
A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles


1
Graduate School of Engineering, Nagoya University, Nagoya, Aichi 464-8603, Japan
2
ESICB, Kyoto University, Katsura, Kyoto 615-8520, Japan
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(6), 191; https://doi.org/10.3390/catal7060191
Submission received: 15 May 2017
/
Revised: 16 June 2017
/
Accepted: 16 June 2017
/
Published: 19 June 2017
(This article belongs to the Special Issue Computational Methods and Their Application in Catalysis)
Abstract
:Recently, it has been reported that the morphology of Au nanoparticles (NPs) affects the catalytic activity of CO oxidation; twin crystal NPs show higher activity for CO oxidation than single-crystal NPs. In this study, density functional calculations have been carried out to investigate the morphology effect of Au NPs using CO as a probe molecule. In the case of Au NPs with a size of more than 2 nm, CO adsorption energy on the Au NPs is mainly determined by a coordination number (CN) of adsorption sites. CO binding to a multiple twin NP with a size of about 1 nm is stronger than that on a single-crystal NP with the same size. A simple CN explanation cannot be applied to the enhancement of CO binding to the small multiple twin NP. This enhancement is related to a deformation of the NP structure before and after CO adsorption. It is suggested that the multiple twin NP with a size of less than 1 nm, which shows the deformation upon CO adsorption, contributes to the higher activity for CO oxidation.
1. Introduction
It is well known that gold nanoparticles (NPs) show high catalytic activity for CO oxidation at low temperature [1,2,3]. The catalytic activity is affected by the size of Au NPs and abruptly increases as its size decreases to less than 3 nm. The size effect is sometimes explained by the reactivity in terms of special reaction sites at the metal–support interface [2,4]. There is another explanation for the size effect [5,6,7]. The percentage of highly-uncoordinated gold atoms on the Au NP surface yields observed differences in the reactivity of Au NPs. Other than the size, the morphology of Au NPs also influences their catalysis [8,9,10,11,12,13,14,15,16]. The ratio of different surface facets depends not only on the size of the nanoparticles, but also their morphology [17]. The morphology, which determines the number of coordination sites available, such as corners and edges, is also important for the catalytic activity. For example, the activity of the water-gas shift (WGS) reaction over Au/CeO2 drastically decreased with the loss of the 2D-layer morphology of Au [12]. Choudhary et al. suggested that quantum size effects, as a result of bilayered Au morphology, were important [8]. However, there is controversy regarding the explanation of the activity based on the thickness of the Au morphology. Lemire et al. [6] studied CO adsorption on gold monolayer islands and found that the adsorption property was no different from that on bulk gold. They suggested that the exceptional activity of gold nanoparticles for the low-temperature CO oxidation arose from the presence of highly-uncoordinated atoms. Twinning of Au NPs shows higher activity for the CO oxidation reaction than single crystal Au NPs [9,14,15]. The morphology effect by twinning was shown to be independent of the size effect. Even though Au NPs with the same particle distribution were synthesized, twin NPs were more active for CO oxidation than single-crystal NPs [14]. Determining the reason why twin NPs are more active will give us a deeper understanding for the design of Au NP catalyst.
In this study, density functional calculations have been carried out to investigate the morphology effect of Au NPs using CO as a probe molecule. This is because CO adsorption has been reported to have a strong correlation with the catalytic activity for CO oxidation [18,19,20]. The morphology effect of NPs ranging from 1 to 3 nm in size is examined. The morphology effect for NPs ranging from 2 to 3 nm in size can be explained by the coordination number of adsorption sites. For five-fold twin NPs with a size of 1 nm, the stability of NPs upon CO adsorption affects CO adsorption energy on the NP. Thus, the stability of NPs upon CO adsorption is also significant for the morphology effect.
2. Computational Details
Spin-polarized density-functional theory (DFT) calculations were performed using the plane wave self consistent field (PWscf) code in Quantum Espresso [21]. We employed the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional [22,23] and Vanderbilt ultra-pseudopotentials [24]. The system wave function was represented by a basis set of plane waves limited by an energy cutoff of 30 Ry, while the cutoff for the electron density representation was set to 300 Ry (1 Ryd ≈ 13.6 eV). All geometry optimizations were performed until the maximum force on atoms was less than 10−3 au. A cubic unit cell of 34 × 34 × 34 Å was used for large NPs and a unit of 21 × 21 × 21 Å was used for the smallest NPs and an isolated CO molecule. The sizes of the unit cells were sufficient to electronically isolate periodic images of the nanoparticles. A gamma k-point sampling of the Brillouin zone was chosen. Adequate spin states of nanoparticles were assigned using total magnetization parameters. The Fermi-Dirac smearing procedure [25] with a smearing parameter of 0.0007 Ry was applied. This smearing width is sufficiently small to gurantee a correct description of the spin states [26]. CO adsorption energy (Ead) on NPs was calculated using the following equation:
Ead = E(CO-NP) − E(NP) − E(CO)
Here, E(CO-NP) is the total energy of the Au NP interacting with CO, E(NP) is the total energy of the Au NP without CO, and E(CO) is the energy of the isolated CO molecule. Thus, a negative value of Ead means an exothermic adsorption.
The point group symmetry determined by the morphology of the NP affects the electronic states in the whole system. In the case of small Au clusters, the spin states do not always have the lowest spin because the high symmetry of Au clusters has a possibility of exhibiting degenerate states at the higest occupied molecular orbital level [26]. For example, a small Ag cluster with a symmetry of Ih has high spin states [27]. Symmetry change by the morphology is also important for the interpretation of observed spectra [28]. We calculated several spin states for Au55 NPs with a high symmetry of Oh. For the sake of comparison, the same spin states were calculated at the M06L/Def2-TZVPP(SDD)//M06L/Def2-SVP(SDD) level using the ORCA code [29]. Table 1 shows the results. The ground state of Au55 NP has the spin state of Sz = 0 and the energy difference to the first excited state is large. The lowest spin state is also expected for the large size of NPs with a lower symmetry than Oh. All spin states of Au NPs in this study were assumed to have the lowest spin. That is to say, the z component of the spin, Sz, was equal to 0 for even numbers of atoms and 1/2 for odd numbers of atoms.
