
Strengths, Weaknesses, Opportunities and Threats: Computational Studies of Mn- and Fe-Catalyzed Epoxidations



Abstract
:1. Introduction and Context
1.1. Scope of the Review
2. Statistical Meta-Analysis
3. Ground State Spin Multiplicity of Mn(Salen) Species
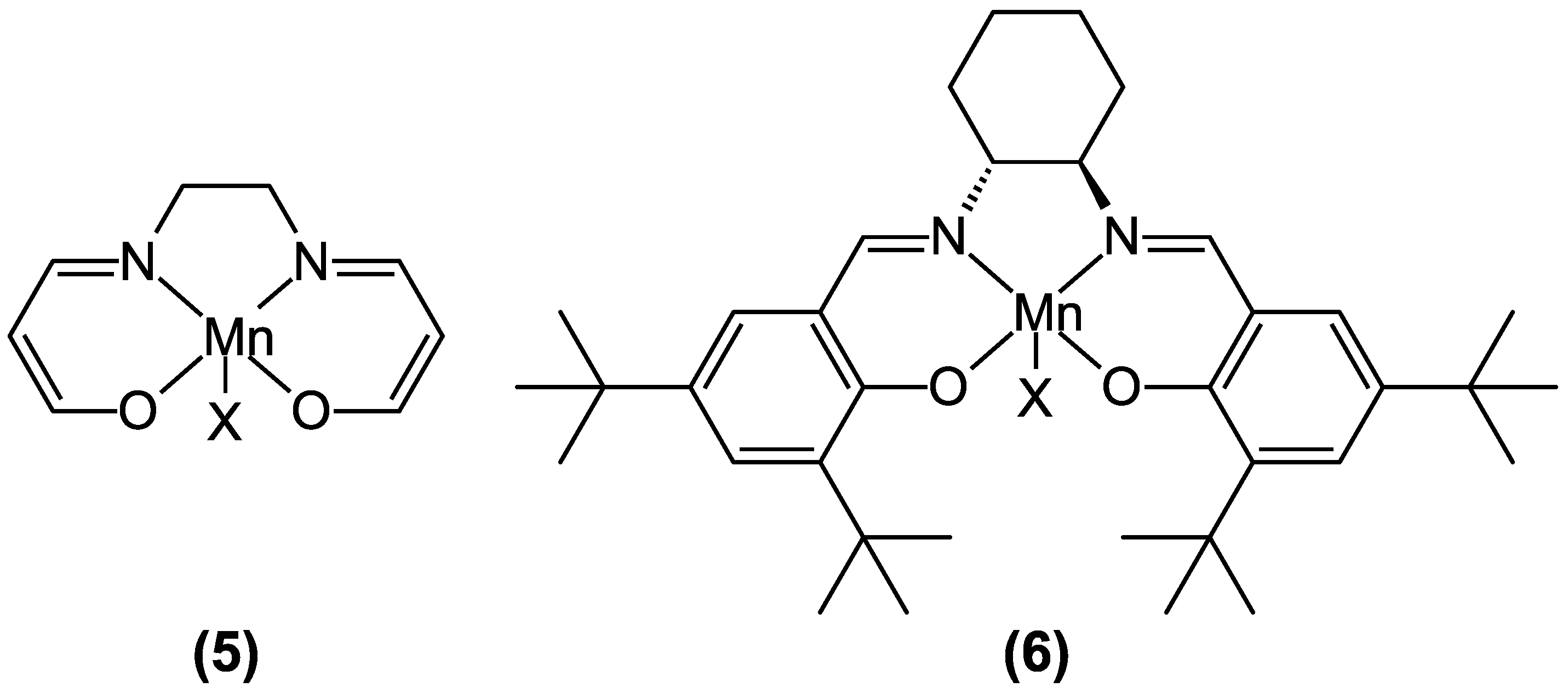
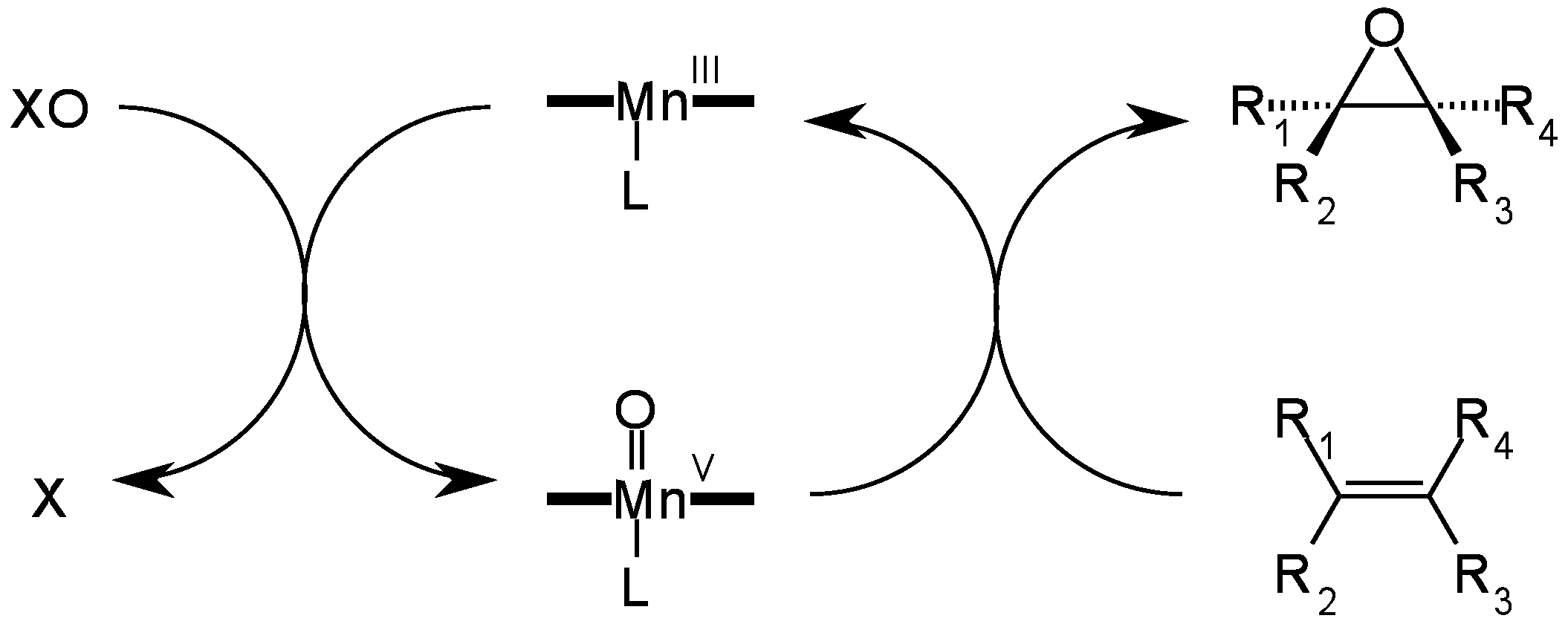
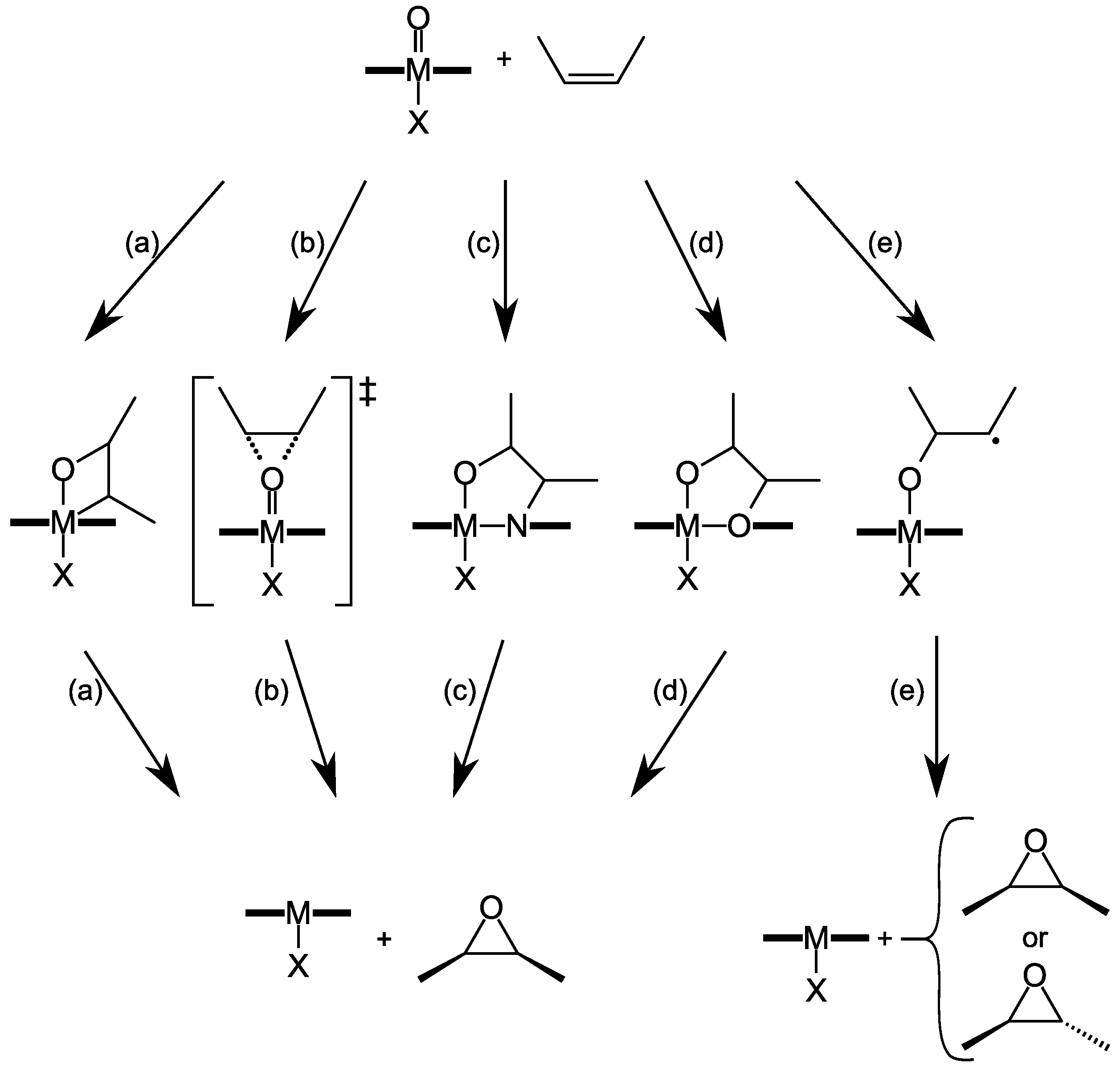
4. Insights on the Reaction Mechanism
5. Comparison with Experimental Data
6. Other Epoxidation Catalysts: A Brief Overview
7. Perspectives for Future Developments: SWOT Analysis
7.1. Strengths
7.2. Weaknesses
7.3. Opportunities
7.4. Threats
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chandrachud, P.P.; Jenkins, D.M. High valent Fe-IV chemistry in sustainable oxidation catalysis. Tetrahedron Lett. 2015, 56, 2369–2376. [Google Scholar] [CrossRef]
- Webb, M.A.; Jung, Y.; Pesko, D.M.; Savoie, B.M.; Yamamoto, U.; Coates, G.W.; Balsara, N.P.; Wang, Z.G.; Miller, T.F. Systematic Computational and Experimental Investigation of Lithium-Ion Transport Mechanisms in Polyester-Based Polymer Electrolytes. ACS Cent. Sci. 2015, 1, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Poater, A.; Childers, M.I.; Widger, P.C.B.; LaPointe, A.M.; Lobkovsky, E.B.; Coates, G.W.; Cavallo, L. Enantioselective Polymerization of Epoxides Using Biaryl-Linked Bimetallic Cobalt Catalysts: A Mechanistic Study. J. Am. Chem. Soc. 2013, 135, 18901–18911. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Pereira, C.; Rebelo, S. Green Oxidation Catalysis with Metal Complexes: From Bulk to Nano Recyclable Hybrid Catalysts. In Catalysis: Volume 24; The Royal Society of Chemistry: London, UK, 2012; Volume 24, pp. 116–203. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-Catalyzed Epoxidations of Alkenes with Hydrogen Peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Murata, K.; Inaba, M. Direct epoxidation of propylene by molecular oxygen over a catalyst system containing palladium and a peroxo-heteropoly compound in methanol. Chem. Commun. 2004, 2004, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.R.; Dawson, J.H. Oxidizing Intermediates in P450 Catalysis: A Case for Multiple Oxidants. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450; Hrycay, G.E., Bandiera, M.S., Eds.; Springer: Cham, Switzerland, 2015; pp. 63–81. [Google Scholar]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Katsuki, T. Mn-salen Catalyst, Competitor of Enzymes, for Asymmetric Epoxidation. J. Mol. Catal. A Chem. 1996, 113, 87–107. [Google Scholar] [CrossRef]
- Dalton, C.T.; Ryan, K.M.; Wall, V.M.; Bousquet, C.; Gilheany, D.G. Recent progress towards the understanding of metal–salen catalyzed asymmetric alkene epoxidation. Top. Catal. 1998, 5, 75–91. [Google Scholar] [CrossRef]
- McGarrigle, E.M.; Gilheany, D.G. Chromium and Manganese-salen Promoted Epoxidation of Alkenes. Chem. Rev. 2005, 105, 1563–1602. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Z.G.; Xu, X.; Gong, L.Z.; Mahmood, M.H.; Liu, H.Y. Reactivity of (oxo)manganese(V) corroles in one-electron redox state: Insights from conceptual DFT and transition state calculations. J. Porphyr. Phthalocyanines 2013, 17, 1196–1203. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Palaniandavar, M. Tuning the olefin epoxidation by manganese(iii) complexes of bisphenolate ligands: Effect of Lewis basicity of ligands on reactivity. Dalton Trans. 2014, 43, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their Structure, Reactivity, and Selectivity Modeled by QM/MM Calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.M.Q.; Neves, C.M.B.; Pires, S.M.G.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mimicking P-450 processes and the use of metalloporphyrins. Pure Appl. Chem. 2013, 85, 1671–1681. [Google Scholar] [CrossRef]
- Groves, J.T. Reactivity and mechanisms of metalloporphyrin- catalyzed oxidations. J. Porphyr. Phthalocyanines 2000, 4, 350–352. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. Transition Metal Mediated Epoxidation as Test Case for the Performance of Different Density Functionals: A Computational Study. J. Phys. Chem. A 2003, 107, 5466–5471. [Google Scholar] [CrossRef]
- Kavousi, H.; Rezaeifard, A.; Raissi, H.; Jafarpour, M. A DFT investigation of axial N -donor ligands effects on the high valent manganese-oxo meso -tetraphenyl porphyrin. J. Porphyr. Phthalocyanines 2015, 19, 651–662. [Google Scholar] [CrossRef]
- Hai-Yang, L.; Li, L.; Xiao, Y.; Xiang-Li, W.; Zhi-Guang, X.; Shi-Jun, L.; Chi-Kwong, C. DFT calculations on manganese(III)5,10,15-tris(pentafluorophenyl)-corrole. Acta Phys. Chim. Sin. 2008, 24, 1602–1608. [Google Scholar]
