
Advances in the Knowledge of N-Heterocyclic Carbenes Properties. The Backing of the Electrochemical Investigation




Abstract
:1. Introduction
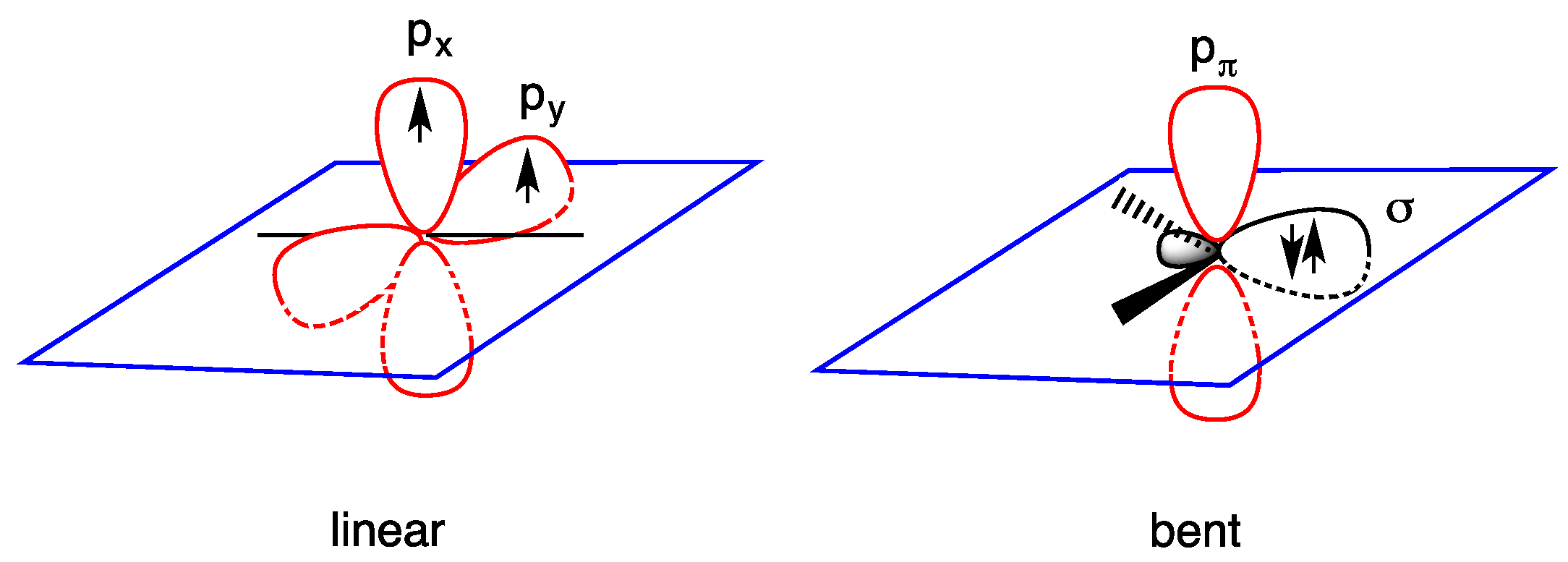
1.1. Carbenes
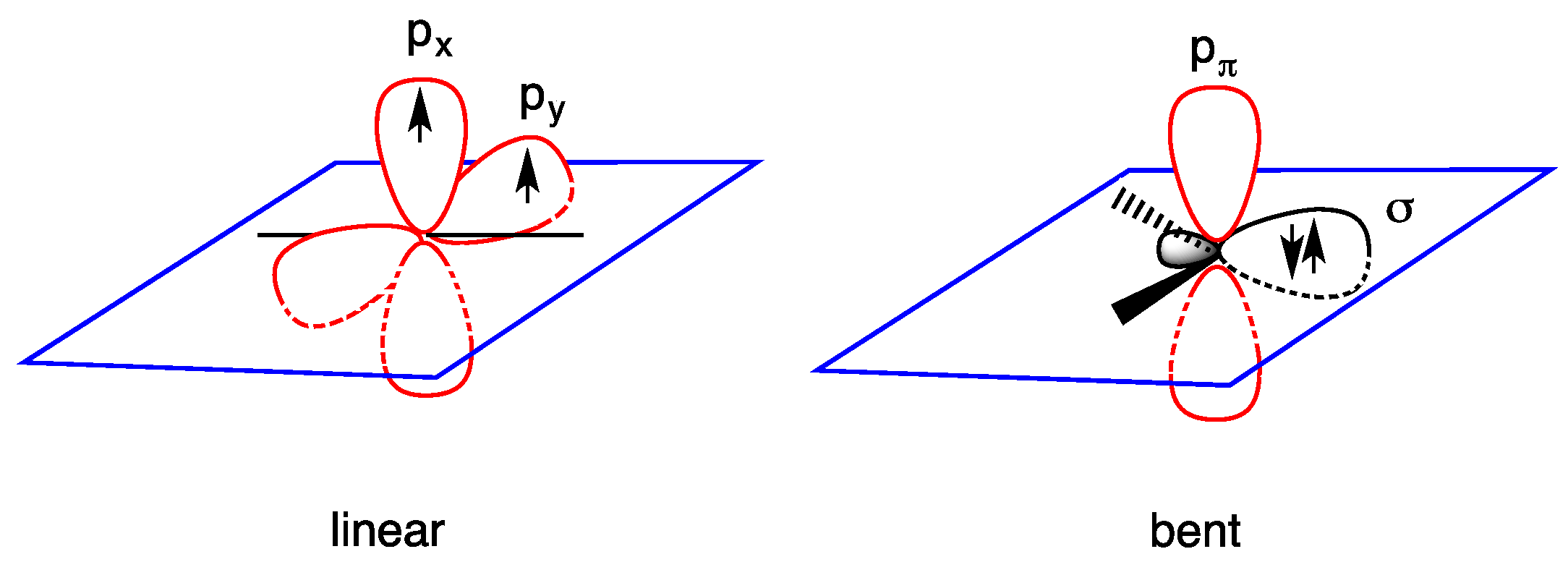
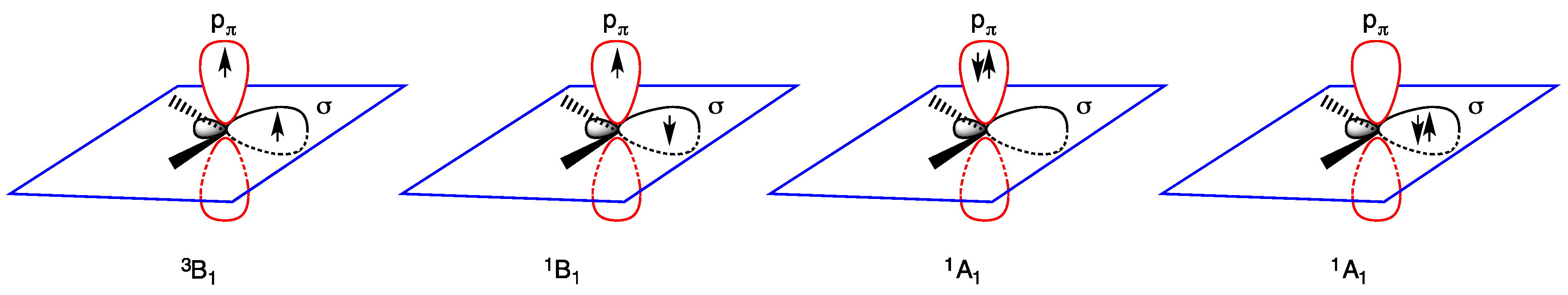
- triplet ground state, the two nonbonding electrons occupy the two empty orbitals δ and pπ with parallel or antiparallel spin orientation: δ1 pπ1 (3B1 or 1B1 state); and
- singlet ground state, the two nonbonding electrons occupy only the empty δ orbital as a lone pair, being empty