3. Results
3.1. Model Nanoparticles
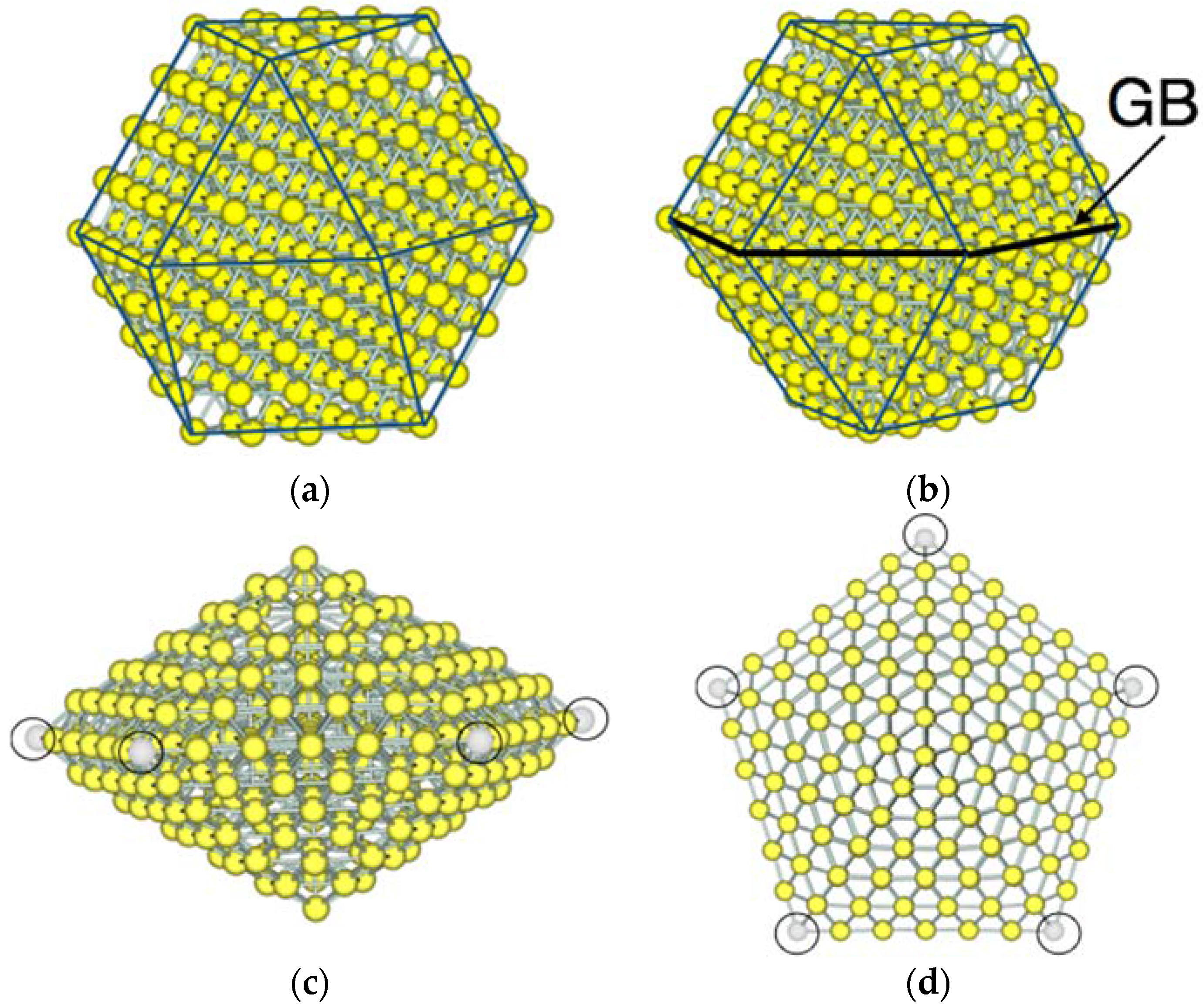
Figure 1 shows examples of the morphology of NPs adopted in this study. A truncated cuboctahedron structure is used for a single-crystal nanoparticle. This structure has a symmetry of Oh. Hereafter, we call the single-crystal NP as “Oh-NP”. In the Oh-NP structure, (111) faces are always located next to (100) faces. For twin NPs, two types of structures are adopted; one is a single twin structure, which has a single grain boundary, and the other is a multiple twin type, which has five-fold twinning. Both types of twin NPs were experimentally observed [13,14]. The single-twin NP has the symmetry of D3h (hereafter, referred to as “D3h-NP”). In the D3h-NP structure, the single twinning results in the grain boundary at which the (111) and (100) faces are next to the (111) and (100) faces, respectively. The five-fold twin NP is a decahedron and has the symmetry of D5h (hereafter, referred to as “D5h-NP”). The D5h-NP is composed of five distorted tetrahedral units, which are joined at a common edge and each of which shares two of their (111) faces as twinning planes.
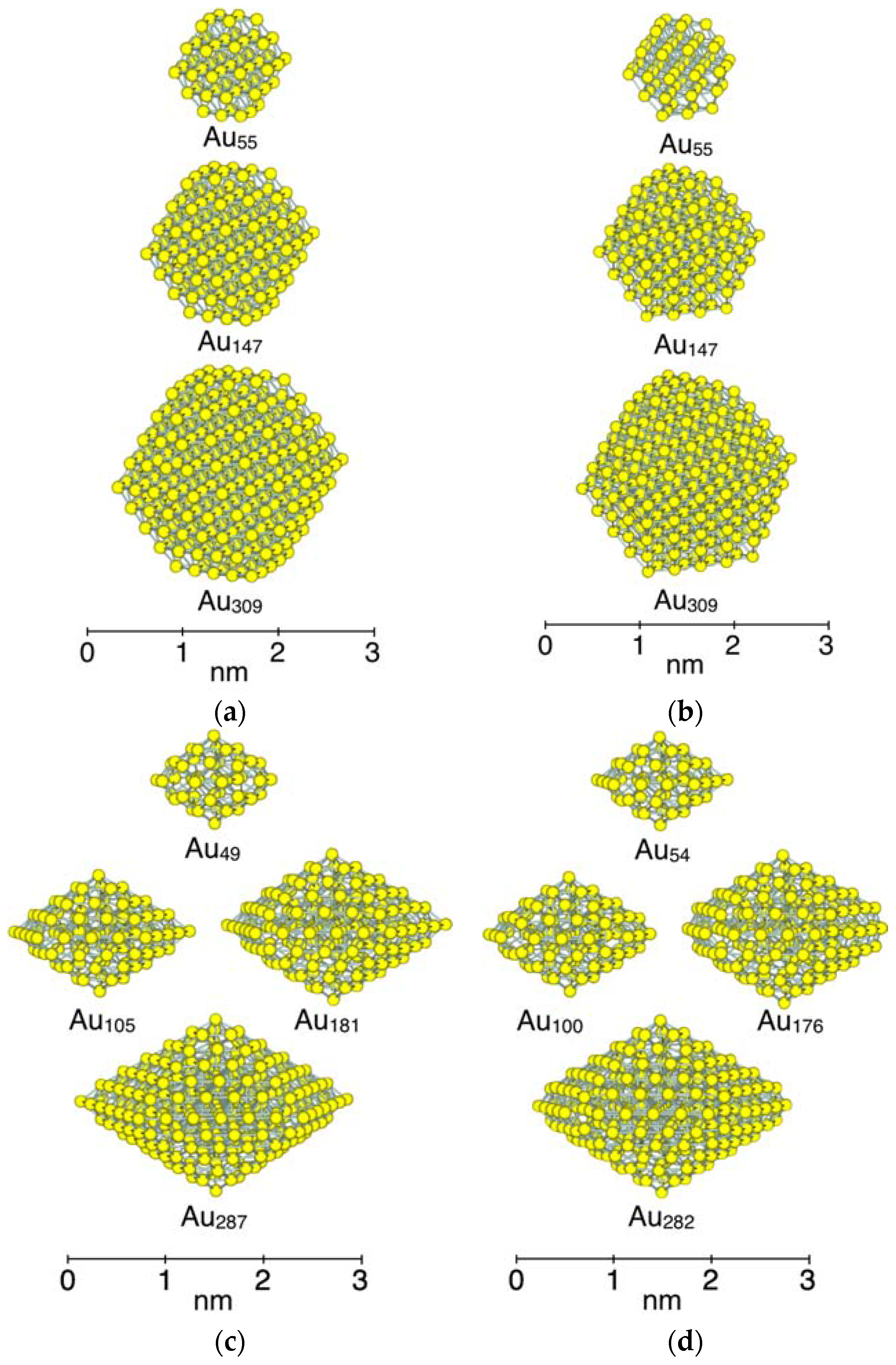
To investigate the size dependency of CO adsorption, several sizes of NPs were created, as shown in Figure 2. The smallest size of NP is Au55 for the Oh-NP and D3h-NP. The size of NP is increased by adding atoms all around the particle. We categorized three generations of different sizes of NP for both structures. As shown in Figure 2a,b, the first generation is Au55 NPs which have a diameter of about 1 nm. The second generation and the third generation are Au147 with a diameter of about 2 nm and Au309 with a diameter of about 3 nm, respectively. In the case of the D5h-NP, the size is increased by alternately adding atoms to the upper side and lower side of the particle. As a result, the spin states change with the increase in NP size because the number of atoms varies to be even and odd, alternatively. To hold the same symmetry and spin state, five atoms at corners (white atoms marked by black circles in Figure 1c,d) are used for adjuster atoms. For example, the removal of the five atoms from Au54 with the spin state of Sz = 0 leads to the D5h-NP of Au49 which has the spin state of Sz = 1/2 with the same D5h symmetry. Although the removal of these atoms elongates a bond length between an atom at a vertex adsorption site and the neighboring atoms by 0.04 Å (2.79 Å to 2.82 Å), the shape around the adsorption sites remains the same. For the D5h-NP of Sz = 1/2, Au49 is the first generation, with a diameter of about 1 nm. Au105 and Au181 are the second generation, which have a diameter of about 2 nm. Au287 is the third generation, with a diameter of about 3 nm. In the case of the spin state of Sz = 0, the five corner atoms (white atoms in Figure 1c,d) in the second and third generations of NPs are removed to make the number of atoms even.
