Effect of Residual Na+ on the Combustion of Methane over Co3O4 Bulk Catalysts Prepared by Precipitation


Chemical Technologies for Environmental Sustainability Group, Department of Chemical Engineering, Faculty of Science and Technology, University of the Basque Country UPV/EHU, P.O. Box 644, E-48080 Bilbao, Spain
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(10), 427; https://doi.org/10.3390/catal8100427
Submission received: 6 September 2018
/
Revised: 21 September 2018
/
Accepted: 28 September 2018
/
Published: 29 September 2018
(This article belongs to the Special Issue Catalytic Concepts for Methane Combustion)
Abstract
:The effect of the presence of residual sodium (0.4 %wt) over a Co3O4 bulk catalyst for methane combustion was studied. Two samples, with and without residual sodium, were synthesized by precipitation and thoroughly characterised by X-ray diffraction (XRD), N2 physisorption, Wavelength Dispersive X-ray Fluorescence (WDXRF), temperature-programmed reduction with hydrogen followed by temperature-programmed reduction with oxygen (H2-TPR/O2-TPO), temperature-programmed reaction with methane (CH4-TPRe), ultraviolet–visible–near-infrared diffuse reflectance spectroscopy (UV-vis-NIR DRS), Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). It was found that during calcination, a fraction of the sodium atoms initially deposited on the surface diffused and migrated into the spinel lattice, inducing a distortion that improved its textural and structural properties. However, surface sodium had an overall negative impact on the catalytic activity. It led to a reduction of surface Co3+ ions in favour of Co2+, thus ultimately decreasing the Co3+/Co2+ molar ratio (from 1.96 to 1.20) and decreasing the amount and mobility of active lattice oxygen species. As a result, the catalyst with residual sodium (T90 = 545 °C) was notably less active than its clean counterpart (T90 = 500 °C). All of this outlined the significance of a proper washing when synthesizing Co3O4 catalyst using a sodium salt as the precipitating agent.
1. Introduction
Methane is a powerful greenhouse effect gas (25 times higher than that of CO2) that appears as a residue or flue gas in many different applications. These include thermal plants, refineries or natural gas engines. To protect the environment from its noxious effect, this pollutant needs to be properly removed [1]. Usually, the off-gas treatment technology must be efficient at low temperatures when operating with large gas flows with small amounts of methane. Under these conditions, the most suitable technique is catalytic oxidation.
The traditional catalysts for oxidation of methane have been based on noble metals, such as platinum or palladium. In the recent years, extensive research has been made with the aim of finding other alternatives. This is mainly due to their high price, low availability and tendency to deactivate and age [2,3]. The most significant part of this research is focused on both single and mixed transition metal oxides. Materials such as perovskites, hexaaluminates or spinels composed of different metals have been studied and proven active for the oxidation of methane [4,5,6,7]. Spinel oxides, specifically, are mixed oxides with general formula AB2O4, where A is a metal with oxidation state +2 and tetrahedral coordination and B is a metal with oxidation state +3 and octahedral coordination [8]. The activity of these types of materials for oxidation reactions lies on the easiness that the metal cations possess to shift between the two oxidation states. This provides oxygen anions in the lattice with high mobility, making them suitable for reactions following the Mars-van Krevelen mechanism [9].
In the particular case of A and B being cobalt cations, the resulting compound is called spinel type cobalt oxide (Co3O4), which is one of the most active materials for the oxidation of methane and other hydrocarbons. This is essentially due to its great redox properties [10]. Nevertheless, the specific properties of a given Co3O4 catalyst greatly depend on many different factors and parameters, with one of the most determinant being the synthesis process. Cobalt oxide catalysts can be prepared by a wide number of methods, including the calcination of a cobalt salt, basic precipitation, combustion synthesis, hard templating, hydrothermal synthesis, mechanical grinding or sol-gel complexation, among others [11,12,13,14,15]. Although all of them lead to the same material, each synthesis methodology provides different properties to the final catalyst, thereby notably affecting its behaviour for a particular reaction or process.
Regarding this point, various research studies can be found that have tried to define the most suitable synthesis process for a certain reaction. For example, Liu et al. found that a cobalt oxide obtained from a citrate precursor prepared by soft reactive grinding was the most active for total oxidation of propane [16]. Monteverde Videla et al. reported that the most active Co3O4 catalyst for the oxygen evolution reaction was that obtained by a hard template method using SBA-15 silica [17]. In this sense, de Rivas et al. [18] observed that a Co3O4 bulk catalyst prepared by a simple basic precipitation process was more efficient for the oxidation of 1.2-dichloroethane compared to samples prepared by sol-gel complexation or mechanical grinding.
Precipitation-based synthesis processes are among the most commonly used methods to prepare Co3O4 and many other mixed oxide catalysts. While being a simple method, they usually produce catalysts with moderately good structural and redox properties, which can be suitable for oxidation reactions. In addition, precipitation methods are easy to set up on an industrial scale [19]. When proposing a precipitation synthesis route, the choice of the precipitating agent is normally crucial, since the same catalyst obtained from different precipitants do not perform similarly [20]. Moreover, it must be noticed that after the precipitation, residual amounts of the precipitating agent, or species derived from it, can remain on the catalytic precursor, generally altering the final properties of the desired catalyst.
Apart from the routes that involve ammonia-based reagents, the most frequently used precipitating substances are sodium and potassium bases, namely NaOH, KOH or Na2CO3. This means that, after the synthesis process, trace amounts of these alkaline metals can be present in the resulting catalyst. In some cases, this can have a beneficial effect. For example, Park et al. [21] and Haneda et al. [22], on different papers, demonstrated that residual sodium and other alkali metals were responsible for the good activity of Co3O4 catalysts for NO decomposition. Meanwhile, Asano et al. [23] found that a low amount of potassium on Co3O4 promoted the decomposition of N2O. Regarding N2O decomposition, numerous papers have reported the promotional effects of sodium and other alkali over Co3O4 catalysts. For instance, Maniak et al. [24] found a significant promotional effect of potassium over Mn3O4 and Co3O4, characterised by the formation of surface K+-O− dipoles that facilitated electron transfer from the surface to N2O molecules. On the other hand, Stelmachowski et al. [25], and Obalová et al. [26] found that the larger promotional effect was achieved by caesium, due to its larger ionic radius that allows for a higher surface coverage and ionization. In all of these studies, the promoting effect was deemed to be due to an increase in the amount of Co2+ cations within the catalyst surface, an increased electron density in the surface and enhanced oxygen desorption from Co3O4. This was all due to the presence of the alkaline metals.
However, regarding the oxidation of methane, there is not, to the best of our knowledge, any research study that deals with the effect of residual alkaline metals on Co3O4 catalysts. Considering the results obtained by the authors mentioned above, it is not clear whether the residual alkaline metals would have a beneficial effect on the catalytic performance of Co3O4, since methane oxidation over Co3O4 seems to be controlled by the population of highly reducible Co3+ species.
For this reason, in the present paper, two Co3O4 catalysts were prepared by a basic precipitation method, intentionally leaving some residual sodium in one of them to study its effect over the catalytic activity for the complete oxidation of methane. The obtained catalysts were thoroughly characterised and the role of the surface cobalt and oxygen species on the oxidation process was ascertained.
2. Results and Discussion
2.1. Synthesis of Co3O4 Precursors
When preparing the two bulk Co3O4 catalysts by precipitation with sodium carbonate, the catalytic precursors were filtrated and washed with varying amounts of deionized water between 1–8 L. The filtrates extracted from the filtration step were recovered and their pH was measured to estimate the extent of the removal of residual sodium from the precursor. The evolution of pH value as a function of the volume of water used for washing is shown in Figure 1.