- Li-Zhen, G.; Zhi-Guang, X.; Xuan, X.; Jing, H.; Qi, W.; Hai-Yang, L. Axial Coordination Behavior of Corrole Mn and MnVO Complexes with N-Based Ligands. Acta Phys. Chim. Sin. 2014, 30, 265–272. [Google Scholar]
- Xiao-Hui, Z.; Zhi-Guang, X.; Li-Zhen, G.; Xuan, X.; Gui-Xian, S.; Hua-Bin, C.; Hai-Yang, L. Stability of trans-Dioxo Manganese(V) Corrole Complex. Acta Phys. Chim. Sin. 2015, 31, 1069–1076. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Abashkin, Y.G.; Collins, J.R.; Burt, S.K. (Salen)Mn(III)-Catalyzed Epoxidation Reaction as a Multichannel Process with Different Spin States. Electronic Tuning of Asymmetric Catalysis: A Theoretical Study. Inorg. Chem. 2001, 40, 4040–4048. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Nemeth, L.; Bare, S.R. Science and Technology of Framework Metal-Containing Zeotype Catalysts. In ADVANCES IN CATALYSIS, VOL 57; Advances in Catalysis; Jentoft, F., Ed.; Elsevier BV: Amsterdam, The Netherlands, 2014; Volume 57, pp. 1–97. [Google Scholar]
- Pereira, C.; Pereira, A.M.; Quaresma, P.; Tavares, P.B.; Pereira, E.; Araújo, J.P.; Freire, C. Superparamagnetic Fe2O3@SiO2 nanoparticles: A novel support for the immobilization of [VO(acac)2]. Dalton Trans. 2010, 39, 2842–2854. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Cepera, R.M.; Pereira, M.C.; Lima, D.Q.; Fabris, J.D.; Oliveira, L.C. Heterogeneous catalyst based on peroxo-niobium complexes immobilized over iron oxide for organic oxidation in water. Appl. Catal. B 2011, 107, 237–244. [Google Scholar] [CrossRef]
- Pospisil, P.J.; Carsten, D.H.; Jacobsen, E.N. X-ray Structural Studies of Highly Enantioselective Mn(salen) Epoxidation Catalysts. Chem. Eur. J. 1996, 2, 974–980. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Babushkin, D.E.; Talsi, E.P. Detection of {EPR} Spectra in S = 2 States of MnIII(salen) Complexes. Mendeleev Commun. 1999, 9, 29–31. [Google Scholar] [CrossRef]
- Campbell, K.A.; Lashley, M.R.; Wyatt, J.K.; Nantz, M.H.; Britt, R.D. Dual-Mode EPR Study of Mn(III) Salen and the Mn(III) Salen-Catalyzed Epoxidation of cis-β-Methylstyrene. J. Am. Chem. Soc. 2001, 123, 5710–5719. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.; Mosquera, R.A.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Charge Distribution in Mn(salen) Complexes. Int. J. Quantum Chem. 2014, 114, 525–533. [Google Scholar] [CrossRef]
- Abashkin, Y.G.; Burt, S.K. (Salen)Mn(III) Compound as a Nonpeptidyl Mimic of Catalase: DFT Study of the Metal Oxidation by a Peroxide Molecule. J. Phys. Chem. B 2004, 108, 2708–2711. [Google Scholar] [CrossRef]
- Curet-Arana, M.C.; Emberger, G.A.; Broadbelt, L.J.; Snurr, R.Q. Quantum chemical determination of stable intermediates for alkene epoxidation with Mn-porphyrin catalysts. J. Mol. Catal. A Chem. 2008, 285, 120–127. [Google Scholar] [CrossRef]
- Bogaerts, T.; Wouters, S.; Voort, P.V.D.; Speybroeck, V.V. Mechanistic Investigation on Oxygen Transfer with the Manganese-Salen Complex. ChemCatChem 2015, 7, 2711–2719. [Google Scholar] [CrossRef] [Green Version]
- Ivanic, J.; Collins, J.R.; Burt, S.K. Theoretical study of the low lying electronic states of oxoX(salen) (X = Mn, Mn−, Fe, and Cr−) complexes. J. Phys. Chem. A 2004, 108, 2314–2323. [Google Scholar] [CrossRef]
- Neese, F. Prediction of Molecular Properties and Molecular Spectroscopy with Density Functional Theory: From Fundamental Theory to Exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Khavrutskii, I.V.; Musaev, D.G.; Morokuma, K. Structure, Stability, and Electronic and NMR Properties of Various Oxo- and Nitrido-Derivatives of [L(Salen)Mn(III)]+, Where L = None and Imidazole. A Density Functional Study. Inorg. Chem. 2003, 42, 2606–2621. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.S.; Sherrill, C.D. The Electronic Structure of oxo-Mn(salen): Single-reference and Multireference Approaches. J. Chem. Phys. 2006, 124, 144314. [Google Scholar] [CrossRef] [PubMed]
- Takatani, T.; Sears, J.S.; Sherrill, C.D. Assessing the Performance of Density Functional Theory for the Electronic Structure of Metal-Salens: The d6-Metals. J. Phys. Chem. A 2009, 113, 9231–9236. [Google Scholar] [CrossRef] [PubMed]
- Takatani, T.; Sears, J.S.; Sherrill, C.D. Assessing the Performance of Density Functional Theory for the Electronic Structure of Metal-Salens: The M06 Suite of Functionals and the d4-Metals. J. Phys. Chem. A 2010, 114, 11714–11718. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, L.; Jacobsen, H. Electronic Effects in (salen)Mn-Based Epoxidation Catalysts. J. Org. Chem. 2003, 68, 6202–6207. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, H.; Cavallo, L. Re-evaluation of the Mn(salen) Mediated Epoxidation of Alkenes by Means of the B3LYP* Density Functional. Phys. Chem. Chem. Phys. 2004, 6, 3747–3753. [Google Scholar] [CrossRef]
