-Phase Ti4O7-Supported PEMFC Catalysts at High Potentials. J. Electrochem. Soc. 2008, 155, B321–B326. [Google Scholar] [CrossRef]
- Ioroi, T.; Siroma, Z.; Fujiwara, N.; Yamazaki, S.; Yasuda, K. Sub-Stoichiometric Titanium Oxide-Supported Platinum Electrocatalyst for Polymer Electrolyte Fuel Cells. Electrochem. Commun. 2005, 7, 183–188. [Google Scholar] [CrossRef]
- Kakinuma, K.; Wakasugi, Y.; Uchida, M.; Kamino, T.; Uchida, H.; Deki, S.; Watanabe, M. Preparation of Titanium Nitride-Supported Platinum Catalysts with Well Controlled Morphology and Their Properties Televant to Polymer Electrolyte Fuel Cells. Electrochim. Acta 2012, 77, 279–284. [Google Scholar] [CrossRef]
- Kakinuma, K.; Uchida, M.; Kamino, T.; Uchida, H.; Watanabe, M. Synthesis and Electrochemical Characterization of Pt Catalyst Supported on Sn0.96Sb0.04O2−δ with a Network Structure. Electrochim. Acta 2011, 56, 2881–2887. [Google Scholar] [CrossRef]
- Kakinuma, K.; Chino, Y.; Senoo, Y.; Uchida, M.; Kamino, T.; Uchida, H.; Deki, S.; Watanabe, M. Characterization of Pt catalysts on Nb-Doped and Sb-Doped SnO2−δ Support Materials with Aggregated Structure by Rotating Disk Electrode and Fuel Cell Measurements. Electrochim. Acta 2013, 110, 316–324. [Google Scholar] [CrossRef]
- Senoo, Y.; Kakinuma, K.; Uchida, M.; Uchida, H.; Deki, S.; Watanabe, M. Improvements in Electrical and Electrochemical Properties of Nb-Doped SnO2 Supports for Fuel Cell Cathodes Due to Aggregation and Pt Loading. RSC Adv. 2014, 4, 32180–32188. [Google Scholar] [CrossRef]
- Senoo, Y.; Taniguchi, K.; Kakinuma, K.; Uchida, M.; Uchida, H.; Deki, S.; Watanabe, M. Cathodic Performance and High Potential Durability of Ta–SnO2−δ-Supported Pt Catalysts for PEFC Cathodes. Electrochem. Commun. 2015, 51, 37–40. [Google Scholar] [CrossRef]
- Oyama, S.T. The Chemistry of Transition Metal Carbides and Nitrides; Blackie Academic and Professional: London, UK, 1996; Chapter 1; pp. 9–14. [Google Scholar]
- Nakao, S.; Yamada, N.; Hitosugi, T.; Hirose, Y.; Shimada, T.; Hasegawa, T. High Mobility Exceeding 80 cm2 V−1 s−1 in Polycrystalline Ta-Doped SnO2 Thin Films on Glass Using Anatase TiO2 Seed Layers. Appl. Phys. Express 2010, 3, 031102. [Google Scholar] [CrossRef]
- La Conti, A.B.; Griffith, A.E.; Cropley, C.C.; Kosek, J.A. Titanium Carbide Bipolar Plate for Electrochemical Devices. U.S. Patent 6,083,641, 4 July 2000. [Google Scholar]
- Jalan, V.; Frost, D.G. Fuel Cell Electrocatalyst Support Comprising an Ultra-Fine Chainy-Structured Titanium Carbide. U.S. Patent 4,795,684, 3 January 1989. [Google Scholar]
- Ma, L.; Sui, S.; Zhai, Y. Preparation and Characterization of Ir/TiC Catalyst for Oxygen Evolution. J. Power Sources 2008, 177, 470–477. [Google Scholar] [CrossRef]
- Watanabe, M.; Uchida, M.; Motoo, S. Application of the Gas Diffusion Electrode to a Backward Feed and Exhaust (BFE) Type Methanol Anode. J. Electroanal. Chem. 1986, 199, 311–322. [Google Scholar] [CrossRef]
- Watanabe, M.; Uchida, M.; Motoo, S. Preparation of Highly Dispersed Pt + Ru Clusters and the Activity for the Electro-Oxidation of Methanol. J. Electroanal. Chem. 1987, 229, 395–406. [Google Scholar] [CrossRef]
- Brambilla, A.; Calloni, A.; Berti, G.; Bussetti, G.; Duò, L.; Ciccacci, F. Growth and Interface Reactivity of Titanium Oxide Thin Films on Fe(001). J. Phys. Chem. C 2013, 117, 9229–9236. [Google Scholar] [CrossRef]