It was observed that the pH of the filtrates remained almost constant (pH = 7.8) after washing the precipitate with four litres of water, which would suggest that all residual sodium from the synthesis process was removed with this amount of water. This residual pH was somewhat above 7 due to the basicity and slight solubility of the precipitate.
2.2. Physico-Chemical Characterisation
The structural characterisation of the catalysts was performed by XRD. The diffractograms in Figure 2a only revealed signals attributable to a pure phase of spinel-type Co3O4 (JCPDS 042-1467). No signals attributable to Na2O, or any other Co-Na mixed oxides, were detected in the catalyst with residual sodium (CC-Na). However, the diffraction peaks displayed by the CC-Na sample were markedly wider than those displayed by the Na-free CC sample and were shifted towards lower diffraction angles, as can be seen in Figure 2b. This revealed that the presence of trace amounts of sodium (0.4 wt% as determined by WDXRF) induced a distortion of the spinel lattice of Co3O4.
The crystallite size of Co3O4 was estimated from the full width half maximum (FWHM) of the characteristic peak located at 37.1°, assignable to the (311) crystalline plane, by applying the Scherrer equation. Besides, the cell size could be calculated by performing a profile matching of the diffractograms. It was found that the crystallite size of the CC-Na catalyst was almost half of that of the CC catalyst, while its cell size was notably larger. This was in agreement with the aforementioned distortion caused by the inclusion of sodium into the spinel lattice. This distortion can be explained in terms of the difference in ionic radius between Co2+ and Co3+ ions (72 and 69 pm, respectively) and Na+ ions (113 pm).
The results from the textural characterisation, also displayed in Table 1, showed that the catalyst with residual sodium exhibited almost twice as much surface area as the Na-free sample, and a pore volume a 25% larger. This suggested that the distortion caused in the spinel lattice also caused an enhancement of the textural properties of the Co3O4. The pore size distributions, shown in Figure 3, also evidenced a shrinking effect over the mean pore diameter when cobalt oxide is contaminated with residual sodium. These effects have already been reported in other studies regarding the doping of Co3O4 catalysts with other metallic elements [27,28]. It must be noticed that all those variations were originated by a relatively low bulk concentration of sodium (0.4 wt%).
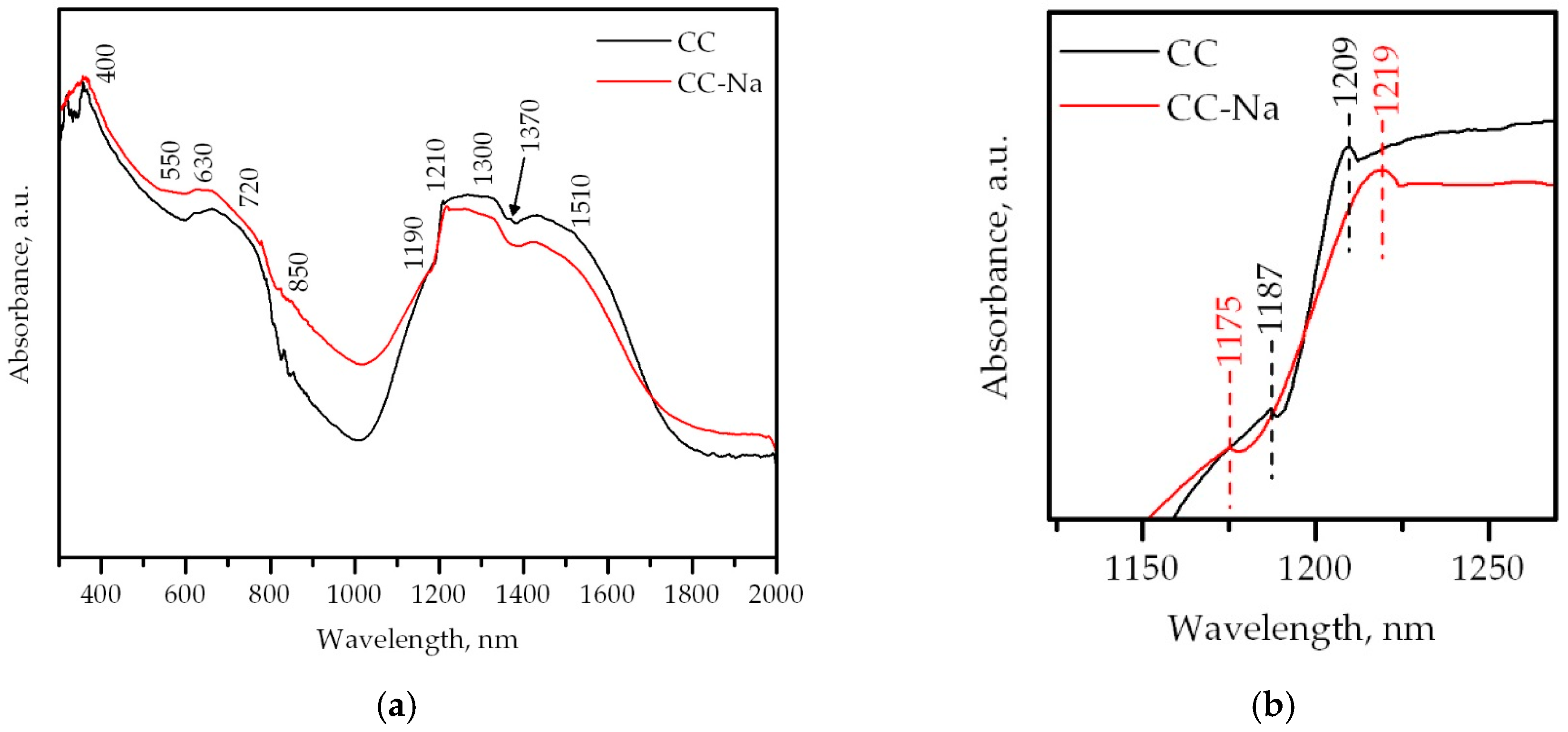
The structure of the samples were additionally studied by UV-vis-NIR DRS, Raman spectroscopy and XPS. Figure 4a shows the UV-vis-NIR diffuse reflectance spectra. Both samples displayed absorption bands at 630, 1210, 1300 and 1510 nm, associated with the presence of tetrahedral Co2+ species. Since the Co3O4 oxide also contains octahedral Co3+ species, their corresponding absorption bands (weak shoulders at 400 and 720 nm) were also expected to be visible [29]. However, the former was not detectable, while the latter was somewhat visible, only for the CC sample. This could suggest that the amount of Co3+ present in this catalyst was larger with respect to the Na-containing oxide. In addition, a band at 1370 nm was only detected for the CC sample. This band was associated with the 2ν harmonics band from residual hydroxyl groups [30].
On the other hand, both samples displayed absorption bands at 550, 850 and 1190 nm, related to Co2+ ions with octahedral coordination as in CoO. Interestingly, these bands were more noticeable for the CC-Na sample. This suggested that the presence of this species was larger with respect to the CC sample. An additional feature could also be seen when examining the spectra in detail, as depicted in Figure 4b. When zooming into the 1150–1250 nm region, where the well-defined absorption signals at around 1190 and 1210 nm were located, it could be noted that both signals for the CC-Na sample were shifted with respect to the CC sample. Specifically, the former was redshifted 12 nm from its original position, while the latter was blueshifted 10 nm from its original position. These findings could further confirm the lattice distortion caused by the residual sodium.
