
Untangling the Role of the Capping Agent in Nanocatalysis: Recent Advances and Perspectives


Abstract
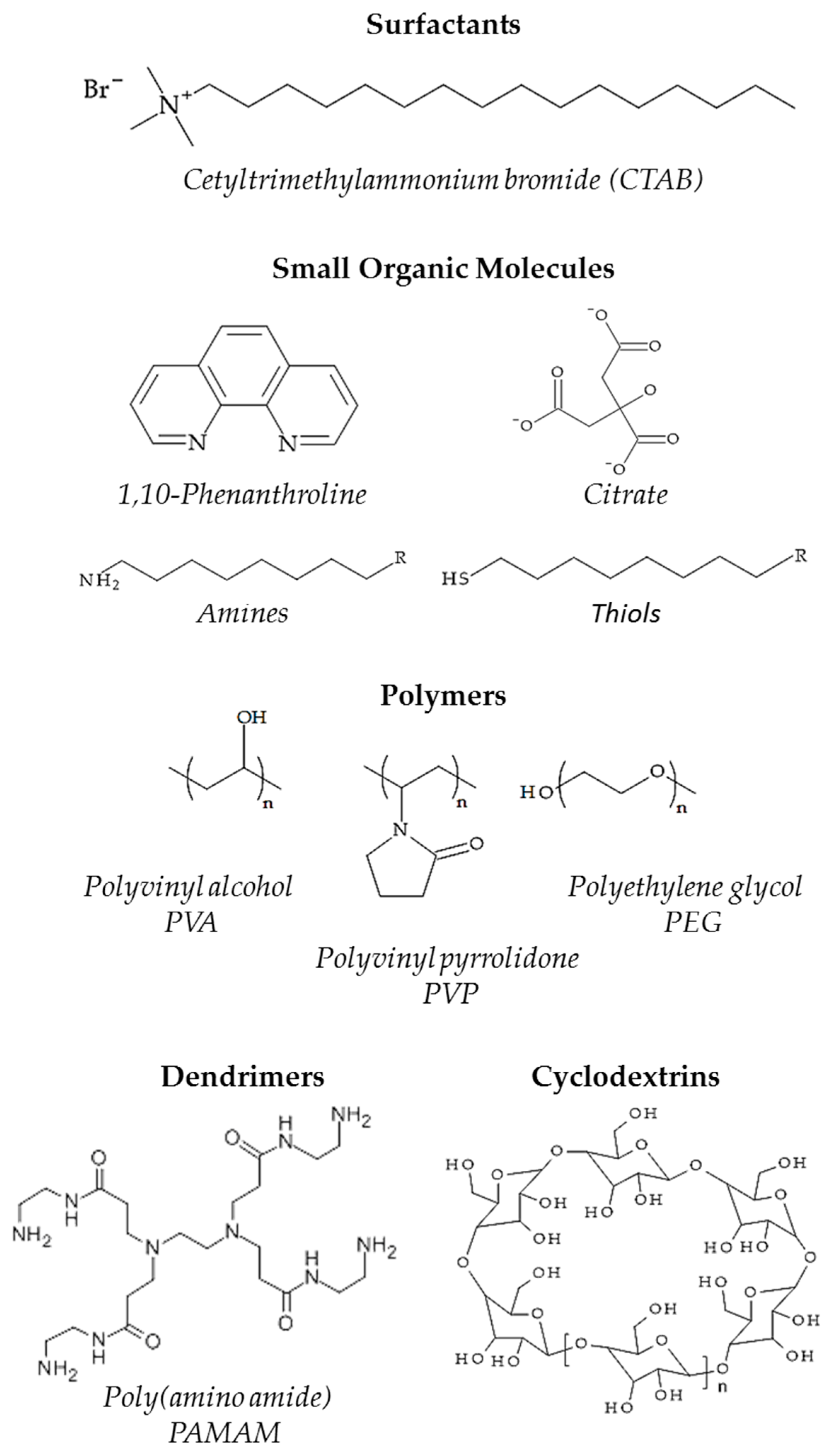
:1. Introduction
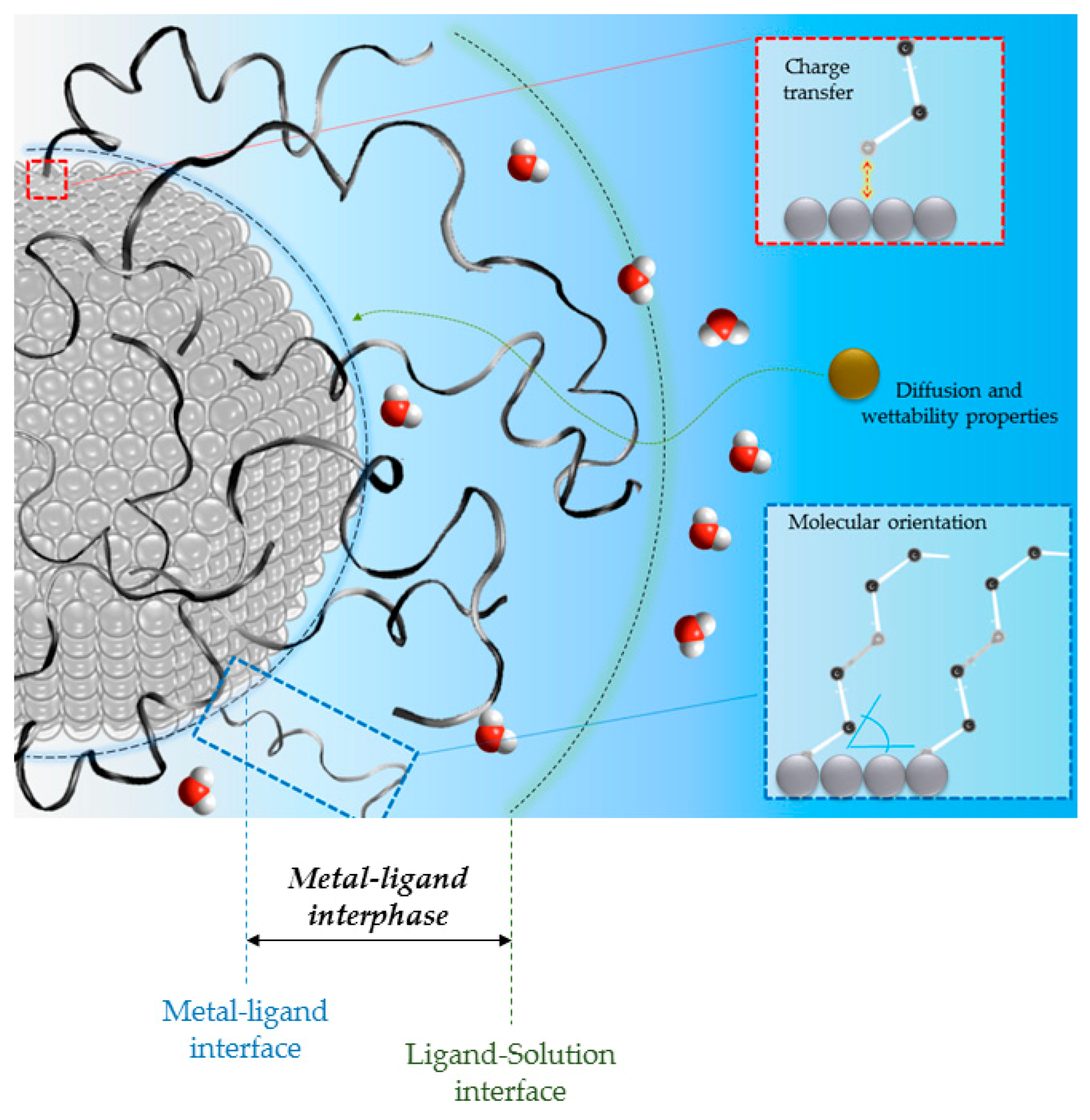
2. The Metal-Ligand Interphase and Its Impact on the Catalytic Performances
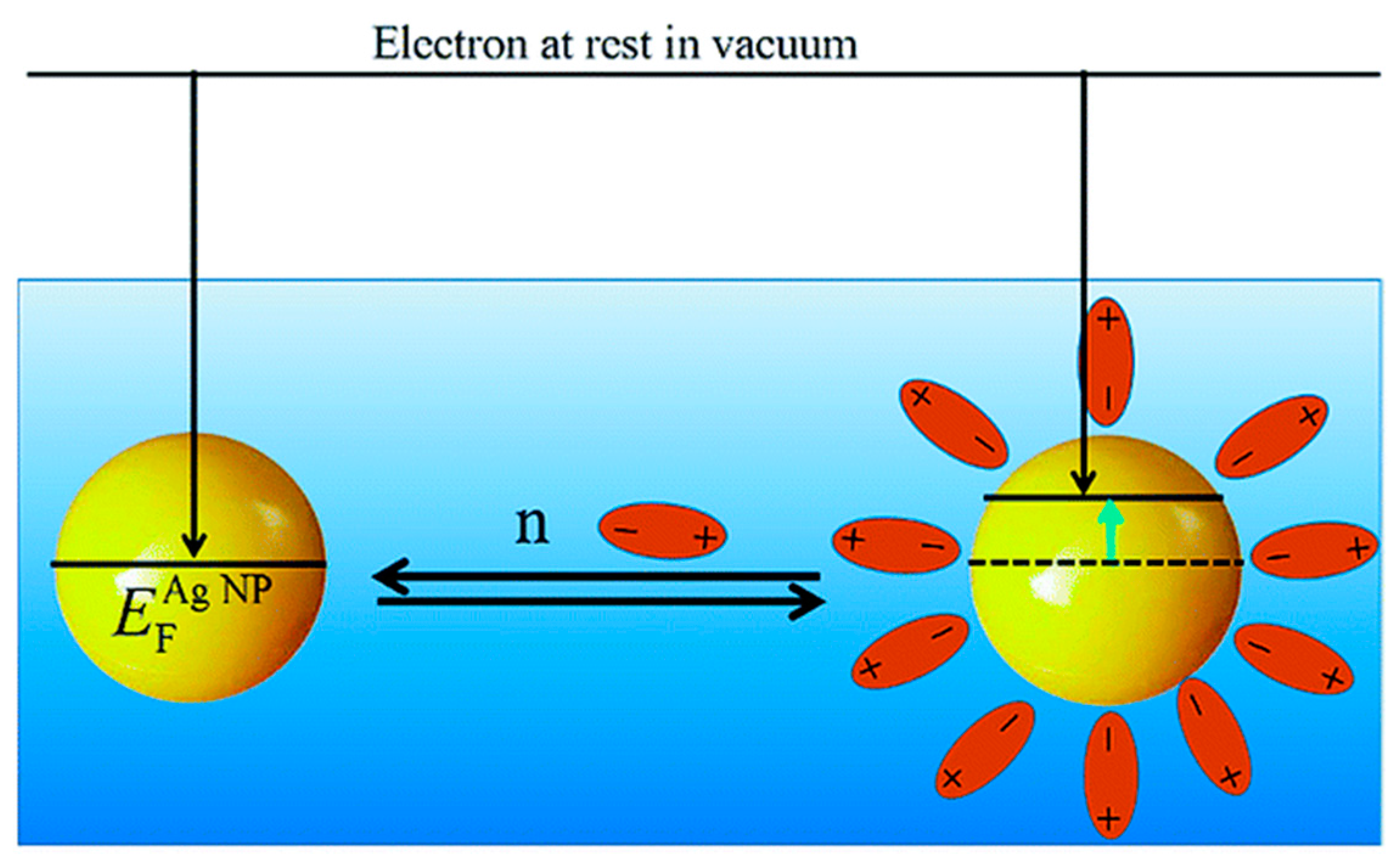