- Ashley, D.C.; Baik, M.H. The Electronic Structure of [Mn(V)=O]: What is the Connection between Oxyl Radical Character, Physical Oxidation State, and Reactivity? ACS Catal. 2016, 6, 7202–7216. [Google Scholar] [CrossRef]
- Teixeira, F.; Mosquera, R.A.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Principal Component Analysis of Mn(salen) Catalysts. Phys. Chem. Chem. Phys. 2014, 16, 25364–25376. [Google Scholar] [CrossRef] [PubMed]
- Linker, T. The Jacobsen-Katsuki Epoxidation and Its Controversial Mechanism. Angew. Chem. Int. Ed. Engl. 1997, 36, 2060–2062. [Google Scholar] [CrossRef]
- Bautz, J.; Comba, P.; Lopezrden, C.; Menzel, M.; Rajaraman, G. Biomimetic High-Valent Non-Heme Iron Oxidants for the cis-Dihydroxylation and Epoxidation of Olefins. Angew. Chem. Int. Ed. 2007, 46, 8067–8070. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Tahsini, L.; de Visser, S.P.; Kang, H.Y.; Kim, S.J.; Nam, W. Effect of Porphyrin Ligands on the Regioselective Dehydrogenation versus Epoxidation of Olefins by Oxoiron(IV) Mimics of Cytochrome P450. J. Phys. Chem. A 2009, 113, 11713–11722. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, L.; Jacobsen, H. Toward a Catalytic Cycle for the Mn-Salen Mediated Alkene Epoxidation: A Computational Approach. Inorg. Chem. 2004, 43, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, J.; Datta, A.; Dasgupta, S.; Chakraborty, A.; Menéndez, M.I.; Chattopadhyay, T. Development of an efficient magnetically separable nanocatalyst: Theoretical approach on the role of the ligand backbone on epoxidation capability. RSC Adv. 2015, 5, 92634–92647. [Google Scholar] [CrossRef]
- Benet-Buchholz, J.; Comba, P.; Llobet, A.; Roeser, S.; Vadivelu, P.; Wadepohl, H.; Wiesner, S. Iron vs. ruthenium—A comparison of the stereoselectivity in catalytic olefin epoxidation. Dalton Trans. 2009, 5910–5923. [Google Scholar] [CrossRef] [PubMed]
- Eshtiagh-Hosseini, H.; Beyramabadi, S.A.; Mirzaei, M.; Morsali, A.; Salimi, A.R.; Naseri, M.A. 3,3′-dihydroxy-4,4′-[1,2-cyclohexanediyl-bis(nitrilomethylidyne)]-bis-phenol schiff-base and its Mn(II) complex: Synthesis, experimental, and theoretical characterization. J. Struct. Chem. 2013, 54, 1063–1069. [Google Scholar] [CrossRef]
- Hull, J.F.; Balcells, D.; Sauer, E.L.O.; Raynaud, C.; Brudvig, G.W.; Crabtree, R.H.; Eisenstein, O. Manganese Catalysts for C-H Activation: An Experimental/Theoretical Study Identifies the Stereoelectronic Factor That Controls the Switch between Hydroxylation and Desaturation Pathways. J. Am. Chem. Soc. 2010, 132, 7605–7616. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Franke, A.; Brindell, M.; Oszajca, M.; Zahl, A.; van Eldik, R. Combined Experimental and Theoretical Study on the Reactivity of Compounds I and II in Horseradish Peroxidase Biomimetics. Chem. Eur. J. 2014, 20, 14437–14450. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, X.; Li, H.; Jalbout, A.F. Metalloporphyrin-Dioxygen Interactions and the Effects of Neutral Axial Ligands. J. Phys. Chem. C 2009, 113, 14316–14323. [Google Scholar] [CrossRef]
- Balland, V.; Charlot, M.F.; Banse, F.; Girerd, J.J.; Mattioli, T.A.; Bill, E.; Bartoli, J.F.; Battioni, P.; Mansuy, D. Spectroscopic characterization of an Fe-IV intermediate generated by reaction of XO− (X = Cl, Br) with an Fe-II complex bearing a pentadentate non-porphyrinic ligand - Hydroxylation and epoxidation activity. Eur. J. Inorg. Chem. 2004, 2004, 301–308. [Google Scholar] [CrossRef]
- Comba, P.; Rajaraman, G. Epoxidation and 1,2-Dihydroxylation of Alkenes by a Nonheme Iron Model System—DFT Supports the Mechanism Proposed by Experiment. Inorg. Chem. 2008, 47, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, T.; Shiota, Y.; Ohta, T.; Yoshizawa, K. Does the Hydroperoxo Species of Cytochrome P450 Participate in Olefin Epoxidation with the Main Oxidant, Compound I? Criticism from Density Functional Theory Calculations. Bull. Chem. Soc. Jpn. 2003, 76, 721–732. [Google Scholar] [CrossRef]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Sharma, P.K.; Shaik, S. Searching for the Second Oxidant in the Catalytic Cycle of Cytochrome P450: A Theoretical Investigation of the Iron(III)-Hydroperoxo Species and Its Epoxidation Pathways. J. Am. Chem. Soc. 2002, 124, 2806–2817. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, M.; Limberg, C.; Herwig, C.; Ziemer, B. Stabilizing the Boat Conformation of Piperazines Coordinated to Iron(II): Iso-Butyl Substituents Lead to Robust Oxidation Catalysts via Hyperconjugation. Z. Anorg. Allg. Chem. 2009, 635, 1823–1830. [Google Scholar] [CrossRef]