Raman spectroscopy results, shown in Figure 5, also pointed out the aforementioned lattice distortion. The resulting spectra for both samples showed five Raman bands. The bands located at 194, 519 and 617 cm−1 could be attributed to the F2g mode. The bands located at 479 and 687 cm−1 could be assigned to the Eg and A1g modes, respectively [16]. However, the CC-Na sample showed these bands slightly shifted and became wider with respect to those of the CC sample. Additionally, the band attributed to the A1g mode also showed a strong asymmetry. This is a clear sign of lattice distortion according to the bibliography [31,32].
Figure 6a,b show the Co 2p and O 1s XPS spectra of the samples, respectively. The Co 2p3/2 spectra of both oxides was composed of five different signals. The two features with the lowest binding energies, located at 779.5 and 780.7 eV, were assigned to Co3+ and Co2+ ions, respectively. The signal centered at 782.2 eV was attributed to the presence of CoO in the surface of the samples [33]. The abundance of this species was higher for the CC-Na sample (14%) than for the CC sample (8%), as also suggested by the results from UV-vis-NIR DRS. The two signals with the highest binding energies (785.7 and 789.3 eV) were identified as the satellite signals from Co3+ and Co2+ ions, respectively. The comparable intensity of both satellite signals pointed out that the surface of the catalysts was mainly composed of Co3O4 [34]. On the other hand, the O 1s spectra exhibited two signals for both samples, located at 529.9 and 531.3 eV, respectively. The low binding energy signal was attributed to lattice oxygen species (Olatt). The high binding energy signal was assigned to weakly adsorbed oxygen species (Oads) [35]. Finally, for the CC-Na catalyst an extra peak at a binding energy of 1071.8 eV was attributed to the presence of Na+ ions.
The deconvolution and integration of the XPS spectra allowed for a quantitative analysis of the composition of the surface, as shown in Table 2. It must be noticed that the concentration of sodium on the surface of the CC-Na sample was several times higher (5.1 wt%) than in the bulk (0.4 wt%). This evidenced that the majority of the sodium atoms present in that sample were located on its surface. A certain amount had likely migrated and diffused into the spinel lattice during the calcination process. In addition to that, the Co3+/Co2+ molar ratio of the catalyst with sodium was notably lower than for its clean counterpart, while the Oads/Olatt molar ratio was higher. This was explained as a consequence of sodium atoms donating electrons to oxygen ions within the spinel lattice. The resulting increase in electronic density transformed Na sites into strong Lewis sites that are able to donate electrons to cobalt cations with a concomitant effective decrease in their overall oxidation state [22,23]. This effect was further confirmed by the slight shift (0.4 eV) of the O 1s spectra of the CC-Na towards lower binding energy values. This indicated an increase in the electron density of oxygen anions and also caused a weakening of Co-O bonds, which is coherent with observed increase in the Oads/Olatt molar ratio.
2.3. Redox Characterisation
Figure 7a shows the H2-TPR profiles of the cobalt oxide samples. As described by other authors, the TPR profile of a Co3O4 catalyst, namely the CC sample, consisted of two different contributions to the H2 uptake [36,37]. A first contribution, located at 310 °C, assignable to the reduction of Co3+ ions into Co2+; and a second contribution, centered at 350–400 °C, attributable to the subsequent reduction of Co2+ into metallic cobalt. The TPR profile of the sample with residual sodium also exhibited these two contributions, but their shape was notably different. This was probably a consequence of the distortion of the spinel lattice caused by sodium. Furthermore, the second H2 uptake did not take the shape of a single peak, but instead was formed of at least two different contributions. This suggested the presence of Co2+ species with different reducibilities, namely free CoO that was detected by both DRS and XPS analysis along with Co2+ from the spinel lattice.
Immediately after the H2-TPR analysis, an O2-TPO run was carried out to check the capacity of reoxidation of the catalysts after their complete reduction to metallic cobalt. The TPO profiles, shown in Figure 7b, clearly evidenced the influence of sodium on the reoxidability of the Co3O4 catalysts. The CC sample showed a high intensity oxidation peak centered at 330 °C with a tail starting around 200 °C. The sample with sodium showed a much lower intensity peak located at about 360 °C, with a tail starting around 250 °C. Furthermore, the total O2 uptake of the Na-containing sample was only 51% than that of the Na-free oxide. These results revealed that not only the sample with sodium was more difficult to reoxidize, but also that it could not be completely reoxidized, after complete reduction, below 600 °C. This was likely due to the reducing effect of sodium ions over cobalt species and the subsequent weakening of Co-O bonds, which in turn made more difficult the reincorporation of oxygen into the spinel lattice.
Nevertheless, in spite of the differences in reducibility between the two catalysts, their reduction started at the same temperature (about 200 °C) as can be seen in Figure 7a. This could be attributable to the high reducing power of hydrogen, that may hide subtle differences in reducibility. For this reason, a CH4-TPRe experiment in the absence of oxygen was conducted over both oxides, since the ability of methane for reducing is lower than that of hydrogen. The results are shown in Figure 8.
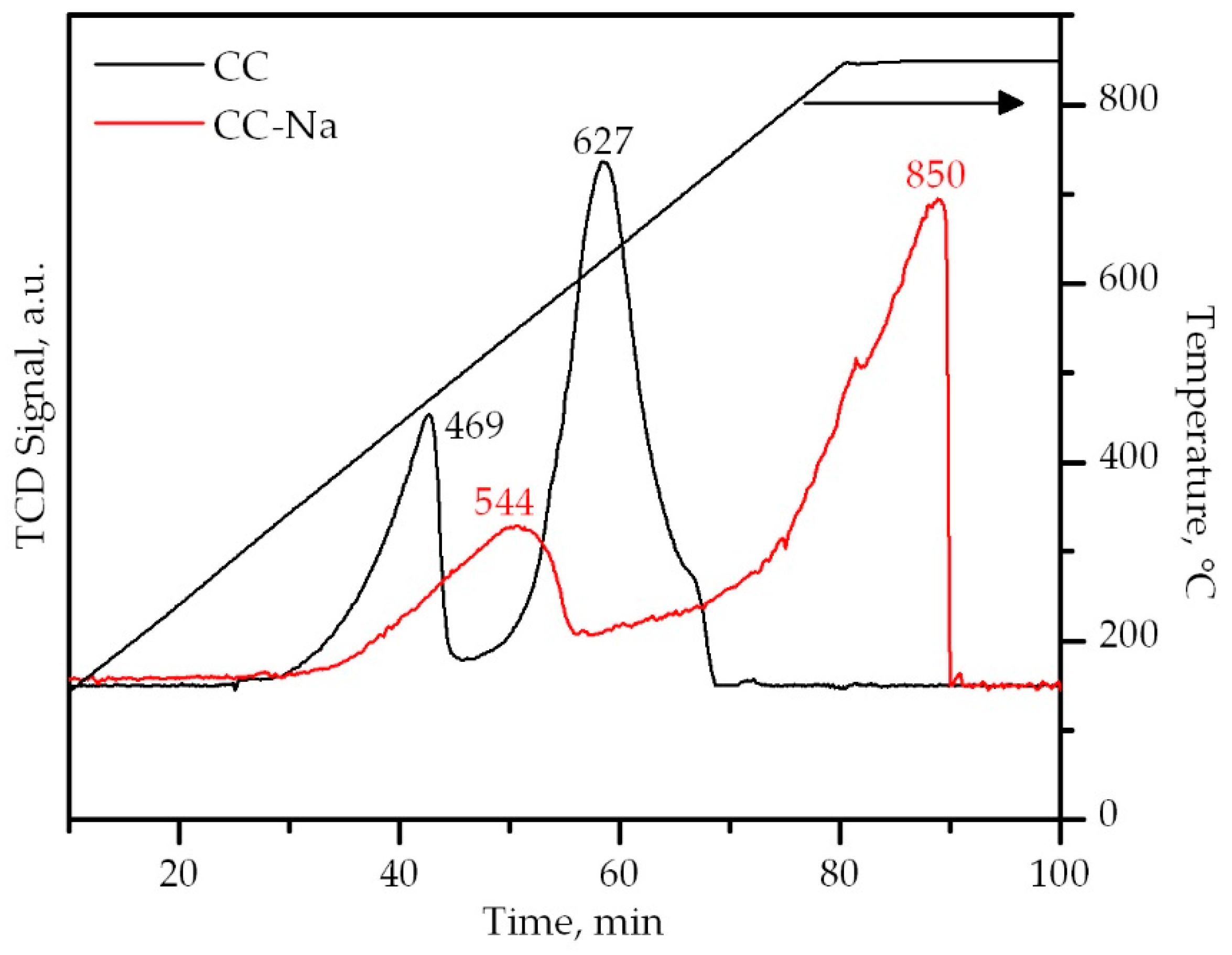
The CH4-TPRe profiles of the Co3O4 catalysts consisted of two reaction peaks, in a similar fashion to the H2-TPR profiles. However, these peaks did not exhibit the same shape as the TPR peaks, nor did they appear as the same temperatures. The CC sample was characterised by a low temperature reduction peak, assigned to the reaction of methane with O2− ions associated with Co3+ ions, centered at 469 °C. The corresponding peak of the CC-Na sample was centered at 544 °C. On the other hand, the second reaction peak, attributed to the reaction of methane with O2− ions associated with Co2+ ions, appeared at 627 °C for the CC sample. For the sample with sodium, it did not appear until the isothermal step at 850 °C. On the basis of these results, it was evident that the presence of sodium in the Co3O4 catalyst remarkably decreased the mobility of the oxygen species of the spinel lattice, as well as the reducibility of the cobalt ions themselves. However, this was only clearly visible when the reduction of the samples was carried out with methane.