- Yano, H.; Akiyama, T.; Bele, P.; Uchida, H.; Watanabe, M. Durability of Pt/Graphitized Carbon Catalysts for the Oxygen Reduction Reaction Prepared by the Nanocapsule Method. Phys. Chem. Chem. Phys. 2010, 12, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Park, Y.-C.; Kakinuma, K.; Yano, H.; Tryk, A.D.; Kamino, T.; Uchida, H.; Watanabe, M. Effect of the State of Distribution of Supported Pt Nanoparticles on Effective Pt Utilization in Polymer Electrolyte Fuel Cells. Phys. Chem. Chem. Phys. 2013, 5, 11236–11247. [Google Scholar] [CrossRef] [PubMed]
- Markovic, N.; Adzic, R.; Cahan, B.; Yeager, E. Structural Effects in Electrocatalysis: Oxygen Reduction on Platinum Low Index Single-Crystal Surfaces in Perchloric Acid Solutions. J. Electroanal. Chem. 1994, 377, 249–259. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity Benchmarks and Requirements for Pt, Pt-alloy, and Non-Pt Oxygen Reduction Catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Lee, M.; Uchida, M.; Tryk, D.A.; Uchida, H.; Watanabe, M. The Effectiveness of Platinum/Carbon Electrocatalysts: Dependence on Catalyst Layer Thickness and Pt Alloy Catalytic Effects. Electrochim. Acta 2011, 56, 4783–4790. [Google Scholar] [CrossRef]
- Okaya, K.; Yano, H.; Kakinuma, K.; Watanabe, M.; Uchida, H. Temperature Dependence of Oxygen Reduction Reaction Activity at Stabilized Pt Skin-PtCo Alloy/Graphitized Carbon Black Catalysts Prepared by a Modified Nanocapsule Method. ACS Appl. Mater. Interfaces 2012, 4, 6982–6991. [Google Scholar] [CrossRef] [PubMed]
- Paulus, U.A.; Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Oxygen Reduction on a High-surface Area Pt/Vulcan Carbon Catalyst: A Thin-film Rotating Ring-Disk Electrode Study. J. Electroanal. Chem. 2001, 495, 134–145. [Google Scholar] [CrossRef]
- Ignaszak, A.; Songa, C.; Zhu, W.; Zhang, J.; Bauer, A.; Baker, R.; Neburchilov, V.; Ye, S.; Campbell, S. Titanium Carbide and Its Core-Shelled Derivative TiC@TiO2 as Catalyst Supports for Proton Exchange Membrane Fuel Cells. Electrochim. Acta 2012, 69, 397–405. [Google Scholar] [CrossRef]
- Higuchi, E.; Uchida, H.; Watanabe, M. Effect of Loading Level in Platinum-Dispersed Carbon Black Electrocatalysts on Oxygen Reduction Activity Evaluated by Rotating Disk Electrode. J. Electroanal. Chem. 2005, 583, 69–76. [Google Scholar] [CrossRef]
- Iiyama, A.; Shinohara, K.; Iguchi, S.; Daimaru, A. Handbook of Fuel Cells: Fundamentals, Technology and Applications; Vielstich, W., Lamm, A., Gasteiger, H.A., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2009; Volume 6. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiwata, M.; Kakinuma, K.; Wakisaka, M.; Uchida, M.; Deki, S.; Watanabe, M.; Uchida, H. Oxygen Reduction Reaction Activity and Durability of Pt Catalysts Supported on Titanium Carbide. Catalysts 2015, 5, 966-980. https://doi.org/10.3390/catal5020966
Chiwata M, Kakinuma K, Wakisaka M, Uchida M, Deki S, Watanabe M, Uchida H. Oxygen Reduction Reaction Activity and Durability of Pt Catalysts Supported on Titanium Carbide. Catalysts. 2015; 5(2):966-980. https://doi.org/10.3390/catal5020966
Chicago/Turabian StyleChiwata, Morio, Katsuyoshi Kakinuma, Mitsuru Wakisaka, Makoto Uchida, Shigehito Deki, Masahiro Watanabe, and Hiroyuki Uchida. 2015. "Oxygen Reduction Reaction Activity and Durability of Pt Catalysts Supported on Titanium Carbide" Catalysts 5, no. 2: 966-980. https://doi.org/10.3390/catal5020966