2.1. Chemical Bonding and Electronic Structure at the Metal-Ligand Interphase
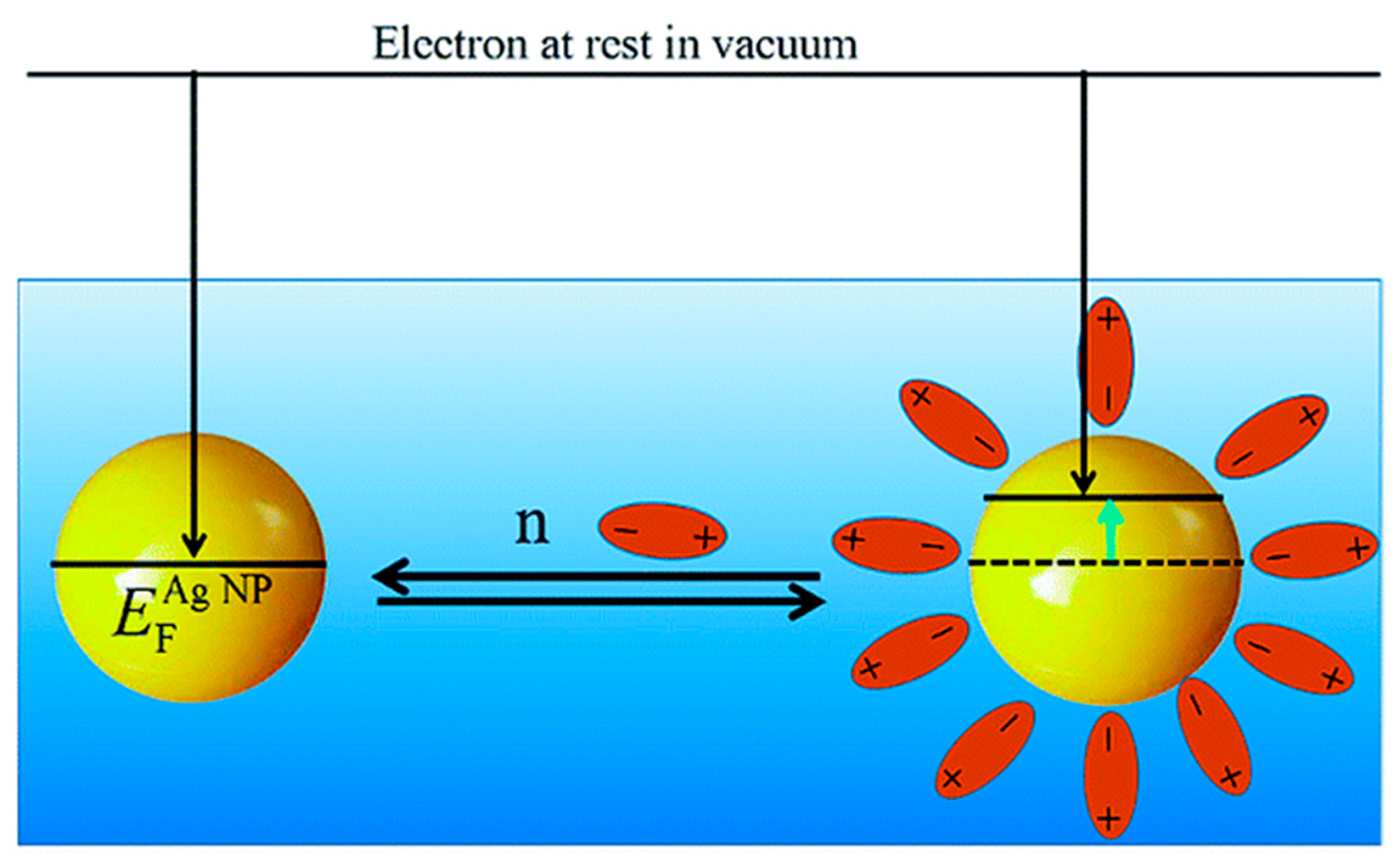
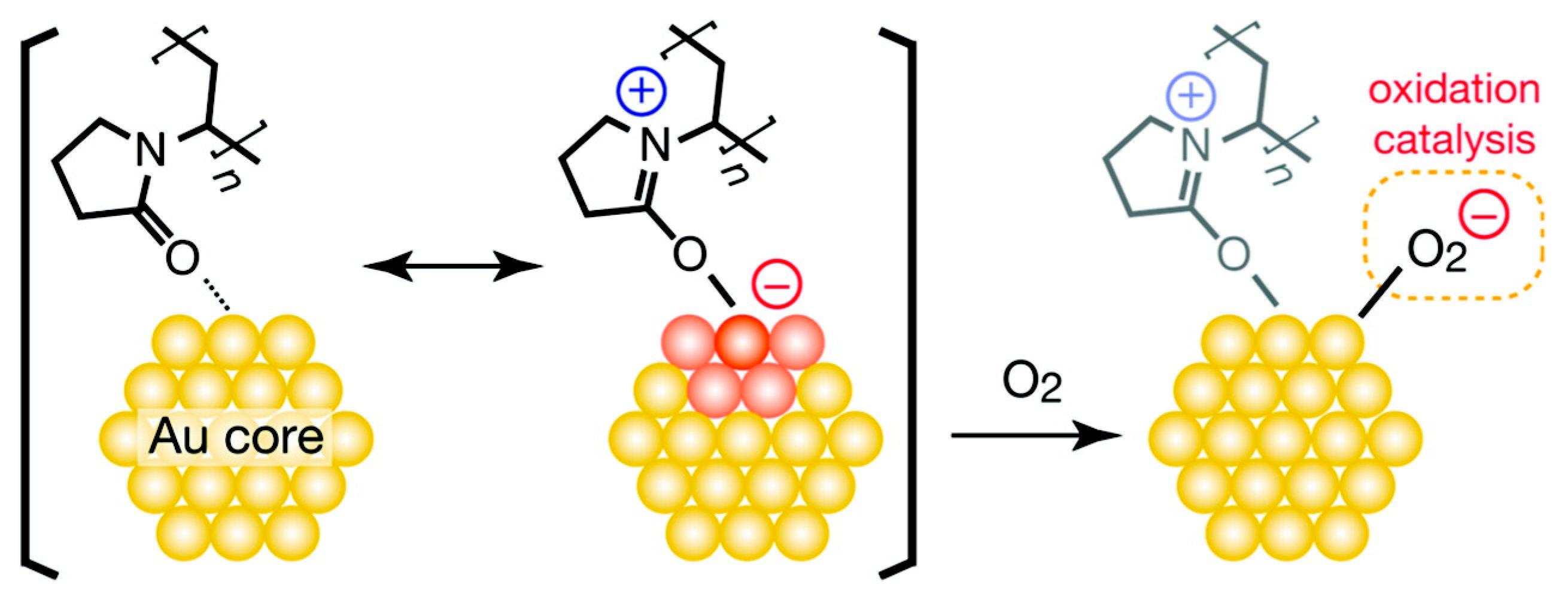
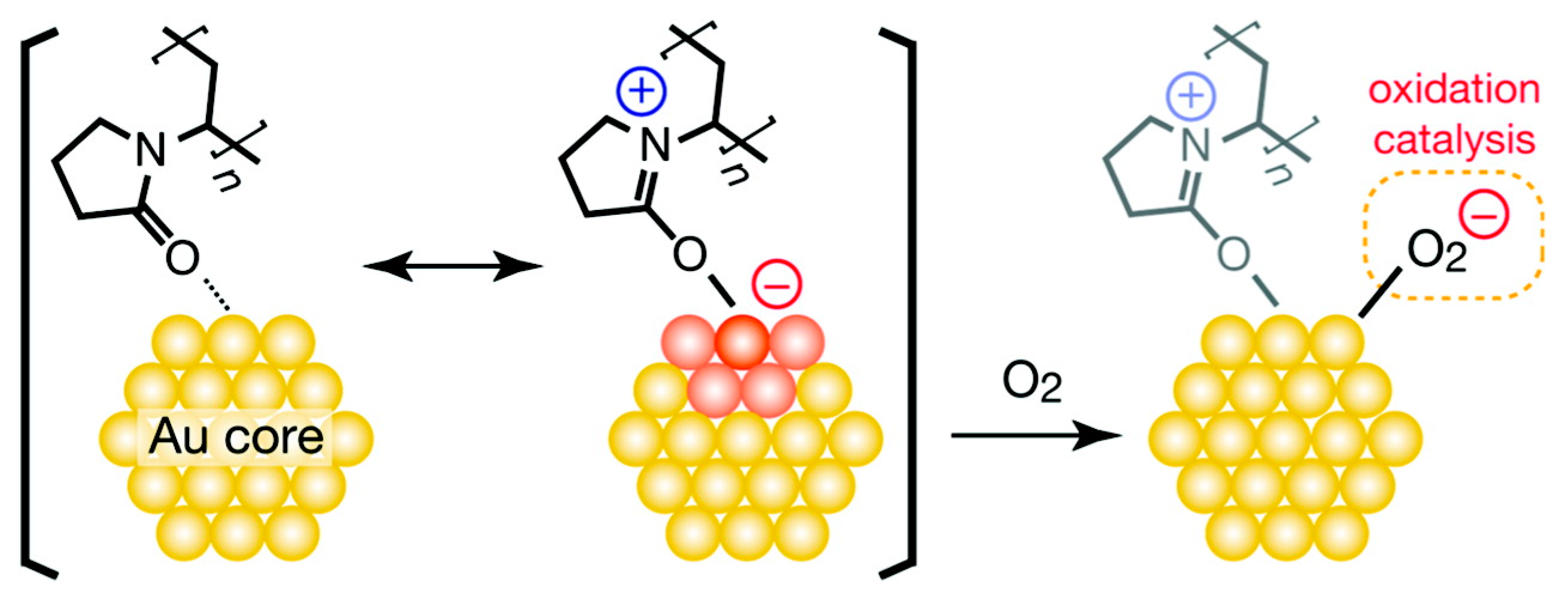
2.1.1. Activity and Selectivity Enhancement Induced by Charge Transfer
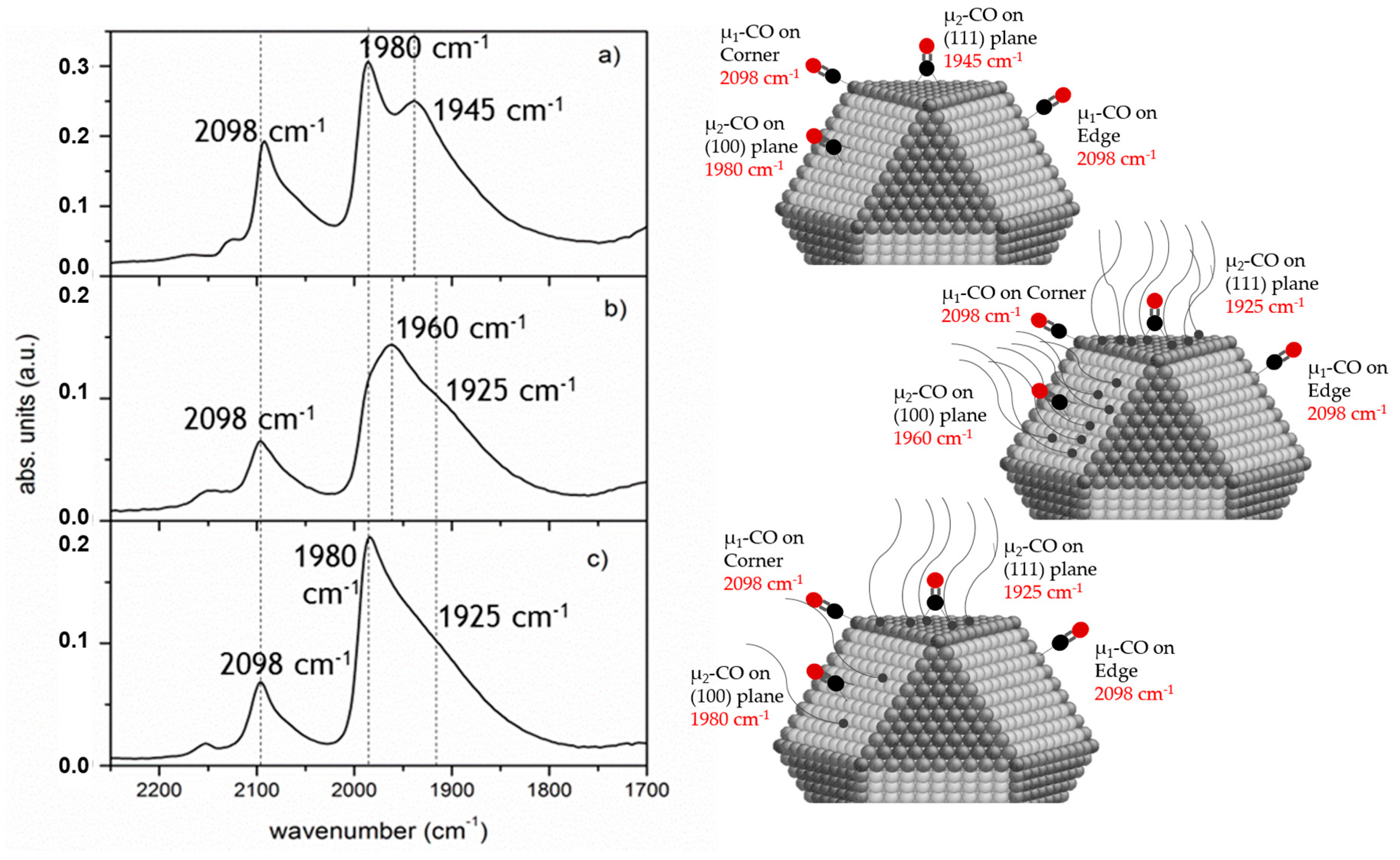
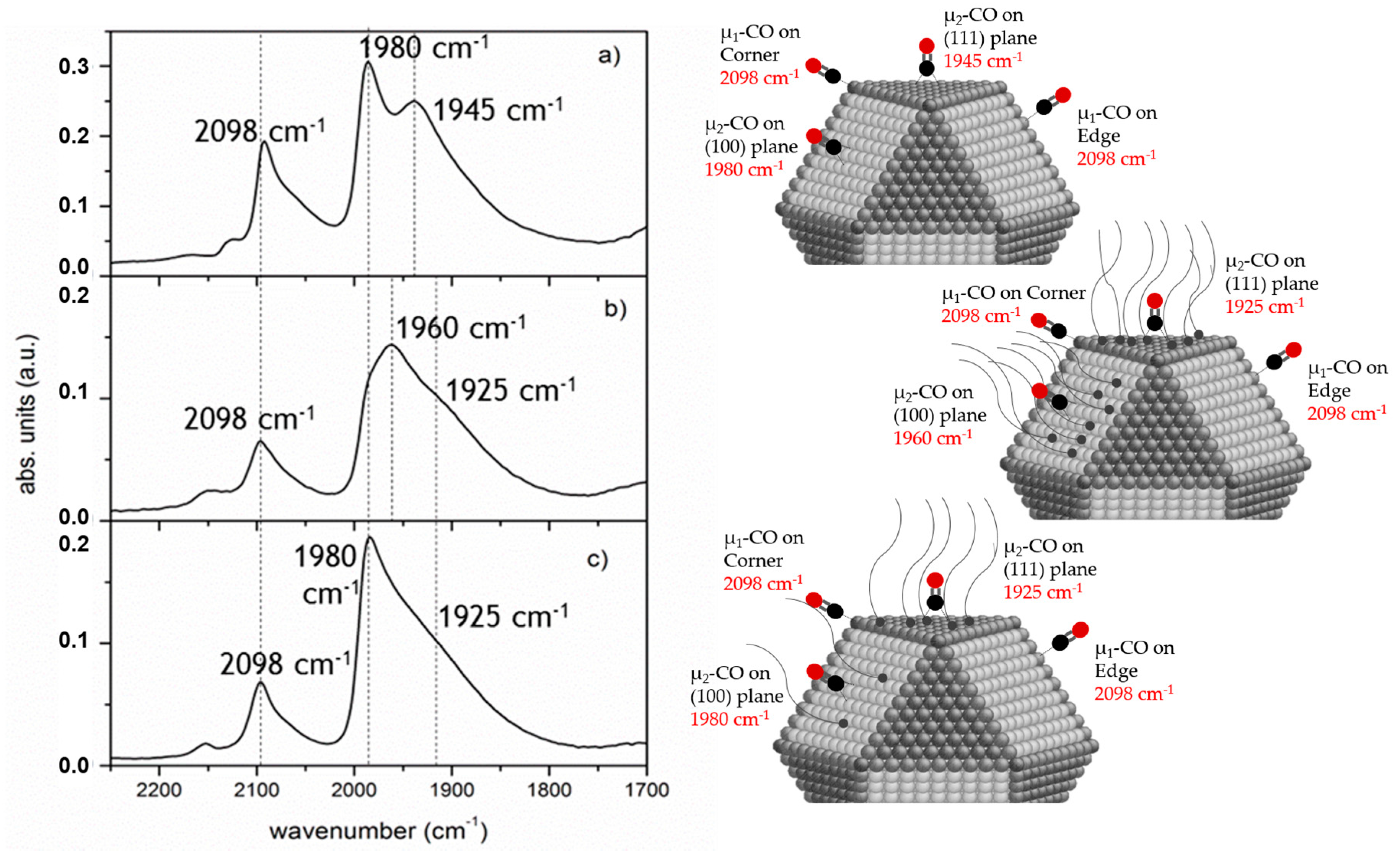
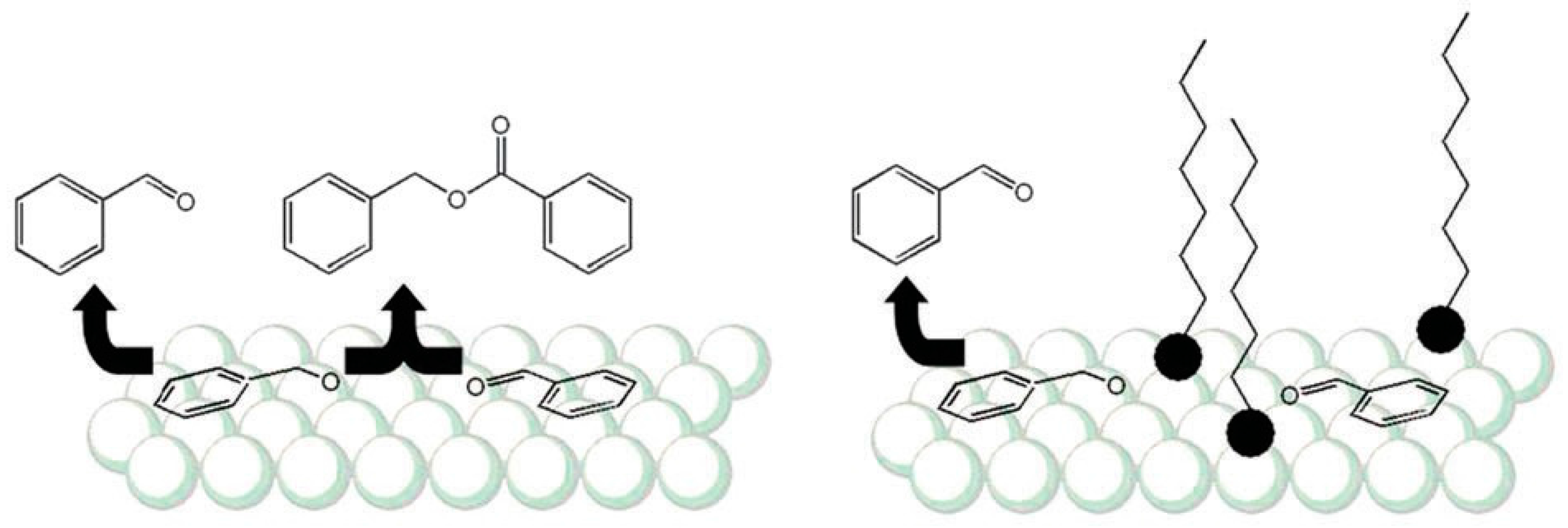