the pπ orbital: δ2 pπ0 (1A1 state). The δ2 pπ0 (1A1 state) is generally regarded as more stable than the δ0 pπ2 (1A1 state) in which the lone pair occupies the pπ orbital (Figure 2).
1.2. N-Heterocyclic Carbenes (NHCs)
1.3. Umpolung and the Peculiar Chemistry of NHCs
- the structure of the cycle and the presence of other hetero-atoms (S, O) in addition to nitrogen atom;
- the nature of the substituents at the hetero and carbon atoms;
- the presence, in the reaction mixture, of the parent azolium cation and of the counter ion; and
- the presence and the nature of the solvent.
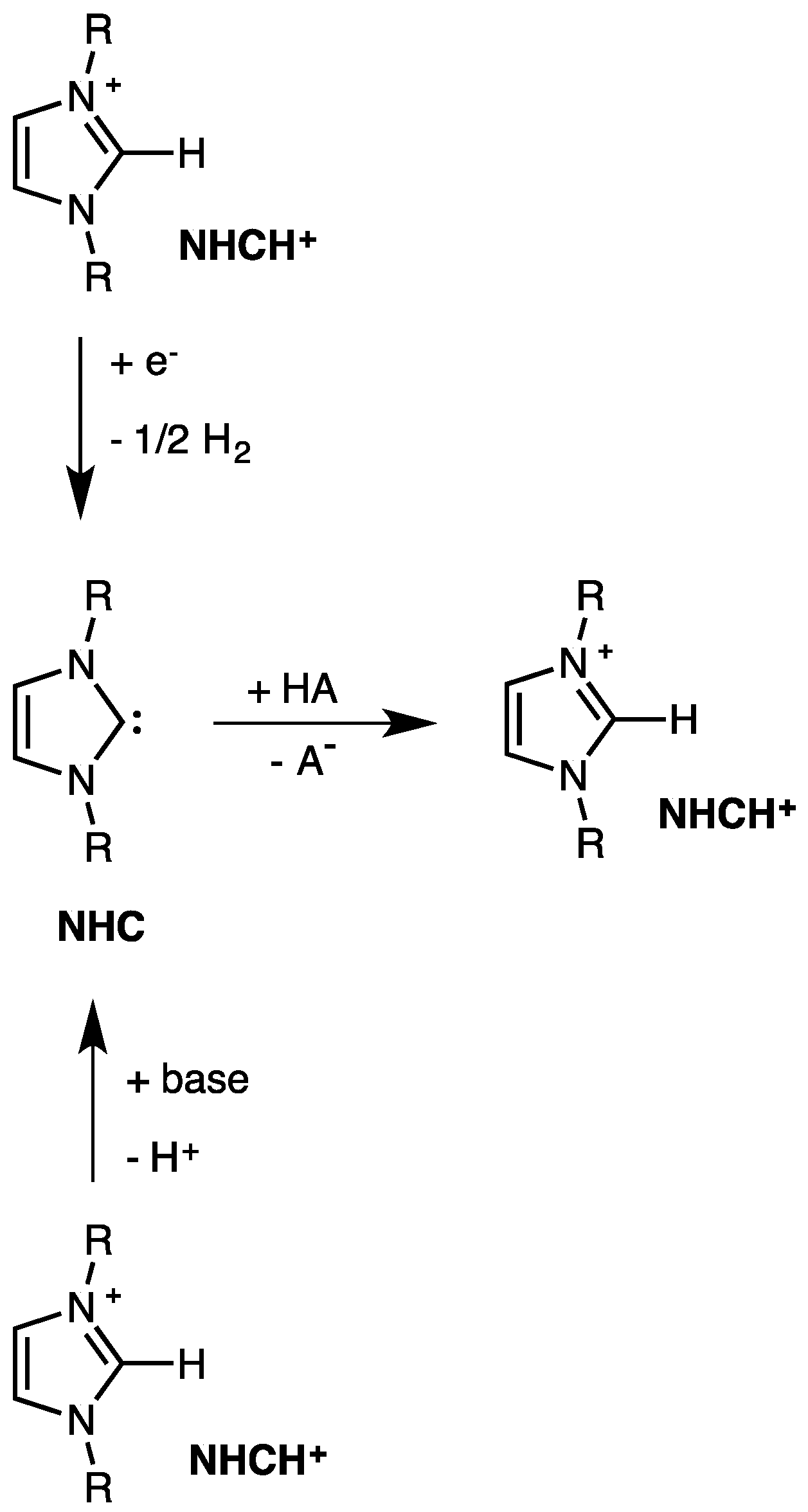
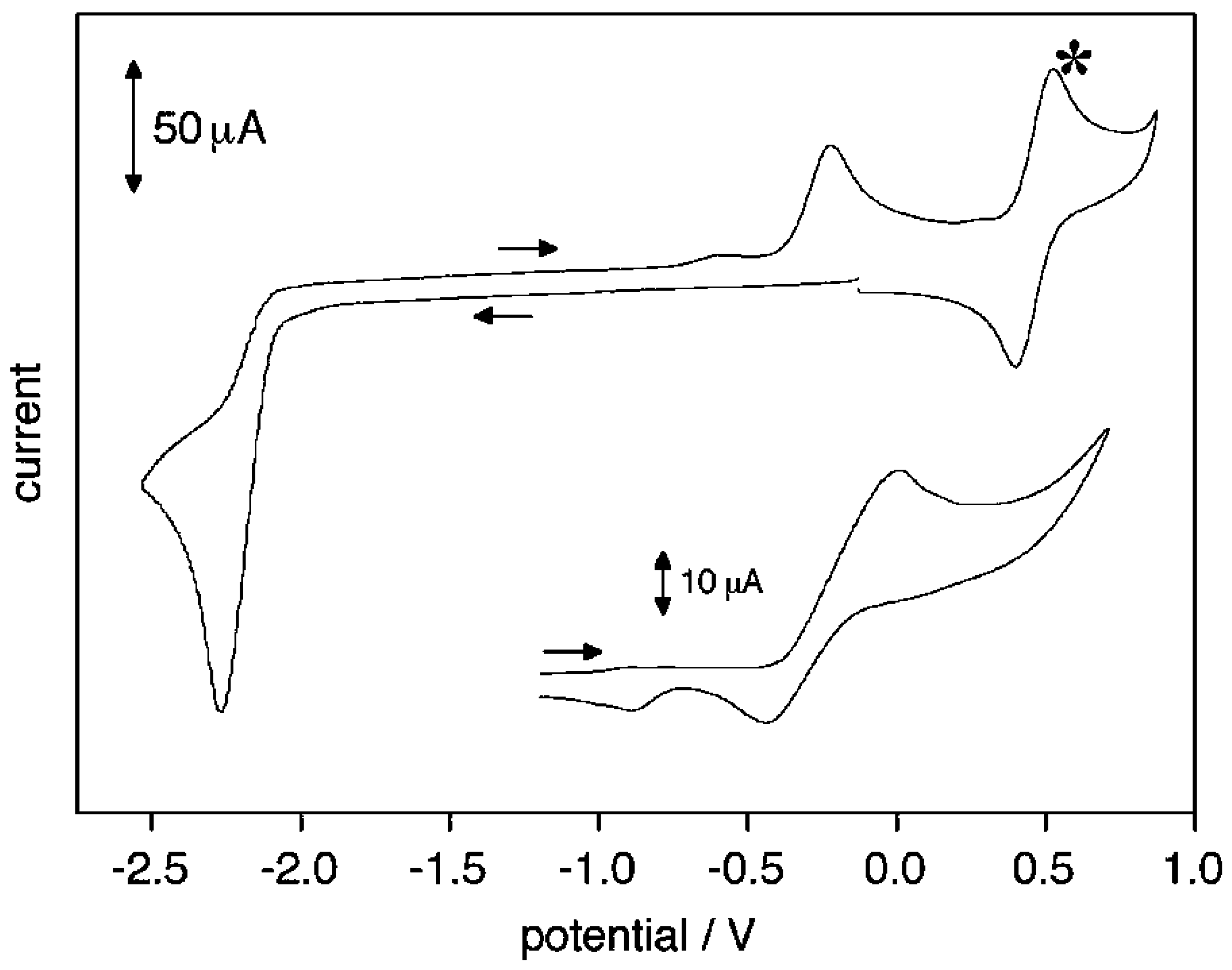
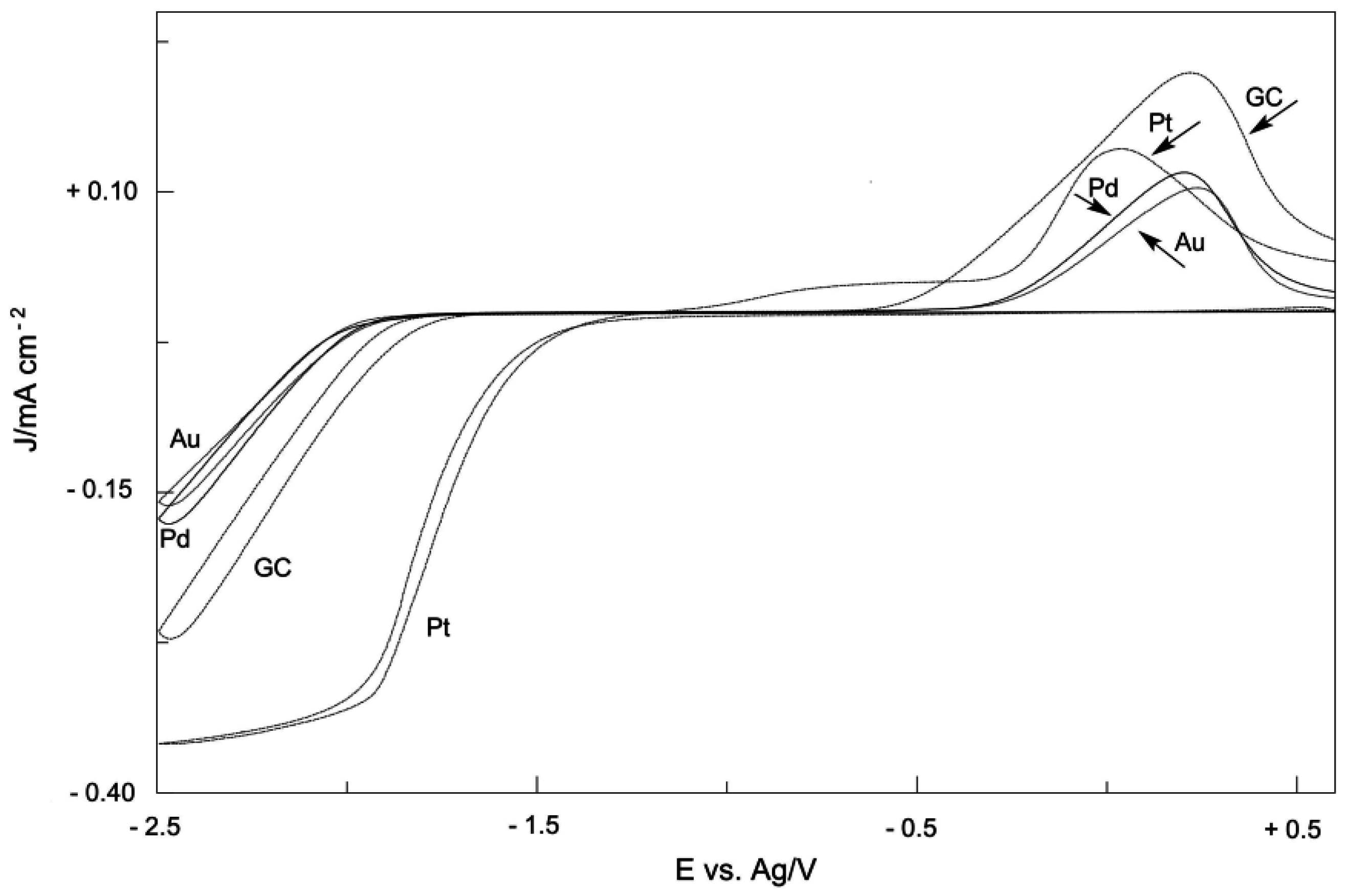
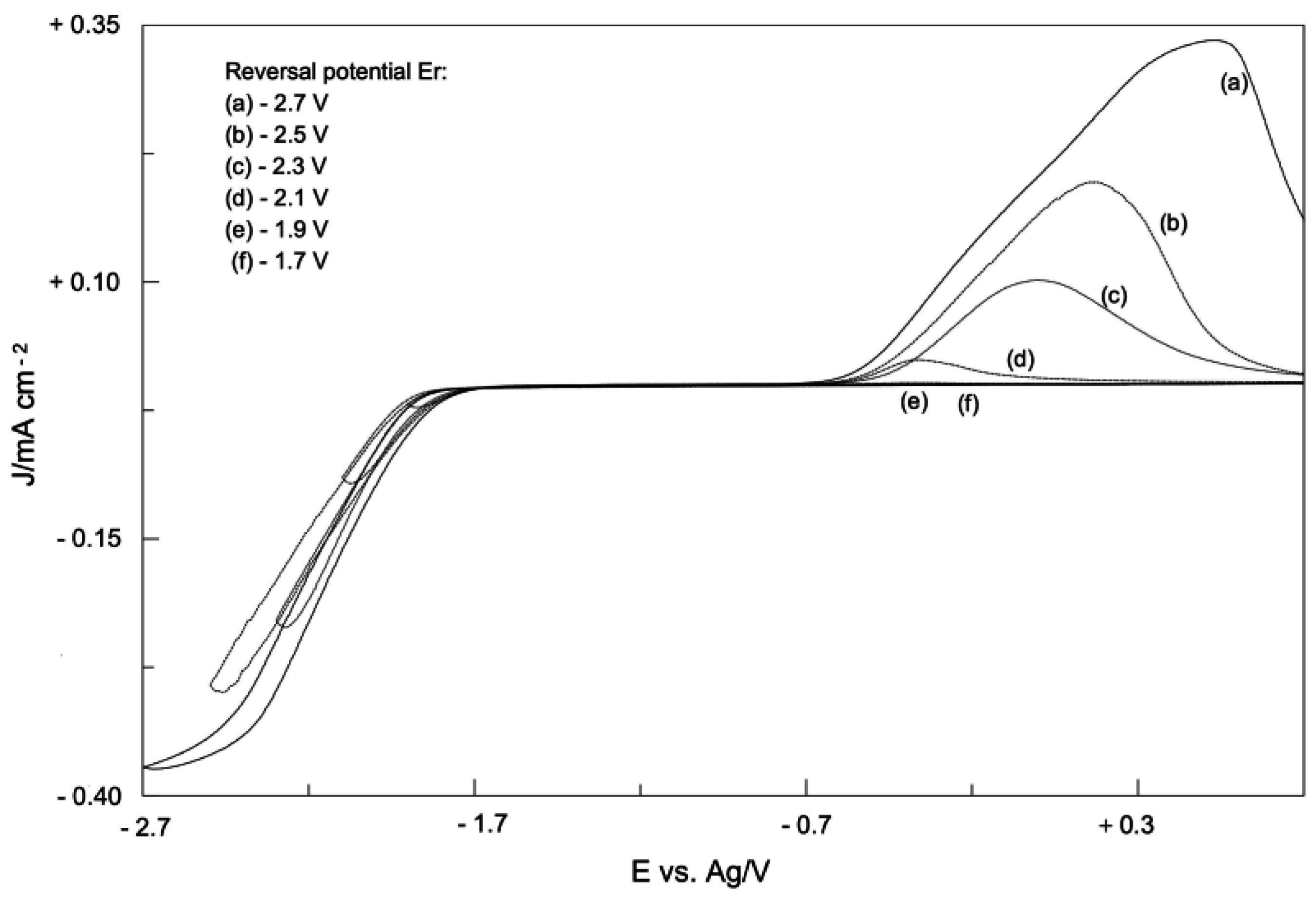
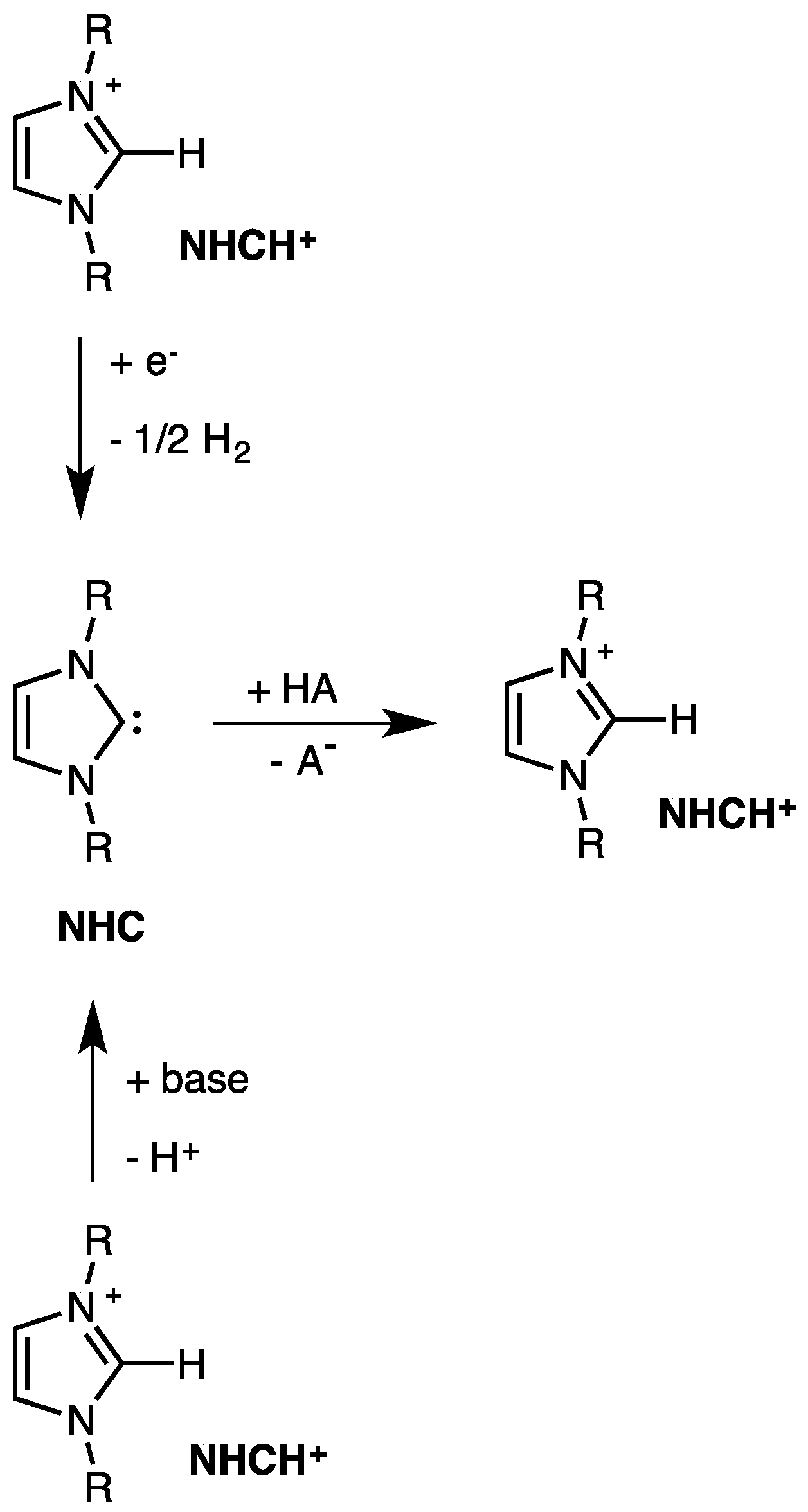
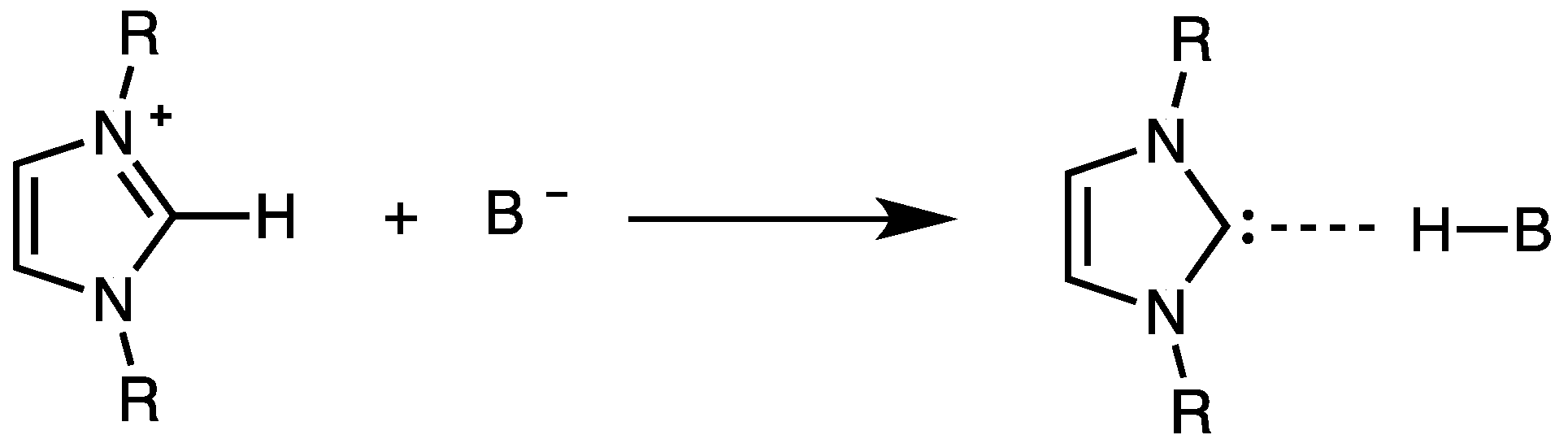
2. Electrodic Activity of the System NHCH+/NHC
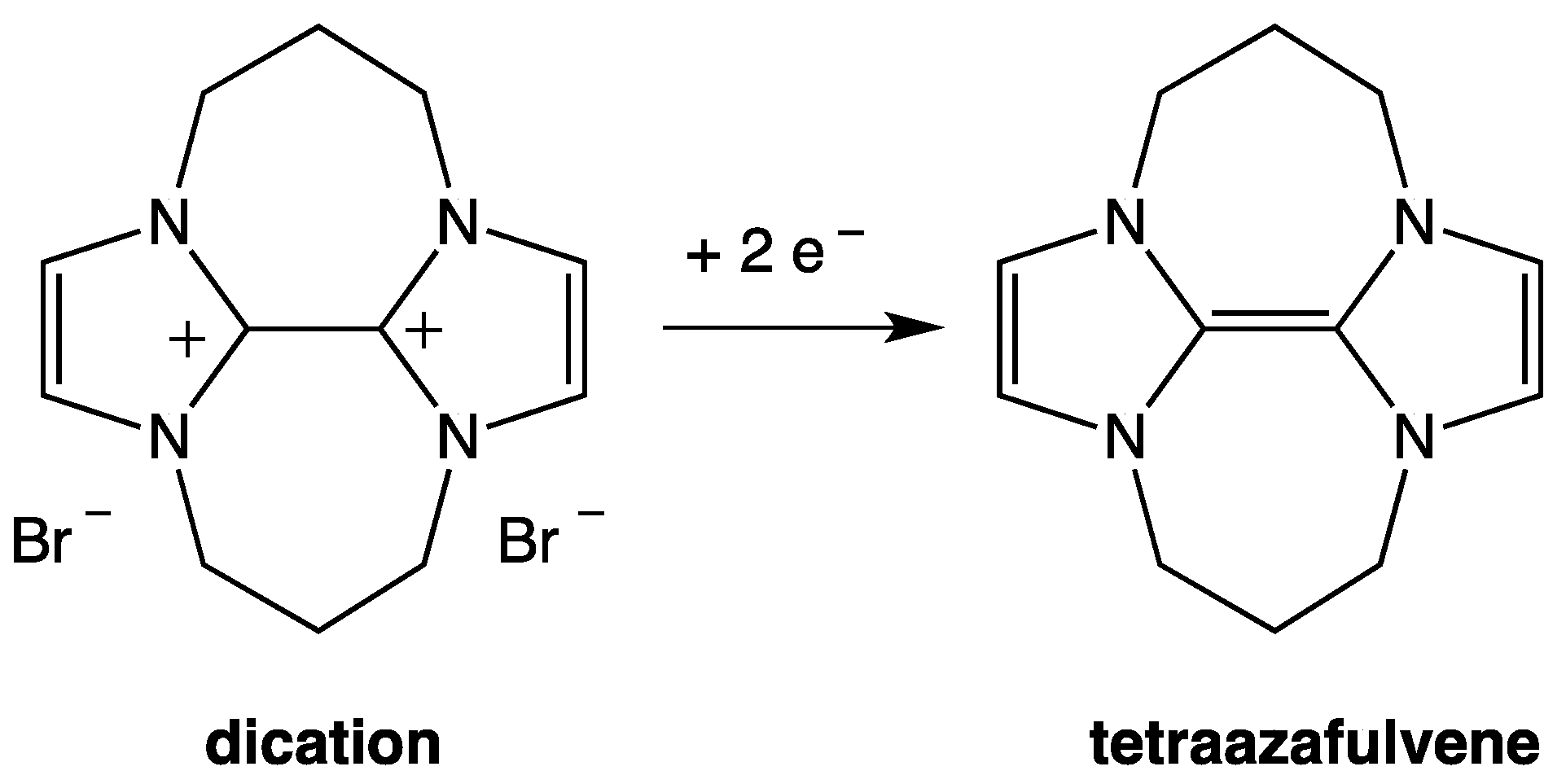
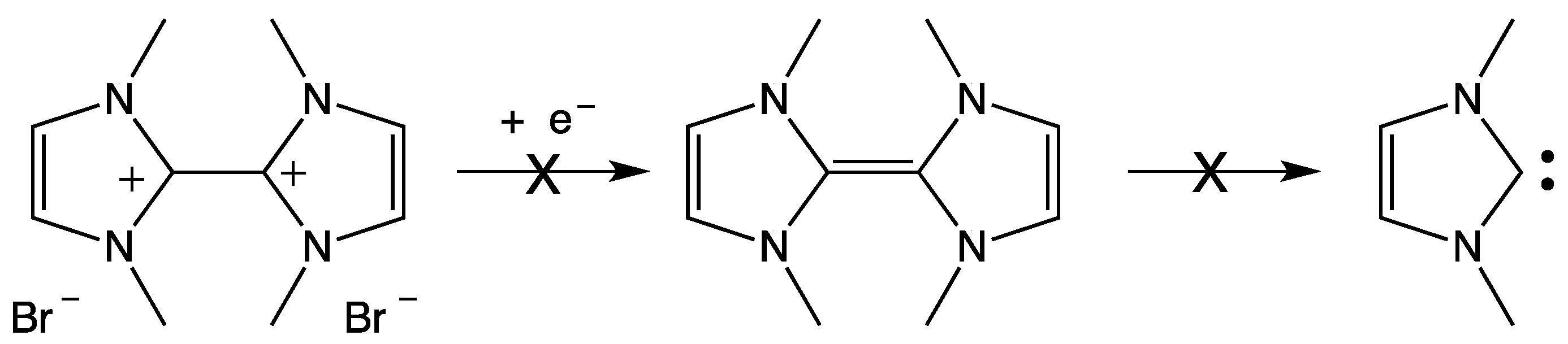
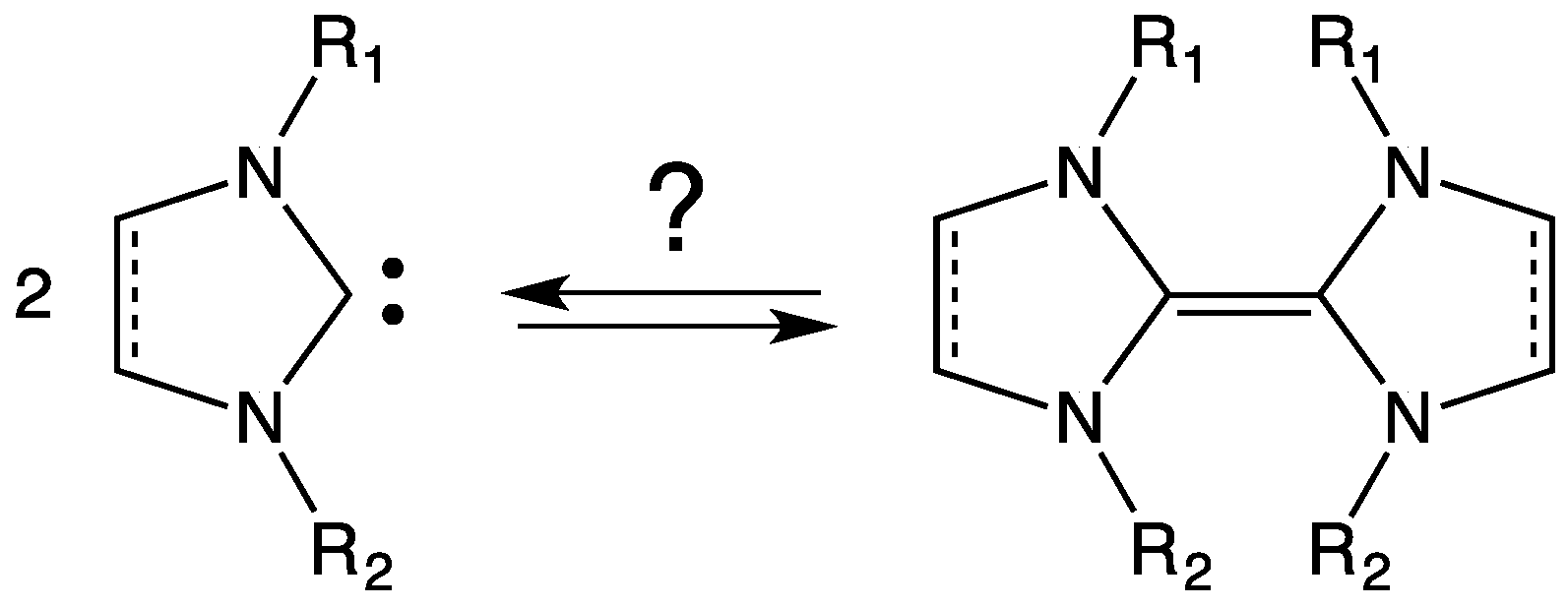
3. NHC Stability
- The investigated NHCs (imidazole-, triazole-, and thiazole-based) are involved in a remarkable degradation process.
