
l-Amino Acid Production by a Immobilized Double-Racemase Hydantoinase Process: Improvement and Comparison with a Free Protein System


Abstract
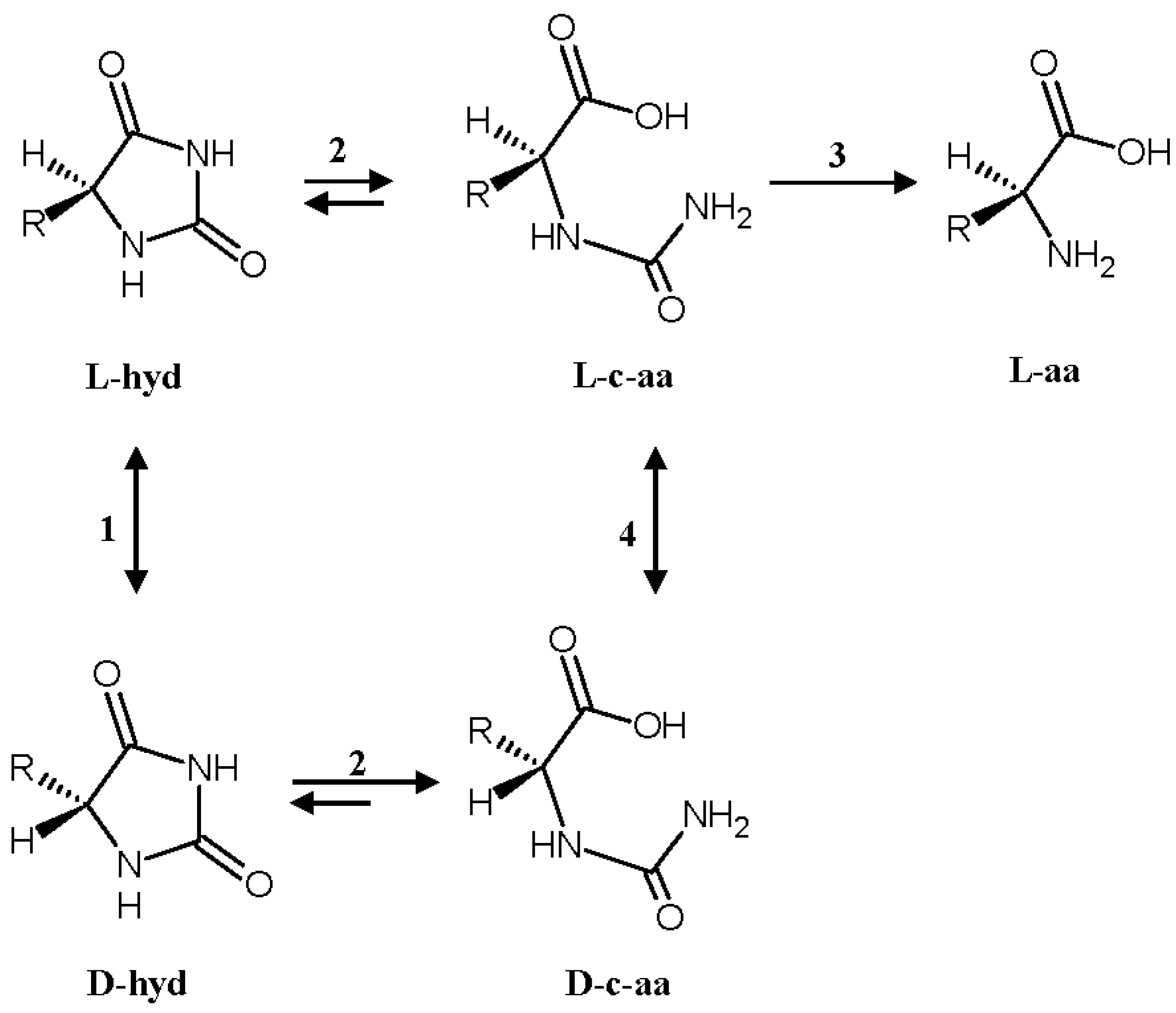
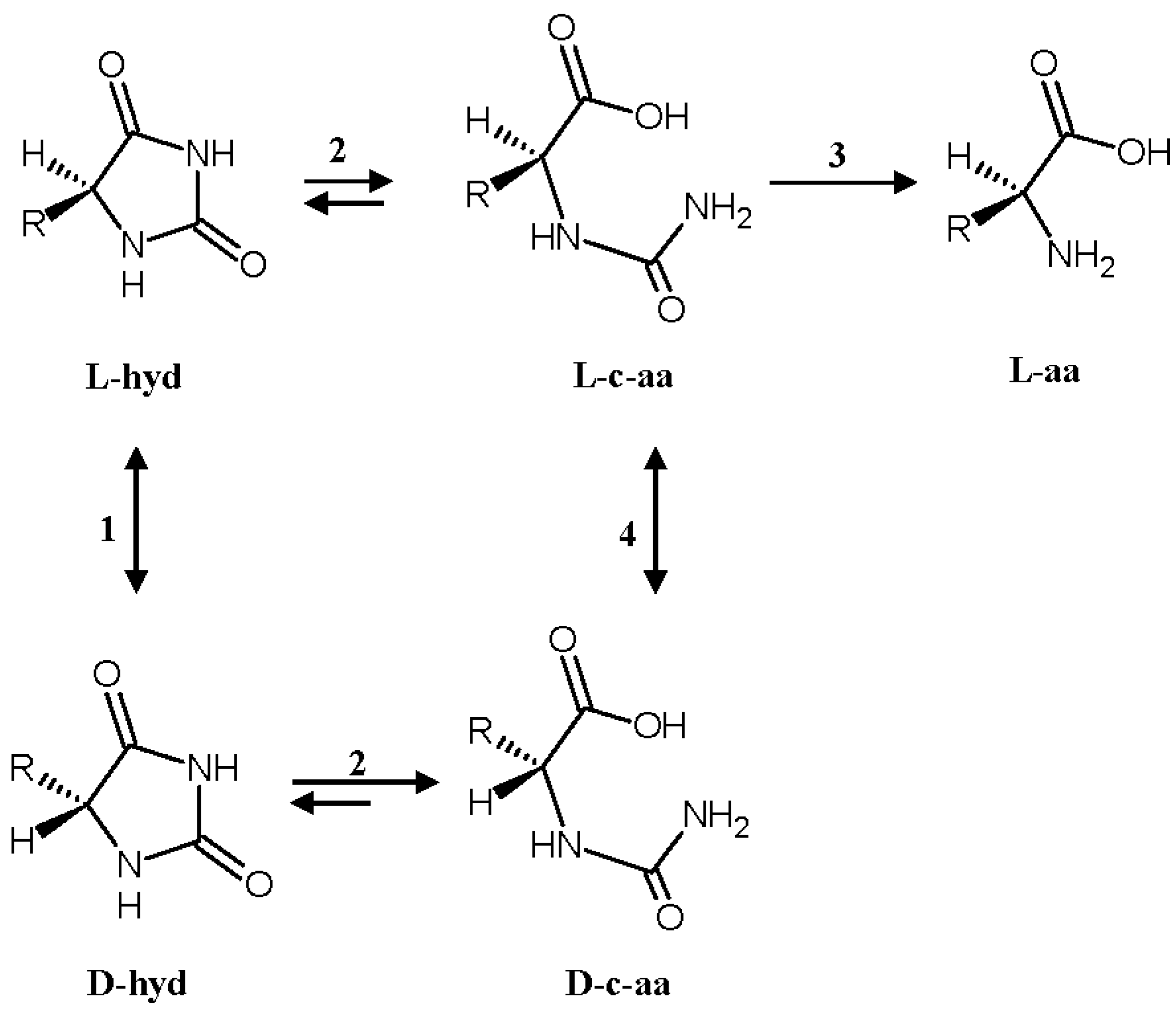
:1. Introduction
2. Results and Discussion
2.1. Protein Loading
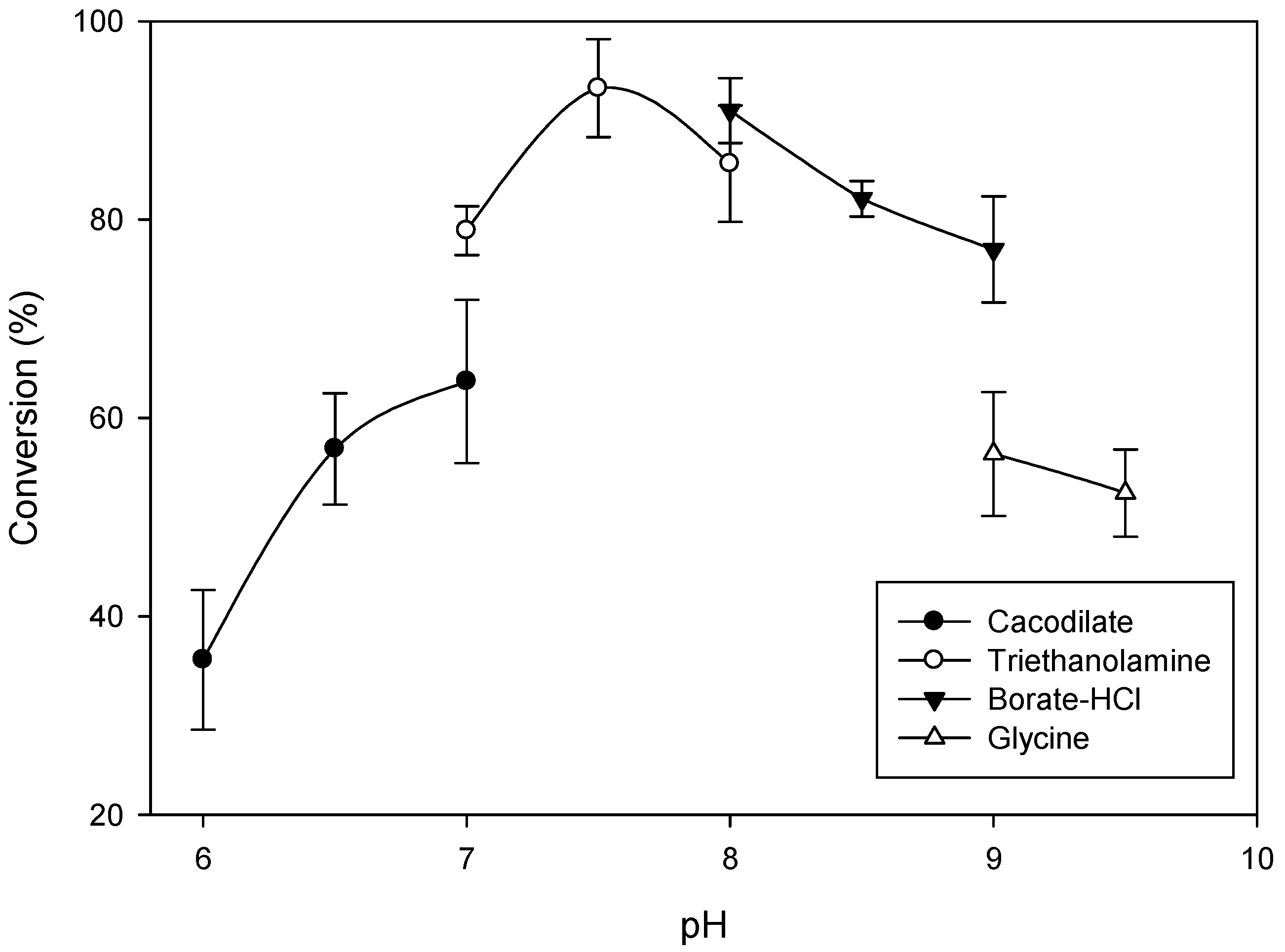
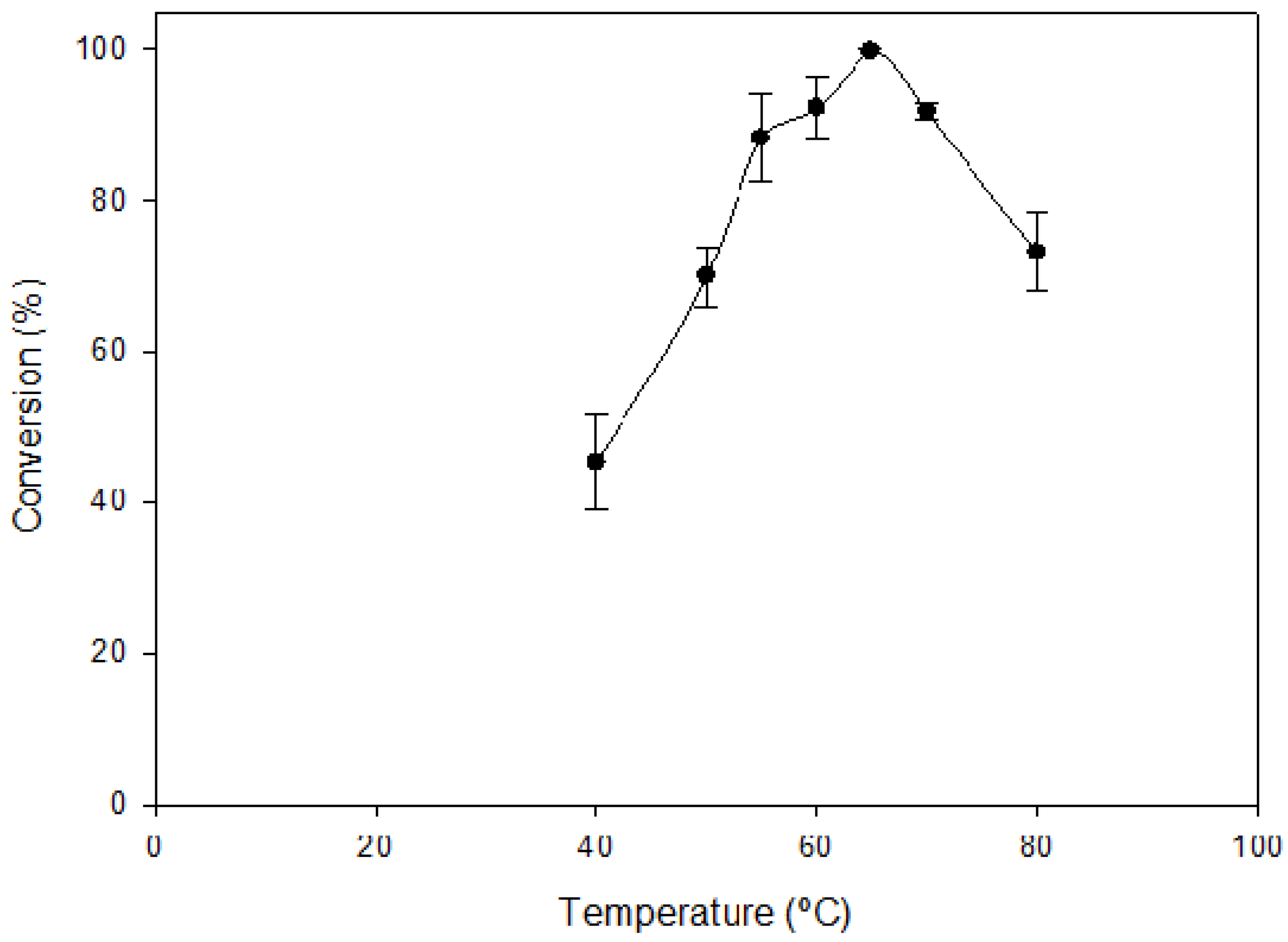
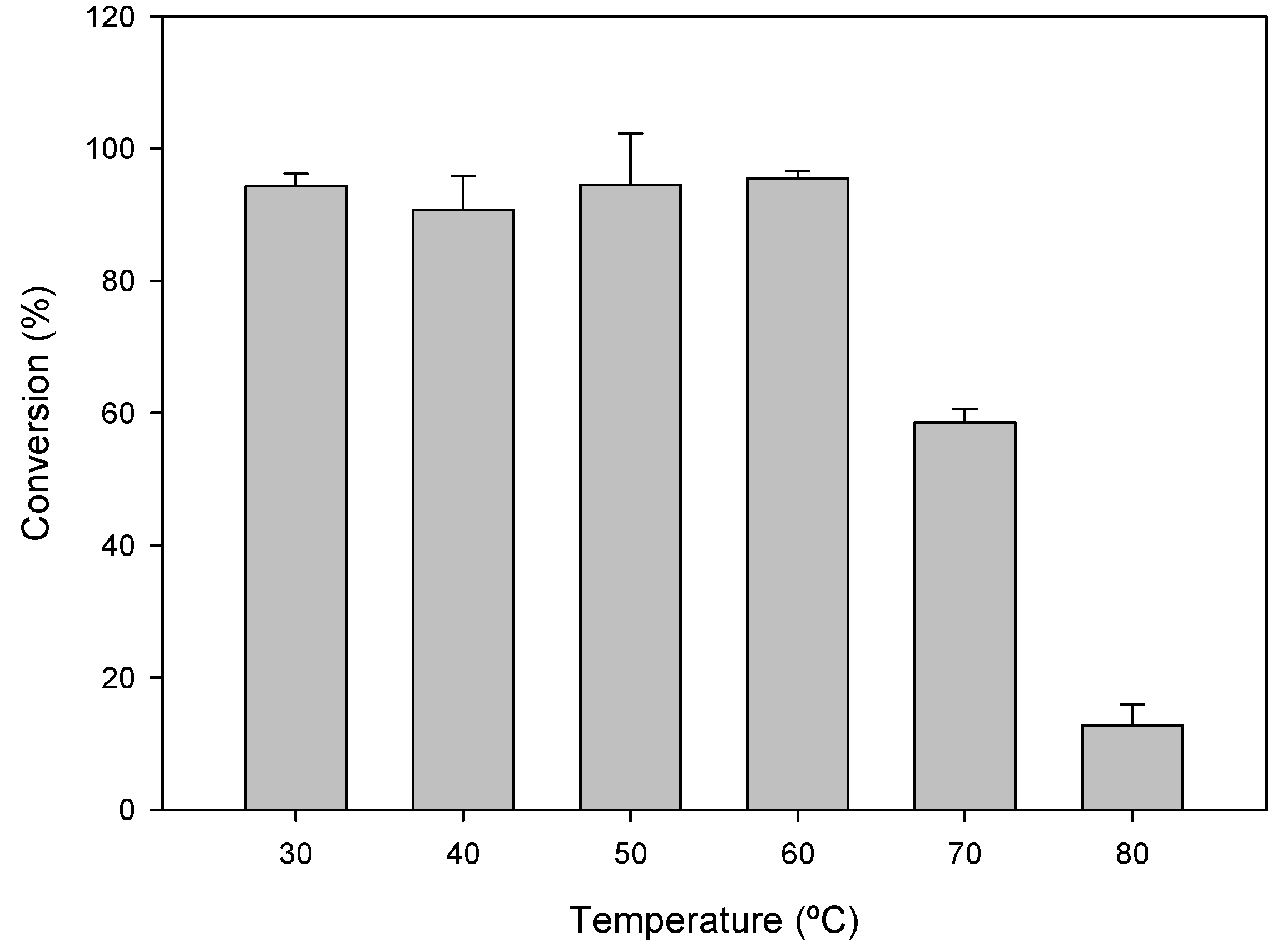
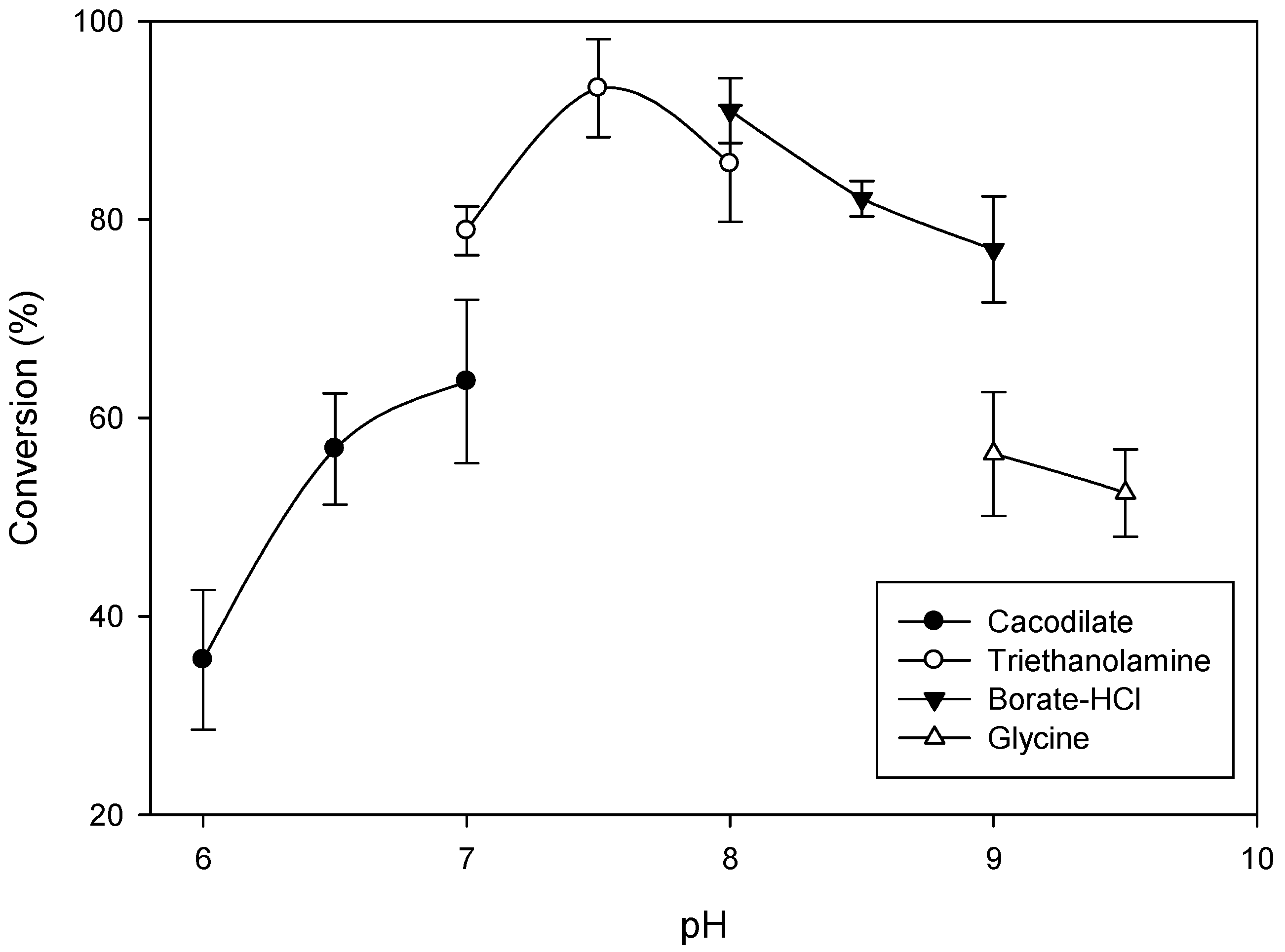
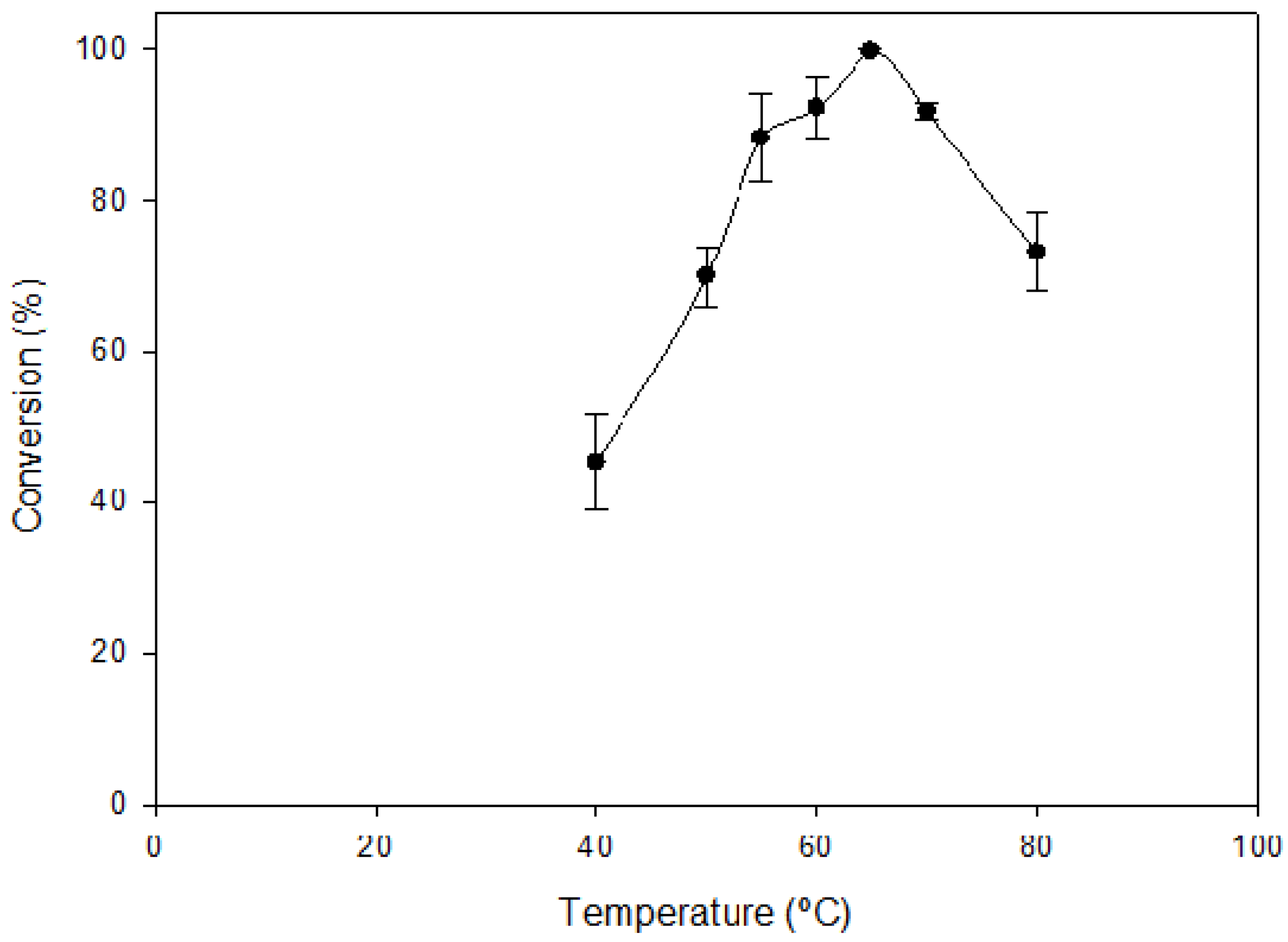
2.2. Optimal Reaction Conditions
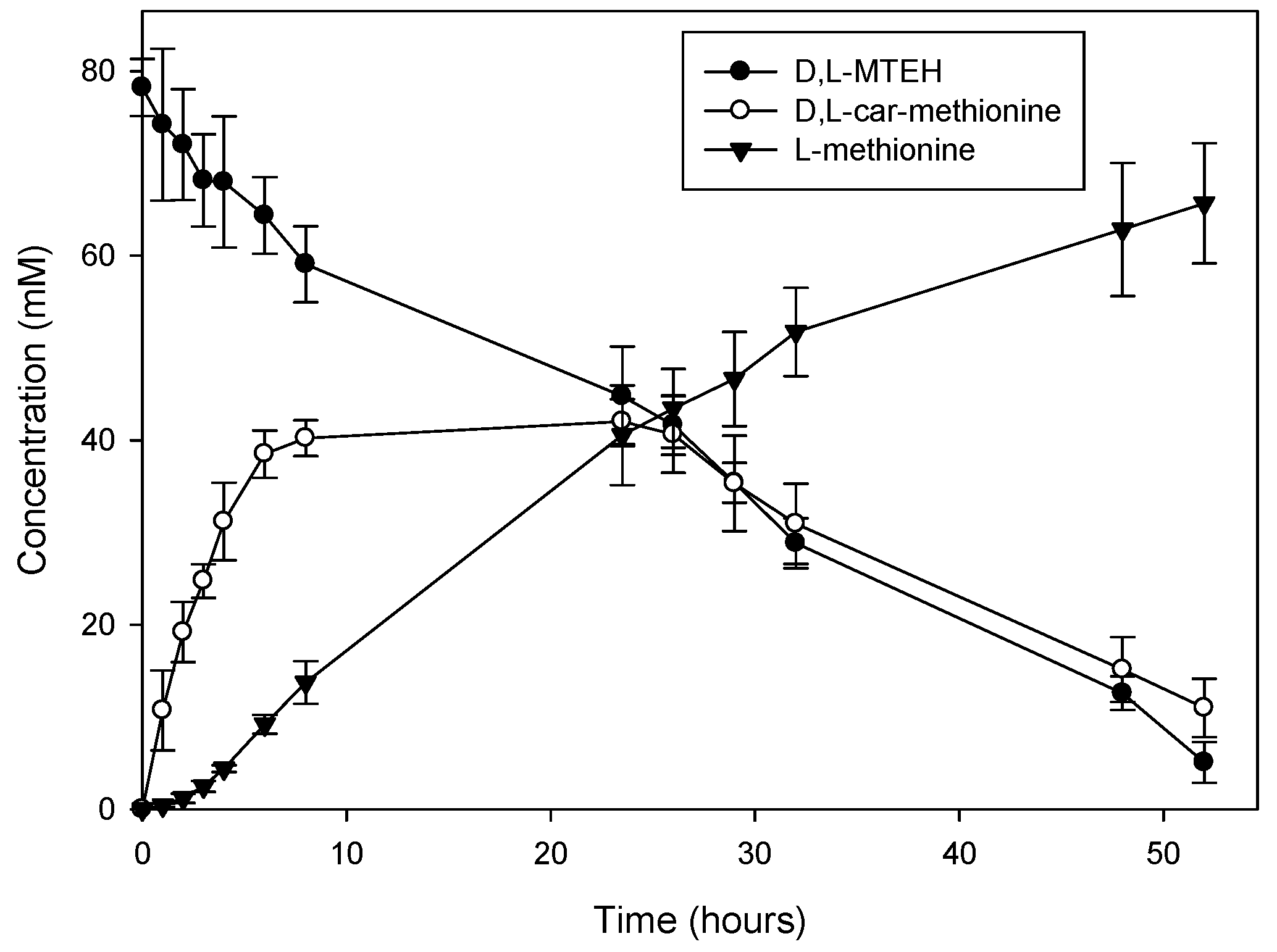
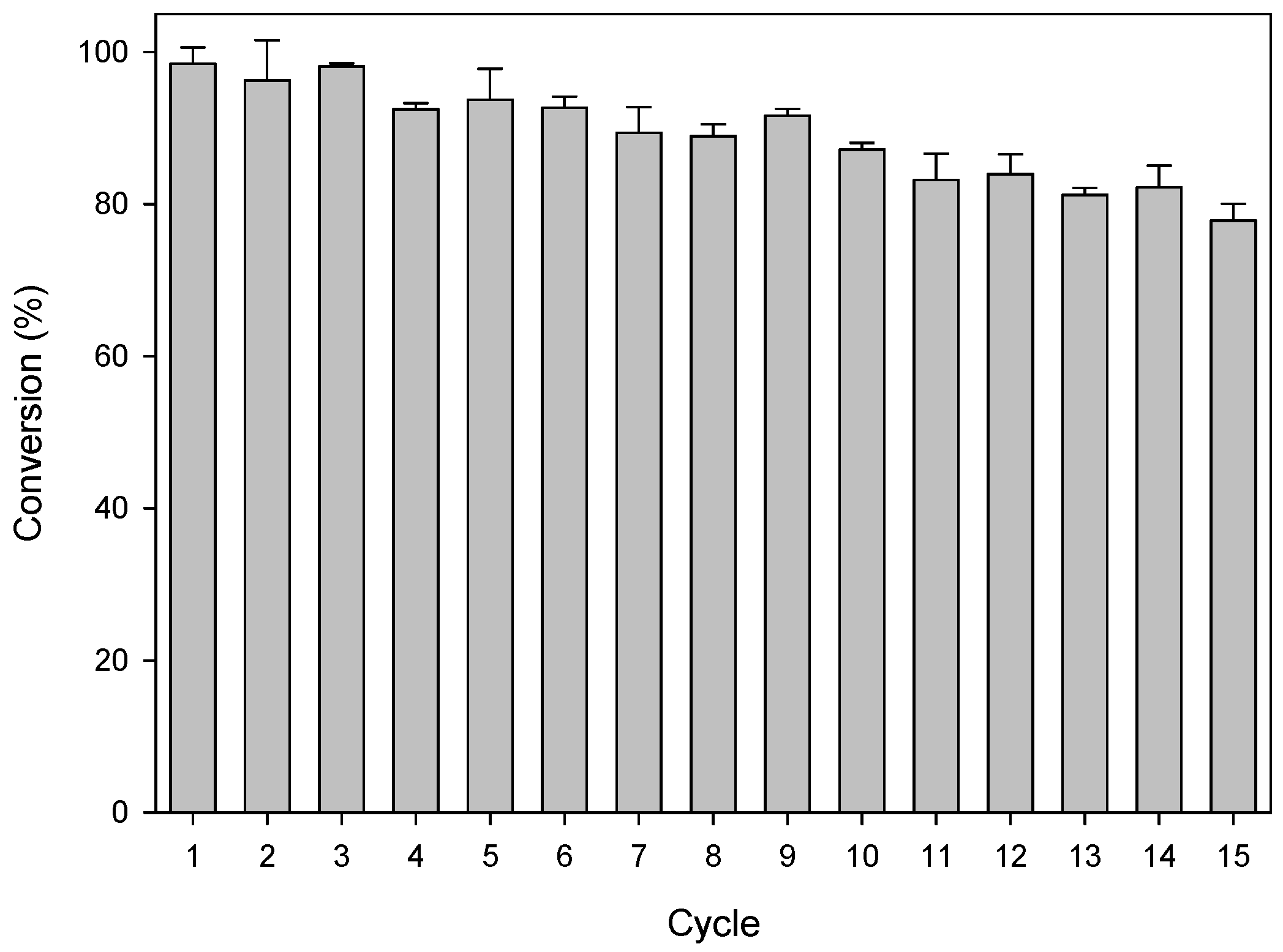
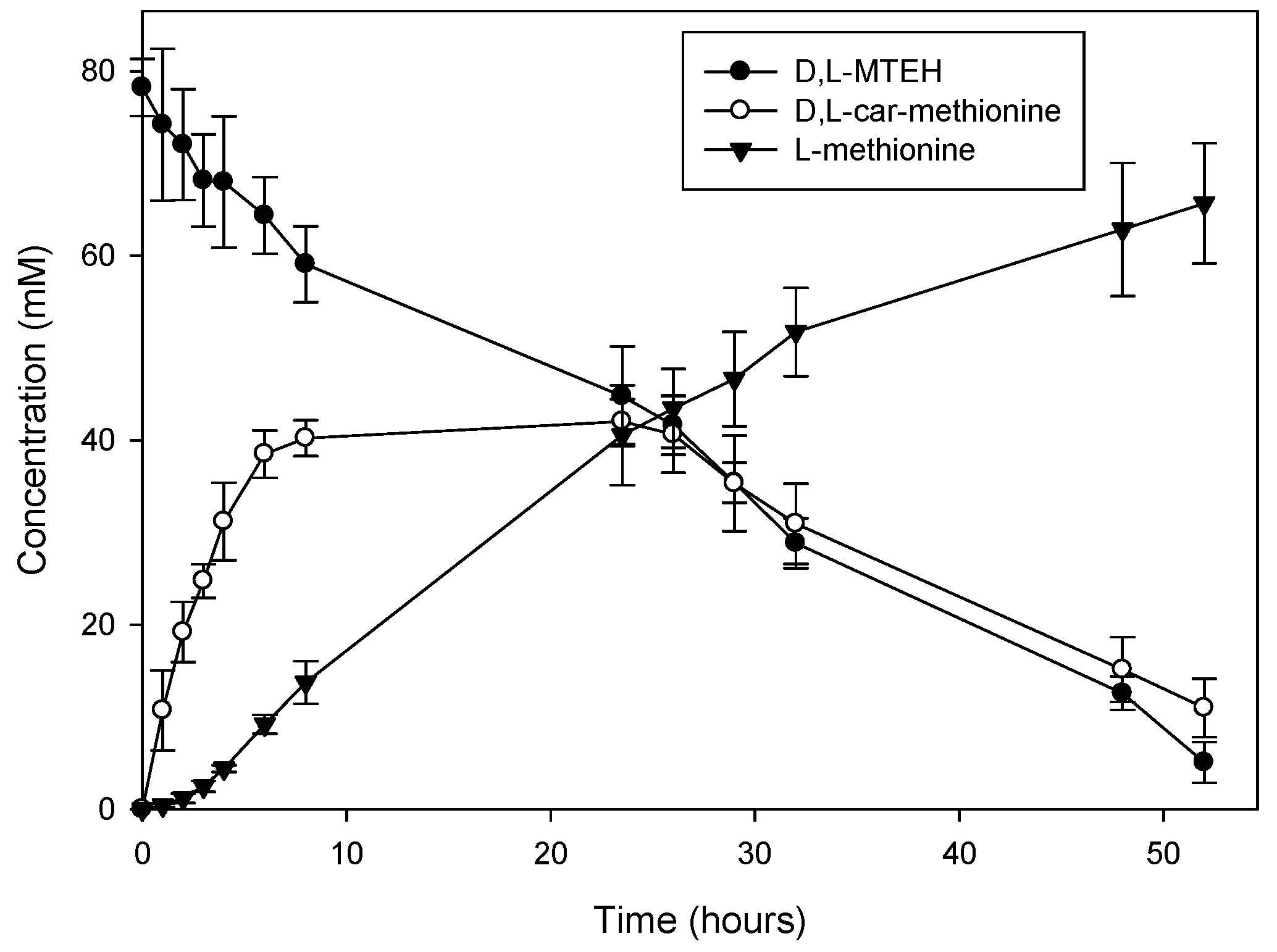
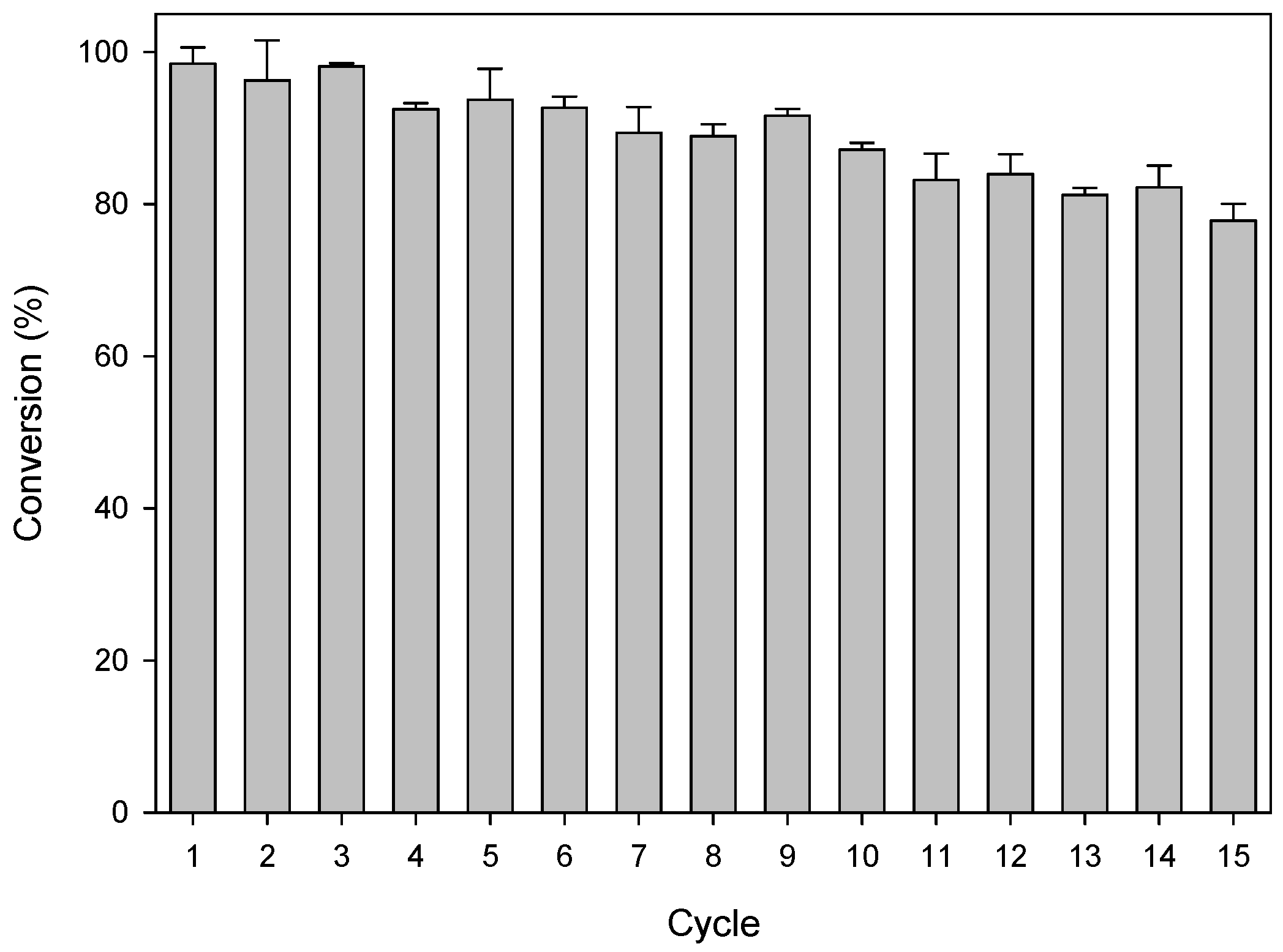
2.3. Efficiency of the System: Substrate Spectra and Reaction Cycles
3. Materials and Methods
3.1. Protein Purification
3.2. Immobilization
3.3. System Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leuchtenberger, W.; Huthmacher, K.; Drauz, K. Biotechnological production of amino acids and derivatives: Current status and prospects. Appl. Microbiol. Biotechnol. 