It must be considered that according to the results from the CH4-TPRe tests, these Co3O4 catalysts were not completely reduced during the reaction process. This is because the temperature levels needed for their complete reduction with methane were higher than 600 °C, and their reoxidation occurred at temperatures around 300 °C. For this reason, the results from the O2-TPO analysis may not be too relevant, but they could serve as an additional indication of the detrimental effect of sodium on the redox properties of Co3O4.
2.4. Catalytic Performance
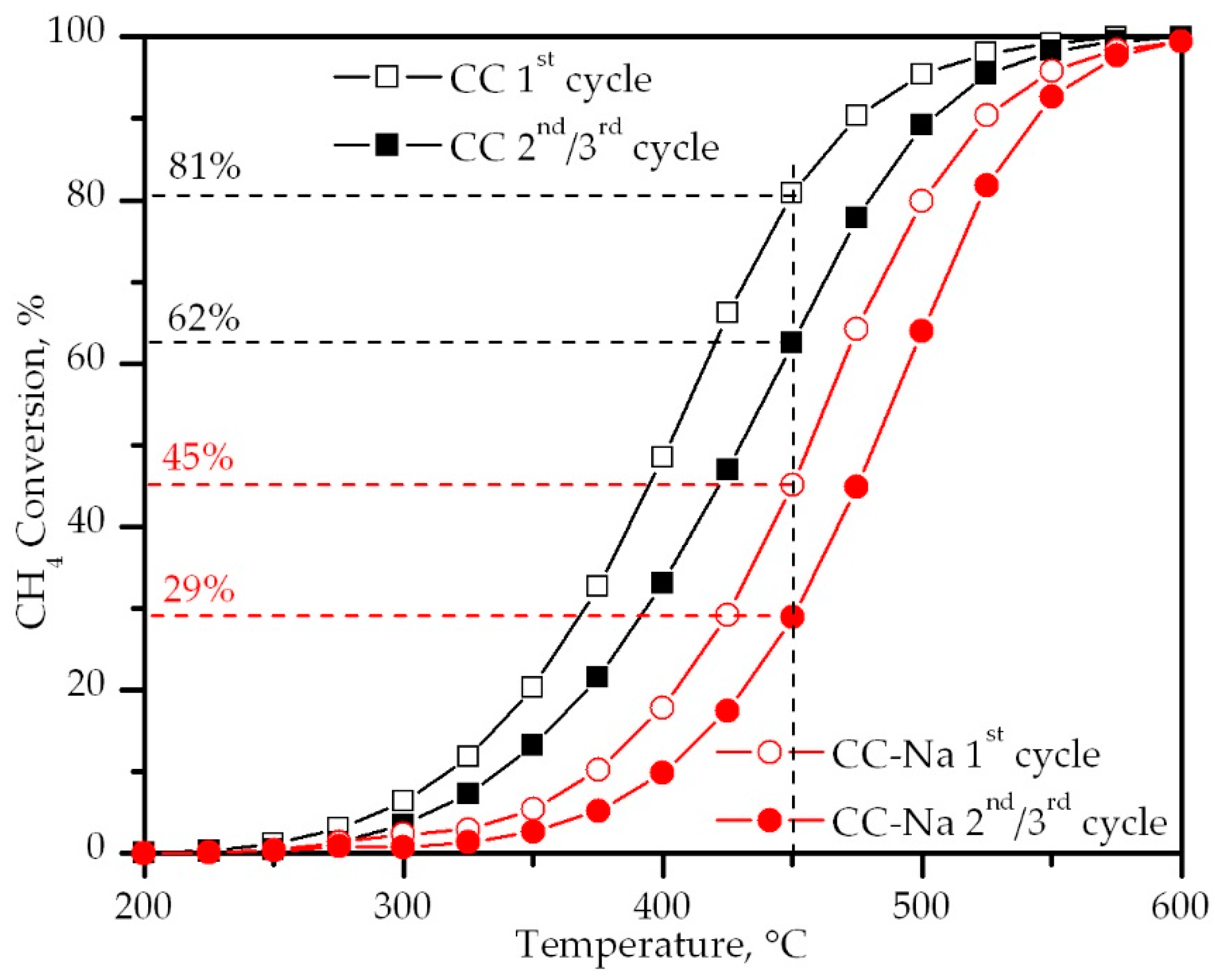
The activity tests were carried out three times consecutively for each catalyst. In both cases, the second and third cycles were characterised by an identical light-off curve. The light-off curves for the first and second/third cycles are shown in Figure 9. It was clear that the sample without sodium was far more active than the sample with sodium, thus evidencing the inhibiting effect caused by this metal. Considering that the primary mechanism of methane oxidation over metallic oxides is the Mars-van Krevelen mechanism, the negative effect of sodium on the activity of the CC-Na catalyst could be explained as a consequence of the reduced oxygen mobility. This was demonstrated by CH4-TPRe, and the decrease on the amount of Co3+ on the surface and the subsequent decrease in the active lattice oxygen species, as shown by the XPS results. However, it must be noticed that both samples were able to achieve a 100% methane conversion at 600 °C. This pointed out that the aforementioned inhibition effect was only apparent at low temperatures, as can be observed from the marked differences between the T10 and T50 values (Table 3).