2.1.2. Shielding Effect and Selective Blocking
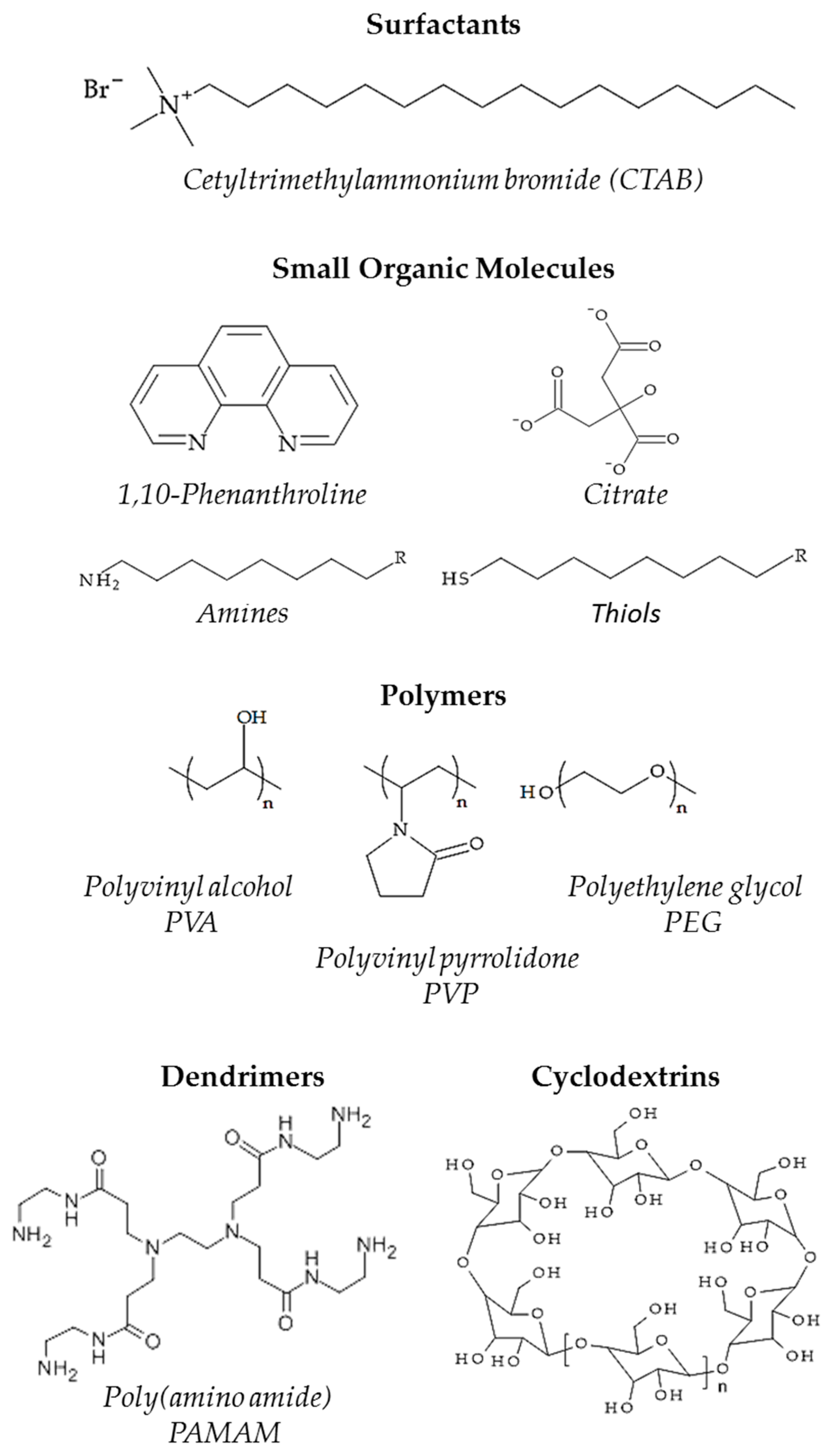
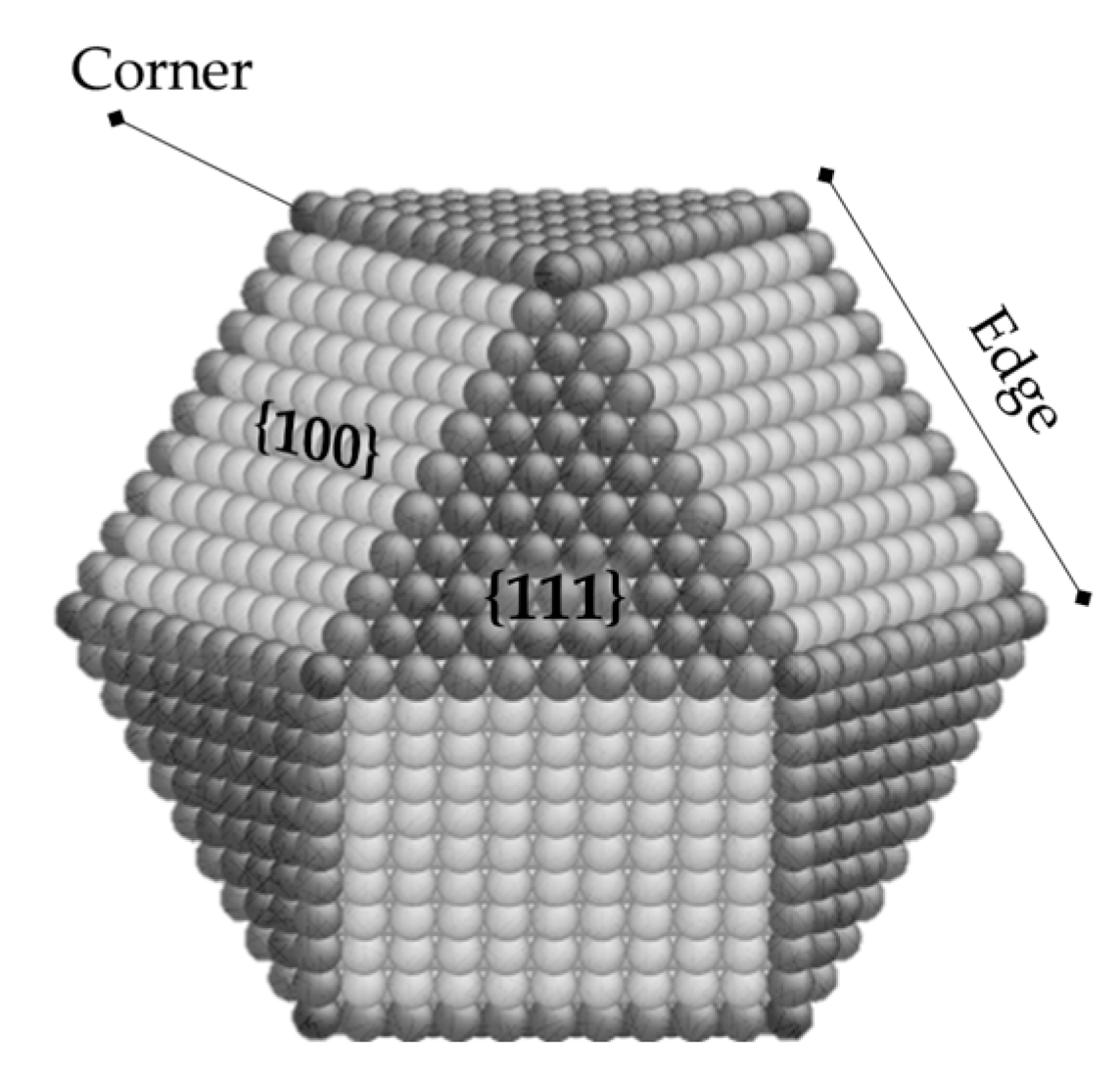
2.2. The Orientation of Molecules and the Morphology of Metal-Ligand Interphase
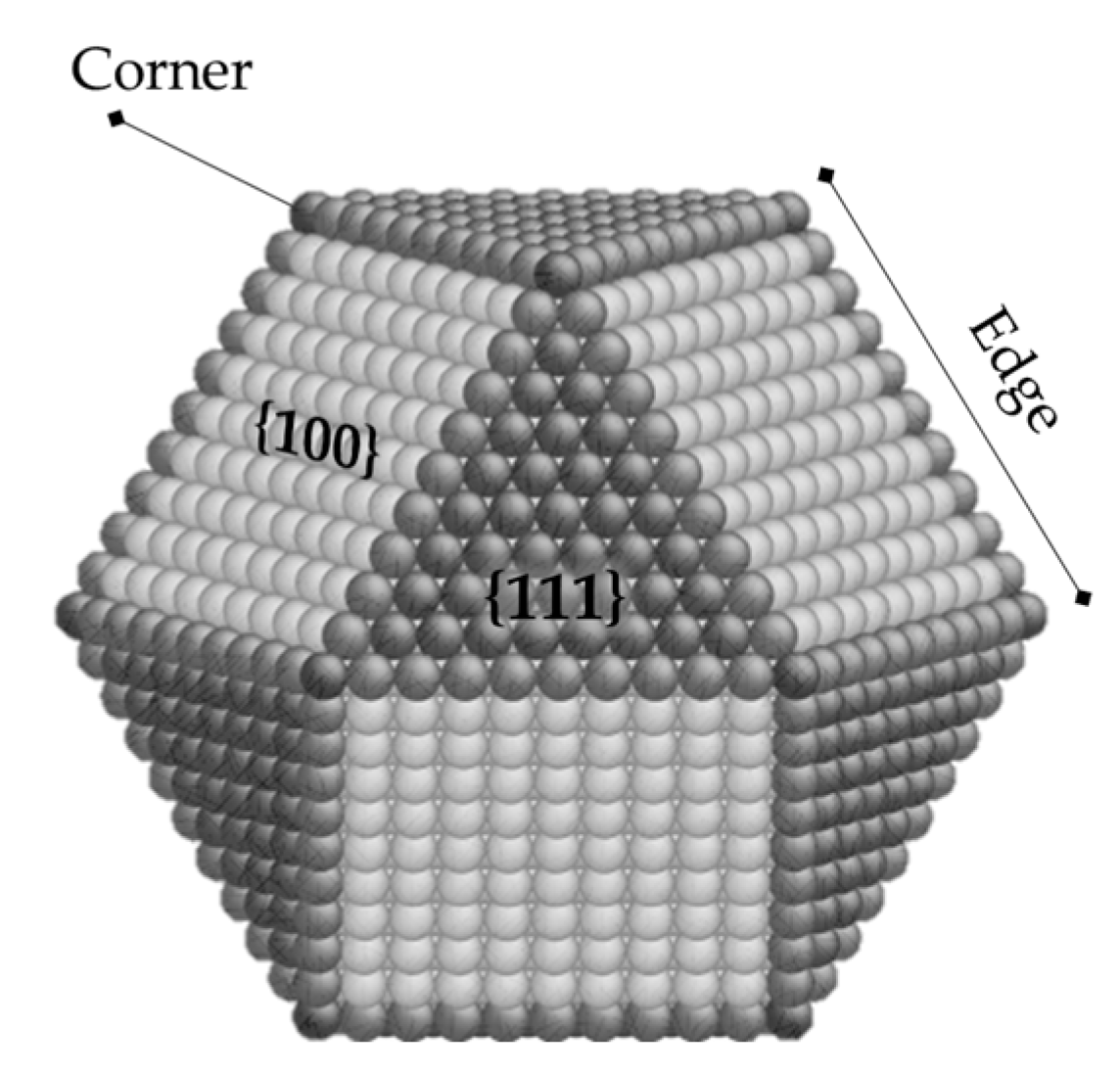
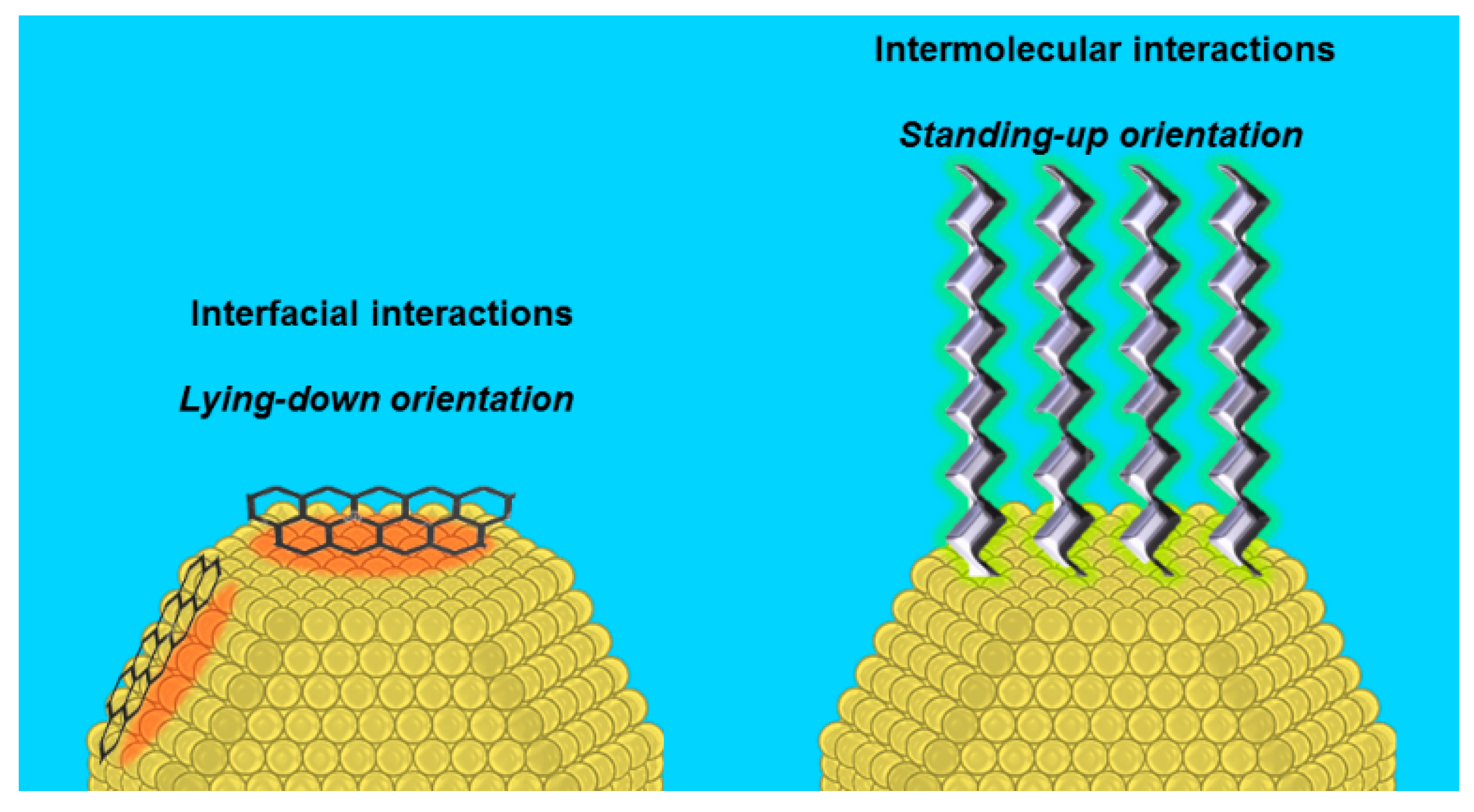
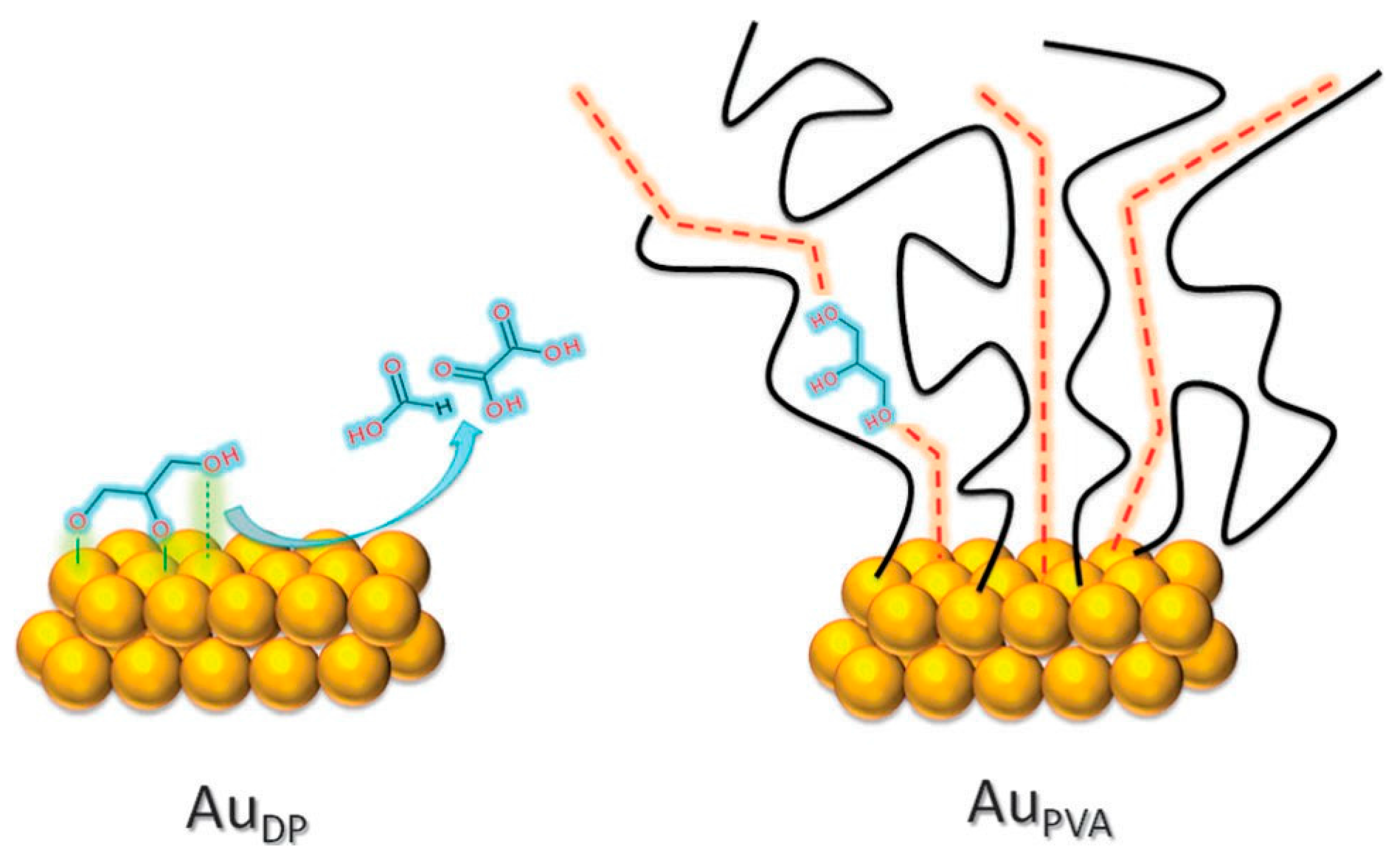