2005, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Jiménez, J.M.; Martínez-Rodríguez, S.; Rodríguez-Vico, F.; Las Heras-Vázquez, F.J. Optically pure α-aminoacids production by the “Hydantoinase Process”. Recent Pat. Biotechnol. 2008, 2, 35–46. [Google Scholar] [PubMed]
- Hummel, W.; Geueke, B.; Osswald, S.; Weckbecker, C.; Hutmacher, K. Method for the preparation of l-amino acids from d-amino acids. US 7217544 B2, 15 May 2007. [Google Scholar]
- May, O.; Verseck, S.; Bommarius, A.; Drauz, K. Development of dynamic kinetic resolution processes for biocatalytic production of natural and non-natural l-amino acids. Org. Process Res. Dev. 2002, 6, 452–457. [Google Scholar] [CrossRef]
- May, O.; Siemann, M.; Pietzsch, M.; Kiess, K.; Mattes, R.; Syldatk, C. Substrate-dependent enantioselectivity of a novel hydantoinase from Arthrobacter aurescens DSM 3745: Purification and characterization as new member of cyclic amidases. J. Biotechnol. 1998, 61, 1–13. [Google Scholar] [CrossRef]
- Kao, C.; Lo, H.; Hsu, S.; Hsu, W. A novel hydantoinase process using recombinant Escherichia coli cells with dihydropyrimidinase and l-N-carbamoylase activities as biocatalyst for the production of l-homophenylalanine. J. Biotechnol. 