- Faponle, A.S.; Quesne, M.G.; Sastri, C.V.; Banse, F.; de Visser, S.P. Differences and Comparisons of the Properties and Reactivities of Iron(III)-hydroperoxo Complexes with Saturated Coordination Sphere. Chem. Eur. J. 2014, 21, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- García-Aguilar, J.; Miguel-García, I.; Juan-Juan, J.; Such-Basáñez, I.; Fabián, E.S.; Cazorla-Amorós, D.; Berenguer-Murcia, Á. One step-synthesis of highly dispersed iron species into silica for propylene epoxidation with dioxygen. J. Catal. 2016, 338, 154–167. [Google Scholar] [CrossRef]
- Quiñonero, D.; Musaev, D.G.; Morokuma, K. Theoretical Studies of the Complex [(BPMEN)Fe(II)(NCCH3)2]2+, Precursor of Non-Heme Iron Catalysts for Olefin Epoxidation and Cis-Dihydroxylation. Inorg. Chem. 2003, 42, 8449–8455. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, D.; Morokuma, K.; Musaev, D.G.; Mas-Ballesté, R.; Que, L. Metal-Peroxo versus Metal-Oxo Oxidants in Non-Heme Iron-Catalyzed Olefin Oxidations: Computational and Experimental Studies on the Effect of Water. J. Am. Chem. Soc. 2005, 127, 6548–6549. [Google Scholar] [CrossRef] [PubMed]
- Kwong, H.K.; Lo, P.K.; Lau, K.C.; Lau, T.C. Epoxidation of alkenes and oxidation of alcohols with hydrogen peroxide catalyzed by a manganese(V) nitrido complex. Chem. Commun. 2011, 47, 4273–4275. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Wang, B.; Wang, Y.; Xia, C.; Lee, Y.M.; Nam, W.; Sun, W. Proton-Promoted and Anion-Enhanced Epoxidation of Olefins by Hydrogen Peroxide in the Presence of Nonheme Manganese Catalysts. J. Am. Chem. Soc. 2016, 138, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, D.; Musaev, D.G.; Morokuma, K. Computational insights to the mechanism of alkene epoxidation by manganese-based catalysts in the presence of bicarbonate. J. Mol. Struct. THEOCHEM 2009, 903, 115–122. [Google Scholar] [CrossRef]
- Rutkowska-Zbik, D.; Tokarz-Sobieraj, R.; Witko, M. Quantum chemical description of oxygen activation process on Co, Mn, and Mo porphyrins. J. Chem. Theory Comput. 2007, 3, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Hirao, H.; Kumar, D. Reactivity patterns of cytochrome P450 enzymes: Multifunctionality of the active species, and the two states-two oxidants conundrum. Nat. Prod. Rep. 2007, 24, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Abashkin, Y.G.; Burt, S.K. (Salen)Mn-Catalyzed Epoxidation of Alkenes: A Two-Zone Process with Different Spin-State Channels as Suggested by DFT Study. Org. Lett. 2004, 6, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Tiwari, M.; Dhar, B.B.; Vanka, K.; Gupta, S.S. Mechanism of Oxygen Atom Transfer from Fe-V(O) to Olefins at Room Temperature. Inorg. Chem. 2015, 54, 6112–6121. [Google Scholar] [CrossRef] [PubMed]
- Norrby, P.O.; Linde, C.; Aakermark, B. On the Chirality Transfer in the Epoxidation of Alkenes Catalyzed by Mn(salen) Complexes. J. Am. Chem. Soc. 1995, 117, 11035–11036. [Google Scholar] [CrossRef]
- Linde, C.; Arnold, M.; Åkermark, B.; Norrby, P.O. Is There a Radical Intermediate in the (salen)Mn-Catalyzed Epoxidation of Alkenes? Angew. Chem. Int. Ed. Engl. 1997, 36, 1723–1725. [Google Scholar] [CrossRef]
- Kürti, L.; Blewett, M.M.; Corey, E.J. Origin of Enantioselectivity in the Jacobsen Epoxidation of Olefins. Org. Lett. 2009, 11, 4592–4595. [Google Scholar] [CrossRef] [PubMed]
- Linde, C.; Akermark, B.; Norrby, P.; Svensson, M. Timing Is Critical: Effect of Spin Changes on the Diastereoselectivity in Mn(salen)-Catalyzed Epoxidation. J. Am. Chem. Soc. 1999, 121, 5083–5084. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. Radical Intermediates in the Jacobsen – Katsuki Epoxidation. Angew. Chem. Int. Ed. 2000, 39, 589–592. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. Manganese-Salen Complexes as Oxygen-Transfer Agents in Catalytic Epoxidations—A Density Functional Study of Mechanistic Aspects. Eur. J. Inorg. Chem. 2003, 2003, 892–902. [Google Scholar] [CrossRef]
- Jacobsen, H.; Cavallo, L. Donor-Ligand Effect on the Product Distribution in the Manganese-Catalyzed Epoxidation of Olefins: A Computational Assessment. Organometallics 2006, 25, 177–183. [Google Scholar] [CrossRef]
- Reiher, M.; Salomon, O.; Artur Hess, B. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Rich, J.; Rodríguez, M.; Romero, I.; Fontrodona, X.; van Leeuwen, P.W.N.M.; Freixa, Z.; Sala, X.; Poater, A.; Solà, M. N-Tetradentate SPANamine Derivatives and Their Mn II -Complexes as Catalysts for Epoxidation of Alkenes. Eur. J. Inorg. Chem. 2012, 2013, 1213–1224. [Google Scholar] [CrossRef]