A differential kinetic analysis was performed for conversion values below 10%, assuming a first order with respect to methane and zeroth with respect to oxygen, which is a suitable model for the Mars-van Krevelen mechanism in the presence of excess oxygen [38,39]. The results shown in Table 3 further confirmed the negative influence of residual sodium over the CC-Na catalyst. The activation energy of this sample was significantly higher with respect to the CC sample. In addition, the pre-exponential factor of the CC-Na sample was about four times higher than that of the CC sample, which could be a consequence of the beneficial effect of residual sodium over the structural properties of this catalyst.
The fact that the first reaction cycle was characterised by a different light-off curve pointed out that it worked as ‘conditioning’ cycle, after which the tested catalysts became stable under the employed reaction conditions. For this reason, the tested catalysts were characterised after the reaction tests to look for differences with respect to fresh counterparts that could be caused by this conditioning process. The results from the textural/structural characterisation, shown in Table 4, revealed a loss of specific surface area and pore volume for both catalysts. The loss of specific surface area was comparable between the two catalysts (around 33% in both cases). The loss of pore volume was more severe for the Na-free catalyst. Additionally, a general enlargement of the average pore size was observed for both catalysts. Interestingly, the loss of surface area and pore volume of the CC catalyst was not accompanied by a marked increase in the crystallite size, as it would be expected if the effect were caused by sintering. Moreover, the shrinkage of the cell size was noticed for both samples (from 8.052 to 8.046 Å over the CC sample and from 8.064 to 8.048 Å over the CC-Na sample). All this suggested that the conditioning cycle caused a moderate surface restructuring of the catalysts that made them slightly less active, but more thermally stable.
The effect of such surface restructuration could be seen through the XPS analysis of the samples after the light-off runs. A decrease of the Co3+/Co2+ molar ratio was found for both samples, from 1.96 to 1.64 for the CC sample and from 1.20 to 1.02 for the CC-Na sample; which was accompanied by an increase in the Oads/Olatt molar ratio from 0.40 to 0.52 for the Na-free oxide and from 0.57 to 0.70 for the sample with sodium. In addition to that, the CC-Na sample suffered an increase in the Na surface concentration with respect to its fresh counterpart, achieving a molar concentration of 11.3% (9.0 wt%) of sodium.
2.5. Stability over a Prolonged Time
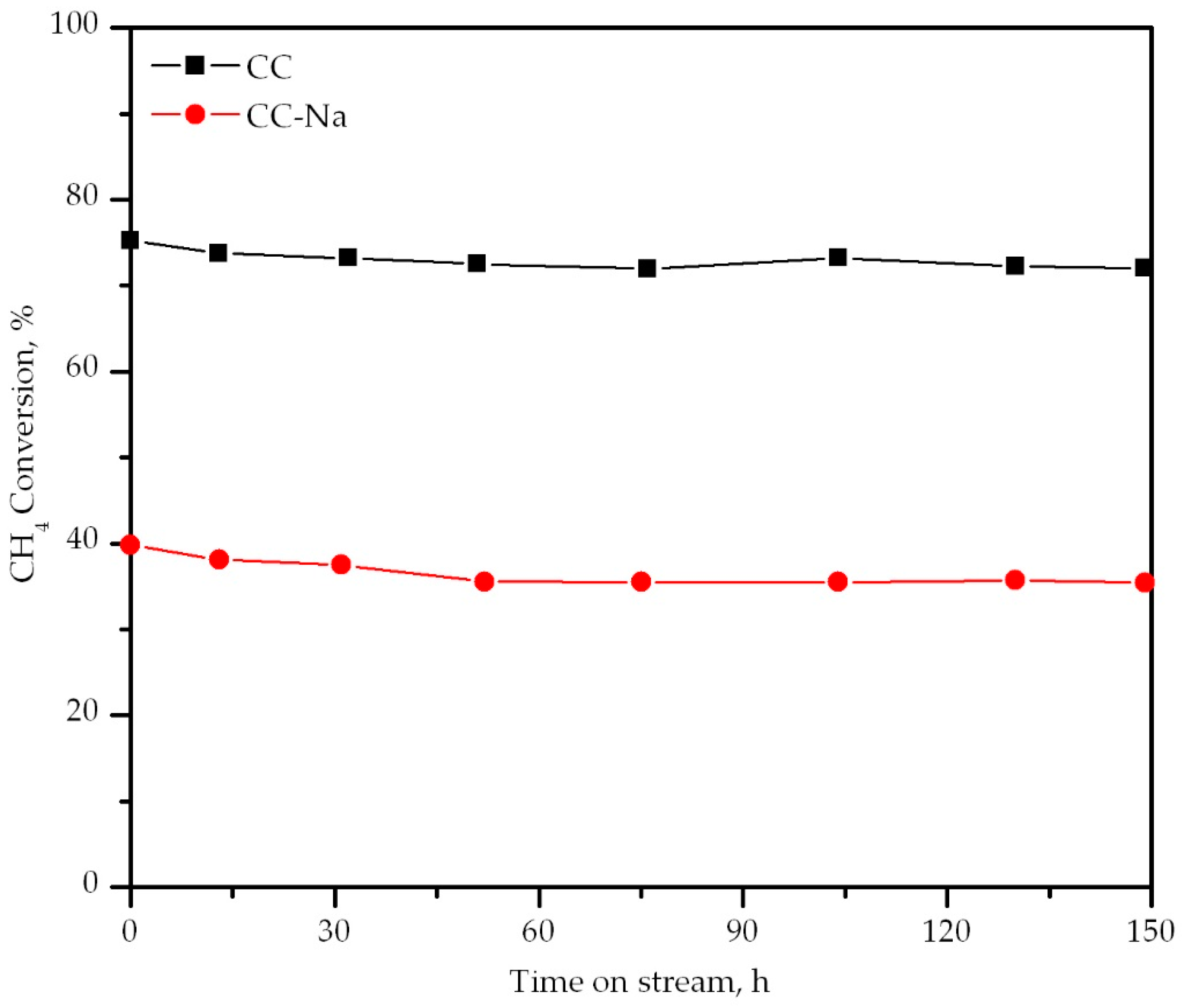
Finally, the stability of the catalysts over a prolonged period of time (150 h) was tested at 450 °C. This was to check if the presence of residual sodium could also affect the performance of the catalysts. It must be pointed out that a fresh sample of both oxide catalysts was used for this run. Both catalysts were thermally stable, although the conversion level they could achieve was remarkably different (Figure 10). The catalyst without sodium achieved a constant methane conversion of around 75%. The CC-Na catalyst could only achieve around 40% conversion for the entire time span of the test. This evidenced that the presence of residual sodium did not affect the thermal stability of the catalysts, but it did influence their activity.
The characterisation of the post-run catalysts revealed the effect of prolonged periods of operation over their properties. As can be seen in Table 4, the loss of properties for the sample without sodium was less marked than for the catalyst with sodium. However, interestingly, none of the catalysts exhibited any crystallite size growth after the stability test, which pointed out that they were structurally stable at temperatures up to 450 °C.
Comparing the impact caused by the light-off cycles with the loss of properties caused by the prolonged operation at constant temperature (450 °C), it became clear that operating at higher temperatures (600 °C) for short periods of time is more critical than operating at lower temperatures for extended periods of time. Indeed, if we compare the conversion levels obtained at 450 °C for the two aforementioned cases, it can be seen that the difference in conversion between the first and the third light-off cycle (around 20%) was much larger than the difference in conversion between the first light-off cycle and the start of the stability tests (around 5%). This evidenced that the loss of properties in the light-off cycles took place during the time the catalysts were operated at high temperatures (above 450 °C). For the stability tests it took place during the first hour of the test.