2008, 134, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Buson, A.; Negro, A.; Grassato, L.; Tagliaro, M.; Basaglia, M.; Grandi, C.; Fontana, A.; Nuti, M.P. Identification; sequencing and mutagenesis of the gene for a d-carbamoylase from Agrobacterium radiobacter. FEMS Microbiol. Lett. 1996, 145, 55–62. [Google Scholar] [CrossRef]
- Pietzsch, M.; Wiese, A.; Ragnitz, K.; Wilms, B.; Altenbuchner, J.; Mattes, R.; Syldatk, C. Purification of recombinant hydantoinase and l-N-carbamoylase from Arthrobacter aurescens expressed in Escherichia coli: comparison of wild-type and genetically modified proteins. J. Chromatogr. B: Biomed. Sci. Appl. 2000, 737, 179–186. [Google Scholar] [CrossRef]
- Rodríguez-Alonso, M.J.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F.; Las Heras-Vázquez, F.J. Rational re-design of the “Double-Racemase Hydantoinase Process” for optically pure production of natural and non-natural l-amino acids. Biochem. Eng. J. 2015, 10, 68–76. [Google Scholar] [CrossRef]
- Clemente-Jiménez, J.M.; Martínez-Rodríguez, S.; Mingorance-Cazorla, L.; De La Escalera-Hueso, S.; Las Heras-Vázquez, F.J.; Rodríguez-Vico, F. Catalytic analysis of a recombinant d-hydantoinase form Agrobacterium tumefaciens. Biotechnol. Lett. 2003, 25, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Las Heras-Vázquez, F.J.; Martínez-Rodríguez, S.; Mingorance-Cazorla, L.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F. Overexpression and characterization of hydantoin racemase from Agrobacterium tumefaciens C58. Biochem. Biophs. Res. Commun. 2003, 303, 541–547. [Google Scholar] [CrossRef]
- Pozo-Dengra, J.; Martínez-Gómez, A.I.; Martínez-Rodríguez, S.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F.; Las Heras-Vázquez, F.J. Evaluation of substrate promiscuity of an l-carbamoyl amino acid amidohydrolase from Geobacillus stearothermophilus CECT43. Biotechnol. Prog. 2010, 26, 954–959. [Google Scholar] [PubMed]
- Pozo-Dengra, J.; Martínez-Gómez, A.I.; Martínez-Rodríguez, S.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F.; Las Heras-Vázquez, F.J. Racemization study on different N-acetylamino acids by a recombinant N-succinylamino acid racemase from Geobacillus kaustophilus CECT4264. Process Biochem. 2009, 44, 835–841. [Google Scholar] [CrossRef]