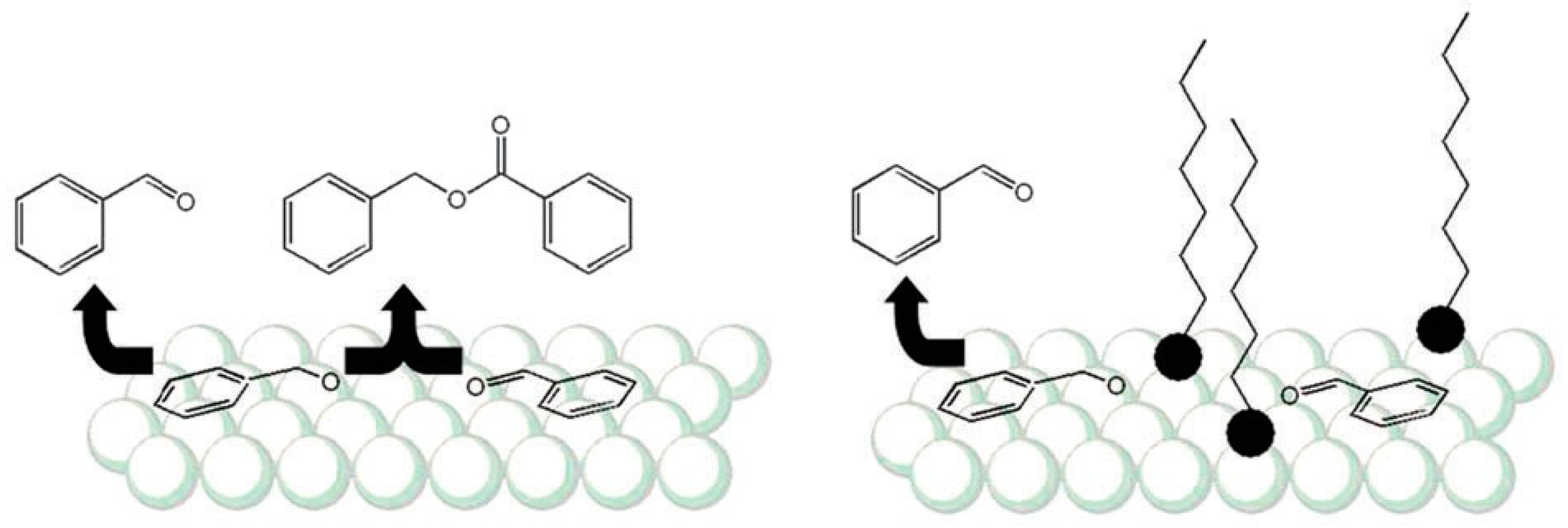
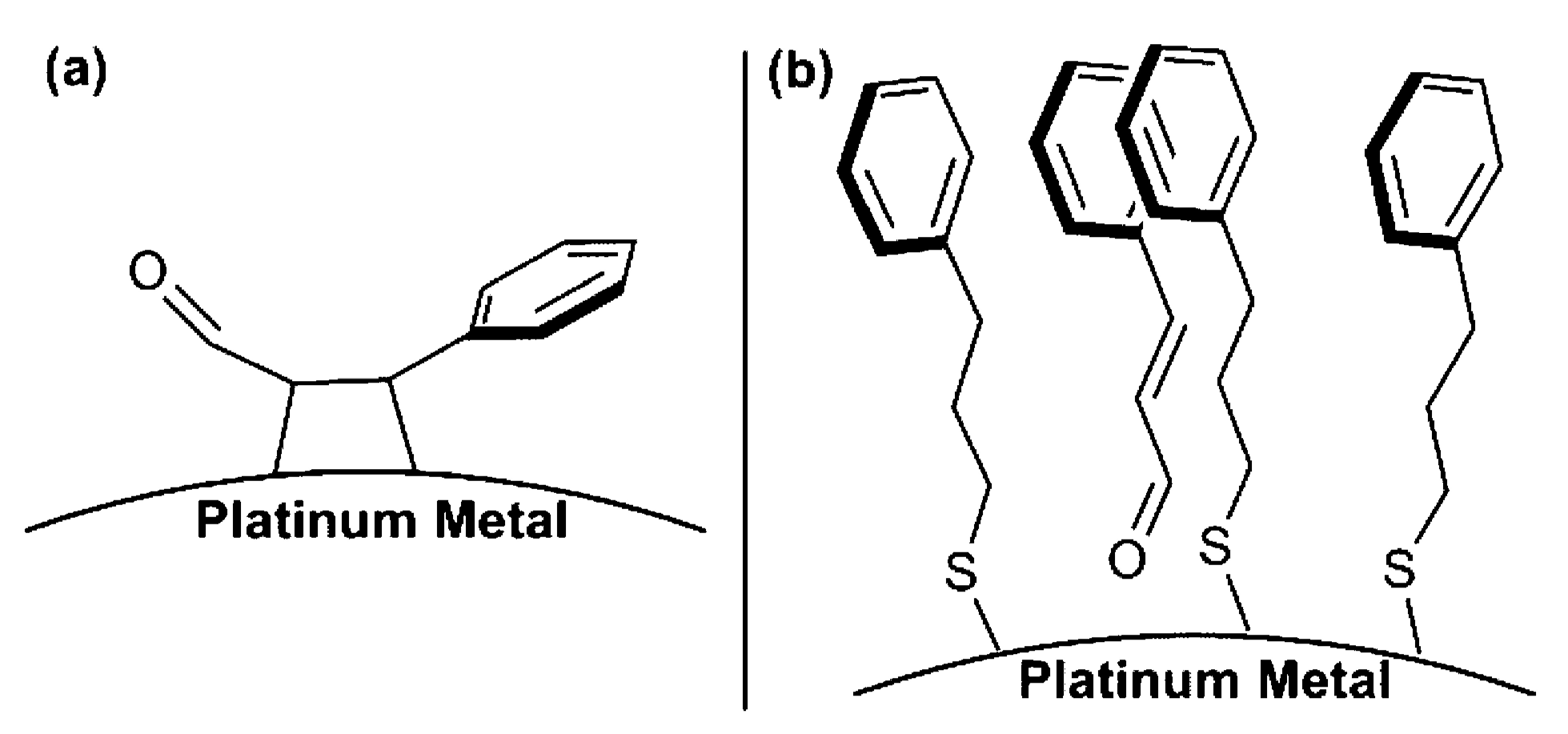
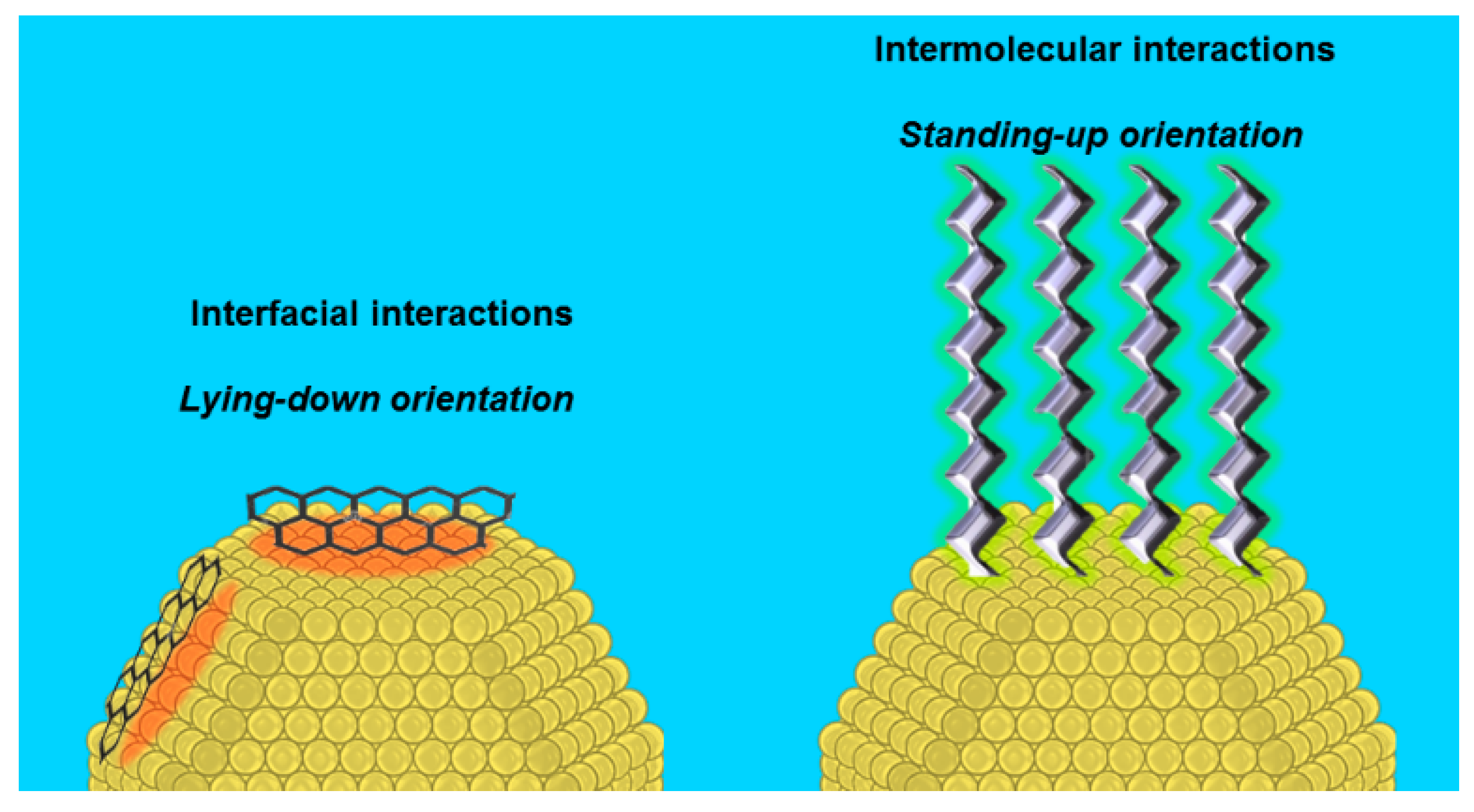
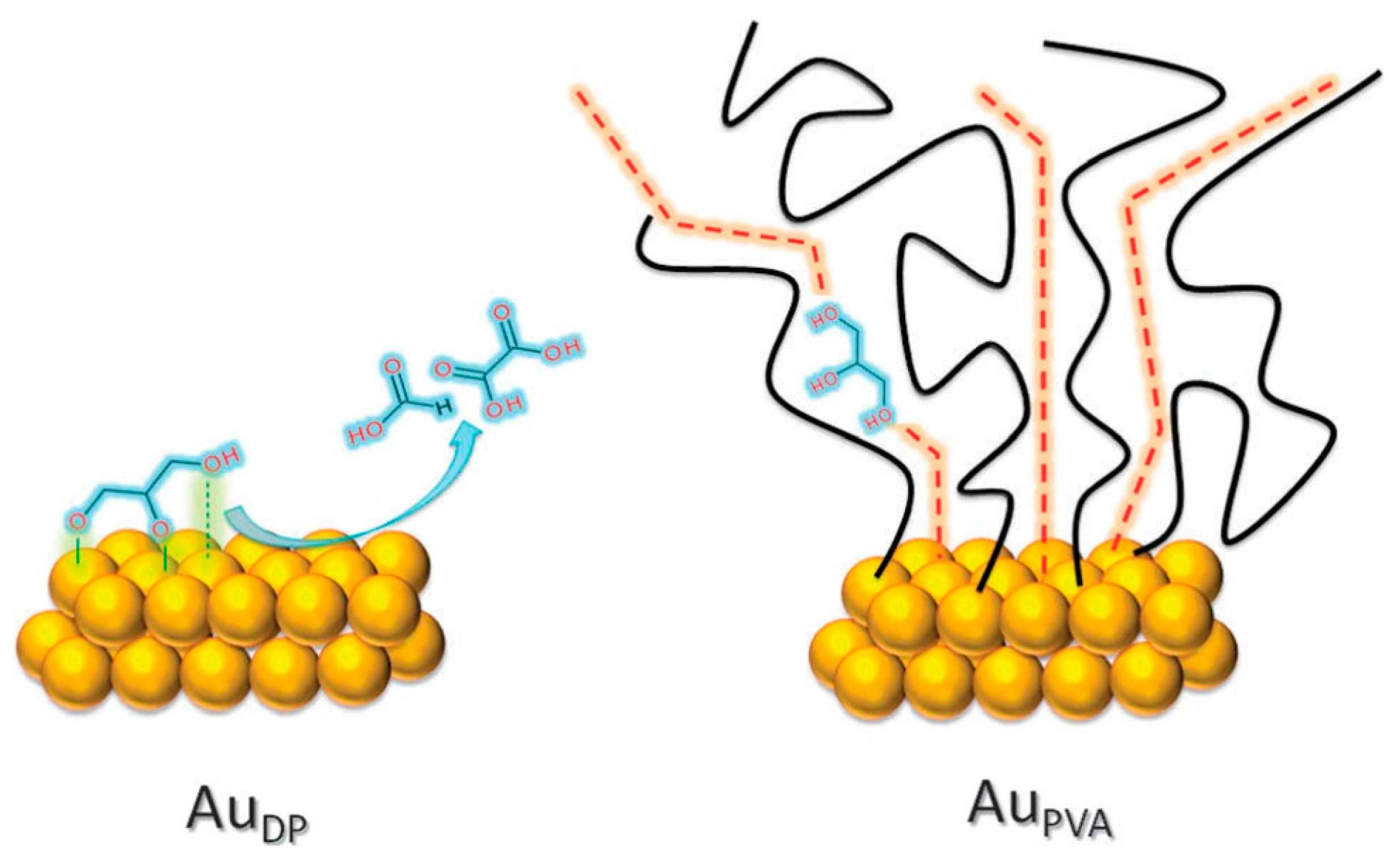
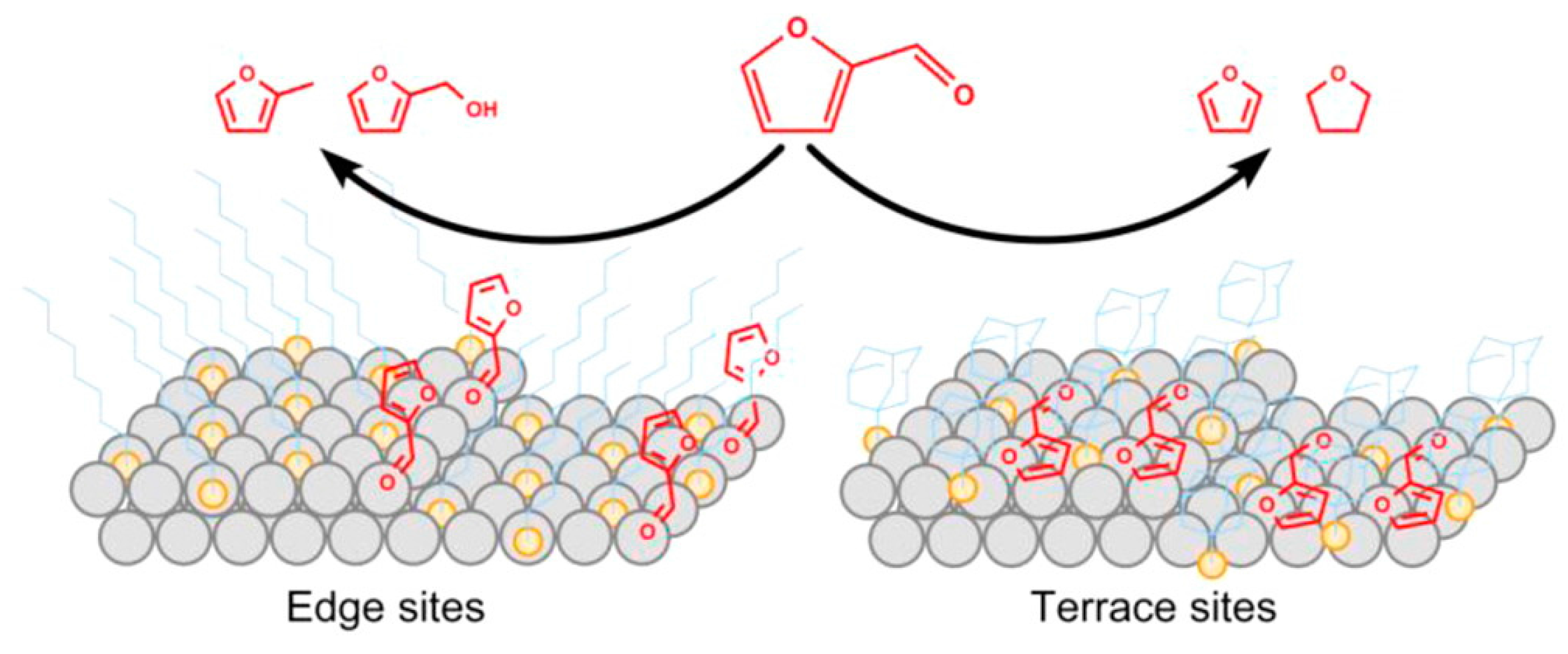
2.2.1. Steric Effects and Surface Crowding
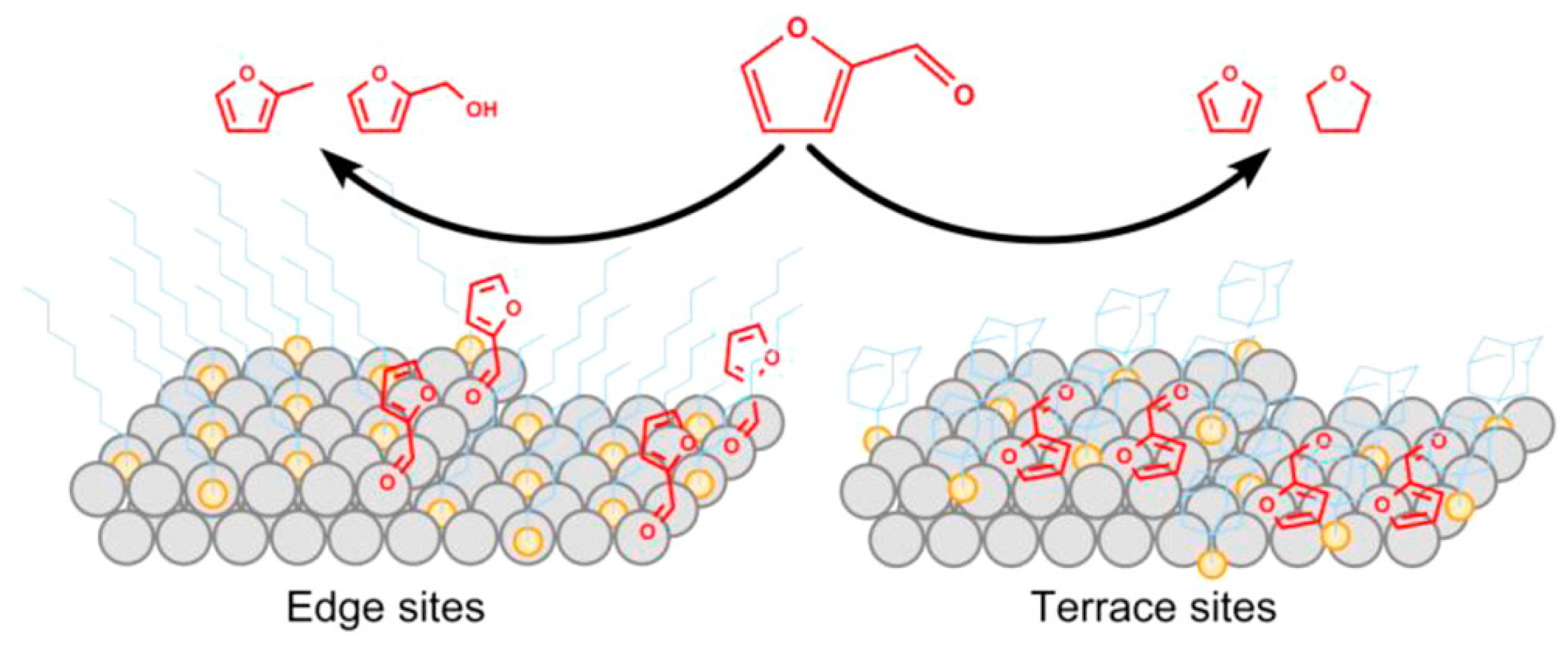
2.2.2. Molecular Recognition
2.2.3. Stereo-Directing Interaction between Ligands and Reactants
