An MM and QM Study of Biomimetic Catalysis of Diels-Alder Reactions Using Cyclodextrins


 and
and
Abstract
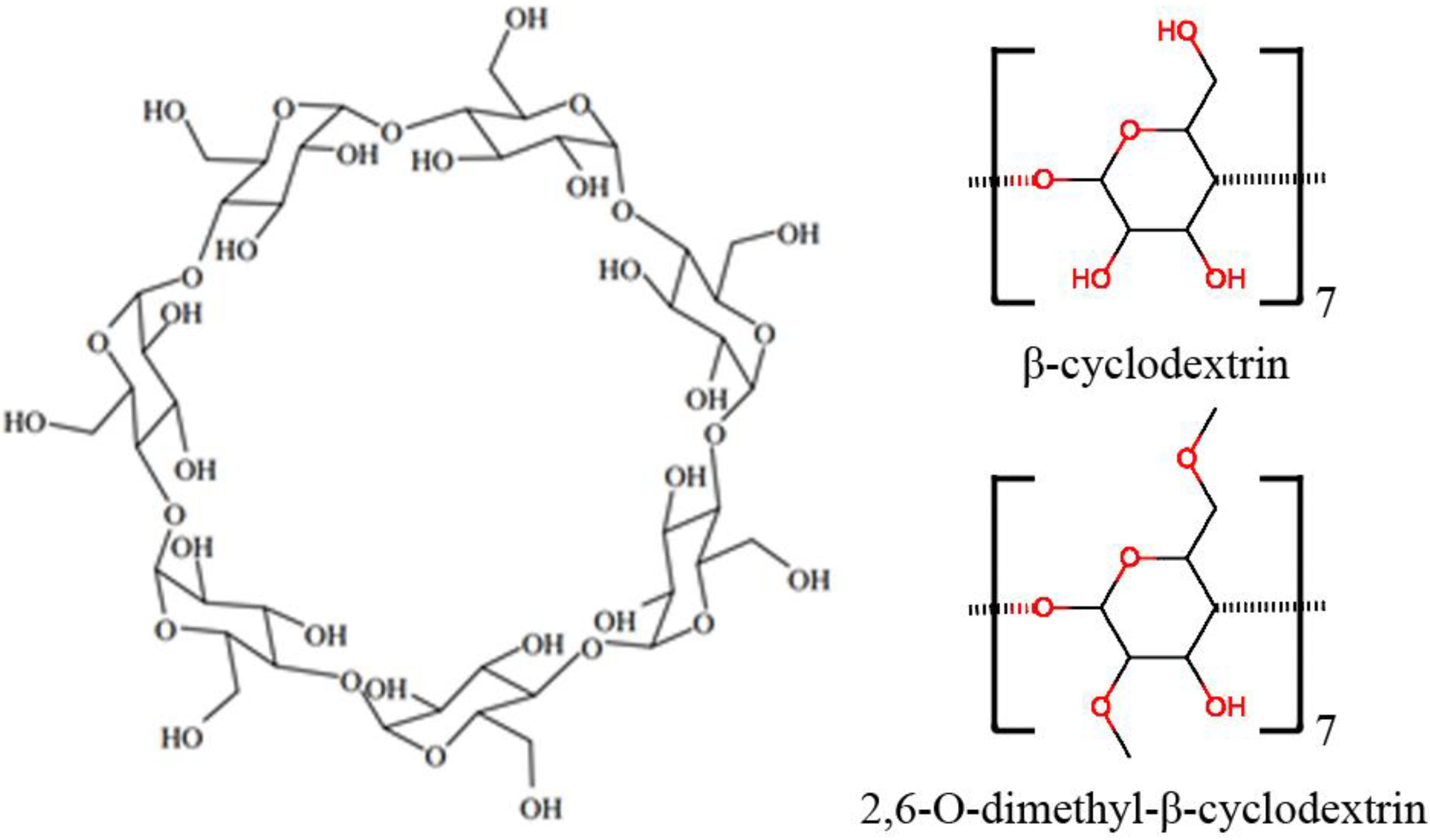
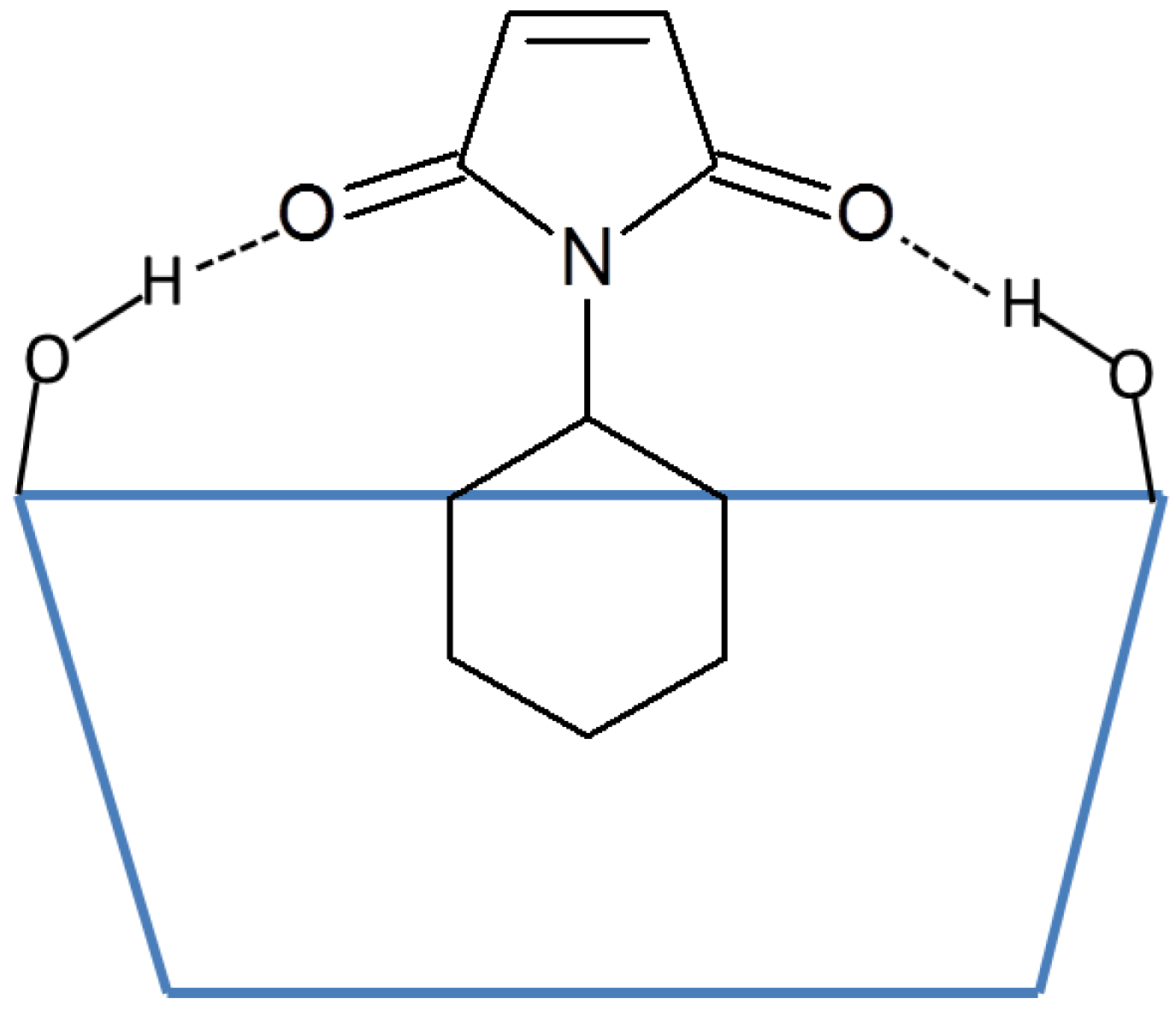
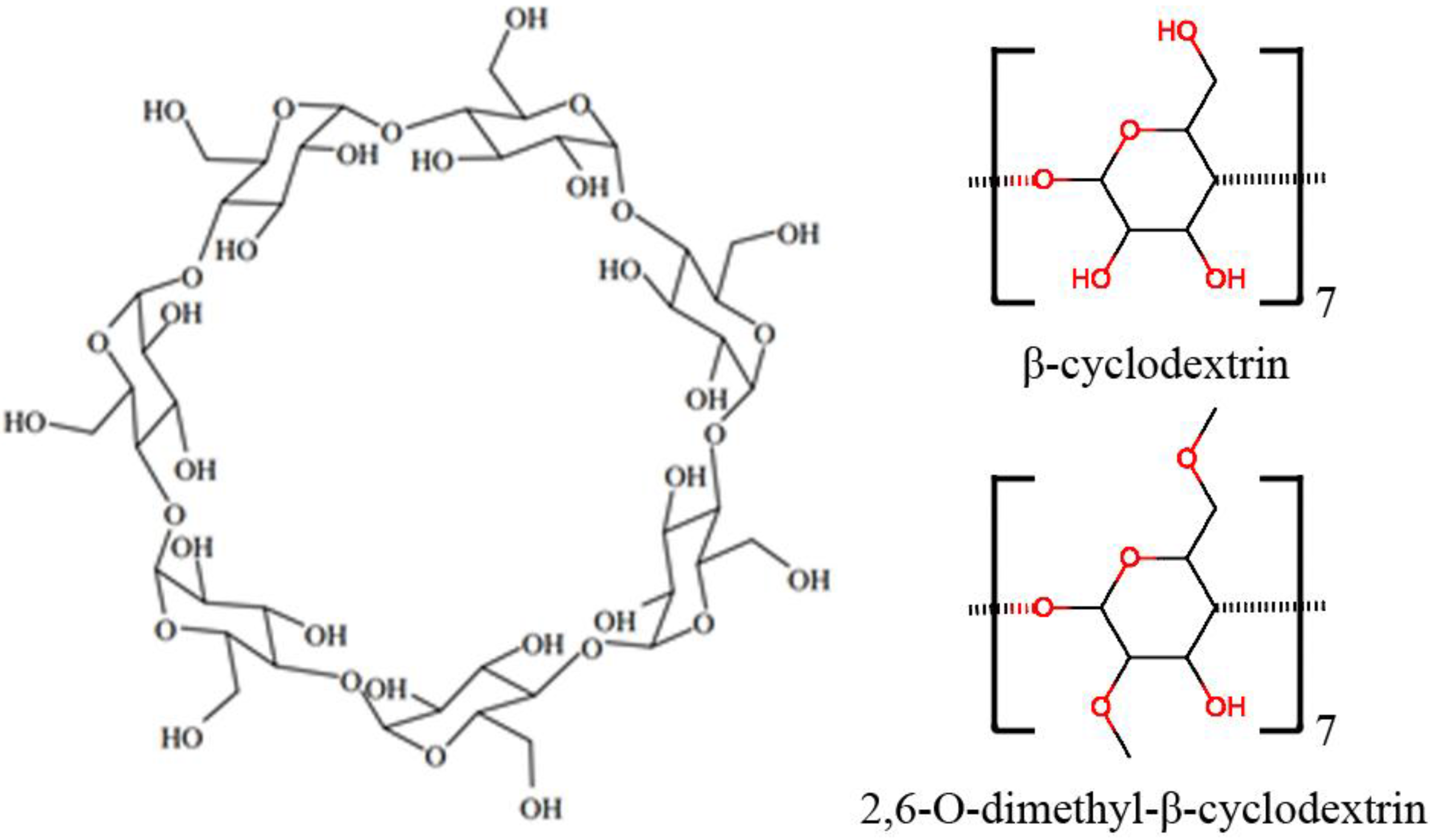
:1. Introduction
2. Results and Discussion
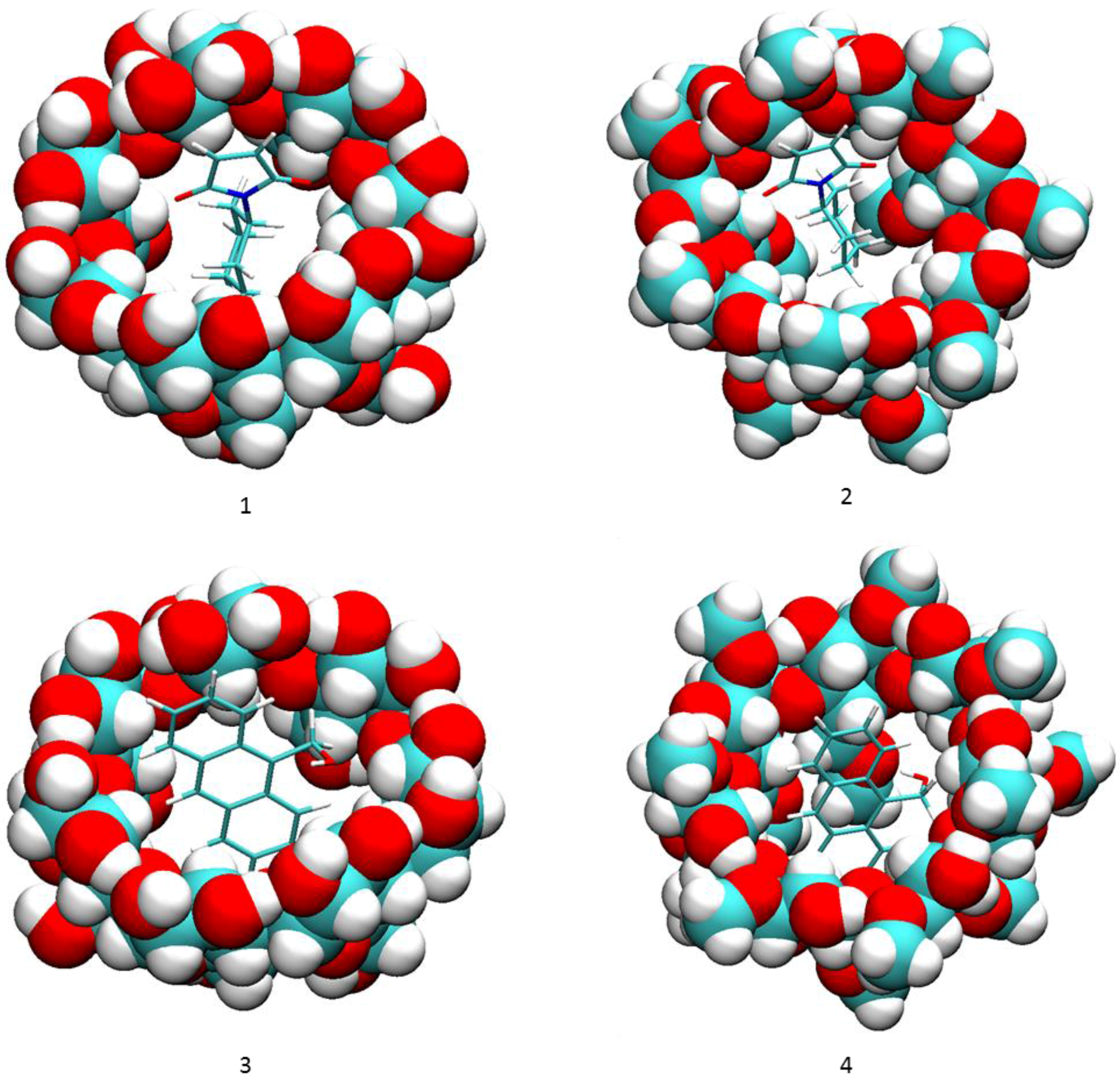
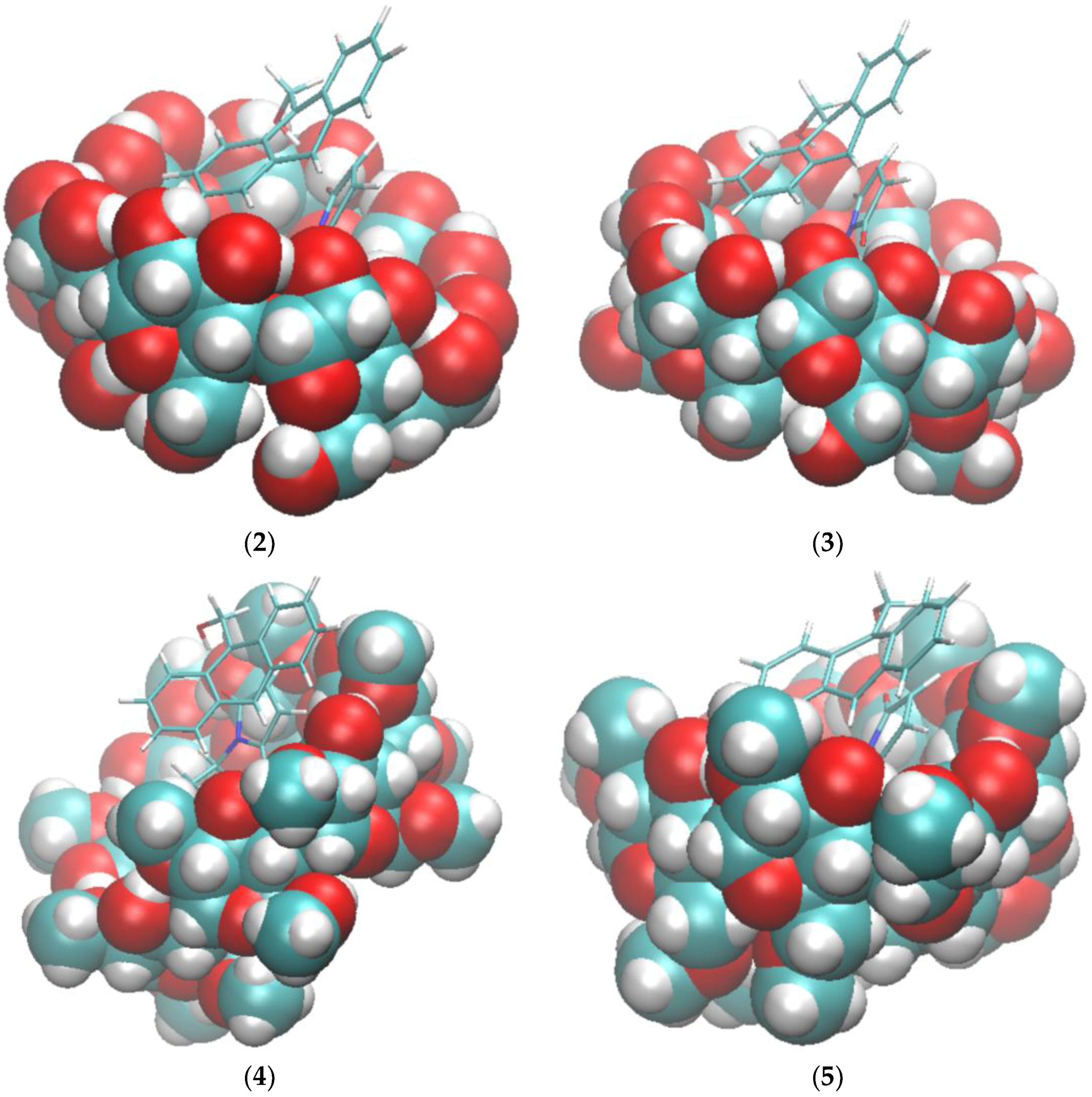
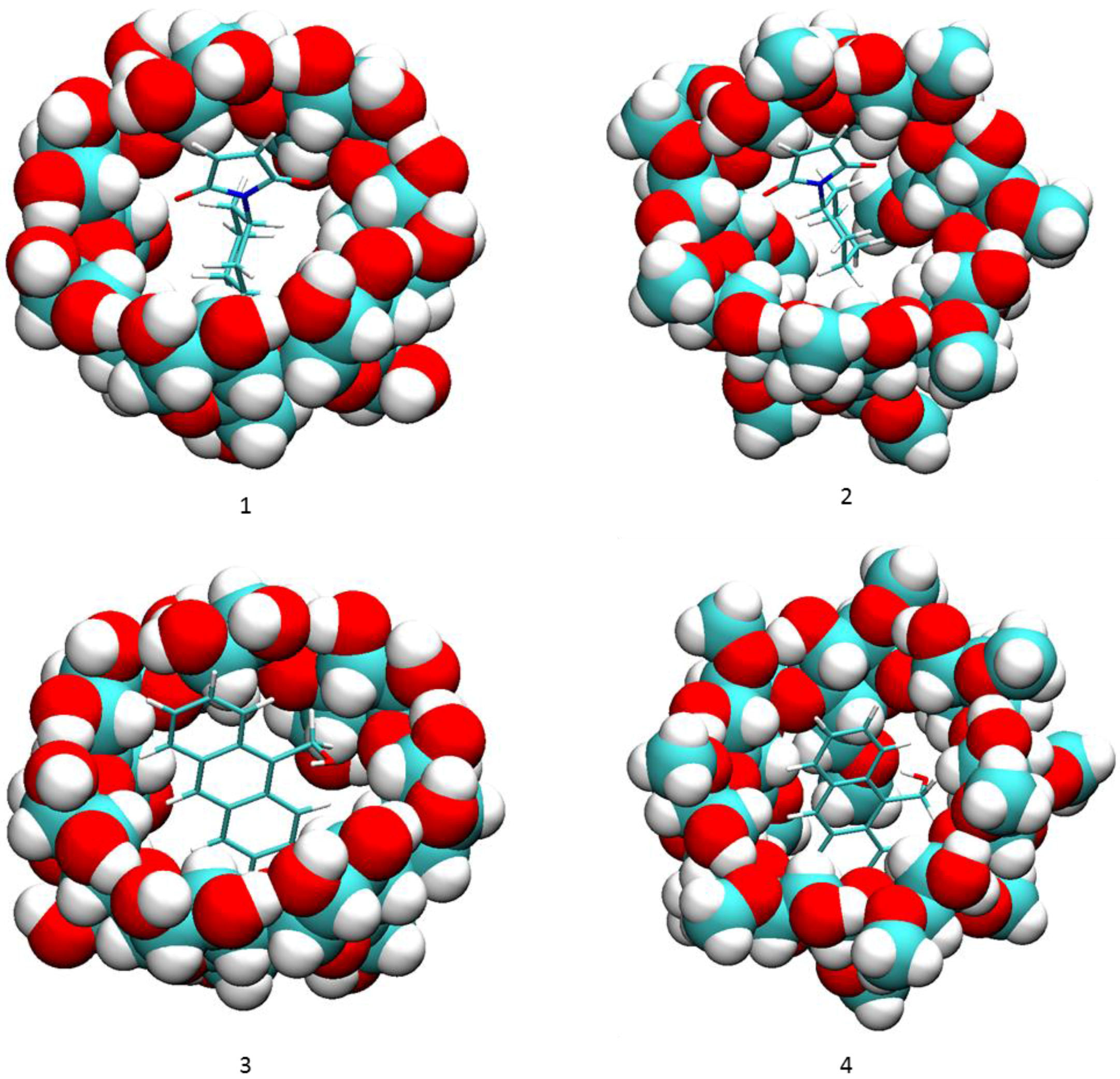
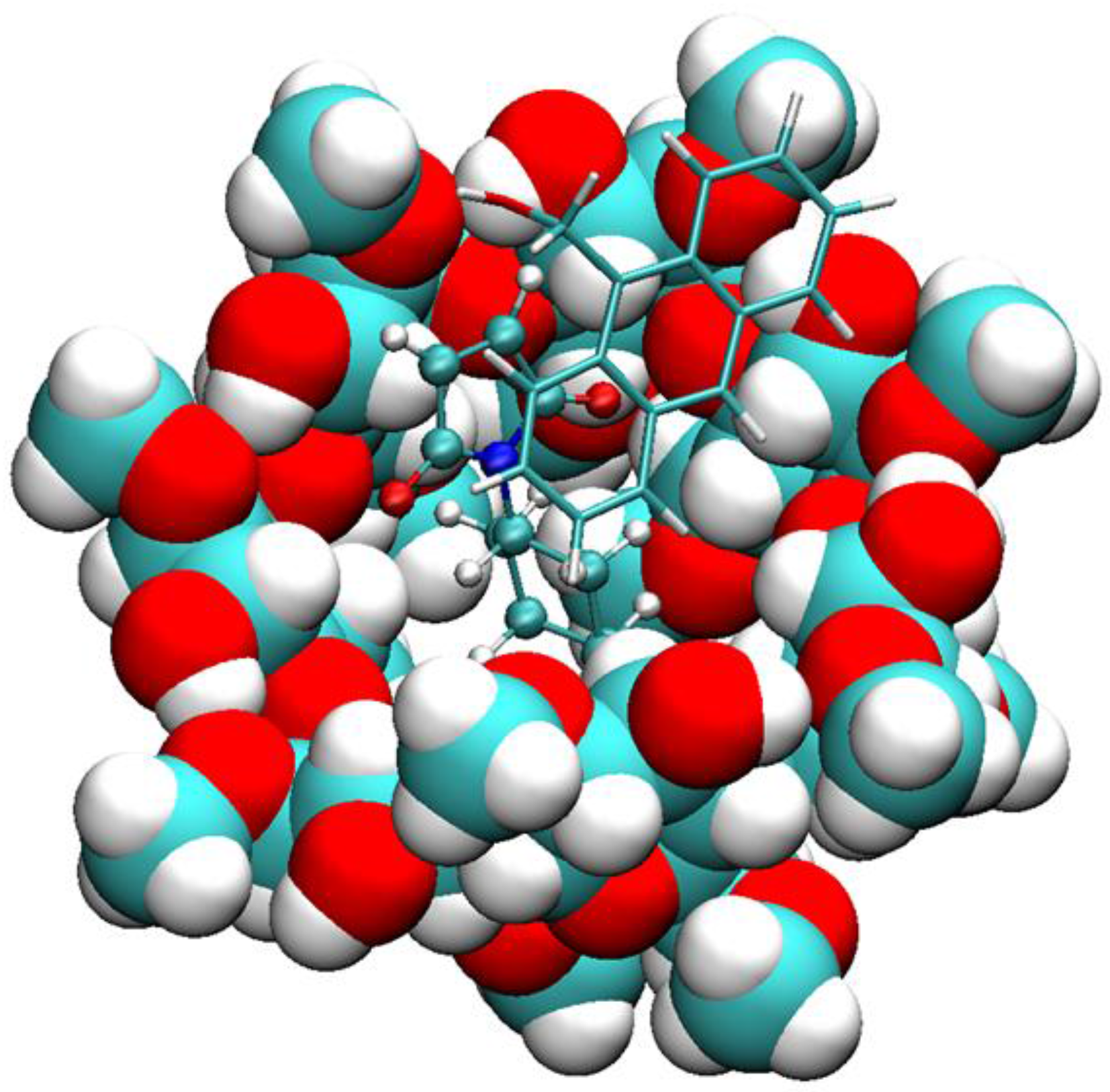
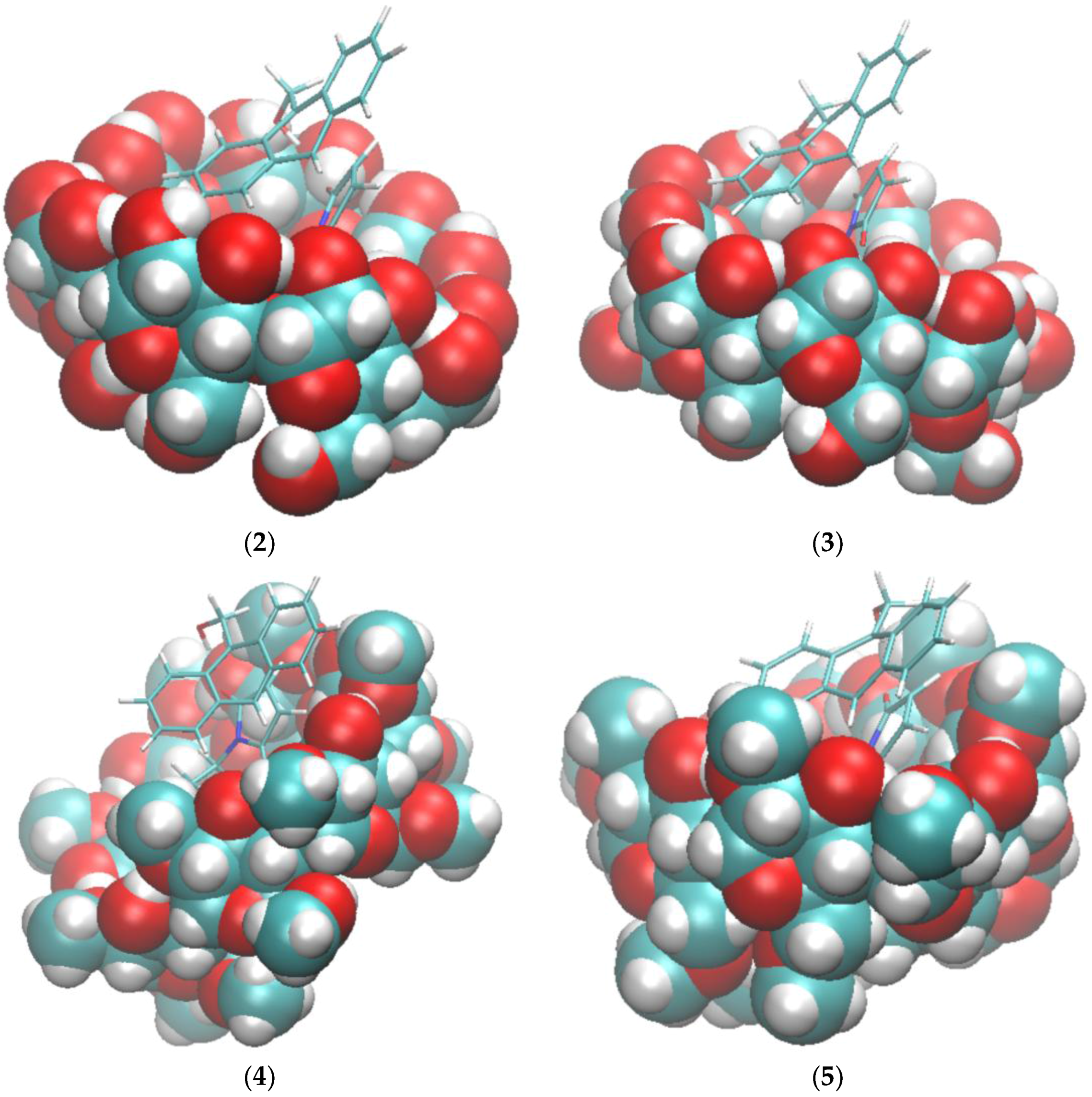
2.1. Free Energy Calculation for Cyclodextrin and Substrate Binding