- Spahn, C.; Minteer, S.D. Enzyme immoblization in biotechnology. Recent. Pat. Eng. 2008, 2, 195–200. [Google Scholar] [CrossRef]
- Rodríguez-Alonso, M.J.; Rodríguez-Vico, F.; Las Heras-Vázquez, F.J.; Clemente-Jiménez, J.M. Immobilization of a Multi-Enzyme system for l-amino acid production. J. Chem. Technol. Biotechol. 2016, 91, 1972–1981. [Google Scholar] [CrossRef]
- Cantone, S.; Ferrario, V.; Corici, L.; Ebert, C.; Fattor, D.; Spizzo, P.; Gardossi, L. Efficient immobilization of industrial biocatalysts: Criteria and constraints for the selection of organic polymeric carriers and immobilisation methods. Chem. Soc. Rev. 2013, 42, 6262–6276. [Google Scholar] [CrossRef] [PubMed]
- Wilchek, M.; Miron, T. Oriented versus random protein immobilization. J. Biochem. Biophys. Methods 2003, 55, 67–70. [Google Scholar] [CrossRef]
- Ragnitz, K.; Syldatk, C.; Pietzsch, M. Optimization of the immobilization parameters and operational stability of immobilized hydantoinase and l-N-carbamoylase from Arthrobacter aurescens for the production of optically pure l-amino acids. Enzyme Microb. Technol. 2001, 28, 713–720. [Google Scholar] [CrossRef]