- The NHC degradation rate increases on increasing NHC concentration. Accordingly, the degradation process could be associated, inter alia, with a coupling reaction between two NHC molecules.
- The NHC half-life is influenced by the nature of the counter ion X− of the parent azolium salt and by the presence and nature of organic solvents (Table 2) [39,40]. In fact, τ1/2 for an NHC is quite different when measured in ionic liquid or in organic solvent. As regards this question, many authors have investigated the role of hydrogen bond between NHC, NHCH+, or X− and the solvent (see Section 6).
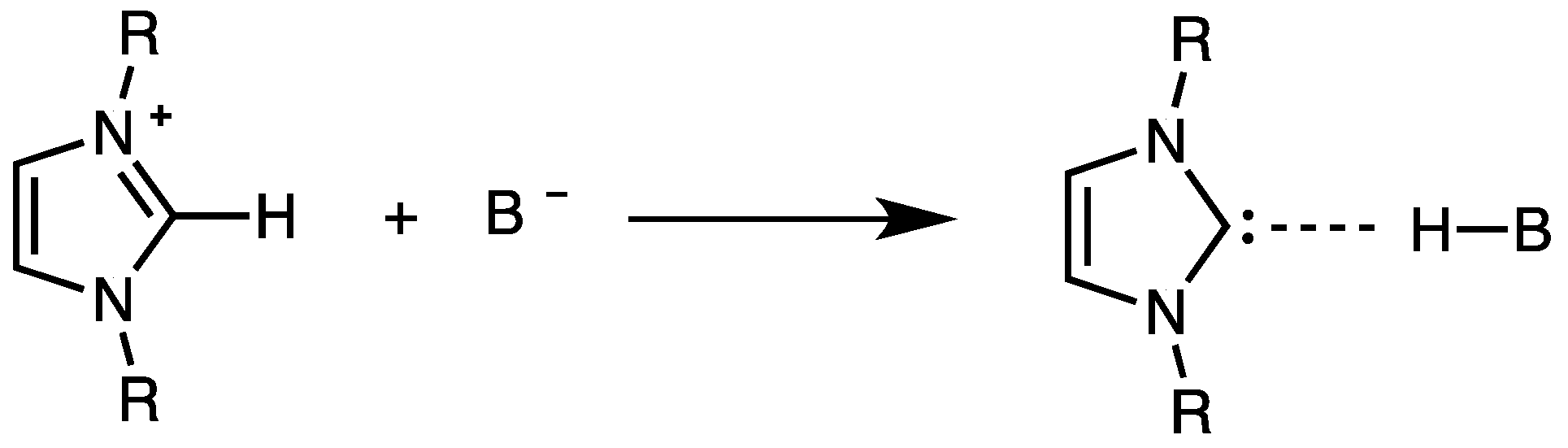
4. The Acidic and Nucleophilic Properties of NHCH+/NHC System
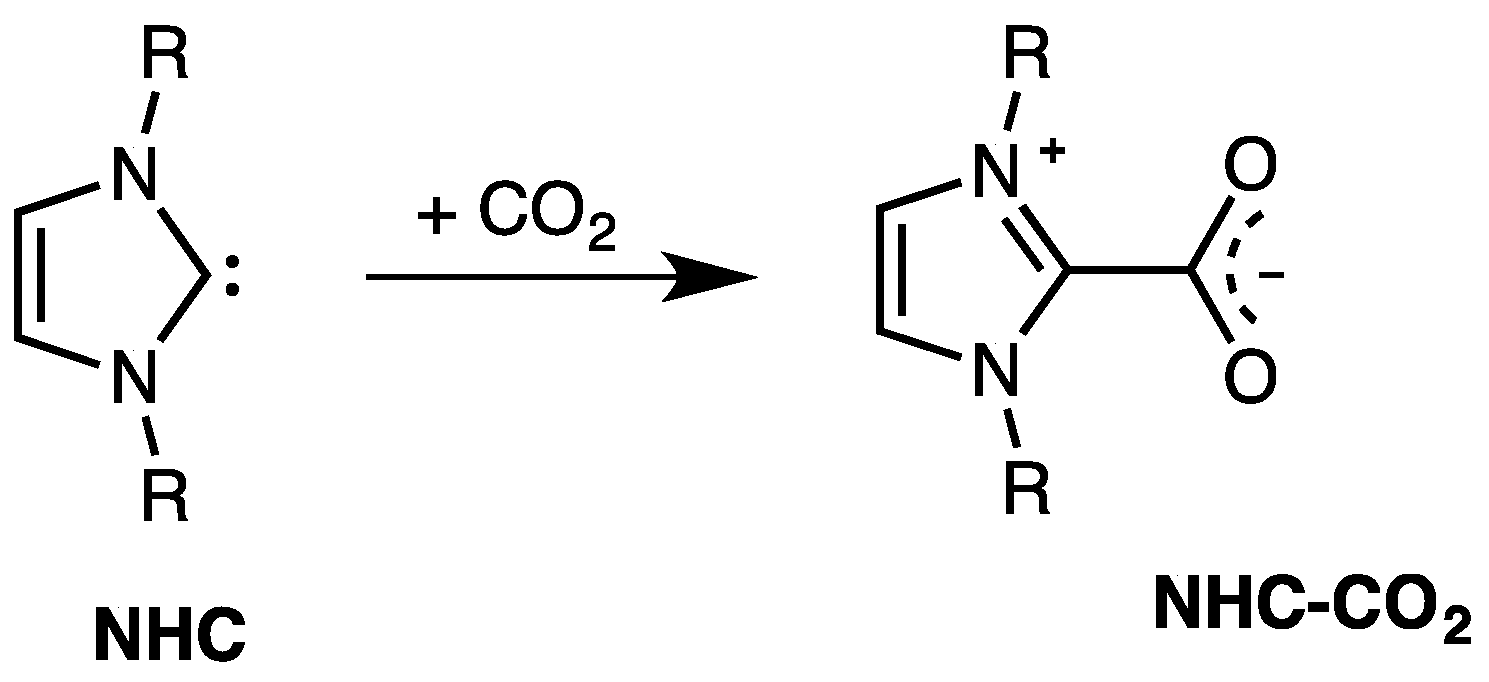
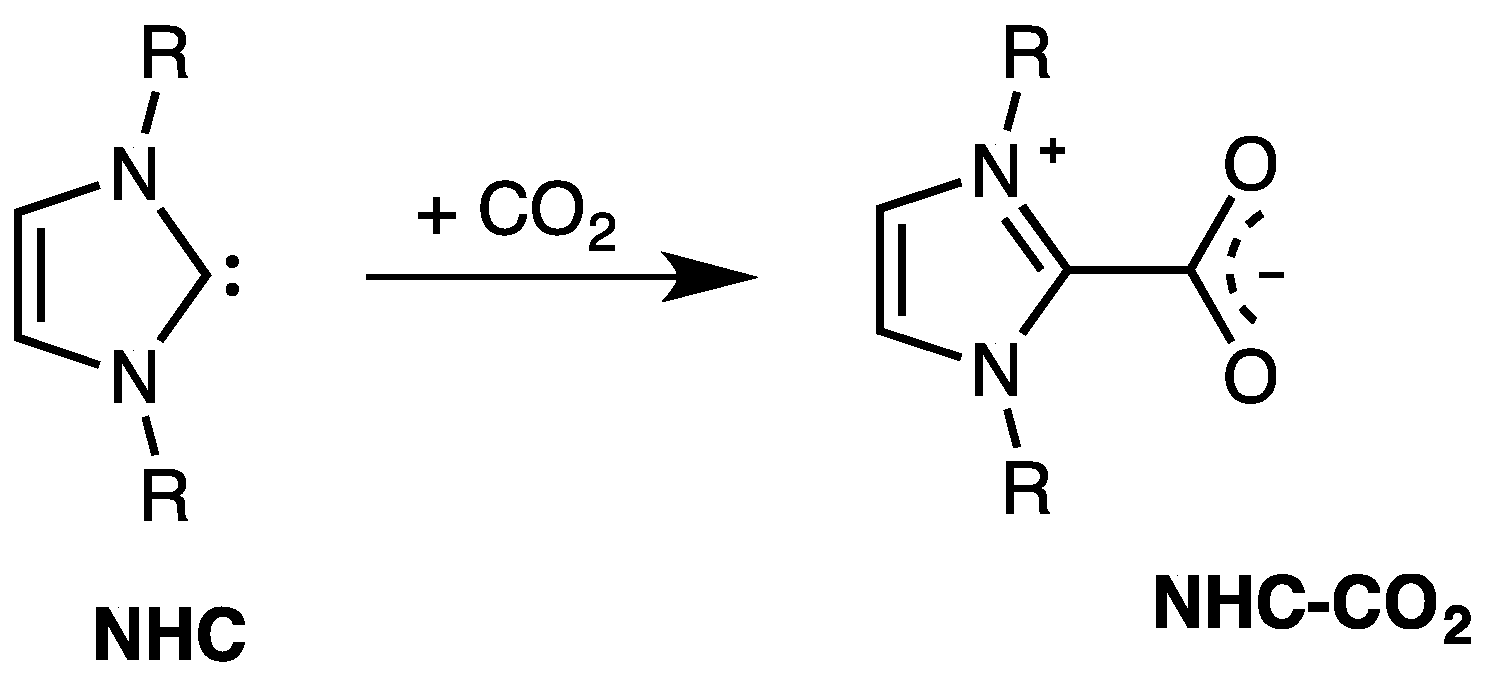
5. NHC and Carbon Dioxide
- the small NHC–CO2 bond dissociation energies suggested an extensive investigation on the possible utilization of NHCs as means to catch and release carbon dioxide; and
- the considerably higher stability of NHC–CO2 adducts than free NHCs spurred many authors to verify the possible use of these adducts as catalysts (or, rather, as latent catalysts) instead of NHCs.