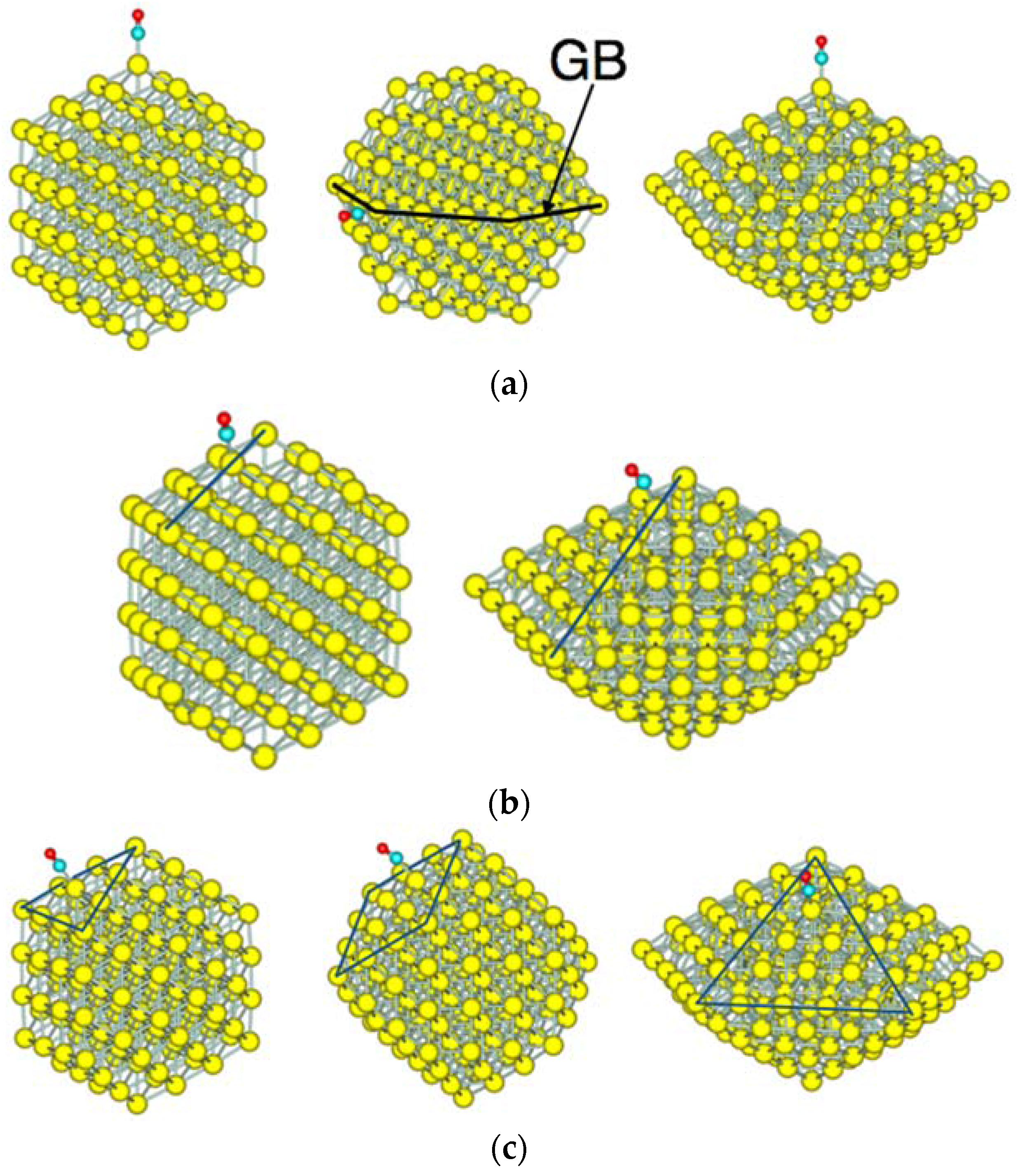
Figure 3 shows the adsorption sites on Au NPs; vertex sites in Figure 3a, edge sites in Figure 3b, and face sites in Figure 3c. For the single-twin NP, the vertex site at the grain boundary was examined. Two types of face sites are shown for the Oh-NP, which has (100) and (111) faces. There is a possibility of a bridge and three-fold-type adsorption on these faces. In our calculation, bridge-type adsorption on the (111) face yielded more stable adsorption than on-top adsorption. When interaction between co-adsorbed molecules can be neglected, multiple bonds between a molecule and surface atoms give rise to more stable adsorption than a single bond. Thus, the bridge-type adsorption for a single molecule, in general, yields a more stable adsorption than the on-top adsorption. Since the objective of this study is to compare the CO adsorption by varying the size and the morphology of NPs, adsorption sites are limited to the on-top adsorption.