3. Materials and Methods
3.1. Catalyst Preparation
Two bulk Co3O4 catalysts were prepared following a simple precipitation route, described elsewhere [18]. This route consisted of an aqueous basic precipitation of a cobalt precursor, resulting in the formation of cobalt hydroxycarbonate. A solution of Na2CO3 (Sigma-Aldrich, St. Louis, MO, USA) 1.2 M was added drop-by-drop to 100 mL of Co(NO3)2·6H2O (ACS reagent, Alfa Aesar, Haverhill, MA, USA) 0.5 M, while the temperature was kept constant at 80 °C, until pH 8.5 was attained. After that, the obtained precipitates were filtrated and washed with different amounts of deionised water between one and eight litres. In this way, the so-called CC-Na precursor was washed with two litres of water to allow some residual sodium to remain within the catalytic precursor. On the contrary, the CC precursor was washed with up to eight litres of water with the aim of removing all the remaining sodium from the precipitation process. The catalyst precursors were dried in static air at 110 °C for 16 h and subjected to calcination in static air at 600 °C to produce the final catalysts.
3.2. Characterisation Techniques
Textural properties of the catalysts were determined from the nitrogen-adsorption isotherms at −196 °C obtained with a Micromeritics TriStar II apparatus (Micromeritics Instrument Corp., Norcross, GA, USA). The specific surface of the samples was obtained by the BET method. All samples were degassed prior to analysis on a Micromeritics SmartPrepapparatus (Micromeritics Instrument Corp., Norcross, GA, USA) at 300 °C for 10 h with a N2 flow.
Wavelength Dispersive X-ray Fluorescence (WDXRF) was used to determine the sodium content of the catalysts. From each sample in powder form, a boron glass pearl was prepared by fusion in an induction micro-furnace, by mixing the sample with the flux agent Spectromelt A12 (Merck KgaA, Darmstadt, Germany) in an approximate proportion of 20:1. The chemical analysis of each pearl was performed under vacuum, using a PANalytical AXIOS sequential WDXRF spectrometer (Malvern Panalytical Ltd., Malvern, UK), equipped with a Rh tube and three different detectors (gas flow, scintillation and Xe sealed).
Structural properties of the catalysts were determined by X-ray diffraction. XRD analysis were performed on a X’PERT-PRO X-ray diffractometer (Malvern Panalytical Ltd., Malvern, UK) using Cu Kα radiation (λ = 1.5406 Å) and a Ni filter. The X-ray tube was operated at 40 kV and 40 mA of current. The samples were scanned from an initial value of 2θ = 5° to a final value of 2θ = 80°, with a step size of 0.026° and a counting time of 2.0 s for each step. The cell size of the Co3O4 phase was obtained by profile matching of the whole XRD patterns using FullProf.2k software (Version 6.30, Institut Laue-Langevin, Grenoble, France, 2018).
Redox properties and Co species distribution were investigated by means of different techniques. Temperature-programmed reduction with hydrogen (H2-TPR) was performed on a Micromeritics Autochem 2920 apparatus (Micromeritics Instrument Corp., Norcross, GA, USA), using a 5%H2/Ar mixture as the reducing gas. The analysis protocol involved an initial pre-treatment step with a 5%O2/He mixture at 300 °C for 30 min. After cooling down to room temperature with flowing He, the TPR experiment was performed, up to 600 °C. Additional information regarding the reducibility of the catalysts was obtained by means of a subsequent temperature-programmed oxidation with a 5%O2/He mixture (O2-TPO) immediately after the H2-TPR run and by temperature-programmed reaction with a 5%CH4/He mixture in the absence of oxygen (CH4-TPRe). The O2-TPO runs were performed just after the H2-TPR runs without exposing the samples to the atmosphere by keeping the samples under an inert gas flow between the two consecutive runs.
X-ray photoelectron spectroscopy (XPS) analysis was performed using a SPECS system (SPECS GmbH, Berlin, Germany) equipped with a Phoibos 150 1D analyzer and a DLD-monochromatic radiation source. UV-vis-NIR diffuse reflectance (UV-vis-NIR DRS) spectra of the samples were obtained on a Jasco V-570 apparatus (JASCO, Easton, MD, USA) in the 200–2300 nm wavelength range.
3.3. Catalytic Activity Determination
Catalytic activity tests were performed in a bench-scale fixed bed reactor, Microactivity modular laboratory system manufactured by PID Eng&Tech S.L. (Process Integral Development S.L., Alcobendas, Spain), operated at atmospheric pressure and monitored by computer. The reactor was a stainless steel (Hasteloy X) tube (Haynes International, Inc., Kokomo, IN, USA), manufactured by Autoclave Engineers (Autoclave Engineers Engineering and Manufacturing, Erie, PA, USA), with an internal diameter of 8.55 mm and a length of 305 mm. The temperature inside the reactor was measured by a multipoint K type thermocouple placed in the catalyst bed.
For each reaction experiment, 1 g of catalyst granulated to a particle size of 0.25–0.3 mm was loaded into the reactor, diluted with 1 g of inert quartz granulated to a particle size of 0.5–0.8 mm to improve gas flow and heat distribution along the bed and avoid diffusional effects affecting the reaction rate. The reaction feed consisted of a gaseous mixture of 1% CH4, 10% O2 and N2 as the balance gas, up to a total gas flow of 500 cm3 min−1. This corresponded to a gas hourly space velocity of approximately 60,000 h−1 (300 mL CH4 g−1 h−1).
Catalytic activity was measured from 200 to 600 °C. Conversion measurements were taken each 25 °C. Additionally, stability tests were carried out for a total time on stream of 150 h at 450 °C. Methane conversion was calculated by the difference between inlet and outlet CH4 concentrations. Inlet and outlet streams were analysed using an on-line Agilent Technologies 7890N gas chromatograph (Agilent Technologies, Santa Clara, CA, USA) equipped with a thermal conductivity detector (TCD) and two columns: a PLOT 5A molecular sieve column (Agilent Technologies, Santa Clara, CA, USA) for the analysis of CH4, O2, N2 and CO; and a PLOT U column (Agilent Technologies, Santa Clara, CA, USA) for CO2 analysis. To ensure that the obtained catalytic results were not affected by both mass and heat transfer limitations, the criteria for intra-granular and extra-granular mass diffusion, energy diffusion and temperature gradients were checked, according to the Eurokin procedure (Eurokin, Delft, The Netherlands) [40].
4. Conclusions
Two bulk Co3O4 catalysts were prepared by a simple precipitation method using sodium carbonate as the precipitating agent. One of the catalysts was properly washed after the precipitation process while the other kept some residual sodium ions.
The characterization of the two catalysts evidenced that the presence of the residual sodium ions had a great impact on the properties of the catalyst. Firstly, the insertion of the sodium atoms into the spinel lattice induced a distortion that improved the structural properties of the Co3O4, inhibiting crystallisation and increasing the specific surface area. However, this distortion was also partially responsible for a decrease in the reducibility of the catalyst. This was in a subtle way when the reducing agent was hydrogen, but severely when the samples were reduced with methane. On the other hand, the concentration of sodium ions on the surface of the catalyst induced a reduction of the Co3+ ions into Co2+, due to their high Lewis acidic properties, thus increasing the electron density within the oxygen ions of the lattice and weakening the Co-O bonds, making it more difficult for the catalyst to reoxidize. This subsequently provoked a decrease in the abundance of lattice oxygen species that were active for the methane oxidation reaction. The overall effect of the presence of residual sodium in the Co3O4 was a notable decrease in the catalytic activity, especially at low temperatures. These results pointed out the importance of properly washing the catalytic precursors after the synthesis process, especially when the precipitating agent was a sodium base. In this sense, the use of ammonia-based precipitants or synthesis routes such as sol-gel complexation may be considered as suitable alternatives for preparing active Co3O4 catalysts for methane combustion.