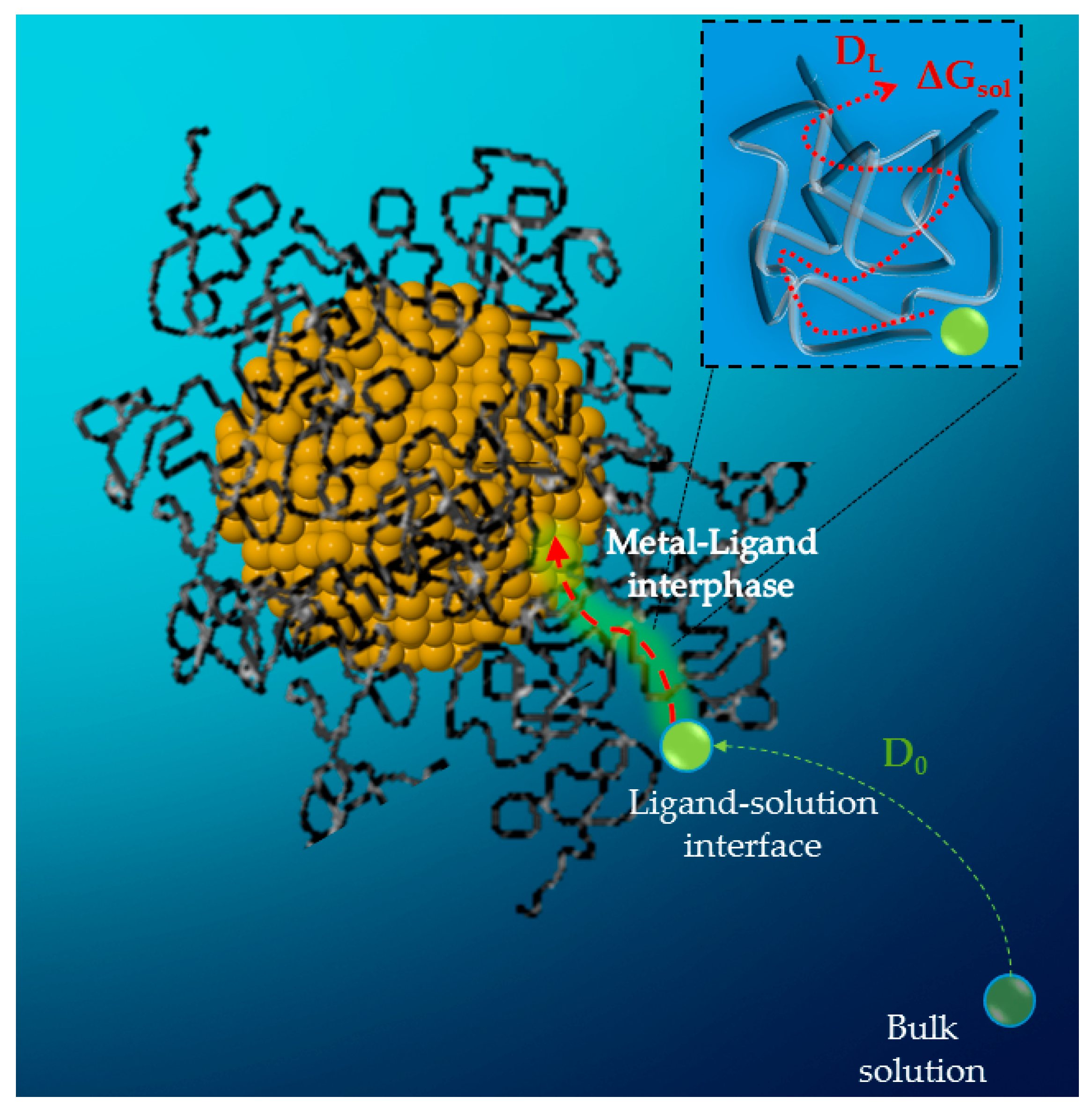
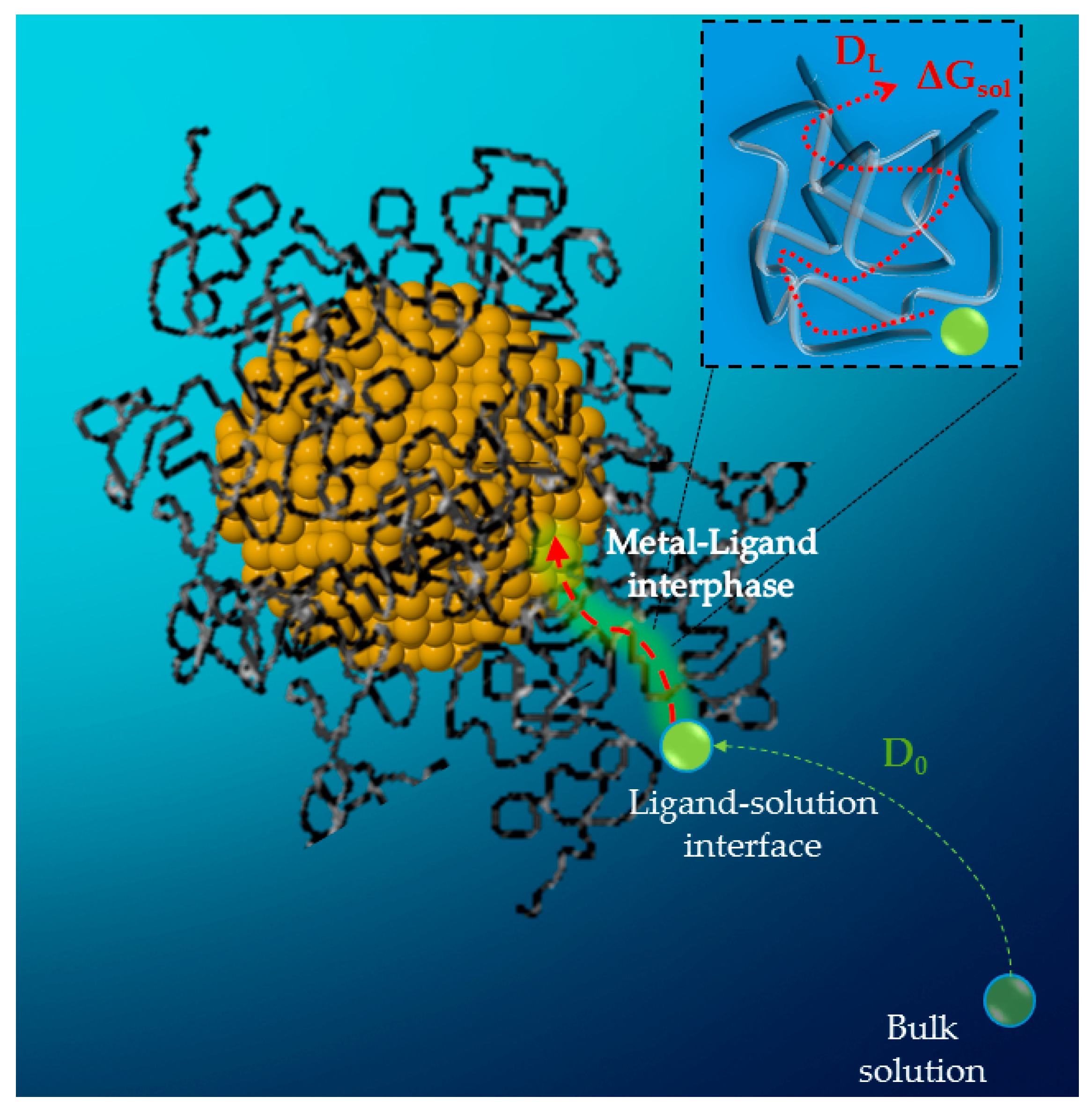
2.3. Modelling the Kinetics and the Diffusion Process in Capped Nanoparticles
3. Conclusions and Perspectives
- The difficulty to synthesize nanocrystals with controlled shape, size and defect distribution in a reproducible manner and independently from the capping agent.
- The challenge of isolating the pure effect of the capping agent from other factors (nanoparticle structure, support, etc.)
- The need to distinguish and to separately control the different contributions (electronic and geometric) on the capping agent behaviour.
- The complexity of real experimental conditions compared to models.