3.2. Size Dependency of CO Adsoprtion
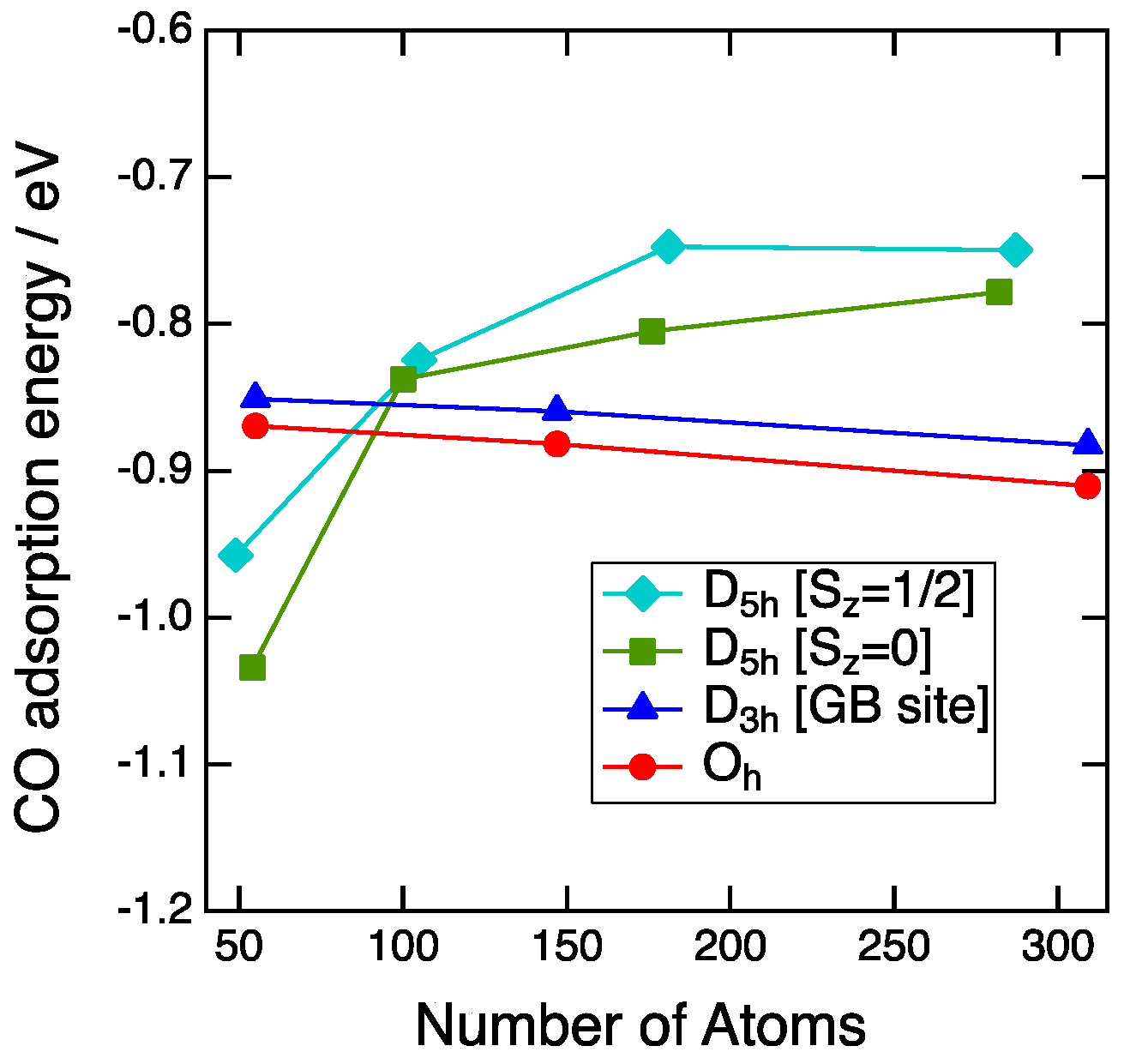
Figure 4 shows the plot of the CO adsorption energy on the vertex sites as a function of the number of Au atoms. For the Oh-NP and D3h-NP, CO adsorption strength gradually decreases as the increase of the number of Au atoms. The differences in energy between the size of about 1 nm and 3 nm are 0.04 eV for the Oh-NP and 0.03 eV for the D3h-NP. Thus, the adsorption energy is mainly determined by the local environment of the adsorption sites, such as the coordination number (CN). The CN of the adsorption sites is a good descriptor for the CO binding to Au NPs [7,19]. Since the CN of the vertex sites of the Oh-NP and D3h-NPs is equal to five, CO adsorption energies of the Oh-NP differ slightly from those of the single-twin NP in all sizes. For the second and the third generations, the D5h-NPs show weaker binding than the Oh-NPs. This is also explained by the coordination number. The CN for the D5h-NP is six, whereas that for the Oh-NP is five. Therefore, the more unsaturated site of the Oh-NP results in the more stable adsorption of CO. The exception occurs in the case of the first generation of the D5h-NP.
The size dependence of the CO adsorption energy for the D5h-NPs is reverse to that of the Oh-NP. Furthermore, the energy abruptly decreases from the second to the first generation and the CO adsorption on the D5h-NP becomes more stable than that of the Oh-NP. This stabilization cannot be explained by the simple CN consideration. This is not due to the strain effect because the local structure around the adsorption sites is not affected by the morphology or the size. The average bond distance between a site atom and the neighboring atoms is 2.82 Å for the D5h-Au54, 2.83 Å for the D5h-Au282, and 2.84 Å for the Oh-Au309. The small difference between these values indicates no strain effect. Next, we focused on the deformation of NPs before and after CO adsorption, since CO adsorption leads to the structural change of the NP itself. Root-mean-square distances (RMSD) between atom positions of NPs were used as a quantitative index for the structural change. At first, the local deformation around the adsorption sites was examined. The average RMSD of the site and neighboring atoms for the D5h-NP (Au54, Sz = 0) and Oh-NP (Au55) were 6.9 pm and 6.8 pm, respectively. Thus, the local changes of both NPs induced by CO adsorption are almost the same. Next, the deformation of the whole NP structure was examined. The average RMSD of all atoms for the D5h-NP (Au54, Sz = 0) and Oh-NP (Au55) were 13.9 pm and 3.1 pm, respectively. The average RMSD increased for the D5h-NP and decreased for the Oh-NP. This means that CO adsorption on the D5h-NP results in the structural change of all of the NP atoms, whereas that on the Oh-NP only affects the local structure around the adsorption sites. Partial optimizations of CO adsorption on the D5h-NPs of Au54 and Au49 with fixing NP geometries led to the adsorption energies of −0.76 eV and −0.84 eV, respectively. Adsorption energies by the full optimizations were −1.03 eV and −0.96 eV for Au54 and Au49, respectively. Thus, the stabilization by deformation was 0.27 eV for Au54 and 0.12 eV for Au49. The average RMSD for the D5h-NPs of Au54 and Au49 were 13.9 pm and 7.2 pm. Thus, the stabilization is related to the deformation degree of the whole NP structure upon CO adsorption. In other words, the morphology effect of the multiple twin NP around the size of about 1 nm arises from the stability of the whole NP structure upon CO adsorption.
3.3. CO Adsorption vs. the Coordination Number
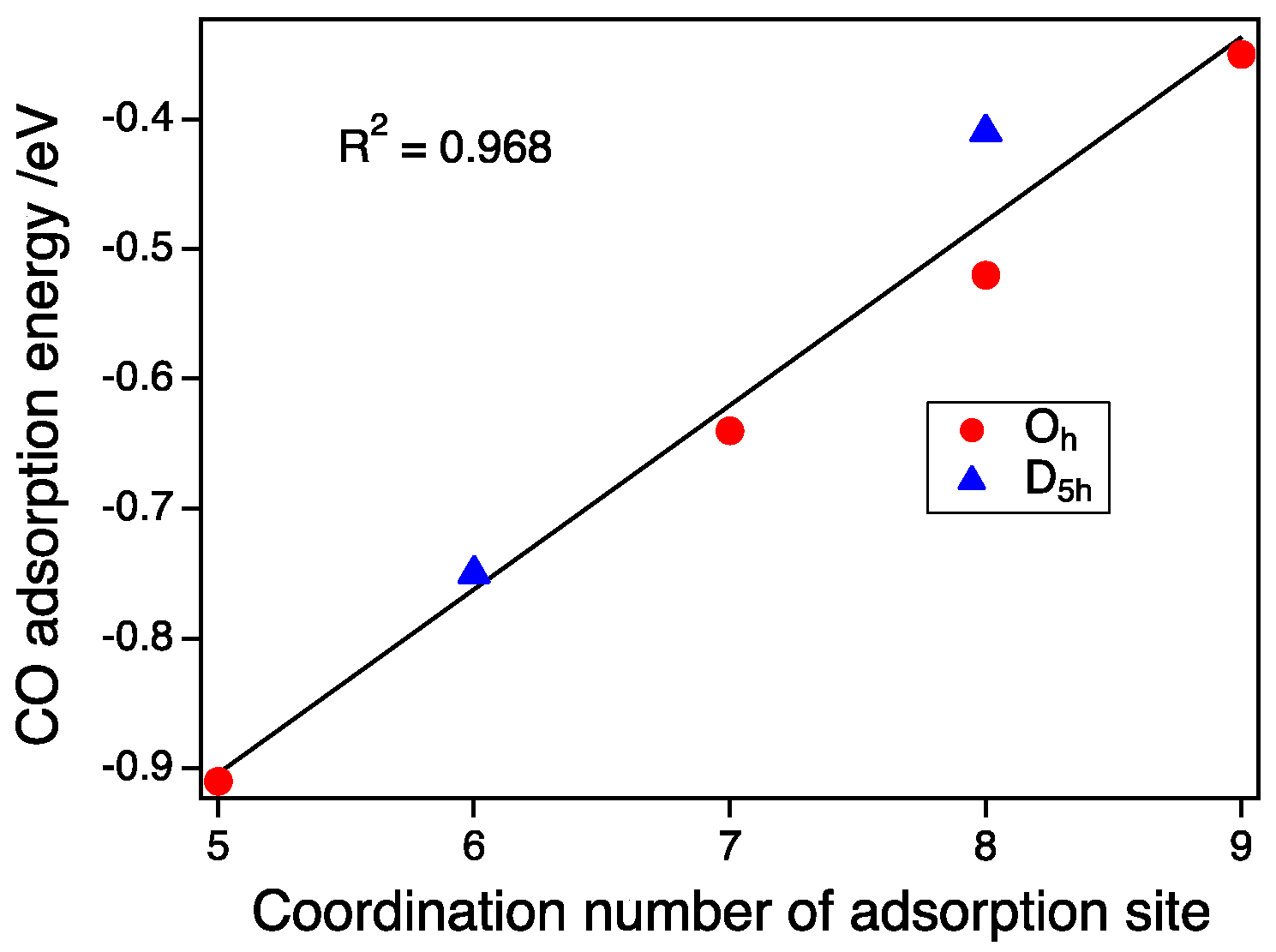
As shown in Figure 4, the size dependence on the CO adsorption for the D5h-NP and Oh-NP with the same spin state of Sz = 1/2 is small between the second and third generations. Therefore, the pure CN dependence without the size effect can be estimated. We calculated the predicted adsorption energy of the Oh-NP of Au287, which had the same number of atoms as the largest D5h-NP, using an interpolation method. Figure 5 shows the plot of the CO adsorption energy as a function of the CN of the on-top sites. Adsorption energies at CN 5, 6, 7, and 9 were obtained from the adsorptions on the vertex site of the Oh-NP, the vertex of the D5h-NP, the edge of the Oh-NP, and the (111) face of the Oh-NP, respectively. Energies at CN 8 were calculated from the adsorption on the edge site of the D5h-NP and the (100) face sites of the Oh-NP. We can clearly establish a linear relationship between the CN and the CO adsorption energy as:
Ead/eV = 0.14 × CN − 1.61.