- Manrique, E.; Poater, A.; Fontrodona, X.; Solà, M.; Rodríguez, M.; Romero, I. Reusable manganese compounds containing pyrazole-based ligands for olefin epoxidation reactions. Dalton Trans. 2015, 44, 17529–17543. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, H.; Cavallo, L. A Possible Mechanism for Enantioselectivity in the Chiral Epoxidation of Olefins with [Mn(salen)] Catalysts. Chem. Eur. J. 2001, 8, 800–807. [Google Scholar] [CrossRef]
- Khavrutskii, I.V.; Musaev, D.G.; Morokuma, K. Epoxidation of unfunctionalized olefins by Mn(salen) catalyst using organic peracids as oxygen source: A theoretical study. Proc. Natl. Acad. Sci. USA 2004, 101, 5743–5748. [Google Scholar] [CrossRef] [PubMed]
- Malek, K.; Jansen, A.; Li, C.; van Santen, R. Enantioselectivity of immobilized Mn-salen complexes: A computational study. J. Catal. 2007, 246, 127–135. [Google Scholar] [CrossRef]
- Malek, K.; Li, C.; van Santen, R.A. New theoretical insights into epoxidation of alkenes by immobilized Mn-salen complexes in mesopores: Effects of substrate, linker and confinement. J. Mol. Catal. A Chem. 2007, 271, 98–104. [Google Scholar] [CrossRef]
- Zabrodsky, H.; Avnir, D. Chirality Continuous Symmetry Measures. 4. Chirality. J. Am. Chem. Soc. 1995, 117, 462–473. [Google Scholar] [CrossRef]
- Oxford, G.A.E.; Snurr, R.Q.; Broadbelt, L.J. Hybrid Quantum Mechanics/Molecular Mechanics Investigation of (salen)Mn for use in Metal-Organic Frameworks. Ind. Eng. Chem. Res. 2010, 49, 10965–10973. [Google Scholar] [CrossRef]
- Oxford, G.A.E.; Dubbeldam, D.; Broadbelt, L.J.; Snurr, R. Elucidating steric effects on enantioselective epoxidation catalyzed by (salen)Mn in metal-organic frameworks. J. Mol. Catal. A Chem. 2011, 334, 89–97. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants for iron(IV)-oxo porphyrin cation radical complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef]
- Yi, W.; Yuan, L.; Kun, Y.; Zhengwen, H.; Jing, T.; Xu, F.; Hong, G.; Yong, W. What factors influence the reactivity of C-H hydroxylation and C=C epoxidation by [Fe-IV(L-ax)(1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane)(O )]n+. J. Biol. Inorg. Chem. 2015, 20, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarzyk, T.; Dziedzic-Kocurek, K.; Rutkowska, I.; Dziliński, K. Mössbauer study of a tetrakis (pentafluorophenyl) porphyrin iron (III) chloride in comparison with the fluorine unsubstituted analogue. Nukleonika 2015, 60, 57–61. [Google Scholar] [CrossRef]
- De Visser, S.P. Predictive Studies of Oxygen Atom Transfer Reactions by Compound I of Cytochrome P450: Aliphatic and Aromatic Hydroxylation, Epoxidation, and Sulfoxidation. In Advances in Inorganic Chemistry Volume 64: Inorganic Bioinorganic Reaction Mechanisms; Advances in Inorganic Chemistry; VanEldik, R., Ed.; Elsevier BV: Amsterdam, The Netherlands, 2012; Volume 64, pp. 1–31. [Google Scholar]
- Shaik, S.; de Visser, S.P.; Kumar, D. One oxidant, many pathways: A theoretical perspective of monooxygenation mechanisms by cytochrome P450 enzymes. J. Biol. Inorg. Chem. 2004, 9, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Woggon, W.D.; Kozuch, S.; Leifels, T.; Meyer, D.; Sbaragli, L. New Synthetic Models of Cytochrome P450: How Different Are They from the Natural Species? Synlett 2005, 675–684. [Google Scholar] [CrossRef]
- Shaik, S.; Lai, W.; Chen, H.; Wang, Y. The Valence Bond Way: Reactivity Patterns of Cytochrome P450 Enzymes and Synthetic Analogs. Acc. Chem. Res. 2010, 43, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Derat, E.; Shaik, S. The intrinsic axial ligand effect on propene oxidation by horseradish peroxidase versus cytochrome P450 enzymes. J. Biol. Inorg. Chem. 2005, 10, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, L.; Liu, W. In Silico Prediction of Cytochrome P450-Mediated Biotransformations of Xenobiotics: A Case Study of Epoxidation. Chem. Res. Toxicol. 2015, 28, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Drzewiecka-Matuszek, A.; Rutkowska-Zbik, D.; Witko, M. Hydrogen peroxide as oxidant in bio-mimetic catalysis by manganese porphyrin: Theoretical DFT studies. Can. J. Chem. 2013, 91, 642–647. [Google Scholar] [CrossRef]
- Teixeira, F.; Melo, A.; Cordeiro, M.N.D.S. Calibration sets and the accuracy of vibrational scaling factors: A case study with the X3LYP hybrid functional. J. Chem. Phys. 2010, 133, 114109. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, T.; Limberg, C. The Reaction of Permanganyl Chloride with Olefins: Intermediates and Mechanism as Derived from Matrix-Isolation Studies and Density Functional Theory Calculations. Chem. Eur. J. 2001, 7, 4674–4685. [Google Scholar] [CrossRef]
- Srour, H.; Maux, P.L.; Chevance, S.; Simonneaux, G. Metal-catalyzed asymmetric sulfoxidation, epoxidation and hydroxylation by hydrogen peroxide. Coord. Chem. Rev. 2013, 257, 3030–3050. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.H.; Ge, H.Q.; Ye, C.P.; Liu, Z.M.; Su, K.X. Advances in Homogeneous and Heterogeneous Catalytic Asymmetric Epoxidation. Chem. Rev. 2005, 105, 1603–1662. [Google Scholar] [CrossRef] [PubMed]