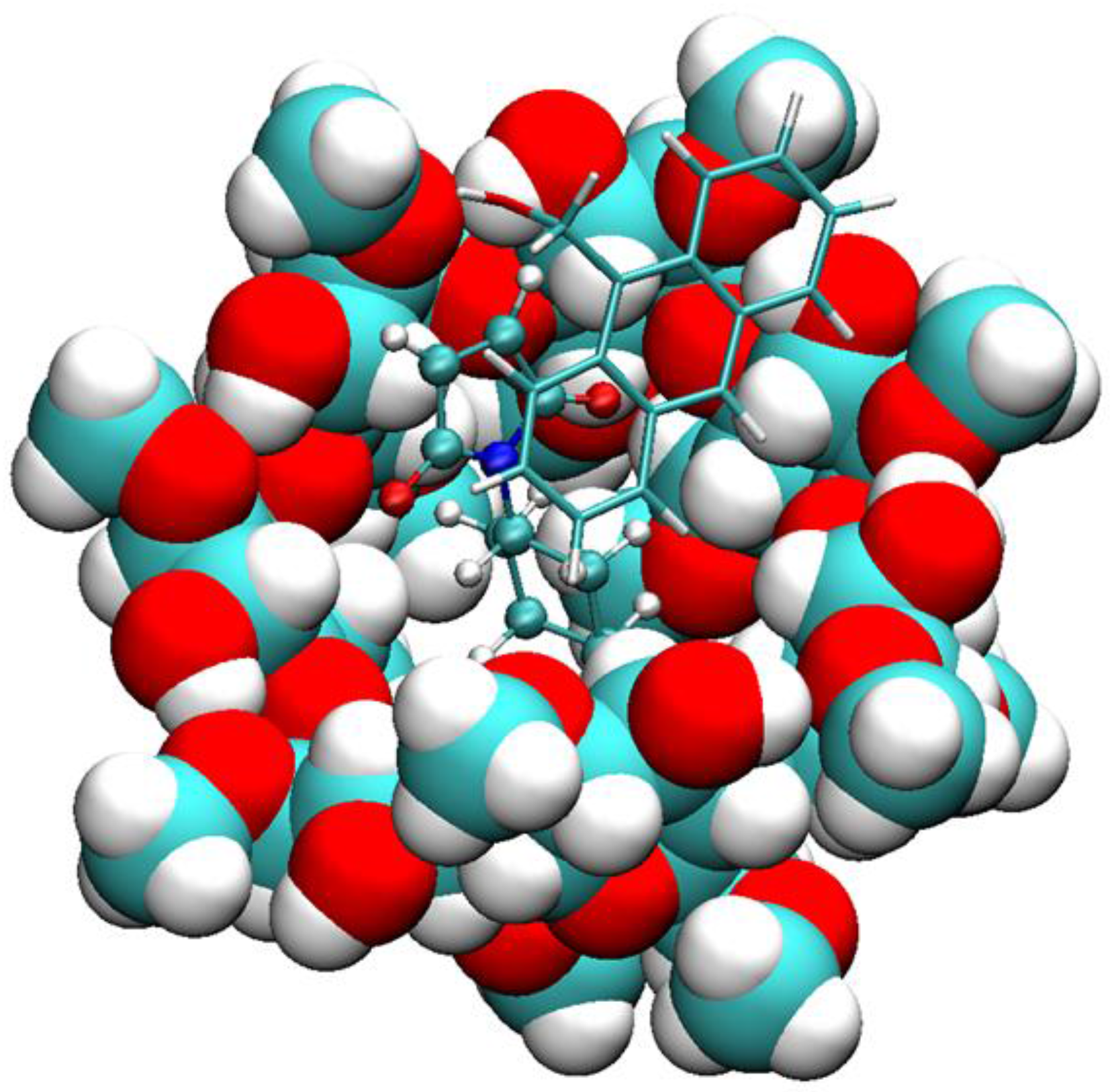
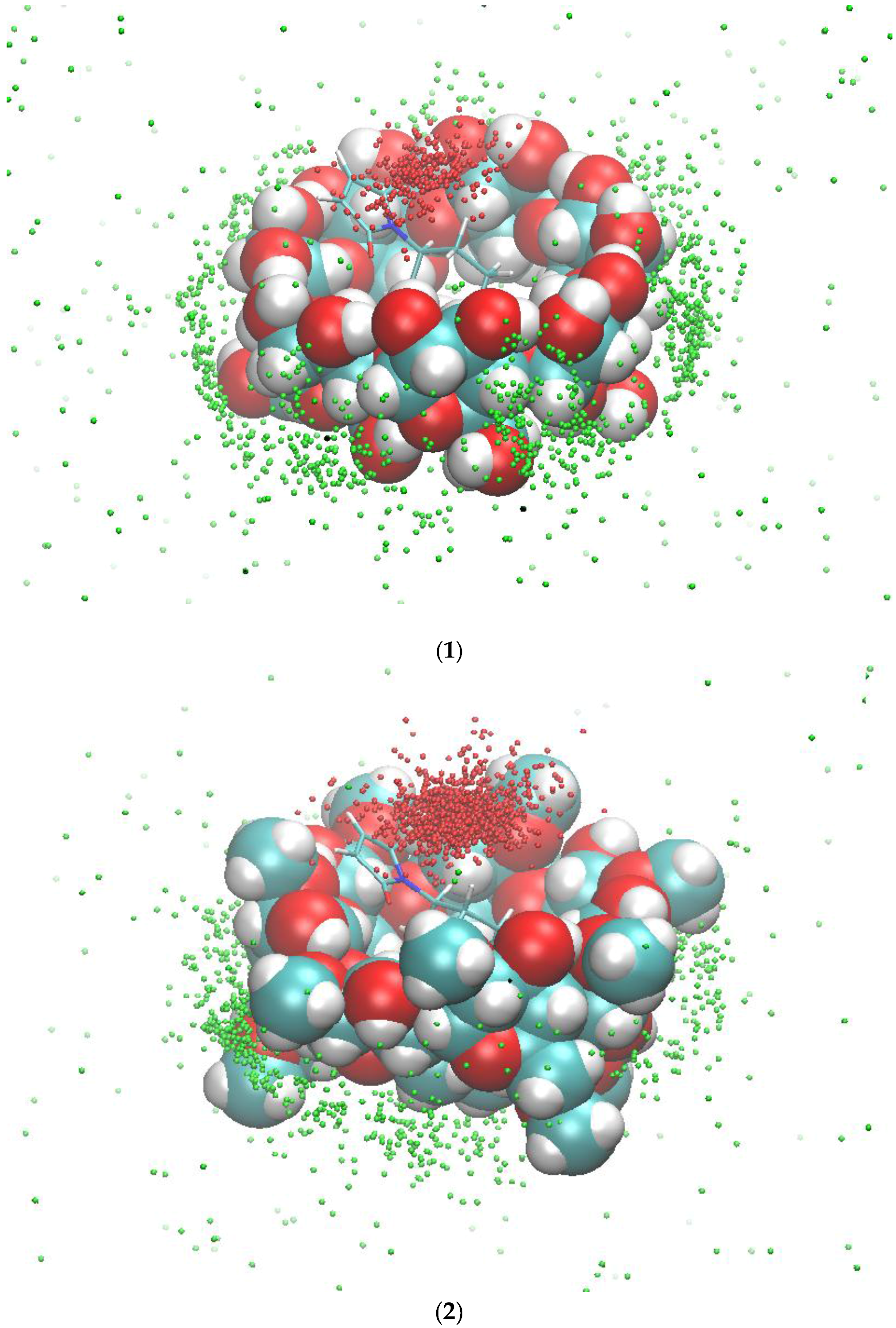
2.2. Conformational Fluctuations Modeled by MD Simulations
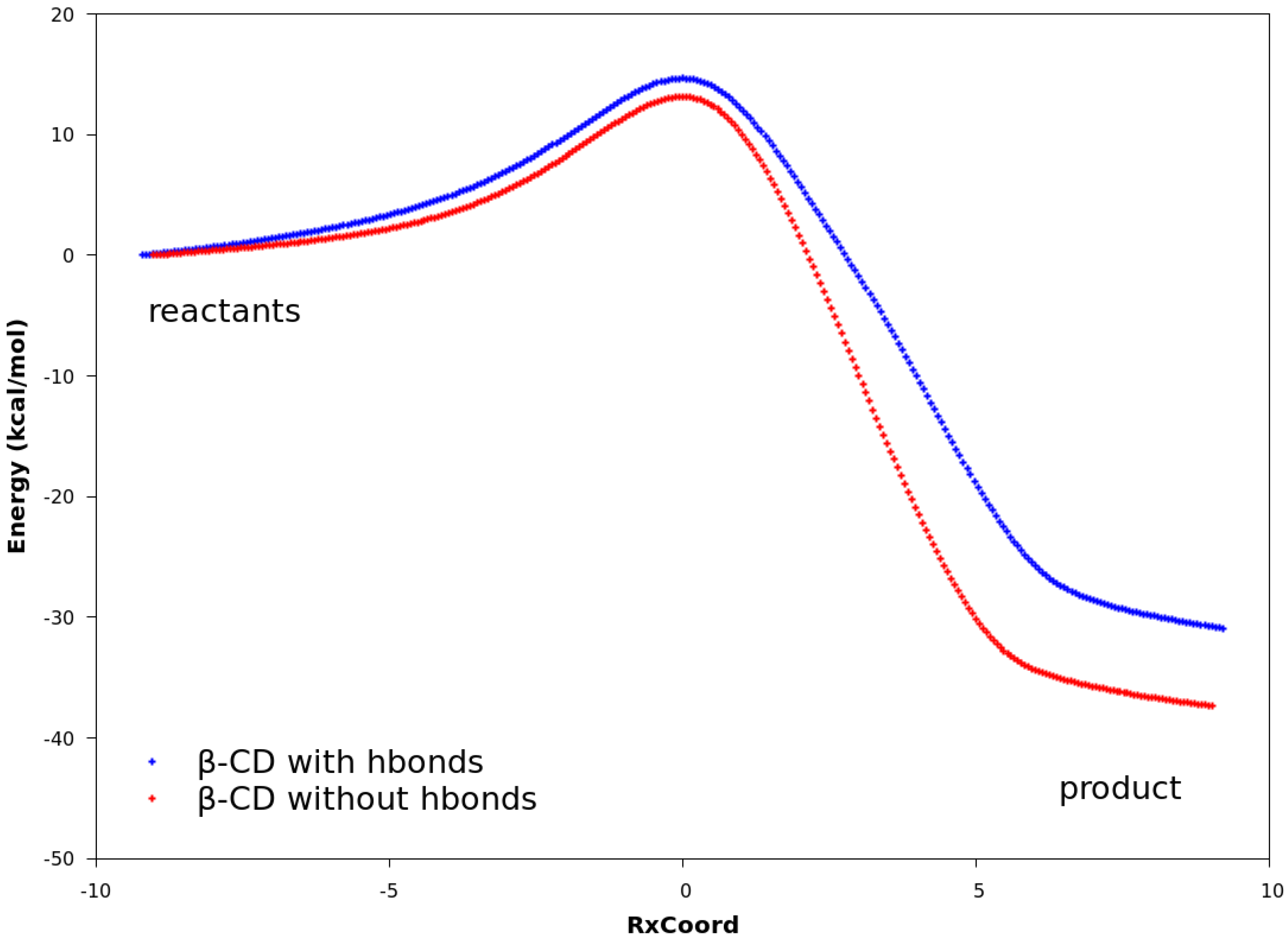
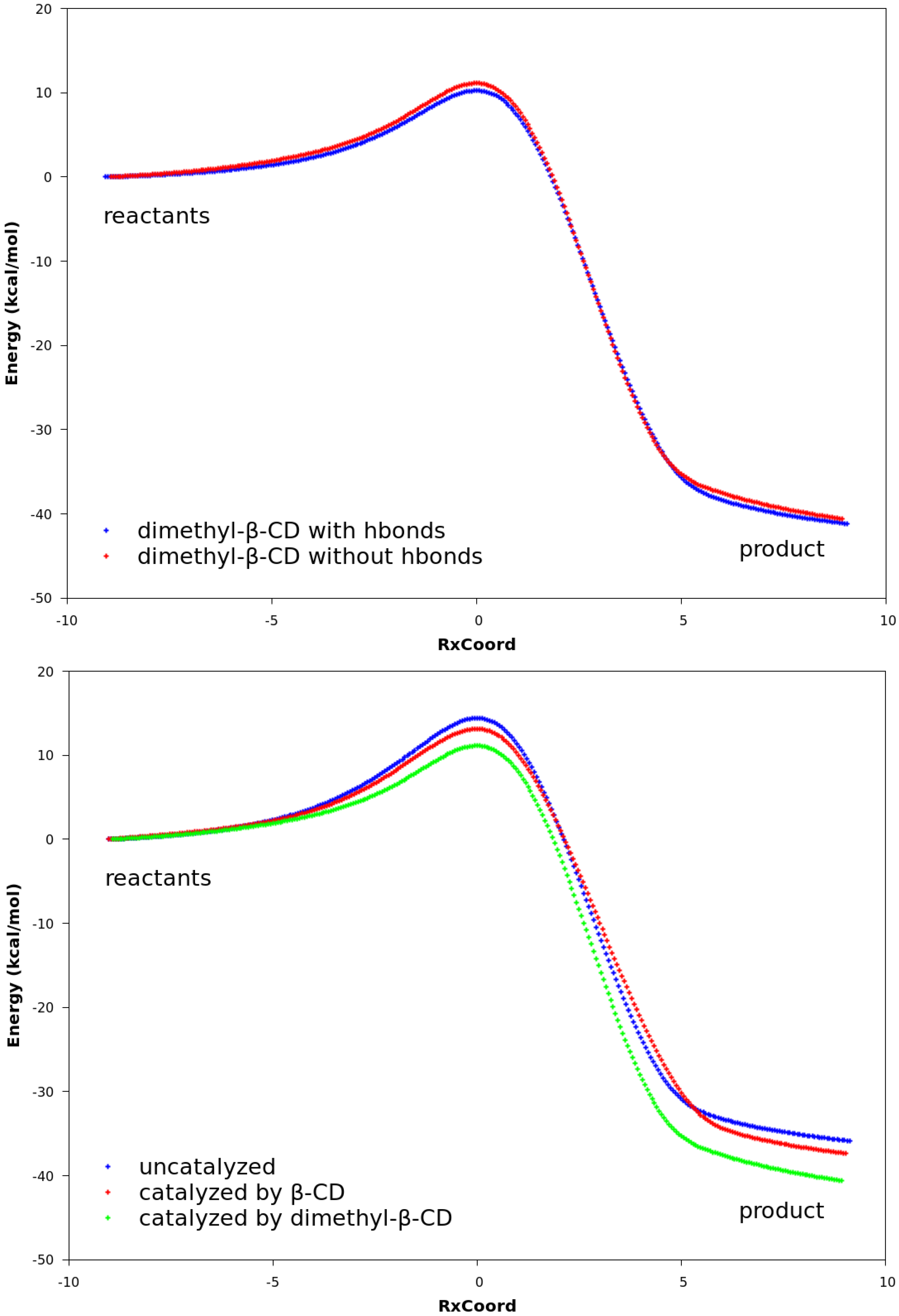
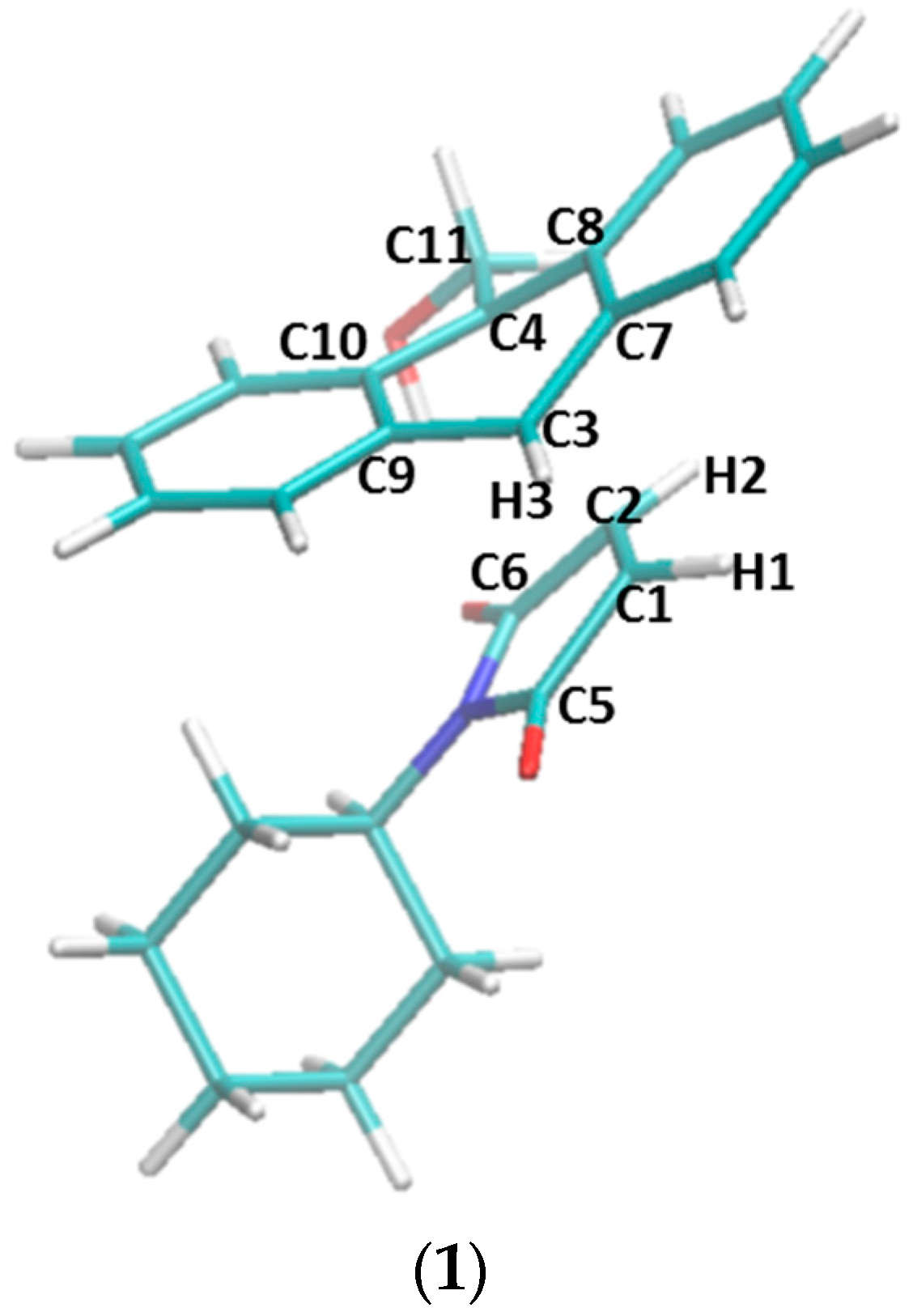
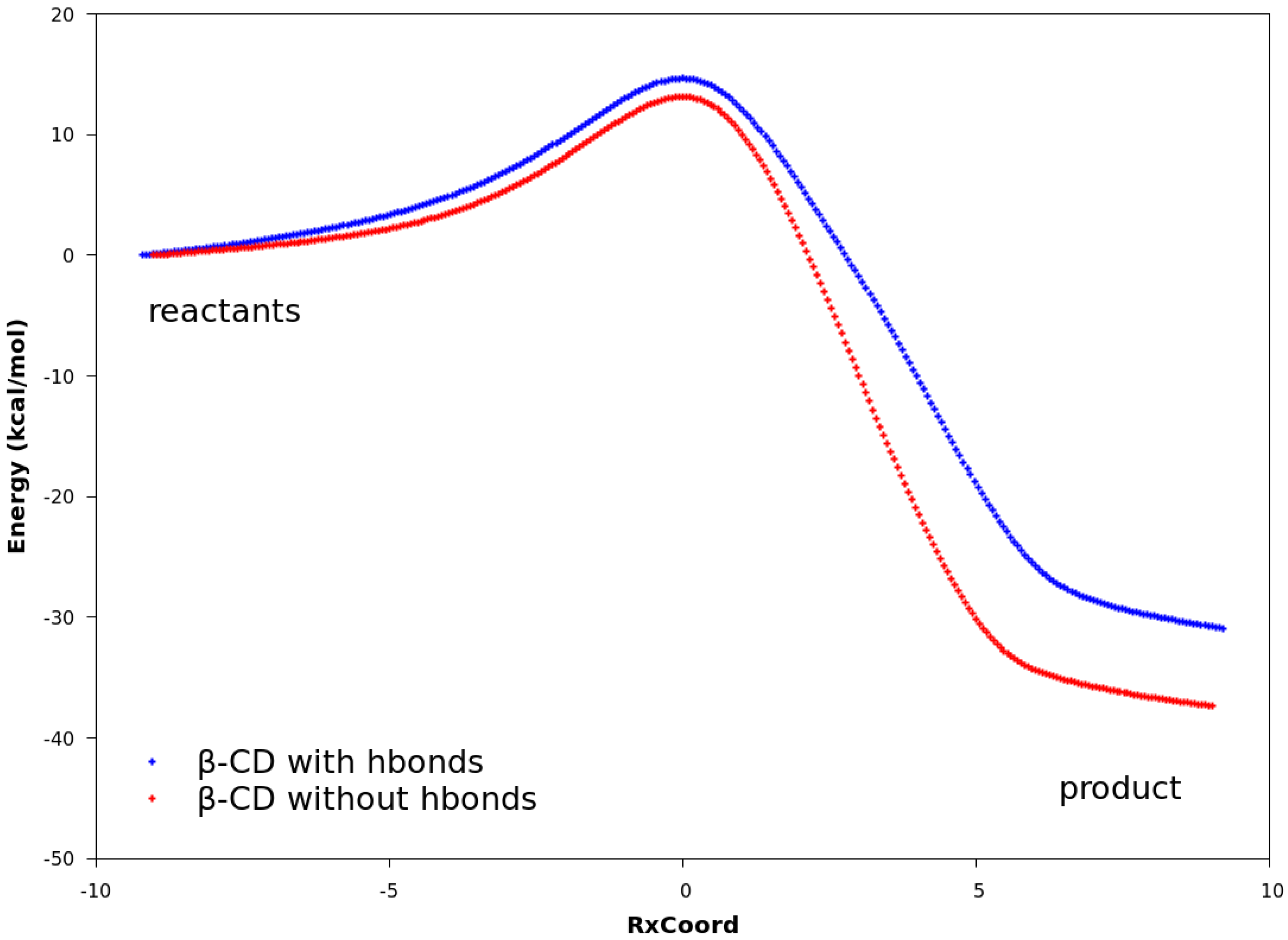
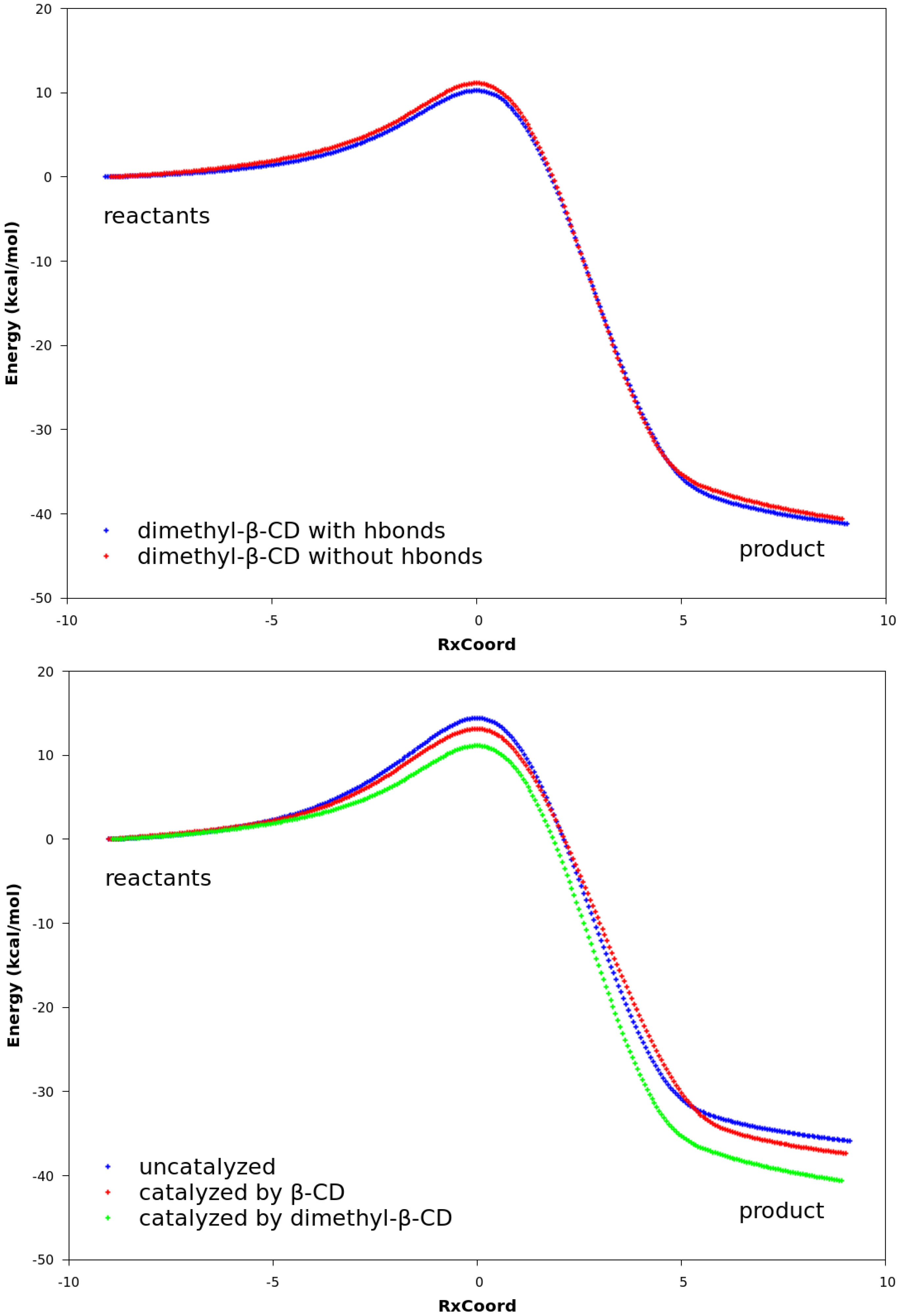
2.3. Explanation of Enhanced Catalysis by QM Calculations
3. Materials and Methods
3.1. Free Energy Calculations with the VM2 Method
3.2. Unbiased MD Simulations
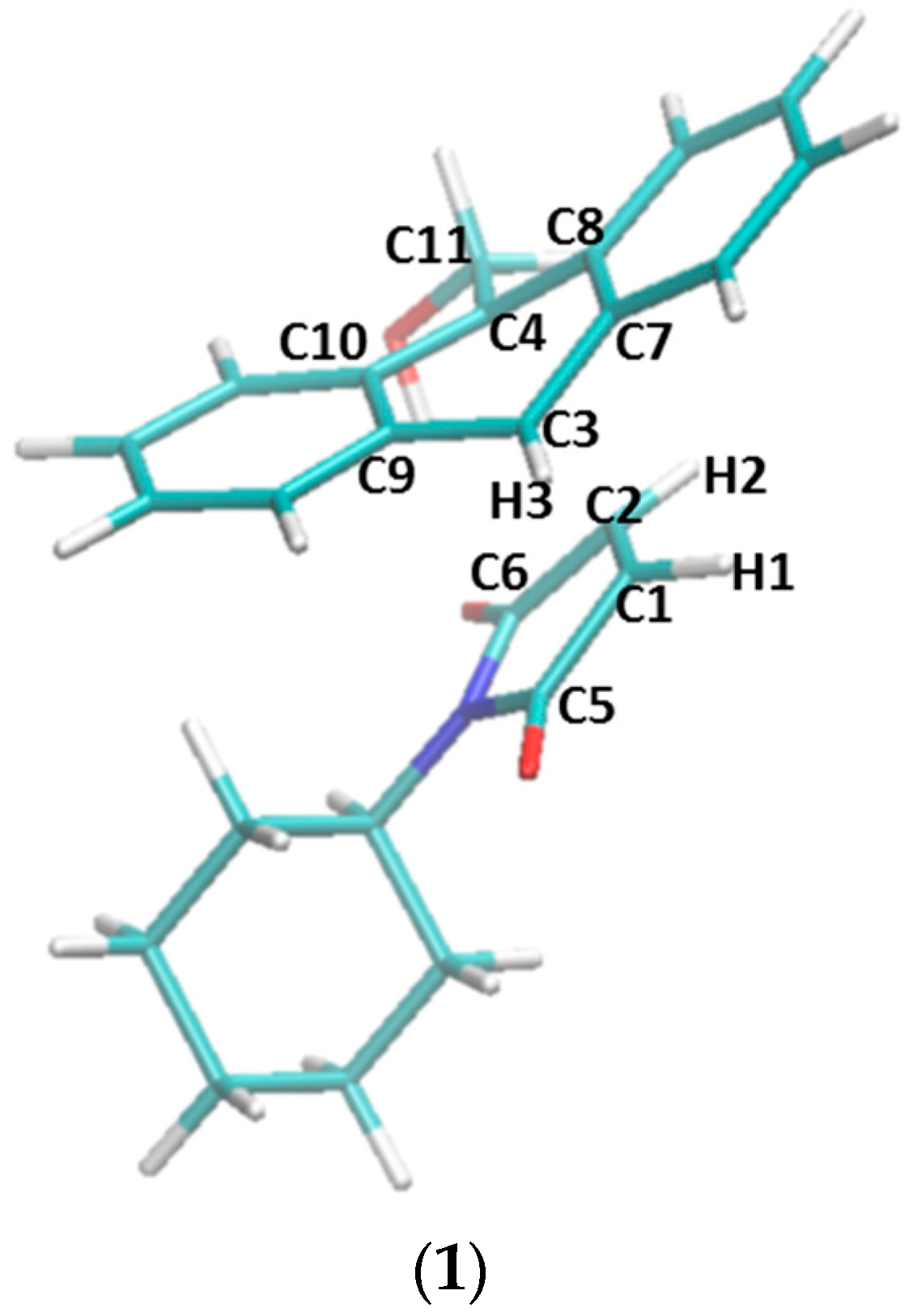