The CO binding dependence on the CN has been already reported [19,30]. Mpourmpakis et al. used bond angles plus the CN as parameters for fitting. The need for bond angles is because they include the size and shape effect into such parameters. The absolute values reported by Lopez et al. [19] are different from our results. The adsorption energy in our study is the expected value in the case of the bulk surface because the size of NPs in our study is large enough to suppress the deformation upon CO adsorption. Experimentally, CO adsorption energies on an Au(211) surface, or 3(111) × (100) in microfacet notation, was determined [31]. The adsorption energy on a step-like (100) facet was 0.52 eV. The adsorption energy on a (111) face was estimated to be −0.28 to −0.39 eV. These experimental values agree with our calculated values. Since the energy dependence on the size is small for the large NPs with a size of more than 2 nm, the adsorption strength can be determined by the CN of the adsorption sites alone. Thus, the morphology effect of such large NPs can be explained by the number of CNs in the surface atoms [7].
4. Discussion
Now, we discuss the catalytic activity of the multiple twin Au-NP. The enhanced binding of CO for Au NPs is central to understanding the enhanced catalytic properties. Meier and Goodman observed the heat of CO adsorption for Au NPs ranging in size from 1.8 to 3.1 nm. They showed that the size of Au NP with the highest heat was very close to that exhibiting the maximum catalytic activity for CO oxidation [20]. DFT works by Lopez et al. [19] and Hvolbæk et al. [32] reported that the coordination number (CN) was the important factor for CO oxidation over Au catalysts in comparison to the role of charge transfer, layer thickness, and interactions with the support. Taylor et al. [7] reported that the relation between the CO oxidation activity and the average CO binding energy was affected by the average surface CN. In the case of the Oh-NPs in our study, the average surface CN was 7.2 for Au147 to 6.6 for Au55. According to Equation (2), the decrease in size leads to the increase in CO binding by 0.84 eV. Thus, the size effect for catalytic activity using the CN can be applied for the Oh-NP. For the second and third generations, this estimate is also applicable for the D5h-NP, which had the average surface CN of more than 7.4 (see Table S1). Since CO binding on the D5h-NP was weak, the D5h-NPs of this size are not active species for enhanced CO oxidation. On the other hand, the CO adsorption energy of the D5h-NP in the first generation was more negative than that of the Oh-NP. Thus, the species showing higher catalytic activity than the single-crystal NPs are expected to be multiple twin structures with a size of less than 1 nm. The enhancement comes from the deformation of the whole NP structure, or the stability of Au-NP, upon gas adsorption. Some theoretical studies [33,34] reported the relation between the stability of metal NPs upon gas adsorption and the activity for CO oxidation. Recently, environmental transmission electron microcopy (ETEM) measurement showed that the fraction of morphology-changeable Au-NPs is not negligible for catalytic activity [35].
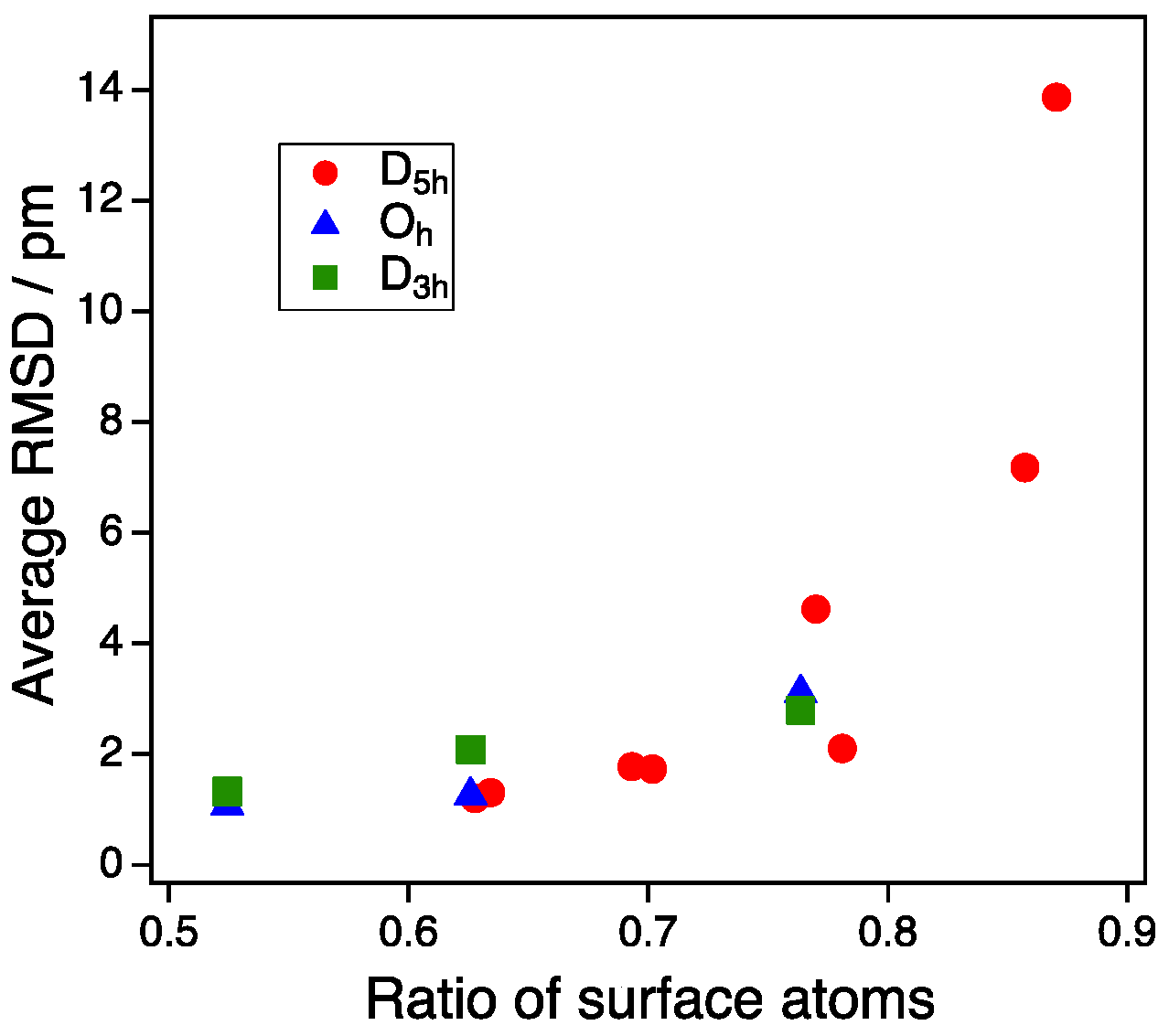
Finally, we suggest a ratio of surface atoms as a simple descriptor of the deformation effect mentioned above. Figure 6 shows the plot of the average RMSD for the vertex adsorption of all NPs as a function of the ratio of surface atoms to all atoms of the NPs. When the ratio of the surface atoms was 0.8 or more, the deformation described by the average RMSD increased sharply. Although this deformation occurred only for the multiple twin NPs (D5h-NPs) in our case, this would also occur for NPs that have a symmetry other than D5h in the case of the smaller size of NPs. Since the average RMSD represents the stability of Au NP upon CO adsorption, there is a possibility that the ratio of surface atoms can be a reaction descriptor for small NPs, in which the stability is significant for gas adsorption.