Author Contributions
R.L.-F. and J.I.G.-O. conceived and designed the experiments; A.C. performed the experiments; B.d.R., R.L.-F. and A.C. analysed the data; A.C. prepared the manuscript and R.L.-F., B.d.R. and J.I.G.-O. revised and corrected the manuscript.
Funding
This research was funded by the Ministry of Economy and Competitiveness grant number [CTQ2016-80253-R] and the University of the Basque Country UPV/EHU (PIF15/335).
Acknowledgments
The author wish to thank the technical and human support provided by SGIker of UPV/EHU.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, B.; Bartlett, K.; Frolking, S.; Hayhoe, K.; Jenkins, J.; Salas, W. Methane and Nitrous Oxide Emissions from Natural Sources; United States Environmental Protection Agency: Washington, DC, USA, 2010. [Google Scholar]
- Li, Z.; Hoflund, G.B. A review on complete oxidation of methane at low temperatures. J. Nat. Gas Chem. 2003, 12, 153–160. [Google Scholar]
- Lampert, J.K.; Kazi, M.S.; Farrauto, R.J. Palladium catalyst performance for methane emissions abatement from lean bum natural gas vehicles. Appl. Catal. B Environ. 1997, 14, 211–223. [Google Scholar] [CrossRef]
- Chen, J.; Arandiyan, H.; Gao, X.; Li, J. Recent advances in catalysts for methane combustion. Catal. Surv. Asia 2015, 19, 140–171. [Google Scholar] [CrossRef]
- Giebeler, L.; Kießling, D.; Wendt, G. LaMnO3 perovskite supported noble metal catalysts for the total oxidation of methane. Chem. Eng. Technol. 2007, 30, 889–894. [Google Scholar] [CrossRef]
- Van Vegten, N.; Baidya, T.; Krumeich, F.; Kleist, W.; Baiker, A. Flame-made MgAl2−xMxO4 (M = Mn, Fe, Co) mixed oxides: Structural properties and catalytic behavior in methane combustion. Appl. Catal. B Environ. 2010, 97, 398–406. [Google Scholar] [CrossRef]
- Yin, F.; Ji, S.; Wu, P.; Zhao, F.; Li, C. Preparation, characterization, and methane total oxidation of AAl12O19 and AMAl11O19 hexaaluminate catalysts prepared with urea combustion method. J. Mol. Catal. A Chem. 2008, 294, 27–36. [Google Scholar] [CrossRef]
- Duan, X.; Pan, M.; Yu, F.; Yuan, D. Synthesis, structure and optical properties of CoAl2O4 spinel nanocrystals. J. Alloy Compd. 2011, 509, 1079–1083. [Google Scholar] [CrossRef]
- Zasada, F.; Janas, J.; Piskorz, W.; Gorczynska, M.; Sojka, Z. Total oxidation of lean methane over cobalt spinel nanocubes controlled by the self-adjusted redox state of the catalyst: Experimental and theoretical account for interplay between the Langmuir-Hinshelwood and Mars-Van Krevelen mechanisms. ACS Catal. 2017, 7, 2853–2867. [Google Scholar] [CrossRef]
- Zasada, F.; Piskorz, W.; Janas, J.; Grybos, J.; Indyka, P.; Sojka, Z. Reactive oxygen species on the (100) facet of cobalt spinel nanocatalyst and their relevance in 16O2/18O2 isotopic exchange, deN2O, and deCH4 processes—A theoretical and experimental account. ACS Catal. 2015, 5, 6879–6892. [Google Scholar] [CrossRef]
- Manouchehri, I.; Kameli, P.; Salamati, H. Facile synthesis of Co3O4/CoO nanoparticles by thermal treatment of ball-milled precursors. J. Supercond. Nov. Magn. 2011, 24, 1907–1910. [Google Scholar] [CrossRef]
- Shi, R.; Chen, G.; Ma, W.; Zhang, D.; Qiu, G.; Liu, X. Shape-controlled synthesis and characterization of cobalt oxides hollow spheres and octahedra. Dalton Trans. 2012, 41, 5981–5987. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Peng, J.; Zhang, L.; Wan, R.; Guo, S.; Zhou, L. Optimization of preparation for Co3O4 by calcination from cobalt oxalate using response surface methodology. Chem. Eng. Res. Des. 2010, 88, 971–976. [Google Scholar] [CrossRef]
- Bai, B.; Arandiyan, H.; Li, J. Comparison of the performance for oxidation of formaldehyde on nano-Co3O4, 2D-Co3O4, and 3D-Co3O4 catalysts. Appl. Catal. B Environ. 2013, 142–143, 677–683. [Google Scholar] [CrossRef]
- Gunnewiek, R.F.K.; Mendes, C.F.; Kiminami, R.H.G.A. Synthesis of spinel cobalt oxide nanoparticles using a modified polymeric precursor method. Adv. Powder Technol. 2016, 27, 1056–1061. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, L.; Chen, M.; Cao, Y.; He, H.; Fan, K. Dry citrate-precursor synthesized nanocrystalline cobalt oxide as highly active catalyst for total oxidation of propane. J. Catal. 2009, 263, 104–113. [Google Scholar] [CrossRef]
- Monteverde, V.; Alessandro, H.A.; Stelmachowski, P.; Ercolino, G.; Specchia, S. Benchmark comparison of Co3O4 spinel-structured oxides with different morphologies for oxygen evolution reaction under alkaline conditions. J. Appl. Electrochem. 2017, 47, 295–304. [Google Scholar] [CrossRef]
- De Rivas, B.; López-Fonseca, R.; Jiménez-González, C.; Gutiérrez-Ortiz, J.I. Synthesis, characterisation and catalytic performance of nanocrystalline Co3O4 for gas-phase chlorinated VOC abatement. J. Catal. 2011, 281, 88–97. [Google Scholar] [CrossRef]
- Singh, M.; Ralhan, N.K.; Singh, S. Synthesis, characterization, scale-up and catalytic behaviour of Co3O4 nanoparticles. Bull. Mater. Sci. 2015, 38, 297–301. [Google Scholar] [CrossRef]
- Xu, R.; Zeng, H.C. Dimensional control of cobalt-hydroxide-carbonate nanorods and their thermal conversion to one-dimensional arrays of Co3O4 nanoparticles. J. Phys. Chem. B 2003, 107, 12643–12649. [Google Scholar] [CrossRef]
- Park, P.W.; Kil, J.K.; Kung, H.H.; Kung, M.C. NO decomposition over sodium-promoted cobalt oxide. Catal. Today 1998, 42, 51–60. [Google Scholar] [CrossRef]
- Haneda, M.; Kintaichi, Y.; Bion, N.; Hamada, H. Alkali metal-doped cobalt oxide catalysts for NO decomposition. Appl. Catal. B Environ. 2003, 46, 473–482. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B Environ. 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Zasada, F.; Piskorz, W.; Kotarba, A.; Sojka, Z. Guidelines for optimization of catalytic activity of 3D transition metal oxide catalysts in N2O decomposition by potassium promotion. Catal. Today 2011, 176, 369–372. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Maniak, G.; Kotarba, A.; Sojka, Z. Strong electronic promotion of Co3O4 towards N2O decomposition by surface alkali dopants. Catal. Commun. 2009, 10, 1062–1065. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Wach, A.; Kustrowski, P.; Mamulová-Kutláková, K.; Michalik, S.; Jirátová, K. Alkali metals as promoters in Co-Mn-Al mixed oxide for N2O decomposition. Appl. Catal. A Gen. 2013, 462–463, 227–235. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Arandiyan, H.; Peng, Y.; Chang, H.; Li, J. Low temperature complete combustion of methane over cobalt chromium oxides catalysts. Catal. Today 2013, 201, 12–18. [Google Scholar] [CrossRef]