- A fine control of the synthesis
- A characterization, able to integrate several ex situ and in situ techniques for probing processes at metal-ligand interface under reaction conditions
- The development of a comprehensive theory for the interpretation
Conflicts of Interest
References
- Schmid, G. Nanoparticles: From Theory to Application, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2011; pp. 1–4. [Google Scholar]
- Rao, C.N.R.; Müller, A.; Cheetham, A.K. The Chemistry of Nanomaterials: Synthesis, Properties and Applications, 1st ed.; Wiley-VCH: Weinheim, Germany, 2006; Volume 1, pp. 1–11. [Google Scholar]
- Daniel, M.C.; Astruc, D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.A.; Contescu, C.I.; Putyera, K. Dekker Encyclopedia of Nanoscience and Nanotechnology, 3rd ed.; CRC Press: New York, NY, USA, 2014; Volume 3, pp. 1813–1904. [Google Scholar]
- Astruc, D. Nanoparticles and Catalysis; Wiley-VCH: Weinheim, Germany, 2008; Volume 1. [Google Scholar]
- Roucoux, A.; Schulz, J.; Patin, H. Reduced transition metal colloids: A novel family of reusable catalysts? Chem. Rev. 2002, 102, 3757–3778. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.F. Metal Nanoparticles for Catalysis: Advances and Applications, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2014. [Google Scholar]
- Stark, W.J.; Stoessel, P.R.; Wohlleben, W.; Hafner, A. Industrial applications of nanoparticles. Chem. Soc. Rev. 2015, 44, 5793–5805. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D.; Lu, F.; Aranzaes, J.R. Nanoparticles as recyclable catalysts: The frontier between homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 2005, 44, 7852–7872. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, R.A.; Neurock, M. Molecular Heterogeneous Catalysis: A Conceptual and Computational Approach, 1st ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 83–160. [Google Scholar]
- Schauermann, S.; Nilius, N.; Shaikhutdinov, S.; Freund, H.J. Nanoparticles for heterogeneous catalysis: New mechanistic insights. Acc. Chem. Res. 2012, 46, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Schlögl, R. Heterogeneous catalysis. Angew. Chem. Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Dimitratos, N.; Chan-Thaw, C.E.; Hammond, C.; Veith, G.M.; Wang, D.; Manzoli, M.; Prati, L.; Hutchings, G.J. Characterisation of gold catalysts. Chem. Soc. Rev. 2016, 45, 4953–4994. [Google Scholar] [CrossRef] [PubMed]
- Morsbach, E.; Spéder, J.; Arenz, M.; Brauns, E.; Lang, W.; Kunz, S.; Baumer, M. Stabilizing Catalytically Active Nanoparticles by Ligand Linking: Toward Three-Dimensional Networks with High Catalytic Surface Area. Langmuir 2014, 30, 5564–5573. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Schiavoni, M.; Prati, L. Material science for the support design: A powerful challenge for catalysis. Catal. Sci. Technol. 2012, 2, 673–682. [Google Scholar] [CrossRef]
- Smith, D.K.; Korgel, B.A. The importance of the CTAB surfactant on the colloidal seed-mediated synthesis of gold nanorods. Langmuir 2008, 24, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Gavia, D.J.; Shon, Y.-S. Catalytic Properties of Unsupported Palladium Nanoparticle Surfaces Capped with Small Organic Ligands. ChemCatChem 2015, 7, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Baygazieva, E.K.; Yesmurzayeva, N.N.; Tatykhanova, G.S.; Mun, G.A.; Khutoryanskiy, V.V. Polymer Protected Gold Nanoparticles: Synthesis, Characterization and Application in Catalysis. Int. J. Biol. Chem. Sci. 2014, 14, 14–23. [Google Scholar]
- Crooks, R.M.; Zhao, M.; Sun, L.; Chechik, V.; Yeung, L.K. Dendrimer-encapsulated metal nanoparticles: Synthesis, characterization, and applications to catalysis. Acc. Chem. Res. 2001, 34, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Noël, S.; Léger, B.; Ponchel, A.; Philippot, K.; Denicourt-Nowicki, A.; Roucoux, A.; Monflier, E. Cyclodextrin-based systems for the stabilization of metallic (0) nanoparticles and their versatile applications in catalysis. Catal. Today 2014, 235, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Raveendran, P.; Fu, J.; Wallen, S.L. Completely “green” synthesis and stabilization of metal nanoparticles. J. Am. Chem. Soc. 2003, 125, 13940–13941. [Google Scholar] [CrossRef] [PubMed]
- Neouze, M.-A.; Schubert, U. Surface Modification and Functionalization of Metal and Metal Oxide Nanoparticles by Organic Ligands. Monatshefte Chem. Chem. Mon. 2008, 139, 183–195. [Google Scholar] [CrossRef]
- Prati, L.; Villa, A. Gold colloids: From quasi-homogeneous to heterogeneous catalytic systems. Acc. Chem. Res. 2014, 47, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Li, D.G.; Wang, C.; Tripkovic, D.; Sun, S.H.; Markovic, N.M.; Stamenkovic, V.R. Surfactant Removal for Colloidal Nanoparticles from Solution Synthesis: The Effect on Catalytic Performance. ACS Catal. 2012, 2, 1358–1362. [Google Scholar] [CrossRef]
- Campisi, S.; Chan-Thaw, C.E.; Wang, D.; Villa, A.; Prati, L. Metal nanoparticles on carbon based supports: The effect of the protective agent removal. Catal. Today 2016, in press. [Google Scholar] [CrossRef]
- Rioux, R.M.; Song, H.; Grass, M.; Habas, S.; Niesz, K.; Hoefelmeyer, J.D.; Yang, P.; Somorjai, G.A. Monodisperse Pt nanoparticles with well-defined surface structure: Synthesis, characterization, catalytic properties, and future prospects. Top. Catal. 2006, 39, 167–174. [Google Scholar] [CrossRef]
- Lopez-Sanchez, J.A.; Dimitratos, N.; Hammond, C.; Brett, G.L.; Kesavan, L.; White, S.; Miedziak, P.; Tiruvalam, R.; Jenkins, R.L.; Carley, A.F.; et al. Facile removal of stabilizer-ligands from supported gold nanoparticles. Nat. Chem. 2011, 3, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, C.; Park, J.Y.; Yamada, Y.; Lee, H.S.; Tsung, C.K.; Yang, P.D.; Somorjai, G.A. Sum Frequency Generation and Catalytic Reaction Studies of the Removal of Organic Capping Agents from Pt Nanoparticles by UV-Ozone Treatment. J. Phys. Chem. C 2009, 113, 6150–6155. [Google Scholar] [CrossRef]
- Niu, Z.; Li, Y. Removal and utilization of capping agents in nanocatalysis. Chem. Mater. 2013, 26, 72–83. [Google Scholar] [CrossRef]
- Wu, B.; Zheng, N. Surface and interface control of noble metal nanocrystals for catalytic and electrocatalytic applications. Nanotoday 2013, 8, 168–197. [Google Scholar] [CrossRef]
- Grubbs, R.B. Roles of polymer ligands in nanoparticle stabilization. Polym. Rev. 2007, 47, 197–215. [Google Scholar] [CrossRef]
- Sonström, P.; Arndt, D.; Wang, X.; Zielasek, V.; Bäumer, M. Ligand Capping of Colloidally Synthesized Nanoparticles—A Way to Tune Metal–Support Interactions in Heterogeneous Gas-Phase Catalysis. Angew. Chem. Int. Ed. 2011, 50, 3888–3891. [Google Scholar] [CrossRef] [PubMed]
- Boudart, M. Principles of Heterogeneous Catalysis. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 1997; p. 1. [Google Scholar]
- Somorjai, G.A.; Li, Y. Introduction to Surface Chemistry and Catalysis, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ertl, G. Catalytic Ammonia Synthesis, 1st ed.; Jennings, J.R., Ed.; Plenum: New York, NY, USA, 1991; p. 109. [Google Scholar]
- Hoffmann, R. Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures, 1st ed.; Wiley-VCH: New York, NY, USA, 1988. [Google Scholar]
- Ponec, V.; Bond, G.C. Catalysis by metals and alloys. In Studies in Surface Science and Catalysis, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 95, pp. 7–72. [Google Scholar]
- Greeley, J.; Nørskov, J.K.; Mavrikakis, M. Electronic structure and catalysis on metal surfaces. Annu. Rev. Phys. Chem. 2002, 53, 319–348. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, M.D.; Peljo, P.; Méndez, M.A.; Smirnov, E.; Girault, H.H. Charging and discharging at the nanoscale: Fermi level equilibration of metallic nanoparticles. Chem. Sci. 2015, 6, 2705–2720. [Google Scholar] [CrossRef]
- Flores, F.; Ortega, J. Basic Theory of the Molecule-Metal Interface. In The Molecule-Metal Interface; Koch, N., Ueno, N., Wee, A.T.S., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 17–50. [Google Scholar]
- Adams, D.M.; Brus, L.; Chidsey, C.E.; Creager, S.; Creutz, C.; Kagan, C.R.; Kamat, P.V.; Lieberman, M.; Lindsay, S.; Marcus, R.A.; et al. Charge Transfer on the Nanoscale: Current Status. J. Phys. Chem. B 2003, 107, 6668–6697. [Google Scholar] [CrossRef]
- Bredas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5003. [Google Scholar] [CrossRef] [PubMed]
- Witte, G.; Lukas, S.; Bagus, P.S.; Wöll, C. Vacuum level alignment at organic/metal junctions: “Cushion” effect and the interface dipole. Appl. Phys. Lett. 2005, 87, 263502. [Google Scholar] [CrossRef]
- Kera, S.; Yabuuchi, Y.; Yamane, H.; Setoyama, H.; Okudaira, K.K.; Kahn, A.; Ueno, N. Impact of an interface dipole layer on molecular level alignment at an organic-conductor interface studied by ultraviolet photoemission spectroscopy. Phys. Rev. B: Condens. Matter Mater. Phys. 2004, 70, 085304. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Nath, S.; Kundu, S.; Esumi, K.; Pal, T. Solvent and Ligand Effects on the Localized Surface Plasmon Resonance (LSPR) of Gold Colloids. J. Phys. Chem. B 2004, 108, 13963–13971. [Google Scholar] [CrossRef]
- Battocchio, C.; Porcaro, F.; Mukherjee, S.; Magnano, E.; Nappini, S.; Fratoddi, I.; Quintiliani, M.; Russo, M.V.; Polzonetti, G. Gold nanoparticles stabilized with aromatic thiols: Interaction at the molecule-metal interface and ligand arrangement in the molecular shell investigated by SR-XPS and NEXAFS. J. Phys. Chem. C 2014, 118, 8159–8168. [Google Scholar] [CrossRef]
- Jiang, P.; Li, S.Y.; Xie, S.S.; Gao, Y.; Song, L. Machinable long PVP-stabilized silver nanowires. Chem. A Eur. J. 2004, 10, 4817–4821. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, H.; Qszwaldowski, R.; Pou, P.; Ortega, J.; Pérez, R.; Flores, F.; Kahn, A. Dipole formation at metal/PTCDA interfaces: Role of the charge neutrality level. Europhys. Lett. 2004, 65, 802–808. [Google Scholar] [CrossRef]
- Borodko, Y.; Humphrey, S.M.; Tilley, T.D.; Frei, H.; Somorjai, G.A. Charge-transfer interaction of poly(vinylpyrrolidone) with platinum and rhodium nanoparticles. J. Phys. Chem. C 2007, 111, 6288–6295. [Google Scholar] [CrossRef]
- Soe, W.H.; Manzano, C.; de Sarkar, A.; Chandrasekhar, N.; Joachim, C. Direct Observation of Molecular Orbitals of Pentacene Physisorbed on Au(111) by Scanning Tunneling Microscope. Phys. Rev. Lett. 2009, 102, 176102. [Google Scholar] [CrossRef] [PubMed]
- Susut, C.; Chen, D.-J.; Sun, S.-G.; Tong, Y.J. Capping polymer-enhanced electrocatalytic activity on Pt nanoparticles: A combined electrochemical and in situ IR spectroelectrochemical study. Phys. Chem. Chem. Phys. 2011, 13, 7467–7474. [Google Scholar] [CrossRef] [PubMed]
- Techane, S.D.; Gamble, L.J.; Castner, D.G. Multitechnique characterization of self-assembled carboxylic acid-terminated alkanethiol monolayers on nanoparticle and flat gold surfaces. J. Phys. Chem. C 2011, 115, 9432–9441. [Google Scholar] [CrossRef] [PubMed]
- Kronik, L.; Yoshitada Morikawa, Y. Understanding the Metal-Molecule Interface from First Principles. In The Molecule-Metal Interface; Koch, N., Ueno, N., Wee, A.T.S., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 51–89. [Google Scholar]
- Lee, L.-H. Molecular Bonding and Adhesion at Polymer-Metal Interphases. J. Adhes. 1994, 46, 15–38. [Google Scholar] [CrossRef]