5.1. Catch and Release of CO2: Role of NHCs
- The control of the global climate change is, at present, a very serious problem [56,57]. Carbon dioxide is the most significant greenhouse gas responsible of this change [58]. Global emissions of carbon dioxide reached 34 billion tons in 2011, up 3% relative to 2010. If the increase in CO2 emissions continues at this rate, within the next two decades global carbon dioxide cumulative emissions will reach levels that will make it impossible to hold the increase in global temperature below 2 °C every year [59]. Therefore, any possible attempt must be oriented to reduce (or at least to stabilize) the levels of carbon dioxide in the atmosphere.
- On the other hand, the reaction of CO2 with organic substrates to form new C–C bonds is a goal pursued by many authors. In fact, carbon dioxide is a non-toxic, abundant, low cost C1 building block in organic synthesis [60].
5.2. NHC–CO2 Adducts as Latent Catalysts
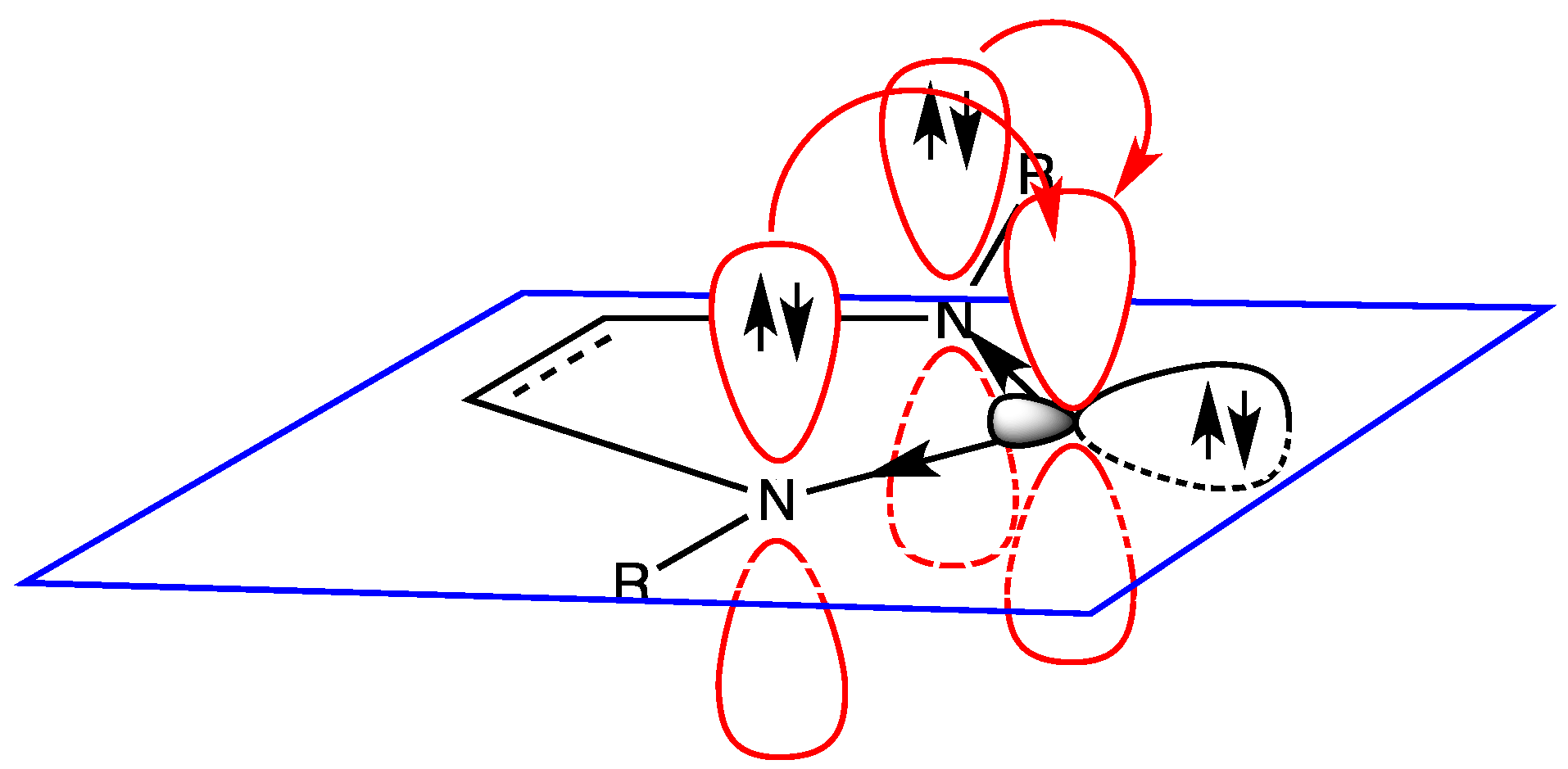
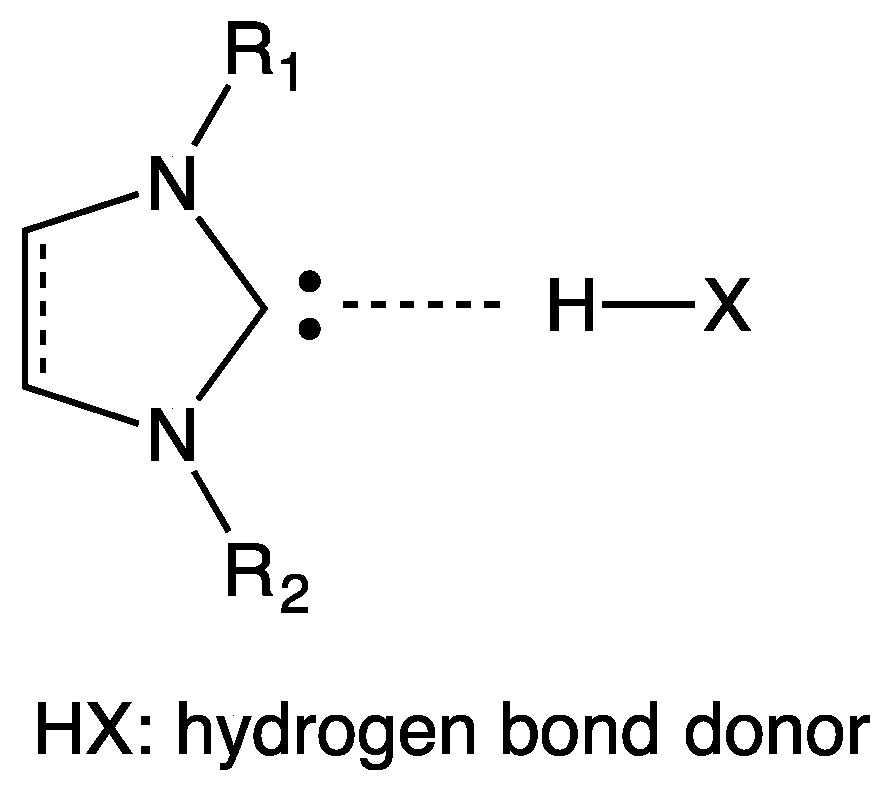
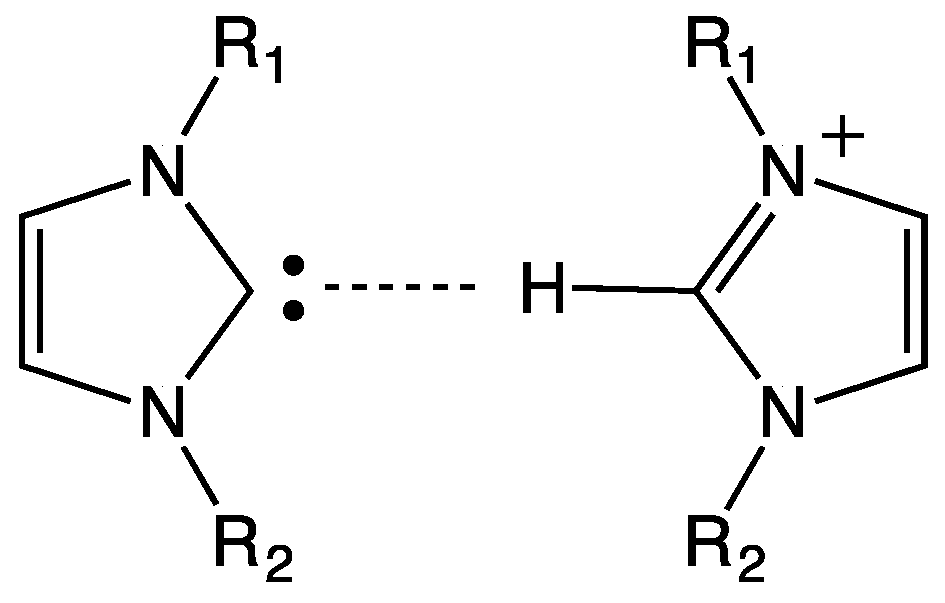
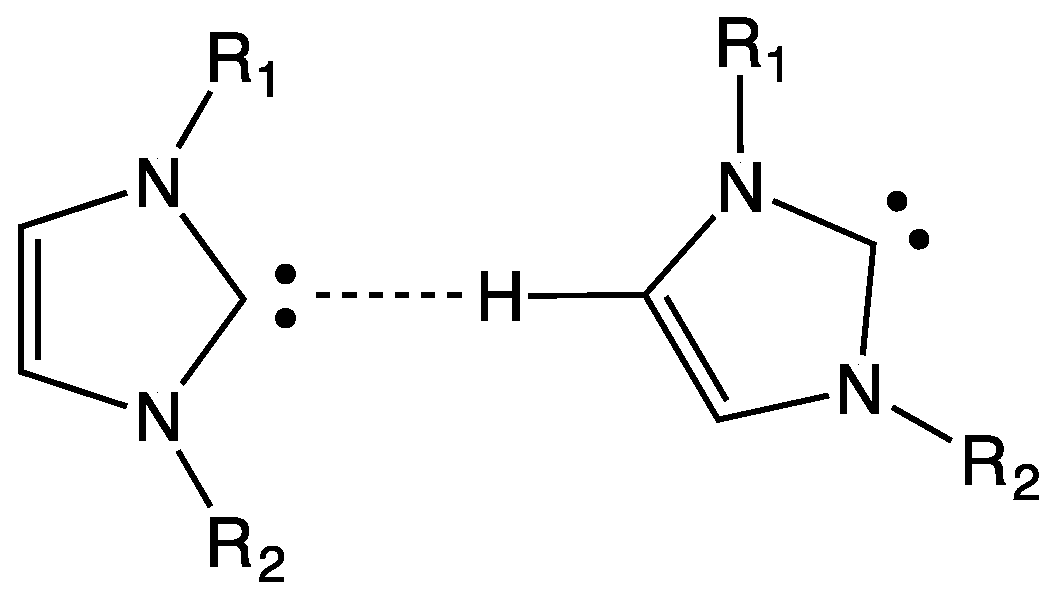
6. Hydrogen Bond and Catalytic Activity of NHC
- the effective presence of dimeric structures instead of (or in addition to) free NHC;
- the higher stability of the dimeric structure than that of free NHC; and
- the nature of the “effective” catalyst, i.e., the attack to the active site of the substrate is carried out by free NHC (being the dimeric structure a precursor of free NHC) or by the dimer? In fact, a direct interaction between substrate and dimeric structure could not be rejected.
- NHCs could form unexpected hydrogen bonds, as weaker interactions, with the alkyl side chain of imidazolium cation; and
7. Spontaneous or Induced Formation of NHC
- The Claisen rearrangement (reaction of kojic acids with ynals yielding dihydropyranones; yields 75%–98%) was carried out using azolium salts as precatalysts in the absence of added base [87]. The authors suggest that the counter-ion X− (Cl− or CH3COO−) plays the role of base generating a trace amount of NHC, which quickly attacks the aldehyde to initiate the catalytic cycle. Azolium salts with less basic counter-ions such as SbF6− or ClO4− are unreactive.
- The benzoin condensation was carried out by adding a benzaldehyde to neat 1-ethyl-3-methyl- or 1-butyl-3-methylimidazolium acetate in excess. Benzoins were isolated in good yields (60%). No products were isolated with ionic liquids containing non-basic anions such as methanesulfonate ion. The efficiency of NHCH+X− as precatalyst was related to the ability of the counter-ion CH3COO− to deprotonate NHCH+ to NHC [86].