- Brandt, P.; Norrby, P.; Daly, A.M.; Gilheany, D.G. Chromium-Salen-Mediated Alkene Epoxidation: A Theoretical and Experimental Study Indicates the Importance of Spin-Surface Crossing and the Presence of a Discrete Intermediate. Chem. Eur. J. 2002, 8, 4299–4307. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Rajagopal, S.; Suvitha, A.; Kawazoe, Y. A combined experimental and theoretical investigation on the oxygenation of organic sulfides by oxo(salen)chromium(V) ion. J. Phys. Org. Chem. 2009, 22, 650–660. [Google Scholar] [CrossRef]
- Vandichel, M.; Leus, K.; der Voort, P.V.; Waroquier, M.; Speybroeck, V.V. Mechanistic insight into the cyclohexene epoxidation with VO(acac)2 and tert-butyl hydroperoxide. J. Catal. 2012, 294, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Adão, P.; Pessoa, J.C.; Henriques, R.T.; Kuznetov, M.L.; Avecilla, F.; Maurya, M.R.; Kumar, U.; Correia, I. Synthesis, Characterization, and Application of Vanadium—Salan Complexes in Oxygen Transfer Reactions. Inorg. Chem. 2009, 48, 3542–3561. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, M.V.; Kuznetov, M.L.; Romakh, V.B.; Shul’pina, L.S.; Silva, J.D.; Pombeiro, A.J.L.; Shul’pin, G.B. Mechanism of oxidations with H2O2 catalyzed by vanadate anion or oxovanadium(V) triethanolaminate (vanadatrane) in combination with pyrazine-2-carboxylic acid (PCA): Kinetic and DFT studies. J. Catal. 2009, 267, 140–157. [Google Scholar] [CrossRef]
- Kuznetov, M.L.; Pessoa, J.C. Epoxidation of olefins catalyzed by vanadium-salan complexes: A theoretical mechanistic study. J. Chem. Soc. Dalton Trans. 2009, 2009, 5460–5468. [Google Scholar] [CrossRef] [PubMed]
- Leus, K.; Muylaert, I.; Vandichel, M.; Marin, G.B.; Waroquier, M.; Van Speybroeck, V.; Van Der Voort, P. The remarkable catalytic activity of the saturated metal organic framework V-MIL-47 in the cyclohexene oxidation. Chem. Commun. 2010, 46, 5085–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschi, M.; Crucianelli, M.; Giuseppe, A.D.; Nicola, C.D.; Marchetti, F. Insights on the mechanistic features of catalytic oxidations of simple and conjugated olefins promoted by VO(acac)2/H2O2 system, in acetonitrile: A computational study. Catal. Today 2012, 192, 56–62. [Google Scholar] [CrossRef]
- Tahmasebi, V.; Grivani, G.; Bruno, G. Synthesis, characterization, crystal structure determination and catalytic activity in epoxidation reaction of two new oxidovanadium(IV) Schiff base complexes. J. Mol. Struct. 2016, 1123, 367–374. [Google Scholar] [CrossRef]
- Sever, R.R.; Root, T.W. DFT Study of Solvent Coordination Effects on Titanium-Based Epoxidation Catalysts. Part Two: Reactivity of Titanium Hydroperoxo Complexes in Ethylene Epoxidation. J. Phys. Chem. B 2003, 107, 4090–4099. [Google Scholar] [CrossRef]
- Limtrakul, J.; Inntam, C.; Truong, T.N. Density functional theory study of the ethylene epoxidation over Ti-substituted silicalite (TS-1). J. Mol. Catal. A Chem. 2004, 207, 139–148. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Hydrogen Peroxide Activation over Ti IV: What Have We Learned from Studies on Ti-Containing Polyoxometalates? Eur. J. Inorg. Chem. 2013, 2013, 1595–1605. [Google Scholar] [CrossRef]
- Antonova, N.S.; Carbo, J.J.; Kortz, U.; Kholdeeva, O.A.; Poblet, J.M. Mechanistic Insights into Alkene Epoxidation with H2O2 by Ti- and other TM-Containing Polyoxometalates: Role of the Metal Nature and Coordination Environment. J. Am. Chem. Soc. 2010, 132, 7488–7497. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.J.; Calhorda, M.J.; Kuhn, F.E. Olefin Epoxidation Catalyzed by eta5-Cyclopentadienyl Molybdenum Compounds: A Computational Study. Organometallics 2010, 29, 303–311. [Google Scholar] [CrossRef]
- Schmidt, A.; Grover, N.; Zimmermann, T.K.; Graser, L.; Cokoja, M.; Pöthig, A.; Kühn, F.E. Synthesis and characterization of novel cyclopentadienyl molybdenum imidazo[1,5-a]pyridine-3-ylidene complexes and their application in olefin epoxidation catalysis. J. Catal. 2014, 319, 119–126. [Google Scholar] [CrossRef]
- Drees, M.; Hauser, S.A.; Cokoja, M.; Kühn, F.E. DFT studies on the reaction pathway of the catalytic olefin epoxidation with CpMoCF3 dioxo and oxo–peroxo complexes. J. Organomet. Chem. 2013, 748, 36–45. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by MoO2(SAP): A New Mode of tert-Butylhydroperoxide Activation. ChemCatChem 2012, 5, 601–611. [Google Scholar] [CrossRef]