3.3. Quantum Mechanics Calculations with the PM3 Method
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, E.M.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Singh, M.; Sharma, R.; Banerjee, U.C. Biotechnological applications of cyclodextrins. Biotechnol. Adv. 2002, 20, 341–359. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.F. Cyclodextrins in non-viral gene delivery. Biomaterials 2014, 35, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Levine, M. Biomimetic Catalysis. ACS Catal. 2011, 1, 1090–1118. [Google Scholar] [CrossRef]
- Tayade, Y.A.; Padvi, S.A.; Wagh, Y.B.; Dalal, D.S. β-Cyclodextrin as a supramolecular catalyst for the synthesis of dihydropyrano[2,3-c]pyrazole and spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] in aqueous medium. Tetrahedron Lett. 2015, 56, 2441–2447. [Google Scholar] [CrossRef]
- Tabushi, I. Cyclodextrin Catalysis as a Model for Enzyme Action. Acc. Chem. Res. 1982, 15, 66–72. [Google Scholar] [CrossRef]
- Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Recent Breakthroughs in Aqueous Cyclodextrin-Assisted Supramolecular Catalysis. Catal. Sci. Technol. 2014, 4, 1899–1908. [Google Scholar] [CrossRef]
- Breslow, R.; Kohn, H.; Siegel, B. Methylated cyclodextrin and a cyclodextrin polymer as catalysts in selective anisole chlorination. Tetrahedron Lett. 1976, 17, 1645–1646. [Google Scholar] [CrossRef]
- DiScenza, D.J.; Gareau, L.; Serio, N.; Roque, J.; Verderame, M.; Levine, M. Cyclodextrin-Promoted Detection of Aromatic Toxicants and Toxicant Metabolites in Urine. Anal. Chem. Lett. 2016, 6, 345–353. [Google Scholar] [CrossRef]