- 1-ethyl-3-methylimidazole-2-thione was isolated from an equimolar mixture of S8 and 1,3-dialkylimidazolium acetate (24 h at 25 °C; yield 50%) [88]. No reaction was observed with S8 using 1,3-dialkylimidazolium salts containing other anions as Cl−, HSO4−, SCN−. According to the opinion of the authors, imidazolium acetate acts as base yielding NHC, which reacts with S8 giving thione.
- the addition of CH3COOH to solutions containing NHC; and
- the addition of CH3COO− to solutions containing NHCH+.
7.1. Effect of the Addition of Acetic Acid to Solutions Containing NHC
7.2. Effect of the Addition of Acetate Anion to Solutions Containing NHCH+
8. Conclusions
- NHC generation by electrolysis of NHCH+X− ionic liquids (neat or in solution), i.e., by cathodic reduction of NHCH+. This procedure, alternative to the deprotonation of NHCH+ via a suitable base B− purposely added to NHCH+X−, allows to avoid the presence of the base conjugate acid BH in the final mixture. The presence of the acid HB could strongly decrease the catalytic activity of NHC, owing the possible formation of the hydrogen-bond adduct NH–HB.
- NHC presence determination, by the appearance in the voltammograms of its oxidation peak. The NHC concentration is evaluated from the current of this peak.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bourissou, D.; Guerret, O.; Gabbai, F.P.; Bertrand, G. Stable carbenes. Chem. Rev. 2000, 100, 39–91. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef] [PubMed]
- Fèvre, M.; Pinaud, J.; Gnanou, Y.; Vignolle, J.; Taton, D. N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis. Chem. Soc. Rev. 2013, 42, 2142–2172. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Vora, H.U.; Wheeler, P.; Rovis, T. Exploiting acyl and enol azolium intermediates via N-heterocyclic carbene-catalyzed reactions of α-reducible aldehydes. Adv. Synth. Catal. 2012, 354, 1617–1639. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-Heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Biju, A.T.; Kuhl, N.; Glorius, F. Extending NHC-catalysis: Coupling aldehydes with unconventional reaction partners. Acc. Chem. Res. 2011, 44, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Marion, N.; Díez-González, S.; Nolan, S.P. N-Heterocyclic carbenes as organocatalysts. Angew. Chem. Int. Ed. 2007, 46, 2988–3000. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-heterocyclic carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.; Hutson, G.E.; Cohen, D.T.; Scheidt, K.A. A continuum of progress: Applications of N-hetereocyclic carbene catalysis in total synthesis. Angew. Chem. Int. Ed. 2012, 51, 11686–11698. [Google Scholar] [CrossRef] [PubMed]
- Wanzlick, H.W. Aspects of nucleophilic carbene chemistry. Angew. Chem. Int. Ed. 1962, 1, 75–80. [Google Scholar] [CrossRef]
- Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G. Analogous α,α′-bis-carbenoid, triply bonded species: Synthesis of a stable λ3-phosphino carbene-λ5-phosphaacetylene. J. Am. Chem. Soc. 1988, 110, 6463–6466. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Breslow, R. On the mechanism of thiamine action. IV. Evidence from studies on model systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Rehbein, J.; Ruser, S.M.; Phan, J. NHC-Catalysed benzoin condensation—Is it all down to the Breslow intermediate? Chem. Sci. 2015, 6, 6013–6018. [Google Scholar] [CrossRef]
- Mahatthananchai, J.; Bode, J.W. On the mechanism of N-heterocyclic carbene-catalyzed reactions involving acyl azoliums. Acc. Chem. Res. 2014, 47, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J.; Candish, L.; Lupton, D.W. Acyl anion feee N-heterocyclic carbene organocatalysis. Chem. Soc. Rev. 2013, 42, 4906–4917. [Google Scholar] [CrossRef] [PubMed]
- Hollóczki, O.; Kelemen, Z.; Nyulászi, L. On the organocatalytic activity of feee N-heterocyclic carbenes: Role of sulfur in thiamine. J. Org. Chem. 2012, 77, 6014–6022. [Google Scholar] [CrossRef] [PubMed]
- Feroci, M.; Chiarotto, I.; Inesi, A. Electrolysis of ionic liquids. A possible keystone for the achievement of green solvent-catalyst systems. Curr. Org. Chem. 2013, 17, 204–219. [Google Scholar] [CrossRef]
- Fuller, J.; Carlin, R.T. Structural and electrochemical characterization of 1,3-bis-(4-methylphenyl) imidazolium chloride. J. Chem. Crystal. 1994, 24, 489–493. [Google Scholar] [CrossRef]
- Shi, Z.; Goulle, V.; Thummel, R.P. An aza-analogue of TTF: 1,1′;3,3′-Bistrimethylene-2,2′-diimidazolinylidine. Tetrahedron Lett. 1996, 37, 2357–2360. [Google Scholar] [CrossRef]
- Suarez, P.A.Z.; Selbach, V.M.; Dullius, J.E.L.; Einloft, S.; Piatnicki, C.M.S.; Azambuja, D.S.; de Souza, R.F.; Dupont, J. Enlarged electrochemical window in dialkyl-imidazolium cation based room-temperature air and water-stable molten salts. Electrochim. Acta 1997, 42, 2533–2535. [Google Scholar] [CrossRef]
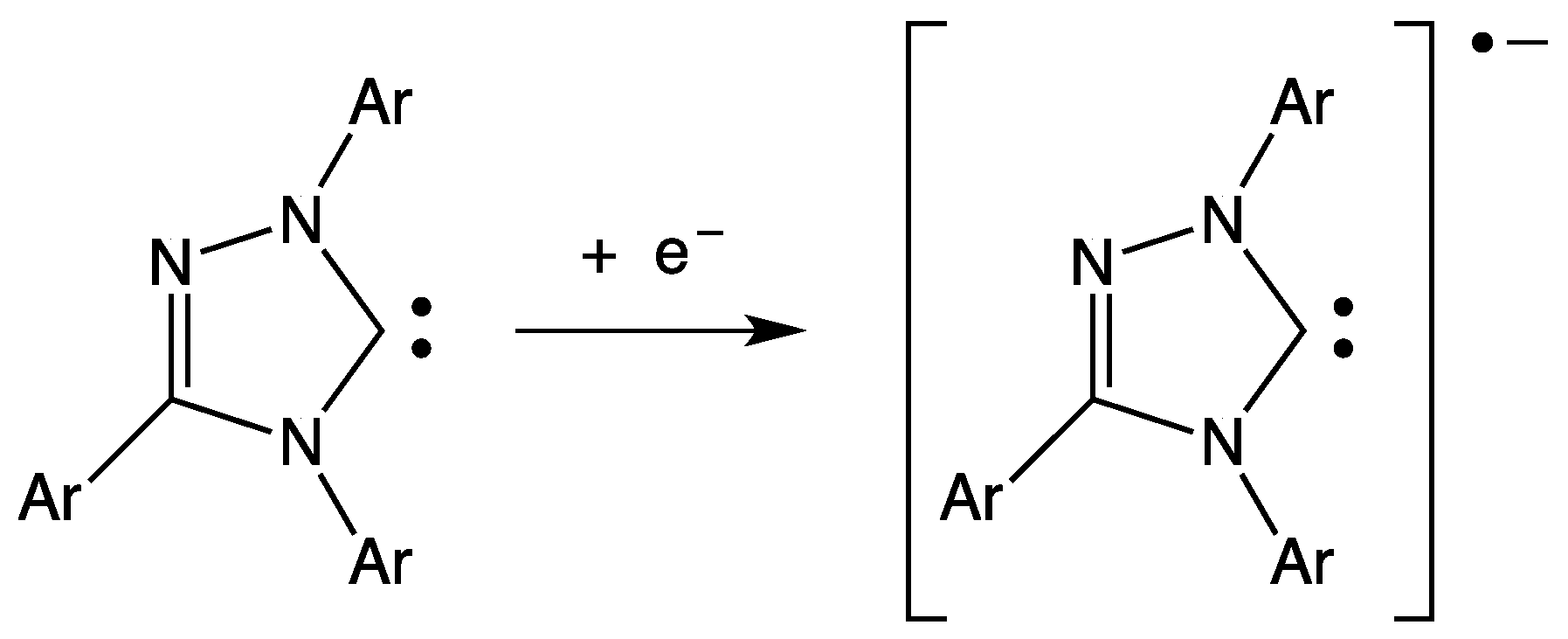
- Enders, D.; Breuer, K.; Raabe, G.; Simonet, J.; Ghanimi, A.; Stegmann, H.B.; Teles, J.H. A stable carbene as π-acceptor electrochemical reduction to the radical anion. Tetrahedron Lett. 1997, 38, 2833–2836. [Google Scholar] [CrossRef]
- Ramnial, T.; McKenzie, I.; Gorodetsky, B.; Tsang, E.M.W.; Clyburne, J.A.C. Reactions of N-heterocyclic carbenes (NHCs) with one-electron oxidants: Possible formation of a carbene cation radical. Chem. Commun. 2004. [Google Scholar] [CrossRef] [PubMed]
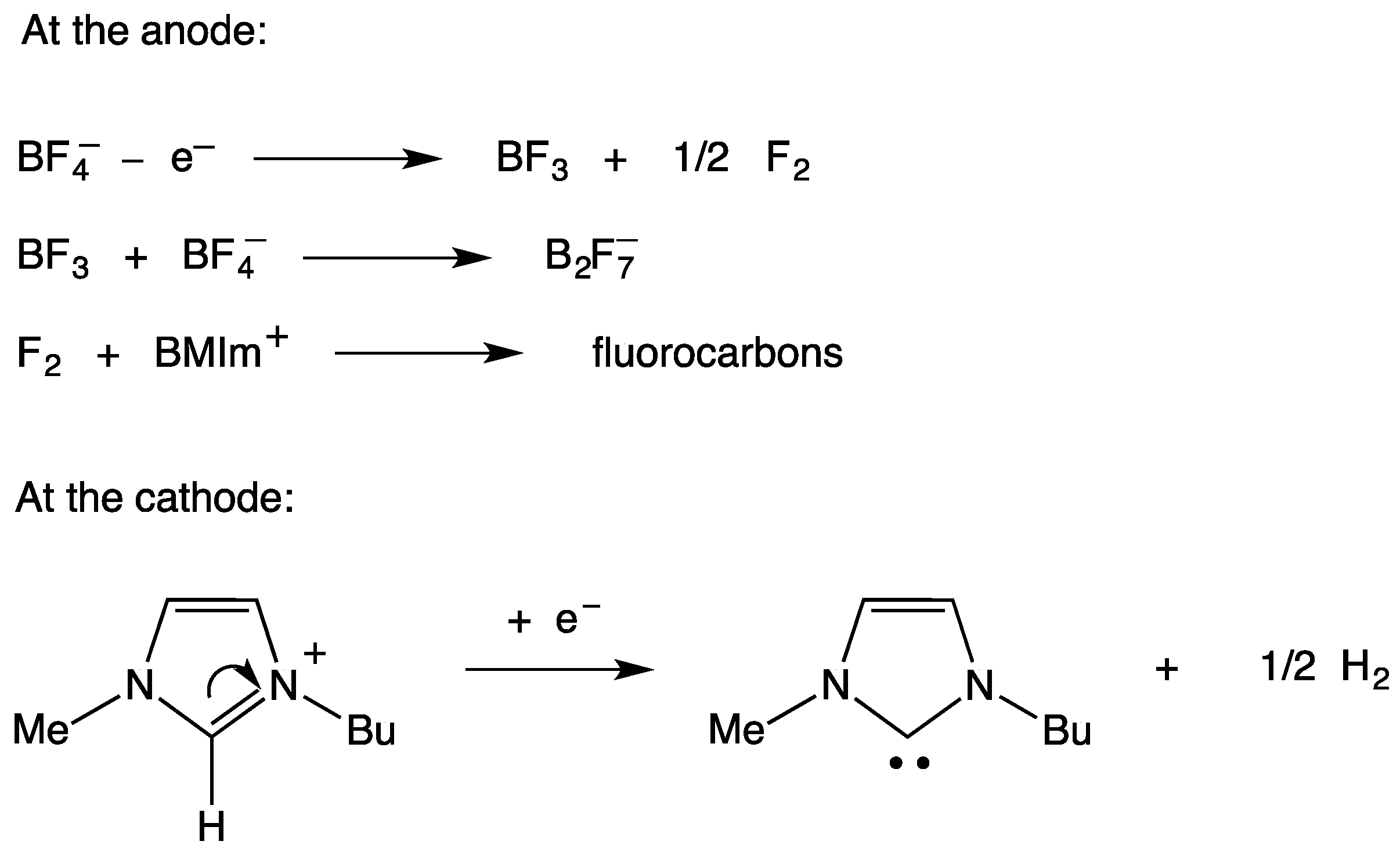
- Xiao, L.; Johnson, K.E. Electrochemistry of 1-butyl-3-methyl-1H-imidazolium tetrafluoroborate ionic liquid. J. Electrochem. Soc. 2003, 150, E307–E311. [Google Scholar] [CrossRef]
- Gorodetsky, B.; Ramnial, T.; Branda, N.R.; Clyburne, J.A.C. Electrochemical reduction of an imidazolium cation: A convenient preparation of imidazol-2-ylidenes and their observation in an ionic liquid. Chem. Commun. 2004, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-X.; Wang, H.; Xiao, Y.; Tu, Z.-Y.; Ding, B.-B.; Lu, J.-X. Synthesis of dialkyl carbonates from CO2 and alcohols via electrogenerated N-heterocyclic carbenes. Electrochem. Commun. 2012, 25, 116–118. [Google Scholar] [CrossRef]
- De Robillard, G.; Devillers, C.H.; Kunz, D.; Cattey, H.; Digard, E.; Andrieu, J. Electrosynthesis of imidazolium carboxylates. Org. Lett. 2013, 15, 4410–4413. [Google Scholar] [CrossRef] [PubMed]
- Feroci, M.; Chiarotto, I.; Vecchio Ciprioti, S.; Inesi, A. On the reactivity and stability of electrogenerated N-heterocyclic carbene in parent 1-butyl-3-methyl-1H-imidazolium tetrafluoroborate: Formation and use of N-heterocyclic carbene–CO2 adduct as latent catalyst. Electrochim. Acta 2013, 109, 95–101. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; Forte, G.; Achille, I. An electrochemical methodology for the cyclic CO2 “catch and release”. The role of the electrogenerated N-heterocyclic carbene in BMIm-BF4. J. CO2 Util. 2013, 2, 29–34. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; D’Anna, F.; Gala, F.; Noto, R.; Ornano, L.; Zollo, G.; Inesi, A. N-Heterocyclic carbenes and parent cations: Acidity, nucleophilicity, stability, and hydrogen bonding-electrochemical study and Ab Initio calculations. ChemElectroChem 2016, 3, 1133–1141. [Google Scholar] [CrossRef]