5. Conclusions
DFT calculations were performed for CO adsorption on three generations of Au NPs with sizes of about 1, 2, and 3 nm. The size effect for the CO adsorption energy on the vertex site was small in the case of the single-crystal structure and the single-twin structure. For NPs larger than 2 nm, the CO adsorption energy was mainly determined by the coordination number of the adsorption sites, regardless of the symmetry of the Au NPs. Under the condition that the size effect was removed, a clear linear relationship between the adsorption energy and the coordination number (CN) was obtained without another parameter, such as bond angles. The size dependence of CO adsorption on the vertex site for the multiple twin structure of D5h was large, and a sharp enhancement occurred as the size decreased from 2 nm to 1 nm. This was not due to the strain effect. The deformation of the whole NP structure before and after CO adsorption was large for the multiple twin structure with a size of 1 nm. This deformation resulted in the enhancement of CO adsorption on the multiple twin NP. Thus, the morphology effect for the small size of multiple twin NPs arises from the deformation upon CO adsorption. Finally, we suggested a ratio of surface atoms as a simple descriptor for the deformation effect. When the ratio of the surface atoms was 0.8 or more, the deformation described by the average RMSD increased sharply. In such cases, the CO adsorption energy must be estimated by the deformation in addition to the CN.
Supplementary Materials
The following is available online at www.mdpi.com/2073-4344/7/6/191/s1, Table S1: CO adsorption energy, average surface CN, ratio of surface atoms, and average RMSD for all Au NPs.
Acknowledgments
This work was supported by JSPS KAKENHI Grant-in-Aid for Young Scientists (A), No. 16H06131. The computations were performed at the Research Center for Computational Science, Okazaki, Japan.
Author Contributions
K.S. conceived the computational study, based on higher-level discussions of Au twin-particle with J.O. and A.S. K.S. performed the simulation and analysis, with Au NP structures for input from T.K. K.S. wrote the paper. All authors contributed to preparing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haruta, M. Chance and necessity: My encounter with gold catalysts. Angew. Chem. Int. Ed. Engl. 2014, 53, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Koga, H.; Okumura, M.; Haruta, M. Advances in Gold Catalysis and Understanding the Catalytic Mechanism. Chem. Rec. 2016, 16, 2278–2293. [Google Scholar] [CrossRef] [PubMed]
- Min, B.K.; Friend, C.M. Heterogeneous gold-based catalysis for green chemistry: Low-temperature CO oxidation and propene oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Tokunaga, T.; Zhang, L.; Li, D.W.; Chen, L.Y.; Arai, S.; Yamamoto, Y.; Hirata, A.; Tanaka, N.; Ding, Y.; et al. Atomic Observation of Catalysis-Induced Nanopore Coarsening of Nanoporous Gold. Nano Lett. 2014, 14, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, S.H.; Grønbjerg, U.; Hvolbæk, B.; Schiøtz, J. Understanding the catalytic activity of gold nanoparticles through multi-scale simulations. J. Catal. 2011, 284, 34–41. [Google Scholar] [CrossRef]
- Lemire, C.; Meyer, R.; Shaikhutdinov, S.; Freund, H.J. Do quantum size effects control CO adsorption on gold nanoparticles? Angew. Chem. Int. Ed. Engl. 2004, 43, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.G.; Austin, N.; Gounaris, C.E.; Mpourmpakis, G. Catalyst Design Based on Morphology- and Environment-Dependent Adsorption on Metal Nanoparticles. ACS Catal. 2015, 5, 6296–6301. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Goodman, D.W. Catalytically active gold: The role of cluster morphology. Appl. Catal. A Gen. 2005, 291, 32–36. [Google Scholar] [CrossRef]
- Cunningham, D.A.H.; Vogel, W.; Sanchez, R.M.T.; Tanaka, K.; Haruta, M. Structural analysis of Au/TiO2 catalysts by Debye function analysis. J. Catal. 1999, 183, 24–31. [Google Scholar] [CrossRef]
- Feng, X.F.; Jiang, K.L.; Fan, S.S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef] [PubMed]
- Kartusch, C.; Krumeich, F.; Safonova, O.; Hartfelder, U.; Makosch, M.; Sa, J.; van Bokhoven, J.A. Redispersion of Gold Multiple Twinned Particles during Liquid-Phase Hydrogenation. ACS Catal. 2012, 2, 1394–1403. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, Z.; Wen, J.; Ding, K.; Yang, X.; Poeppelmeier, K.R.; Marks, L.D. Adhesion and Atomic Structures of Gold on Ceria Nanostructures: The Role of Surface Structure and Oxidation State of Ceria Supports. Nano Lett. 2015, 15, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.; Hofmeister, H.; Claus, P. The influence of real structure of gold catalysts in the partial hydrogenation of acrolein. J. Catal. 2003, 213, 86–94. [Google Scholar] [CrossRef]
- Ohyama, J.; Koketsu, T.; Yamamoto, Y.; Arai, S.; Satsuma, A. Preparation of TiO2-supported twinned gold nanoparticles by CO treatment and their CO oxidation activity. Chem. Commun. 2015, 51, 15823–15826. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.D.; Guttel, R.; Leoni, M.; Schuth, F.; Weidenthaler, C. Influence of the Microstructure of Gold-Zirconia Yolk-Shell Catalysts on the CO Oxidation Activity. J. Phys. Chem. C 2010, 114, 19386–19394. [Google Scholar] [CrossRef]
- Quintanilla, A.; Butselaar-Orthlieb, V.C.L.; Kwakernaak, C.; Sloof, W.G.; Kreutzer, M.T.; Kapteijn, F. Weakly bound capping agents on gold nanoparticles in catalysis: Surface poison? J. Catal. 2010, 271, 104–114. [Google Scholar] [CrossRef]
- Marks, L. Experimental studies of small particle structures. Rep. Prog. Phys. 1994, 57, 603. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. 2008, 120, 4913–4917. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Meier, D.C.; Goodman, D.W. The influence of metal cluster size on adsorption energies: CO adsorbed on Au clusters supported on TiO(2). J. Am. Chem. Soc. 2004, 126, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Gruber, M.; Heimel, G.; Romaner, L.; Brédas, J.-L.; Zojer, E. First-principles study of the geometric and electronic structure of Au13 clusters: Importance of the prism motif. Phys. Rev. B 2008, 77, 165411. [Google Scholar] [CrossRef]
- Pereiro, M.; Baldomir, D.; Arias, J.E. Unexpected magnetism of small silver clusters. Phys. Rev. A 2007, 75, 063204. [Google Scholar] [CrossRef]
- Sawabe, K.; Hiro, T.; Shimizu, K.-i.; Satsuma, A. Density functional theory calculation on the promotion effect of H2 in the selective catalytic reduction of NOx over Ag–MFI zeolite. Catal. Today 2010, 153, 90–94. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Mpourmpakis, G.; Andriotis, A.N.; Vlachos, D.G. Identification of descriptors for the CO interaction with metal nanoparticles. Nano Lett. 2010, 10, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Samano, E.; Koel, B.E. CO adsorption and reaction on clean and oxygen-covered Au(211) surfaces. J. Phys. Chem. B 2006, 110, 17512–17517. [Google Scholar] [CrossRef] [PubMed]
- Hvolbæk, B.; Janssens, T.V.W.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Nørskov, J.K. Catalytic activity of Au nanoparticles. Nano Today 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Shimizu, K.-i.; Sawabe, K.; Satsuma, A. Self-Regenerative Silver Nanocluster Catalyst for CO Oxidation. ChemCatChem 2011, 3, 1290–1293. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, D.; Zhu, J. Theoretical study of CO catalytic oxidation on free and defective graphene-supported Au–Pd bimetallic clusters. RSC Adv. 2014, 4, 42554–42561. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yoshida, H.; Kamiuchi, N. Correlation of catalytic activity with the morphology change of supported Au nanoparticles in gas. Surf. Sci. 2017, 659, 16–19. [Google Scholar] [CrossRef]
Figure 1.