- Fernández-Lorente, G.; Ortiz, C.; Segura, R.L.; Fernández-Lafuente, R.; Guisán, J.M.; Palomo, J.M. Immobilization of proteins on glyoxyl activated supports: Dramatic stabilization of enzymes by multlipoint covalent attachment on pre-existing supports. Curr. Org. Chem. 2015, 19, 1–13. [Google Scholar] [CrossRef]
- Soriano-Maldonado, P.; Las Heras-Vázquez, F.J.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F.; Martínez-Rodríguez, S. Enzymatic dynamic kinetic resolution of racemic N-formyl- and N-carbamoyl-amino acids using immobilized l-N-carbamoylase and N-succinyl-amino acid racemase. Appl. Microbiol. Biotechnol. 2015, 99, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Martin, J.; Acosta, A.; Guisán, J.M.; López-Gallego, F. Immobilizing systems biocatalysis for the selective oxidation of glycerol coupled to in situ cofactor recycling and hydrogen peroxide elimination. ChemCatChem 2015, 7, 1939–1943. [Google Scholar] [CrossRef]
- Las Heras-Vazquez, F.J.; Clemente-Jimenez, S.; Martinez-Rodriguez, S.; Rodriguez-Vico, F. Engineering cyclic amidases for non-natural amino acid synthesis. Methods Mol. Biol. 2003, 303, 87–104. [Google Scholar]
- Soriano-Maldonado, P.; Rodríguez-Alonso, M.J.; Hernández-Cervantes, I.; Rodriguez-García, I.; Clemente-Jiménez, J.M.; Rodríguez-Vico, F.; Martínez-Rodríguez, S.; Las Heras-Vázquez, F.J. Amidohydrolase Process: Expanding the use of l-N-carbamoylase/N-succinnyl-amino acid racemase tandem for the production of different optically pure l-amino acid racemase tandem for the production of different opically pure l-amino acids. Process Biochem. 2014, 49, 1281–1287. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AtDHyd | AthyuA1 | BsLcar | GkNSAAR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Prot * | % I * | Act * | Prot * | % I * | Act * | Prot * | % I * | Act * | Prot * | % I * | Act * |
| 0.84 | 98 | 6.80 ± 0.71 | 0.52 | 88 | 0.47 ± 0.03 | 0.83 | 99 | 0.73 ± 0.06 | 0.84 | 100 | 0.12 ± 0.01 |
| 1.70 | 85 | 4.02 ± 0.37 | 1.04 | 66 | 0.67 ± 0.07 | 1.66 | 98 | 0.64 ± 0.05 | 1.68 | 99 | 0.09 ± 0.01 |