- Gronert, S.; Keeffe, J.R.; O’Ferral, R.A.M. Carbene stability. In Contemporary Carbene Chemistry, 1st ed.; Moss, R.A., Doyle, M.P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 3–39. [Google Scholar]
- Moerdyk, J.P.; Bielawski, C.W. Stable carbenes. In Contemporary Carbene Chemistry, 1st ed.; Moss, R.A., Doyle, M.P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 40–74. [Google Scholar]
- Dröge, T.; Glorius, F. The measure of all rings—N-Heterocyclic carbenes. Angew. Chem. Int. Ed. 2010, 49, 6940–6952. [Google Scholar] [CrossRef] [PubMed]
- Denk, M.K.; Hezarkhani, A.; Zheng, F.-L. Steric and electronic effects in the dimerization of Wanzlick carbenes: The alkyl effect. Eur. J. Inorg. Chem. 2007, 2007, 3527–3534. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-heterocyclic carbene dimerization: The balance of sterics and electronics. Organomet 2008, 27, 2679–2681. [Google Scholar] [CrossRef]
- Liu, Y.; Lemal, D.M. Concerning the ‘Wanzlick equilibrium’. Tetrahedron Lett. 2000, 41, 599–602. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; Forte, G.; Vecchio Ciprioti, S.; Inesi, A. Stability and CO2 capture ability of electrogenerated N-heterocyclic carbene in the parent 1-butyl-3-methylimidazoliun ionic liquid (BMIm-X): The role of the anion X−. ChemElectroChem 2014, 1, 1407–1414. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; D’Anna, F.; Forte, G.; Noto, R.; Inesi, A. Stability and organocatalytic efficiency of N-heterocyclic carbenes electrogenerated in organic solvents from imidazolium ionic liquids. Electrochim. Acta 2015, 153, 122–129. [Google Scholar] [CrossRef]
- Chiarotto, I.; Feroci, M.; Forte, G.; Inesi, A. Stability of electrogenerated 1-butyl-3-methyl imidazol-2-ylidene in DMF. Part 2. Role of acid substrates. Electrochim. Acta 2015, 176, 627–635. [Google Scholar] [CrossRef]
- O’Donoghue, A.-M.C.; Massey, R.S. Acid-base chemistry of carbenes. In Contemporary Carbene Chemistry, 1st ed.; Moss, R.A., Doyle, M.P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 75–106. [Google Scholar]
- Bordwell, F.G.; Satish, A.V. Acidities of C2 hydrogen atoms in thiazolium cations and reactivities of their conjugate bases. J. Am. Chem. Soc. 1991, 113, 985–990. [Google Scholar] [CrossRef]
- Alder, R.W.; Allen, P.R.; Williams, S.J. Stable carbenes as strong bases. J. Chem. Soc. Chem. Commun. 1995. [Google Scholar] [CrossRef]
- Chu, Y.; Deng, H.; Cheng, J.-P. An acidity scale of 1,3-dialkylimidazolium salts in dimethyl sulfoxide solution. J. Org. Chem. 2007, 72, 7790–7793. [Google Scholar] [CrossRef] [PubMed]
- Grishina, A.A.; Polyakova, S.M.; Kunetskiy, R.A.; Císařová, I.; Lyapkalo, I.M. 4,5-Disubstituted N,N′-di-tert-alkyl imidazolium salts: New synthesis and structural features. Chem. Eur. J. 2011, 17, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Barhdadi, R.; Troupel, M.; Comminges, C.; Laurent, M.; Doherty, A. Electrochemical determination of pKa of N-bases in ionic liquid media. J. Phys. Chem. 2012, 116, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lang, Q.; Zeng, L.; Wei, M.; Liu, A. Substituent effect on the oxidation peak potentials of phenol derivatives at ordered mesoporous carbons modified electrode and its application in determination of acidity coefficients (pKa). Electrochim. Acta 2014, 115, 283–289. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [PubMed]
- Maji, B.; Breugst, M.; Mayr, H. N-Heterocyclic carbenes: Organocatalysts with moderate nucleophilicity but extraordinarily high lewis basicity. Angew. Chem. Int. Ed. 2011, 50, 6915–6919. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, N.; Steimann, M.; Weyers, G. Synthesis and properties of l,3-diisopropyl-4,5-dimethylimidazolium-2-carboxylate. A stable carbene adduct of carbon dioxide. Z. Naturforsch. 1999, 54, 427–433. [Google Scholar] [CrossRef]
- Duong, H.A.; Tekavec, T.N.; Arif, A.M.; Louie, J. Reversible carboxylation of N-heterocyclic carbenes. Chem. Commun. 2004, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Delaude, L. Betaine adducts of N-heterocyclic carbenes: Synthesis, properties and reactivity. Eur. J. Inorg. Chem. 2009. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H. Recent advances in carbon dioxide capture, fixation, and activation by using N-heterocyclic carbenes. ChemSusChem 2014, 7, 962–998. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.J.; Robertson, K.N.; Kemp, R.A.; Tuononen, H.M.; Clyburne, J.A.C. Structurally simple complexes of CO2. Chem. Commun. 2015, 51, 3942–3956. [Google Scholar] [CrossRef] [PubMed]
- Rogelj, J.; den Elzen, M.; Höhne, N.; Fransen, T.; Fekete, H.; Winkler, H.; Schaeffer, R.; Sha, F.; Riahi, K.; Meinshausen, M. Paris agreement climate proposals need a boost to keep warming well below 2 °C. Nature 2016, 534, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carleton, T.A.; Hsiang, S.M. Social and economic impacts of climate. Science 2016. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.S.; Mantripragada, H.; Marks, A.; Versteeg, P.; Kitchin, J. The outlook for improved carbon capture technology. Prog. Energy Combust. Sci. 2012, 38, 630–671. [Google Scholar] [CrossRef]
- Espinal, L.; Poster, D.L.; Wong-Ng, W.; Allen, A.J.; Green, M.L. Measurement, standards, and data needs for CO2 capture materials: A critical review. Environ. Sci. Technol. 2013, 47, 11960–11975. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels, technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Ramdin, M.; de Loos, T.W.; Vlugt, T.J.H. State-of-the-art of CO2 capture with ionic liquids. Ind. Eng. Chem. Res. 2012, 51, 8149–8177. [Google Scholar] [CrossRef]
- Yang, H.; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R.B.; Bland, A.E.; Wright, I. Progress in carbon dioxide separation and capture: A review. J. Environ. Sci. 2008, 20, 14–27. [Google Scholar] [CrossRef]
- Rao, A.B.; Rubin, E.S. A technical, economic, and environmental assessment of amine-based CO2 capture technology for power plant greenhouse gas control. Environ. Sci. Technol. 2002, 36, 4467–4475. [Google Scholar] [CrossRef] [PubMed]
- Kittel, J.; Idem, R.; Gelowitz, D.; Tontiwachwuthikul, P.; Parrain, G.; Bonneau, A. Corrosion in MEA units for CO2 capture: Pilot plant studies. Energy Procedia 2009, 1, 791–797. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Dong, H.; Zhao, Z.; Zhang, S.; Huang, Y. Carbon capture with ionic liquids: Overview and progress. Energy Environ. Sci. 2012, 5, 6668–6681. [Google Scholar] [CrossRef]
- Shannon, M.S.; Bara, J.E. Properties of alkylimidazoles as solvents for CO2 capture and comparisons to imidazolium-based ionic liquids. Ind. Eng. Chem. Res. 2011, 50, 8665–8677. [Google Scholar] [CrossRef]
- Shannon, M.S.; Hindman, M.S.; Danielsen, S.P.O.; Tedstone, J.M.; Gilmore, R.D.; Bara, J.E. Properties of alkylbenzimidazoles for CO2 and SO2 capture and comparisons to ionic liquids. Sci. China Chem. 2012, 55, 1638–1647. [Google Scholar] [CrossRef]