Morphology of model Au nanoparticles (NPs). (a) A single-crystal structure with the symmetry of Oh (Oh-NP). The (100) faces depicted by a rectangle are always next to the (111) faces depicted by a triangle; (b) a single twin structure with the symmetry of D3h (D3h-NP) which has a grain boundary (GB). At the grain boundary, the (100) and (111) faces are always next to the (100) and (111) faces, respectively; (c) side view and (d) top view of the multiple twin structure with the symmetry of D5h (D5h-NP). This structure exhibits five-fold twinning which share the neighboring (111) faces.
Figure 1.
Morphology of model Au nanoparticles (NPs). (a) A single-crystal structure with the symmetry of Oh (Oh-NP). The (100) faces depicted by a rectangle are always next to the (111) faces depicted by a triangle; (b) a single twin structure with the symmetry of D3h (D3h-NP) which has a grain boundary (GB). At the grain boundary, the (100) and (111) faces are always next to the (100) and (111) faces, respectively; (c) side view and (d) top view of the multiple twin structure with the symmetry of D5h (D5h-NP). This structure exhibits five-fold twinning which share the neighboring (111) faces.
Figure 2.
Size of model Au NPs. (a) Single-crystal Oh-NPs; (b) simple twin D3h-NPs; (c) multiple twin D5h-NPs with the spin state of Sz = 1/2; and (d) multiple twin D5h-NPs with the spin state of Sz = 0. Three generations are categorized for these NPs. The first generation includes NPs of less than 100 atoms and their diameters are about 1 nm. The second generation includes NPs composed of 100 to 200 atoms and their diameters are about 2 nm. The third generation includes NPs of around 300 atoms, and their diameters are about 3 nm.
Figure 2.
Size of model Au NPs. (a) Single-crystal Oh-NPs; (b) simple twin D3h-NPs; (c) multiple twin D5h-NPs with the spin state of Sz = 1/2; and (d) multiple twin D5h-NPs with the spin state of Sz = 0. Three generations are categorized for these NPs. The first generation includes NPs of less than 100 atoms and their diameters are about 1 nm. The second generation includes NPs composed of 100 to 200 atoms and their diameters are about 2 nm. The third generation includes NPs of around 300 atoms, and their diameters are about 3 nm.
Figure 3.
CO adsorption sites of NPs. All adsorption sites are the on-top type. (a) Vertex sites of the Oh-NP, D3h-NP, and D5h-NP. On the D3h-NP, the vertex site is located at the grain boundary (GB); (b) edge sites of the Oh-NP and D5h-NP; and (c) face sites of the Oh-NP and D5h-NP. The Oh-NP has both the (111) face (left) and the (100) face (middle). The D5h-NP has only the (111) face. Yellow, cyan, and red spheres represent Au, C, and O atoms, respectively.
Figure 3.
CO adsorption sites of NPs. All adsorption sites are the on-top type. (a) Vertex sites of the Oh-NP, D3h-NP, and D5h-NP. On the D3h-NP, the vertex site is located at the grain boundary (GB); (b) edge sites of the Oh-NP and D5h-NP; and (c) face sites of the Oh-NP and D5h-NP. The Oh-NP has both the (111) face (left) and the (100) face (middle). The D5h-NP has only the (111) face. Yellow, cyan, and red spheres represent Au, C, and O atoms, respectively.

Figure 4.
CO adsorption energy on the vertex sites plotted with respect to the number of atoms of NPs. Aqua diamonds, green squares, blue triangles, and red circles represent the CO adsorption energies on the D5h-NPs with spin states of Sz = 1/2, the D5h-NPs with spin states of Sz = 0, the D3h-NPs and the Oh-NPs, respectively.
Figure 4.
CO adsorption energy on the vertex sites plotted with respect to the number of atoms of NPs. Aqua diamonds, green squares, blue triangles, and red circles represent the CO adsorption energies on the D5h-NPs with spin states of Sz = 1/2, the D5h-NPs with spin states of Sz = 0, the D3h-NPs and the Oh-NPs, respectively.