- Stowell, C.A.; Korgel, B.A. Iridium Nanocrystal Synthesis and Surface Coating-Dependent Catalytic Activity. Nano Lett. 2005, 5, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Tsunoyama, H.; Ichikuni, N.; Sakurai, H.; Tsukuda, T. Effect of electronic structures of au clusters stabilized by poly(N-vinyl-2-pyrrolidone) on aerobic oxidation catalysis. J. Am. Chem. Soc. 2009, 131, 7086–7093. [Google Scholar] [CrossRef] [PubMed]
- Udumula, V.; Tyler, J.H.; Davis, D.A.; Wang, H.; Linford, M.R.; Minson, P.S.; Michaelis, D.J. Dual optimization approach to bimetallic nanoparticle catalysis: Impact of M1/M2 ratio and supporting polymer structure on reactivity. ACS Catal. 2015, 5, 3457–3462. [Google Scholar] [CrossRef]
- Chen, G.; Xu, C.; Huang, X.; Ye, J.; Gu, L.; Li, G.; Tang, Z.; Wu, B.; Yang, H.; Zhao, Z.; et al. Interfacial electronic effects control the reaction selectivity of platinum catalysts. Nat. Mater. 2016, 15, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.G.; Krylova, G.; Sumer, A.; Schwartz, M.M.; Bunel, E.E.; Marshall, C.L.; Chattopadhyay, S.; Lee, B.; Jellinek, J.; Shevchenko, E.V. Capping ligands as selectivity switchers in hydrogenation reactions. Nano Lett. 2012, 12, 5382–5388. [Google Scholar] [CrossRef] [PubMed]
- Mitsudome, T.; Kaneda, K. Advanced core-shell nanoparticle catalysts for efficient organic transformations. ChemCatChem 2013, 5, 1681–1691. [Google Scholar] [CrossRef]
- Marshall, S.T.; O’Brien, M.; Oetter, B.; Corpuz, A.; Richards, R.M.; Schwartz, D.K.; Medlin, J.W. Controlled selectivity for palladium catalysts using self-assembled monolayers. Nat. Mater. 2010, 9, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Kahsar, K.R.; Schwartz, D.K.; Medlin, J.W. Liquid- and vapor-phase hydrogenation of 1-epoxy-3-butene using self-assembled monolayer coated palladium and platinum catalysts. Appl. Catal. A Gen. 2012, 445–446, 102–106. [Google Scholar] [CrossRef]
- Zhong, R.Y.; Sun, K.Q.; Hong, Y.C.; Xu, B.Q. Impacts of organic stabilizers on catalysis of Au nanoparticles from colloidal preparation. ACS Catal. 2014, 4, 3982–3993. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Veith, G.M.; Vindigni, F.; Prati, L. Sol immobilization technique: A delicate balance between activity, selectivity and stability of gold catalysts. Catal. Sci. Technol. 2013, 3, 3036–3041. [Google Scholar] [CrossRef]
- Tao, A.R.; Habas, S.; Yang, P. Shape Control of Colloidal Metal Nanocrystals. Small 2008, 4, 310–325. [Google Scholar] [CrossRef]
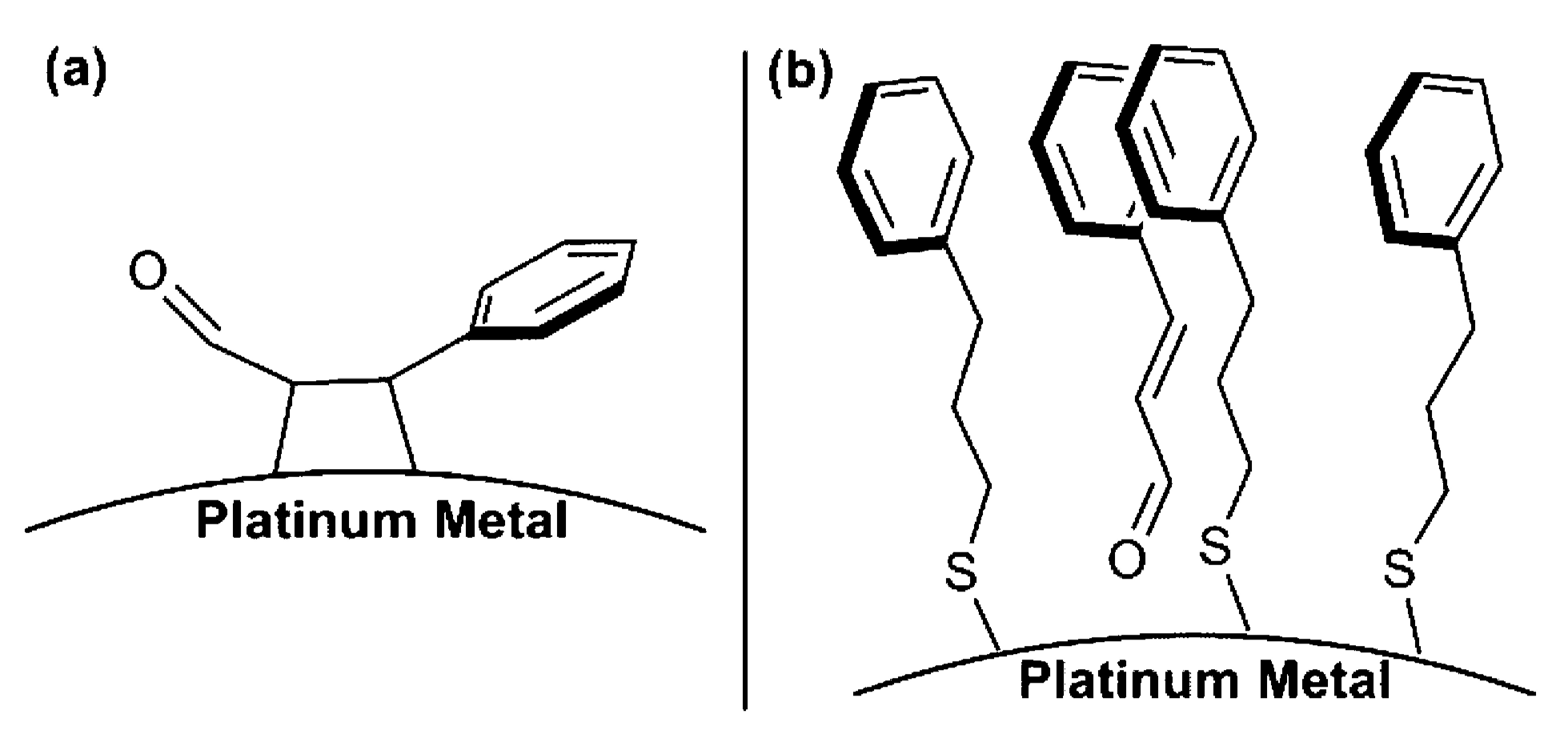
- Haider, P.; Urakawa, A.; Schmidt, E.; Baiker, A. Selective blocking of active sites on supported gold catalysts by adsorbed thiols and its effect on the catalytic behavior: A combined experimental and theoretical study. J. Mol. Catal. A Chem. 2009, 305, 161–169. [Google Scholar] [CrossRef]
- Campisi, S.; Ferri, D.; Villa, A.; Wang, W.; Wang, D.; Kröcher, O.; Prati, L. Selectivity Control in Palladium-Catalyzed Alcohol Oxidation through Selective Blocking of Active Sites. J. Phys. Chem. C 2016, 120, 14027–14033. [Google Scholar] [CrossRef]
- Ferri, D.; Mondelli, C.; Krumeich, F.; Baiker, A. Discrimination of Active Palladium Sites in Catalytic Liquid-Phase Oxidation of Benzyl Alcohol. J. Phys. Chem. B 2006, 110, 22982–22986. [Google Scholar] [CrossRef] [PubMed]
- Centrone, A.; Penzo, E.; Sharma, M.; Myerson, J.W.; Jackson, A.M.; Marzari, N.; Stellacci, F. The role of nanostructure in the wetting behavior of mixed-monolayer-protected metal nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 9886–9891. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Helbig, W.; Quaiser, S.A.; Stimming, U.; Breuer, N.; Vogel, R. Visualization of Surfactants on Nanostructured Palladium Clusters by a Combination of STM and High-Resolution TEM. Science 1995, 267, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Shih, C.W.; Chen, C.D.; Lai, W.C.; Wang, C.C. The shape transition of gold nanorods. Langmuir 1999, 15, 701–709. [Google Scholar] [CrossRef]
- Kano, S.; Tada, T.; Majima, Y. Nanoparticle characterization based on STM and STS. Chem. Soc. Rev. 2015, 44, 970–987. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.M.; Hu, Y.; Silva, P.J.; Stellacci, F. From Homoligand- to Mixed-Ligand- Monolayer-Protected Metal Nanoparticles: A Scanning Tunneling Microscopy Investigation. J. Am. Chem. Soc. 2006, 128, 11135–11149. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Jaeger, R. Absorption-edge resonances, core-hole screening, and orientation of chemisorbed molecules: CO, NO, and N2 on Ni (100). Phys. Rev. B 1982, 26, 4111. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Rogers, S.M.; Dimitratos, N.; Jones, W.; Bowker, M.; Kanaras, A.G.; Wells, P.P.; Catlow, C.R.A.; Parker, S.F. The adsorbed state of a thiol on palladium nanoparticles. Phys. Chem. Chem. Phys. 2016, 18, 17265–17271. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Huang, H.; Yang, J.; Zheng, N.; Fu, G. Selective hydrogenation of α,β-unsaturated aldehydes catalyzed by amine-capped platinum-cobalt nanocrystals. Angew. Chem. Int. Ed. 2012, 51, 3440–3443. [Google Scholar] [CrossRef] [PubMed]
- Makosch, M.; Lin, W.I.; Bumbálek, V.; Sá, J.; Medlin, J.W.; Hungerbühler, K.; van Bokhoven, J.A. Organic thiol modified Pt/TiO2 catalysts to control chemoselective hydrogenation of substituted nitroarenes. ACS Catal. 2012, 2, 2079–2081. [Google Scholar] [CrossRef]
- Castillejos, E.; Jahjah, M.; Favier, I.; Orejón, A.; Pradel, C.; Teuma, E.; Masdeu-Bultò, A.M.; Serp, P.; Gómez, M. Synthesis of platinum-ruthenium nanoparticles under supercritical CO2 and their confinement in carbon nanotubes: Hydrogenation applications. ChemCatChem 2012, 4, 118–122. [Google Scholar] [CrossRef]
- McKenna, F.M.; Anderson, J.A. Selectivity enhancement in acetylene hydrogenation over diphenyl sulphide-modified Pd/TiO2 catalysts. J. Catal. 2011, 281, 231–240. [Google Scholar] [CrossRef]
- Sadeghmoghaddam, E.; Gu, H.; Shon, Y.S. Pd nanoparticle-catalyzed isomerization vs hydrogenation of allyl alcohol: Solvent-dependent regioselectivity. ACS Catal. 2012, 2, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wu, H.; Hua, Q.; Chang, S.; Huang, W. Enhancing catalytic selectivity of supported metal nanoparticles with capping ligands. Phys. Chem. Chem. Phys. 2013, 15, 2273–2277. [Google Scholar] [CrossRef] [PubMed]
- Schoenbaum, C.A.; Schwartz, D.K.; Medlin, J.W. Controlling the surface environment of heterogeneous catalysts using self-assembled monolayers. Acc. Chem. Res. 2014, 47, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Isozaki, K.; Miki, K. Enhanced catalytic activity of self-assembled-monolayer-capped gold nanoparticles. Adv. Mater. 2012, 24, 6462–6467. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.H.; Schoenbaum, C.A.; Schwartz, D.K.; Medlin, J.W. Directing reaction pathways by catalyst active-site selection using self-assembled monolayers. Nat. Commun. 2013, 4, 2448. [Google Scholar] [CrossRef] [PubMed]
- Kahsar, K.R.; Schwartz, D.K.; Medlin, J.W. Control of Metal Catalyst Selectivity through Specific Noncovalent Molecular Interactions. J. Am. Chem. Soc. 2014, 134, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Mallat, T.; Orglmeister, E.; Baiker, A. Asymmetric Catalysis at Chiral Metal Surfaces Asymmetric Catalysis at Chiral Metal Surfaces. Chem. Rev. 2007, 107, 4863–4890. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Sugimura, T. Chiral Catalyst Immobilization and Recycling; de Vos, D.E., Vankelecom, I.F.J., Jacobs, P.A., Eds.; Wiley-VCH: Weinheim, Germany, 2000; p. 173. [Google Scholar]
- Mallat, T.; Baiker, A. Selectivity enhancement in heterogeneous catalysis induced by reaction modifiers. Appl. Catal. A Gen. 2000, 200, 3–22. [Google Scholar] [CrossRef]
- Vargas, A.; Bürgi, T.; Baiker, A. Adsorption of cinchonidine on platinum: A DFT insight in the mechanism of enantioselective hydrogenation of activated ketones. J. Catal. 2004, 226, 69–82. [Google Scholar] [CrossRef]
- Jansat, S.; Gómez, M.; Philippot, K.; Muller, G.; Guiu, E.; Claver, C.; Castillón, S.; Chaudret, B. A case for enantioselective allylic alkylation catalyzed by palladium nanoparticles. J. Am. Chem. Soc. 2004, 126, 1592–1593. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Miyamura, H.; Kobayashi, S. Polymer-incarcerated chiral Rh/Ag nanoparticles for asymmetric 1,4-addition reactions of arylboronic acids to enones: Remarkable effects of bimetallic structure on activity and metal leaching. J. Am. Chem. Soc. 2012, 134, 16963–16966. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Liu, J.H.; Alayoglu, S.; Marcus, M.A.; Fakra, S.C.; Toste, F.D.; Somorjai, G.A. Asymmetric catalysis at the mesoscale: Gold nanoclusters embedded in chiral self-assembled monolayer as heterogeneous catalyst for asymmetric reactions. J. Am. Chem. Soc. 2013, 135, 3881–3886. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Zaera, F. Increase in activity and selectivity in catalysis via surface modification with self-assembled monolayers. J. Phys. Chem. C 2014, 118, 3672–3679. [Google Scholar] [CrossRef]
- Angioletti-Uberti, S.; Lu, Y.; Ballauff, M.; Dzubiella, J. Theory of Solvation-Controlled Reactions in Stimuli-Responsive Nanoreactors. J. Phys. Chem. C 2015, 119, 15723–15730. [Google Scholar] [CrossRef] [Green Version]
- Amsden, B. Solute Diffusion within Hydrogels. Mechanisms and Models. Macromolecules 1998, 31, 8382–8395. [Google Scholar] [CrossRef]
- Horecha, M.; Kaul, E.; Horechyy, A.; Stamm, M. Polymer microcapsules loaded with Ag nanocatalyst as active microreactors. J. Mater. Chem. A 2014, 2, 7431–7438. [Google Scholar] [CrossRef]
- Wu, S.; Dzubiella, J.; Kaiser, J.; Drechsler, M.; Guo, X.; Ballauff, M.; Lu, Y. Thermosensitive Au-PNIPA yolk-shell nanoparticles with tunable selectivity for catalysis. Angew. Chem. Int. Ed. 2012, 51, 2229–2233. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Liu, S.; Zheng, N. C2H2 Treatment as a Facile Method to Boost the Catalysis of Pd Nanoparticulate Catalysts. J. Am. Chem. Soc. 2014, 136, 5583–5586. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst a | Selectivity c (%) | ||||||
|---|---|---|---|---|---|---|---|
| Activity mol b (Au mol)−1·h−1 | Glyc | Gly | Tar | F | Lac | ||
| 1 | AuPVA (1:1) | 236 | 81 | 3 | 5 | 2 | 9 |
| 2 | AuPVA (1:0.5) | 282 | 78 | 5 | 4 | 3 | 10 |
| 3 | AuPVA (1:0.25) | 356 | 75 | 4 | 5 | 5 | 11 |
| 4 | AuPVA (1:0.125) | 434 | 70 | 12 | 2 | 9 | 7 |
| 5 | AuPVA (1:1) washed at 298 K | 355 | 77 | 6 | 3 | 4 | 10 |
| 6 | AuPVA (1:1) washed at 333 K | 138 | 68 | 14 | 3 | 7 | 8 |
| 7 | AuDP | 390 | 69 | 12 | 2 | 9 | 8 |
| 8 | AuDP + PVA (1:1) | 255 | 76 | 5 | 7 | 3 | 9 |
| 9 | AuDP + PVA (1:0.125) | 401 | 70 | 10 | 5 | 7 | 8 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campisi, S.; Schiavoni, M.; Chan-Thaw, C.E.; Villa, A. Untangling the Role of the Capping Agent in Nanocatalysis: Recent Advances and Perspectives. Catalysts 2016, 6, 185. https://doi.org/10.3390/catal6120185
Campisi S, Schiavoni M, Chan-Thaw CE, Villa A. Untangling the Role of the Capping Agent in Nanocatalysis: Recent Advances and Perspectives. Catalysts. 2016; 6(12):185. https://doi.org/10.3390/catal6120185
Chicago/Turabian StyleCampisi, Sebastiano, Marco Schiavoni, Carine Edith Chan-Thaw, and Alberto Villa. 2016. "Untangling the Role of the Capping Agent in Nanocatalysis: Recent Advances and Perspectives" Catalysts 6, no. 12: 185. https://doi.org/10.3390/catal6120185