- Zou, G.; Xu, Y.; Wang, S.; Chen, M.; Shangguan, W. The synergistic effect in Co-Ce oxides for catalytic oxidation of diesel soot. Catal. Sci. Technol. 2015, 5, 1084–1092. [Google Scholar] [CrossRef]
- Das, T.; Sengupta, S.; Deo, G. Effect of calcination temperature during the synthesis of Co/Al2O3 catalyst used for the hydrogenation of CO2. React. Kinet. Mech. Catal. 2013, 110, 147–162. [Google Scholar] [CrossRef]
- Brik, Y.; Kacimi, M.; Ziyad, M.; Bozon-Verduraz, F. Titania-supported Cobalt and cobalt-phosphorus catalysts: Characterization and performances in ethane oxidative dehydrogenation. J. Catal. 2001, 202, 118–128. [Google Scholar] [CrossRef]
- Lopes, I.; El Hassan, N.; Guerba, H.; Wallez, G.; Davidson, A. Size-induced structural modifications affecting Co3O4 nanoparticles patterned in SBA-15 silicas. Chem. Mater. 2006, 18, 5826–5828. [Google Scholar] [CrossRef]
- Windisch, C.F., Jr.; Exarhos, G.J.; Owings, R.R. Vibrational spectroscopic study of the site occupancy distribution of cations in nickel cobalt oxides. J. Appl. Phys. 2004, 95, 5435–5442. [Google Scholar] [CrossRef]
- González-Prior, J.; López-Fonseca, R.; Gutiérrez-Ortiz, J.I.; de Rivas, B. Oxidation of 1,2-dichloroethane over nanocube-shaped Co3O4 catalysts. Appl. Catal. B Environ. 2016, 199, 384–393. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Dupin, J.; Gonbeau, D.; Vinatier, P.; Levasseur, A. Systematic XPS studies of metal oxides, hydroxides and peroxides. Phys. Chem. Chem. Phys. 2000, 2, 1319–1324. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G. Co3O4/CeO2 composite oxides for methane emissions abatement: Relationship between Co3O4-CeO2 interaction and catalytic activity. Appl. Catal. B Environ. 2006, 66, 217–227. [Google Scholar] [CrossRef]
- Pu, Z.; Zhou, H.; Zheng, Y.; Huang, W.; Li, X. Enhanced methane combustion over Co3O4 catalysts prepared by a facile precipitation method: Effect of aging time. Appl. Surf. Sci. 2017, 410, 14–21. [Google Scholar] [CrossRef]
- Bahlawane, N. Kinetics of methane combustion over CVD-made cobalt oxide catalysts. Appl. Catal. B Environ. 2006, 67, 168–176. [Google Scholar] [CrossRef]
- Stefanov, P.; Todorova, S.; Naydenov, A.; Tzaneva, B.; Kolev, H.; Atanasova, G.; Stoyanova, D.; Karakirova, Y.; Aleksieva, K. On the development of active and stable Pd-Co/γ-Al2O3 catalyst for complete oxidation of methane. Chem. Eng. J. 2015, 266, 329–338. [Google Scholar] [CrossRef]
- EUROKIN Spreadsheet on Requirements for Measurement of Intrinsic Kinetics in the Gas-Solid Fixed-Bed Reactor. Available online: http://eurokin.org/ (accessed on 20 February 2018).
Figure 1.
pH of the filtrate from the washing step of the catalytic precursors (CC and CC-Na).

Figure 2.
(a) XRD diffractograms of the catalysts; (b) detail of the most intense diffraction signal.
Figure 2.
(a) XRD diffractograms of the catalysts; (b) detail of the most intense diffraction signal.

Figure 3.
Pore size distributions of the catalysts.

Figure 4.
(a) UV-vis-NIR DRS spectra of the samples; (b) detail of the region around 1200 nm.

Figure 5.
Raman spectra of the catalysts.

Figure 6.
(a) Co 2p XPS spectra of the samples; (b) O 1s XPS spectra of the samples.

Figure 7.
(a) H2-TPR profiles of the samples; (b) O2-TPO profiles of the samples.

Figure 8.
CH4-TPRe profiles of the samples.

Figure 9.
Light-off curves of the catalysts.

Figure 10.
Stability of the catalysts over time.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Results from physico-chemical characterisation.
| Catalyst | BET Surface Area, m2 g−1 | Vp, cm3 g−1 | D, nm | Dpore, Å | Cell Size, Å | Na Content, wt% |
|---|---|---|---|---|---|---|
| CC | 14 | 0.09 | 63 | 148 | 8.052 | 0.00 |
| CC-Na | 26 | 0.12 | 34 | 257 | 8.064 | 0.39 |
Table 2.
Surface composition results from XPS.
| Catalyst | O, %at. | Co, %at. | Na, %at. | Co3+/Co2+ | Oads/Olatt |
|---|---|---|---|---|---|
| CC | 66.5 | 33.5 | 0.0 | 1.96 | 0.40 |
| CC-Na | 57.6 | 35.4 | 7.0 | 1.20 | 0.57 |
Table 3.
Results from the kinetic analysis of activity results.
| Catalyst | T10, °C | T50, °C | T90, °C | Activation Energy, kJ mol−1 | k0 × 10−5, L min−1 gcat−1 |
|---|---|---|---|---|---|
| CC | 335 | 430 | 500 | 74 | 1.1 |
| CC-Na | 400 | 480 | 545 | 99 | 25 |
Table 4.
Comparison of textural/structural properties between fresh and used catalysts.
| CC | CC-Na | |||||
|---|---|---|---|---|---|---|
| Fresh Sample | 3 Light-Off Runs | 150 h at 450 °C | Fresh Sample | 3 Light-Off Runs | 150 h at 450 °C | |
| SBET, m2 g−1 | 14 | 9 | 12 | 26 | 18 | 19 |
| Vp, cm3 g−1 | 0.09 | 0.02 | 0.07 | 0.12 | 0.08 | 0.12 |
| Dpore, Å | 257 | 319 | 302 | 148 | 142 | 190 |
| D, nm | 63 | 61 | 64 | 34 | 46 | 33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Choya, A.; De Rivas, B.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Effect of Residual Na+ on the Combustion of Methane over Co3O4 Bulk Catalysts Prepared by Precipitation. Catalysts 2018, 8, 427. https://doi.org/10.3390/catal8100427
AMA Style
Choya A, De Rivas B, Gutiérrez-Ortiz JI, López-Fonseca R. Effect of Residual Na+ on the Combustion of Methane over Co3O4 Bulk Catalysts Prepared by Precipitation. Catalysts. 2018; 8(10):427. https://doi.org/10.3390/catal8100427
Chicago/Turabian StyleChoya, Andoni, Beatriz De Rivas, Jose Ignacio Gutiérrez-Ortiz, and Rubén López-Fonseca. 2018. "Effect of Residual Na+ on the Combustion of Methane over Co3O4 Bulk Catalysts Prepared by Precipitation" Catalysts 8, no. 10: 427. https://doi.org/10.3390/catal8100427
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.