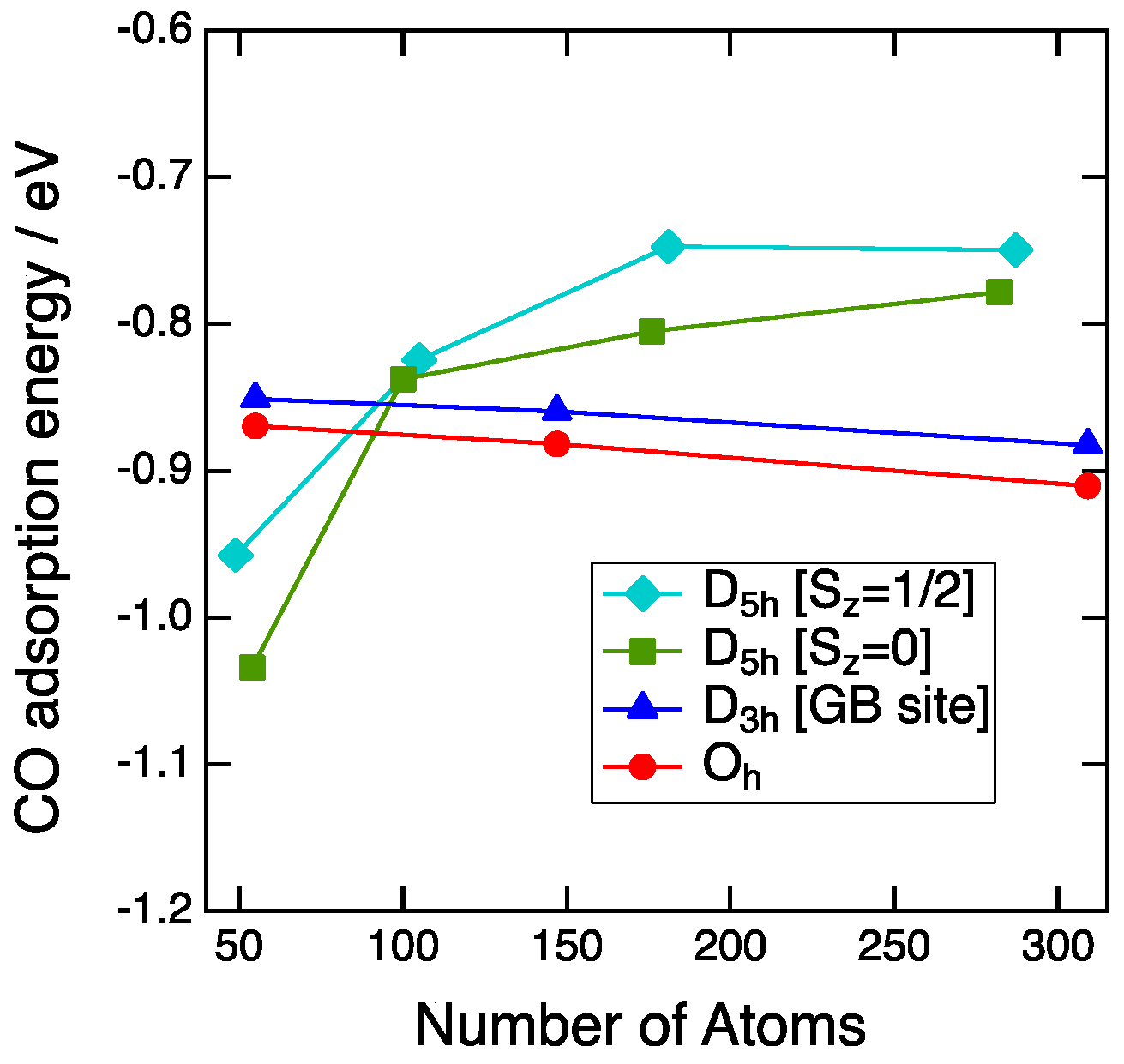
Figure 5.
CO adsorption energy as a function of the coordination number of adsorption sites. Red circles and blue triangles represent the CO adsorption energies on the Oh-NPs and D5h-NPs (Sz = ½), respectively.
Figure 5.
CO adsorption energy as a function of the coordination number of adsorption sites. Red circles and blue triangles represent the CO adsorption energies on the Oh-NPs and D5h-NPs (Sz = ½), respectively.
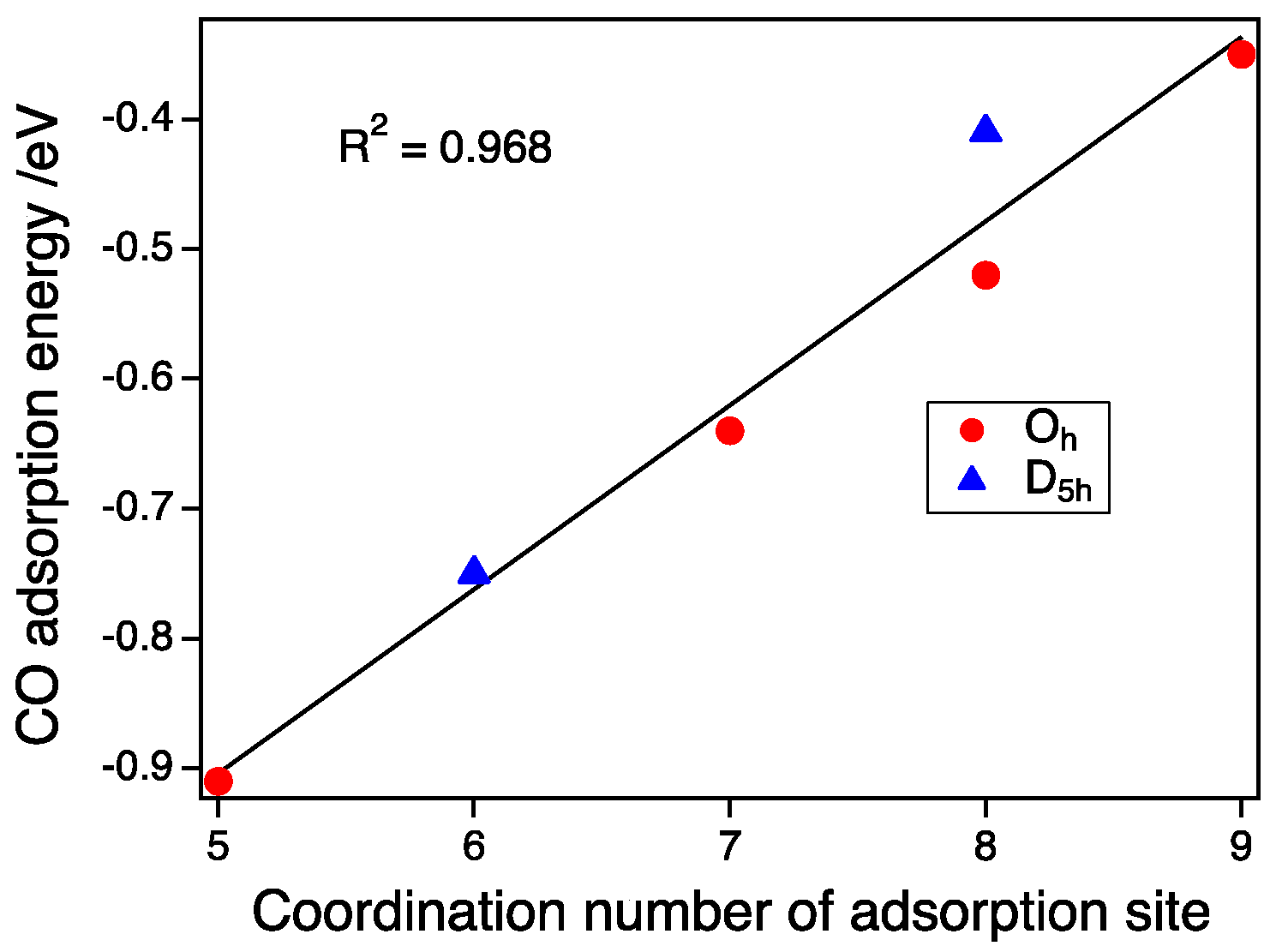
Figure 6.
Average RMSD as a function of the ratio of surface atoms. Average RMSD represents an average value of the root mean square distances (RMSDs) of all NP atom coordinates before and after CO adsorption.
Figure 6.
Average RMSD as a function of the ratio of surface atoms. Average RMSD represents an average value of the root mean square distances (RMSDs) of all NP atom coordinates before and after CO adsorption.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Energy in eV of Au55 (Oh) relative to that of the spin state of Sz = 0.
| Sz | PBE/Plane Wave | M06L/Def2-TZVPP(SDD) * |
|---|---|---|
| 1/2 | - | - |
| 3/2 | 0.26 | 0.25 |
| 5/2 | 1.00 | 0.99 |
* Geometry optimizations were carried out at the M06L/Def2-SVP(SDD) level.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sawabe, K.; Koketsu, T.; Ohyama, J.; Satsuma, A. A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles. Catalysts 2017, 7, 191. https://doi.org/10.3390/catal7060191
AMA Style
Sawabe K, Koketsu T, Ohyama J, Satsuma A. A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles. Catalysts. 2017; 7(6):191. https://doi.org/10.3390/catal7060191
Chicago/Turabian StyleSawabe, Kyoichi, Taiki Koketsu, Junya Ohyama, and Atsushi Satsuma. 2017. "A Theoretical Insight into Enhanced Catalytic Activity of Au by Multiple Twin Nanoparticles" Catalysts 7, no. 6: 191. https://doi.org/10.3390/catal7060191
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.