- Poater, A.; Falivene, L.; Cavallo, L.; Llobet, A.; Rodríguez, M.; Romero, I.; Solà, M. Simple and cheap steric and electronic characterization of the reactivity of Ru(II) complexes containing oxazoline ligands as epoxidation catalysts. Chem. Phys. Lett. 2013, 577, 142–146. [Google Scholar] [CrossRef]
- Coquet, R.; Tada, M.; Iwasawa, Y. Energy-gaining formation and catalytic behavior of active structures in a SiO2-supported unsaturated Ru complex catalyst for alkene epoxidation by DFT calculations. Phys. Chem. Chem. Phys. 2007, 9, 6040–6046. [Google Scholar] [CrossRef] [PubMed]
- Aguiló, J.; Francàs, L.; Bofill, R.; Gil-Sepulcre, M.; García-Antón, J.; Poater, A.; Llobet, A.; Escriche, L.; Meyer, F.; Sala, X. Powerful Bis-facially Pyrazolate-Bridged Dinuclear Ruthenium Epoxidation Catalyst. Inorg. Chem. 2015, 54, 6782–6791. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, C.; Poater, A.; Benet-Buchholz, J.; Cavallo, L.; Solà, M.; Llobet, A. Dinuclear Ru-Aqua Complexes for Selective Epoxidation Catalysis Based on Supramolecular Substrate Orientation Effects. Chem. Eur. J. 2014, 20, 3898–3902. [Google Scholar] [CrossRef] [PubMed]
- Cavazzini, M.; Quici, S.; Pozzi, G. Hydrolytic kinetic resolution of terminal epoxides catalyzed by fluorous chiral Co(salen) complexes. Tetrahedron 2002, 58, 3943–3949. [Google Scholar] [CrossRef]
- Grivani, G.; Vakili, M.; Khalaji, A.D.; Bruno, G.; Rudbari, H.A.; Taghavi, M.; Tahmasebi, V. Synthesis, characterization, crystal structure determination, computational study, and thermal decomposition into NiO nano-particles of a new NiIIL2 Schiff base complex (L=2-{(E)-[2-chloroethyl)imino]methylphenolate). J. Mol. Struct. 2014, 1072, 77–83. [Google Scholar] [CrossRef]
- Düzenli, D.; Atmaca, D.O.; Gezer, M.G.; Onal, I. A density functional theory study of partial oxidation of propylene on Cu2O(001) and CuO(001) surfaces. Appl. Surf. Sci. 2015, 355, 660–666. [Google Scholar] [CrossRef]
- Salman, A.W.; Rehman, G.U.; Abdullah, N.; Budagumpi, S.; Endud, S.; Abdallah, H.H. Synthesis, characterization, density function theory and catalytic performances of palladium(II)–N-heterocyclic carbene complexes derived from benzimidazol-2-ylidenes. Inorg. Chim. Acta 2015, 438, 14–22. [Google Scholar] [CrossRef]
- Salman, A.W.; Rehman, G.U.; Abdullah, N.; Budagumpi, S.; Endud, S.; Abdallah, H.H.; Wong, W.Y. Sterically modulated palladium(II)-N-heterocyclic carbene complexes for the catalytic oxidation of olefins: Synthesis, crystal structure, characterization and DFT studies. Polyhedron 2014, 81, 499–510. [Google Scholar] [CrossRef]
- Dellamorte, J.; Lauterbach, J.; Barteau, M. Palladium-silver bimetallic catalysts with improved activity and selectivity for ethylene epoxidation. Appl. Catal. A 2011, 391, 281–288. [Google Scholar] [CrossRef]
- Robinson, J.R.; Yadav, J.; Fan, X.; Stanton, G.R.; Schelter, E.J.; Pericàs, M.A.; Walsh, P.J. Non-Covalent Immobilization of Rare Earth Heterobimetallic Frameworks and their Reactivity in an Asymmetric Michael Addition. Adv. Synth. Catal. 2014, 356, 1243–1254. [Google Scholar] [CrossRef]
- Yang, B.; Manz, T.A. Hafnium catalysts for direct alkene epoxidation using molecular oxygen as oxidant. RSC Adv. 2015, 5, 12311–12322. [Google Scholar] [CrossRef]
- Yan, W.; Ramanathan, A.; Ghanta, M.; Subramaniam, B. Towards highly selective ethylene epoxidation catalysts using hydrogen peroxide and tungsten- or niobium-incorporated mesoporous silicate (KIT-6). Catal. Sci. Technol. 2014, 4, 4433–4439. [Google Scholar] [CrossRef]
- Abashkin, Y.G.; Burt, S.K. (salen)Mn III Compounds as Nonpeptidyl Mimics of Catalase. Mechanism-Based Tuning of Catalase Activity: A Theoretical Study. Inorg. Chem. 2005, 44, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Hirao, H.; Kumar, D.; Thiel, W.; Shaik, S. Two States and Two More in the Mechanisms of Hydroxylation and Epoxidation by Cytochrome P450. J. Am. Chem. Soc. 2005, 127, 13007–13018. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.; Stillman, M. Challenging Density Functional Theory Calculations with Hemes and Porphyrins. Int. J. Mol. Sci. 2016, 17, 519. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.; Mosquera, R.A.; Melo, A.; Freire, C.; Cordeiro, M.N.D.S. Effects of Axial Coordination on Immobilized Mn(salen) Catalysts. J. Phys. Chem. A 2014, 118, 10788–10796. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K.; Rassolov, V.; Pople, J.A. Gaussian-3 theory using reduced Moller-Plesset order. J. Chem. Phys. 1999, 110, 4703–4709. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Martiínez, T.J. Graphical Processing Units for Quantum Chemistry. Comput. Sci. Eng. 2008, 10, 26–34. [Google Scholar] [CrossRef]