| 2.50 | 76 | 2.86 ± 0.24 | 2.60 | 48 | 0.73 ± 0.08 | 2.49 | 97 | 0.78 ± 0.07 | 2.63 | 99 | 0.12 ± 0.01 |
| 5.00 | 69 | 1.77 ± 0.11 | 5.20 | 48 | 0.49 ± 0.03 | 3.57 | 98 | 0.37 ± 0.04 | - | - | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Alonso, M.J.; Rodríguez-Vico, F.; Las Heras-Vázquez, F.J.; Clemente-Jiménez, J.M. l-Amino Acid Production by a Immobilized Double-Racemase Hydantoinase Process: Improvement and Comparison with a Free Protein System. Catalysts 2017, 7, 192. https://doi.org/10.3390/catal7060192
Rodríguez-Alonso MJ, Rodríguez-Vico F, Las Heras-Vázquez FJ, Clemente-Jiménez JM. l-Amino Acid Production by a Immobilized Double-Racemase Hydantoinase Process: Improvement and Comparison with a Free Protein System. Catalysts. 2017; 7(6):192. https://doi.org/10.3390/catal7060192
Chicago/Turabian StyleRodríguez-Alonso, María José, Felipe Rodríguez-Vico, Francisco Javier Las Heras-Vázquez, and Josefa María Clemente-Jiménez. 2017. "l-Amino Acid Production by a Immobilized Double-Racemase Hydantoinase Process: Improvement and Comparison with a Free Protein System" Catalysts 7, no. 6: 192. https://doi.org/10.3390/catal7060192