- Zhu, X.; Lu, Y.; Peng, C.; Hu, J.; Liu, H.; Hu, Y. Halogen bonding interactions between brominated ion pairs and CO2 molecules: Implications for design of new and efficient ionic liquids for CO2 absorption. J. Phys. Chem. B 2011, 115, 3949–3958. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guo, Y.; Zhu, X.; Cui, G.; Li, H.; Dai, S. Highly efficient CO2 capture by tunable alkanolamine-based ionic liquids with multidentate cation coordination. Chem. Commun. 2012, 48, 6526–6528. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Luo, H.; Luo, X.; Li, G.H.; Dai, S. Equimolar CO2 capture by imidazolium-based ionic liquids and superbase systems. Green Chem. 2010, 12, 2019–2023. [Google Scholar] [CrossRef]
- Lo, R.; Ganguly, B. Efficacy of carbenes for CO2 chemical fixation and activation by their superbasicity/alcohol: A DFT study. New J. Chem. 2012, 36, 2549–2554. [Google Scholar] [CrossRef]
- Xiong, Y.-B.; Wang, H.; Wang, Y.-J.; Wang, R.-M. Novel imidazolium-based poly(ionic liquid)s: Preparation, characterization, and absorption of CO2. Polym. Adv. Technol. 2012, 23, 835–840. [Google Scholar] [CrossRef]
- Bara, J.E.; Camper, D.E.; Gin, D.L.; Noble, R.D. Room-temperature ionic liquids and composite materials: Platform technologies for CO2 capture. Acc. Chem. Res. 2010, 43, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Ajitha, M.J.; Suresh, C.H. Assessment of stereoelectronic factors that influence the CO2 fixation ability of N-heterocyclic carbenes: A DFT study. J. Org. Chem. 2012, 77, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Sanda, F. Design of latent catalysts and their application to polymer synthesis. Macromol. Symp. 1996, 107, 237–242. [Google Scholar] [CrossRef]
- Norris, B.C.; Sheppard, D.G.; Henkelman, G.; Bielawski, C.W. Kinetic and thermodynamic evaluation of the reversible N-heterocyclic carbene-isothiocyanate coupling reaction: Application in latent catalysis. J. Org. Chem. 2011, 76, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Hans, M.; Delaude, L.; Rodriguez, J.; Coquerel, Y. N-Heterocyclic carbene catalyzed carba-, sulfa-, and phospha-Michael additions with NHC–CO2 adducts as precatalysts. J. Org. Chem. 2014, 79, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Brehm, M.; Hollóczki, O.; Kirchner, B. How can a carbene be active in an ionic liquid? Chem. Eur. J. 2014, 20, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Hollóczki, O. Unveiling the peculiar hydrogen bonding behavior of solvated N-heterocyclic carbenes. Phys. Chem. Chem. Phys. 2016, 18, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Chiappe, C. Ionic liquids in organic synthesis: Effect on rate and selectivity. In Ionic liquids in Synthesis, 2nd ed.; Wasserscheidt, P., Welton, T., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 265–292. [Google Scholar]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.A.; Ashworth, C.R.; Matthews, R.P. Hydrogen bonding in ionic liquids. Chem. Soc. Rev. 2015, 44, 1257–1288. [Google Scholar] [CrossRef] [PubMed]
- Hollóczki, O.; Terleczky, P.; Szieberth, D.; Mourgas, G.; Gudat, D.; Nyulászi, L. Hydrolysis of Imidazole-2-ylidenes. J. Am. Chem. Soc. 2011, 133, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, Z.; Hollóczki, O.; Oláh, J.; Nyulászi, L. Oxazol-2-ylidenes. A new class of stable carbenes? RSC Adv. 2013, 3, 7970–7978. [Google Scholar] [CrossRef]
- Dunn, M.H.; Cole, M.L.; Harper, J.B. Effects of an ionic liquid solvent on the synthesis of γ-butyrolactones by conjugate addition using NHC organocatalysts. RSC Adv. 2012, 2, 10160–10162. [Google Scholar] [CrossRef]
- Kelemen, Z.; Hollóczki, O.; Nagy, J.; Nyulászi, L. An organocatalytic ionic liquid. Org. Biomol. Chem. 2011, 9, 5362–5364. [Google Scholar] [CrossRef] [PubMed]
- Kaeobamrung, J.; Mahatthananchai, J.; Zheng, P.; Bode, J.W. An enantioselective Claisen rearrangement catalyzed by N-heterocyclic carbenes. J. Am. Chem. Soc. 2010, 132, 8810–8812. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, H.; Gurau, G.; Holbrey, J.D.; Rogers, R.D. Reaction of elemental chalcogens with imidazolium acetates to yield imidazole-2-chalcogenones: Direct evidence for ionic liquid as proto-carbenes. Chem. Commun. 2011, 47, 3222–3224. [Google Scholar] [CrossRef] [PubMed]
- Hollóczki, O.; Gerhard, D.; Massone, K.; Szarvas, L.; Németh, B.; Veszprémi, T.; Nyulászi, L. Carbenes in ionic liquids. New J. Chem. 2010, 34, 3004–3009. [Google Scholar] [CrossRef]
- Kar, B.D.; Sander, W. Reversible carbine formation in the ionic liquid 1-ethyl-3-methylimidazolium acetate by vaporization and condensation. ChemPhysChem 2015, 16, 3603–3606. [Google Scholar] [CrossRef] [PubMed]
- Hollóczki, O.; Firaha, D.S.; Friedrich, J.; Brehm, M.; Cybik, R.; Wild, M.; Stark, A.; Kirchner, B. Carbene formation in ionic liquids: Spontaneous, induced, or prohibited? J. Phys. Chem. B 2013, 117, 5898–5907. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, Z.; Péter-Szabó, B.; Székely, E.; Hollóczki, O.; Firaha, D.S.; Kirchner, B.; Nagy, J.; Nyulászi, L. An abnormal N-heterocyclic carbene–carbon dioxide adduct from imidazolium acetate ionic liquids: The importance of basicity. Chem. Eur. J. 2014, 20, 13002–13008. [Google Scholar] [CrossRef] [PubMed]
- Feroci, M.; Chiarotto, I.; D’Anna, F.; Ornano, L.; Rizzo, C.; Inesi, A. Azolium and acetate ions in DMF: Formation of free N-heterocyclic carbene. A voltammetric analysis. Electrochem. Commun. 2016, 67, 55–58. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
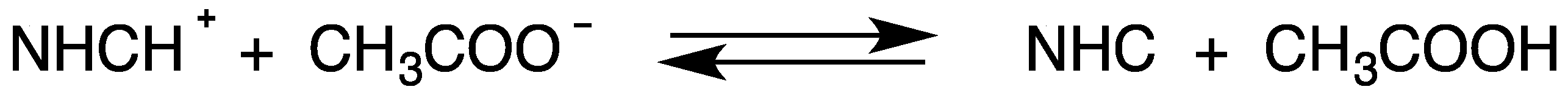{kind=link}
| Entry | NHC | τ1/2 1 (min) | τ1/2/CH3COOH 2 (min) |
|---|---|---|---|
| 1 |  | 70 | 8 |
| 2 |  | 133 | 17 |
| 3 | 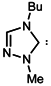 | >>300 | >>300 |
| 4 | 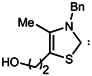 | >>300 | >300 |
| 5 |  | >>300 | >>300 |
| 6 | 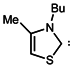 | 175 | 275 |
| Entry | Anion X | Solvent | τ1/2/min 1 |
|---|---|---|---|
| 1 | MeOSO3 | DMF | 14 |
| 2 | Cl | DMF | 30 |
| 3 | OTf | DMF | 37 |
| 4 | PF6 | DMF | 43 |
| 5 | CF3CO2 | DMF | 52 |
| 6 | BF4 | DMF | 69 |
| 7 | I | DMF | 194 |
| 8 | NTf2 | DMF | 262 |
| 9 | BF4 | BMIm-BF4 | 20 |
| 10 | CF3CO2 | BMIm-CF3CO2 | 50 |
| 11 | PF6 | BMIm-PF6 | 80 |
| 12 | NTf2 | BMIm-NTf2 | 220 |
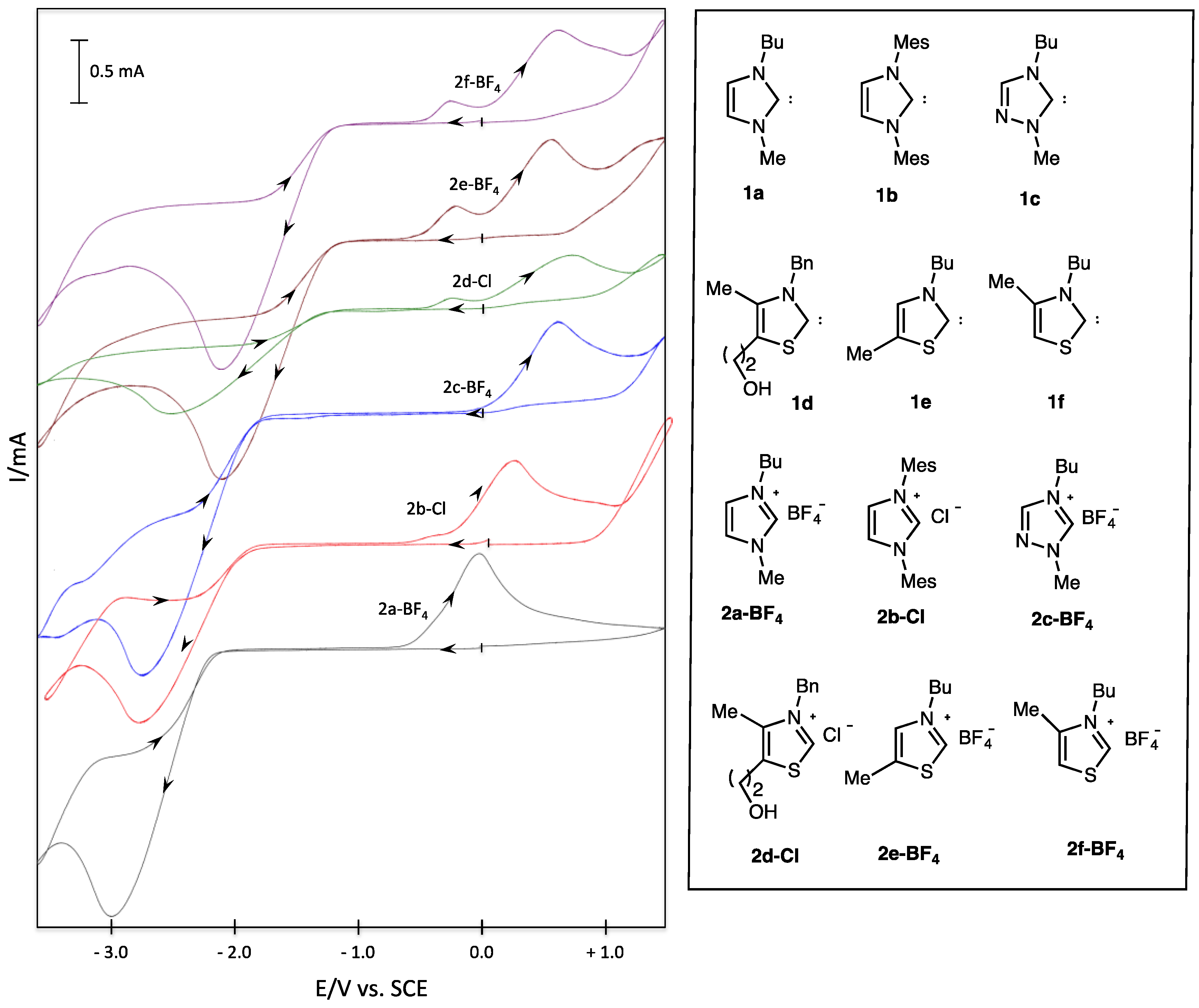
| Entry | Salt | Epred/V | Epox/V |
|---|---|---|---|
| 1 | 2a-BF4 | −3.00 | 0.00 |
| 2 | 2b-Cl | −2.87 | +0.22 |
| 3 | 2c-BF4 | −2.76 | +0.60 |
| 4 | 2d-Cl | −2.47 | +0.75 |
| 5 | 2e-BF4 | −2.11 | +0.58 |
| 6 | 2f-BF4 | −2.09 | +0.64 |
| NHCH+/NHC | Binding Energies (kJ/mol) | NHCH+–NHC 1 H-Bond Distance 2 (Å) | |
|---|---|---|---|
| NHCH+–DMF | NHCH+–NHC | ||
| 2a/1a | −76.75 | −88.41 | 2.00 |
| 2c/1c | −84.88 | −88.51 | 1.96 |
| 2e/1e | −78.65 | −92.59 | 2.03 |
| 2f/1f | −79.78 | −93.34 | 2.03 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feroci, M.; Chiarotto, I.; Inesi, A. Advances in the Knowledge of N-Heterocyclic Carbenes Properties. The Backing of the Electrochemical Investigation. Catalysts 2016, 6, 178. https://doi.org/10.3390/catal6110178
Feroci M, Chiarotto I, Inesi A. Advances in the Knowledge of N-Heterocyclic Carbenes Properties. The Backing of the Electrochemical Investigation. Catalysts. 2016; 6(11):178. https://doi.org/10.3390/catal6110178
Chicago/Turabian StyleFeroci, Marta, Isabella Chiarotto, and Achille Inesi. 2016. "Advances in the Knowledge of N-Heterocyclic Carbenes Properties. The Backing of the Electrochemical Investigation" Catalysts 6, no. 11: 178. https://doi.org/10.3390/catal6110178