- Titov, A.V.; Ufimtsev, I.S.; Luehr, N.; Martsíinez, T.J. Generating Efficient Quantum Chemistry Codes for Novel Architectures. J. Chem. Theor. Comput. 2013, 9, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Martínez, T.J. Atomic orbital-based SOS-MP2 with tensor hypercontraction. I. GPU-based tensor construction and exploiting sparsity. J. Chem. Phys. 2016, 144, 174111. [Google Scholar] [CrossRef] [PubMed]
- Fales, B.S.; Levine, B.G. Nanoscale Multireference Quantum Chemistry: Full Configuration Interaction on Graphical Processing Units. J. Chem. Theor. Comput. 2015, 11, 4708–4716. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, R.; Scuseria, G.E.; Frisch, M.J. Achieving linear scaling in exchange-correlation density functional quadratures. Chem. Phys. Lett. 1996, 257, 213–223. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, H.J.J.; Vishnu, A.; de Jong, W.A. A Case for Soft Error Detection and Correction in Computational Chemistry. J. Chem. Theor. Comput. 2013, 9, 3995–4005. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional | N | % |
|---|---|---|
| B3LYP | 89 | 84.0 |
| BP86 | 27 | 25.5 |
| LDAor LSD | 15 | 14.2 |
| PW91 | 9 | 8.5 |
| BLYP | 8 | 7.5 |
| PBE | 8 | 7.5 |
| mPBE | 8 | 7.5 |
| OPBE | 7 | 6.6 |
| B3PW91 | 4 | 3.8 |
| PBE0 | 3 | 2.8 |
| M06 | 3 | 2.8 |
| B97 | 2 | 1.9 |
| PW91B95 | 2 | 1.9 |
| Functional | Basis Set | Complex | Singlet | Triplet | Quintuplet | Reference |
|---|---|---|---|---|---|---|
| BP86 | DZVP | Mn(acacen)Cl | +37.2 | 0.0 | +33.0 | [32] |
| BP86 | BS1 | Mn(acacen) | NA | +62.0 | 0.0 | [17] |
| B3LYP | BS1 | Mn(acacen) | NA | +112.0 | 0.0 | [17] |
| PW91 | BS2 | Mn(porphyrin) | +80.0 | +51.0 | 0.0 | [33] |
| LSD | BS3 e | Mn(salen) | −66.0 | 0.0 | +104.6 | [31] |
| PW91 | BS3 | Mn(salen) | −42.7 | 0.0 | +2.8 | [31] |
| TPSS | BS3 | Mn(salen) | −43.9 | 0.0 | +5.0 | [31] |
| X3LYP | BS3 | Mn(salen) | −18.7 | 0.0 | +82.3 | [31] |
| X3LYP | BS4 | Mn(salen) | −19.5 | 0.0 | +80.5 | [31] |
| X3LYP | BS5 | Mn(salen) | −21.7 | 0.0 | +84.1 | [31] |
| LSD | BS3 | Mn(salen)Cl | +6.1 | 0.0 | +240.5 | [31] |
| PW91 | BS3 | Mn(salen)Cl | +17.9 | 0.0 | +225.7 | [31] |
| TPSS | BS3 | Mn(salen)Cl | +22.7 | 0.0 | +224.9 | [31] |
| X3LYP | BS3 | Mn(salen)Cl | +16.5 | 0.0 | +238.6 | [31] |
| X3LYP | BS4 | Mn(salen)Cl | +24.1 | 0.0 | +236.8 | [31] |
| X3LYP | BS5 | Mn(salen)Cl | +20.0 | 0.0 | +237.3 | [31] |
| Method | Functional | Basis Set | Species | Singlet | Triplet | Quintet | Reference |
|---|---|---|---|---|---|---|---|
| DFT | B3LYP | DZVP | Oxo-Mn(acacen) | 0.0 | −6.7 | +20.9 | [23] |
| DFT | B3LYP | DZVP | Oxo-Mn(acacen)Cl | 0.0 | –33.9 | −39.3 | [23] |
| CCSD(T) | — | DZVP | Oxo-Mn(acacen) | 0.0 | +5.9 | +72.3 | [23] |
| CCSD(T) | — | DZVP | Oxo-Mn(acacen)Cl | 0.0 | +60.6 | +45.6 | [23] |
| DFT | BP86 | DZVP | Oxo-Mn(acacen)Cl | −59.8 | –32.6 | 0.0 | [32] |
| DMRG-SCF | — | 6-31G(d) | Oxo-Mn(acacen)Cl | 0.0 | −20.0 | +60.0 | [34] |
| DFT | BP86 | BS1 | Oxo-Mn(acacen) | 0.0 | +27.0 | +110.0 | [17] |
| DFT | B3LYP | BS1 | Oxo-Mn(acacen) | 0.0 | +11.0 | +40.0 | [17] |
| DFT | PW91 | BS2 e | Oxo-Mn(porphyrin) | 0.0 | +1.0 | +29.0 | [33] |
| CASSCF | — | 6-31G(d) | Oxo-Mn(acacen) | 0.0 | −12.1 | +165.9 | [35] |
| MRMP2 | — | 6-31G(d) | Oxo-Mn(acacen) | 0.0 | −9.6 | +188.1 | [35] |
| DFT | LSD | BS3 | Oxo-Mn(salen) | −16.0 | 0.0 | +99.2 | [31] |
| DFT | PW91 | BS3 | Oxo-Mn(salen) | +2.8 | 0.0 | +69.6 | [31] |
| DFT | TPSS | BS3 | Oxo-Mn(salen) | +5.0 | 0.0 | +62.7 | [31] |
| DFT | X3LYP | BS3 | Oxo-Mn(salen) | +23.5 | 0.0 | +45.0 | [31] |
| DFT | X3LYP | BS4 | Oxo-Mn(salen) | +21.8 | 0.0 | +40.4 | [31] |
| DFT | X3LYP | BS5 | Oxo-Mn(salen) | +24.9 | 0.0 | +46.5 | [31] |
| DFT | LSD | BS3 | Oxo-Mn(salen)Cl | −11.9 | 0.0 | +232.4 | [31] |
| DFT | PW91 | BS3 | Oxo-Mn(salen)Cl | +5.8 | 0.0 | +218.4 | [31] |
| DFT | TPSS | BS3 | Oxo-Mn(salen)Cl | +7.4 | 0.0 | +219.9 | [31] |
| DFT | X3LYP | BS3 | Oxo-Mn(salen)Cl | +7.8 | 0.0 | +232.3 | [31] |
| DFT | X3LYP | BS4 | Oxo-Mn(salen)Cl | +13.5 | 0.0 | +229.3 | [31] |
| DFT | X3LYP | BS5 | Oxo-Mn(salen)Cl | +7.7 | 0.0 | +231.4 | [31] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, F.; Cordeiro, M.N.D.S. Strengths, Weaknesses, Opportunities and Threats: Computational Studies of Mn- and Fe-Catalyzed Epoxidations. Catalysts 2017, 7, 2. https://doi.org/10.3390/catal7010002
Teixeira F, Cordeiro MNDS. Strengths, Weaknesses, Opportunities and Threats: Computational Studies of Mn- and Fe-Catalyzed Epoxidations. Catalysts. 2017; 7(1):2. https://doi.org/10.3390/catal7010002
Chicago/Turabian StyleTeixeira, Filipe, and M. Natália D. S. Cordeiro. 2017. "Strengths, Weaknesses, Opportunities and Threats: Computational Studies of Mn- and Fe-Catalyzed Epoxidations" Catalysts 7, no. 1: 2. https://doi.org/10.3390/catal7010002