- Breslow, R.; Dong, S.D. Biomimetic Reactions Catalyzed by Cyclodextrins and Their Derivatives. Chem. Rev. 1998, 98, 1997–2011. [Google Scholar] [CrossRef] [PubMed]
- Rideout, D.C.; Breslow, R. Hydrophobic Acceleration of Diels-Alder Reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar] [CrossRef]
- Schneider, H.J.; Sangwan, N.K. Diels-Alder Reactions in Hydrophobic Cavities: A Quantitative Correlation with Solvophobicity and Rate Enhancements by Macrocycles. J. Chem. Soc. Chem. Commun. 1986, 1787–1789. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Phelan, T.; Levine, M. Cyclodextrin-Promoted Diels-Alder Reactions of a Polycyclic Aromatic Hydrocarbon under Mild Reaction Conditions. Tetrahedron Lett. 2015, 56, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Leach, A.G.; Houk, K.N. The Origins of Noncovalent Catalysis of Intermolecular Diels-Alder Reactions by Cyclodextrins, Self-Assembling Capsules, Antibodies, and RNAses. J. Org. Chem. 2002, 67, 4250–4260. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L. Modern Organic Synthesis: Lecture Notes; The Scripps Research Institute Press: San Diego, CA, USA, 1999; pp. 213–272. [Google Scholar]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; St. Clair, J.L.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 239, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Gouverneur, V.E.; Houk, K.N.; de Pascual-Teresa, B.; Beno, B.; Janda, K.D.; Lerner, R.A. Control of the exo and endo Pathways of the Diels-Alder Reaction by Antibody Catalysis. Science 1993, 262, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Luzhkov, V.B.; Venanzi, G.A. Computer Modeling of Phenyl Acetate Hydrolysis in Water and in Reaction with .beta.-Cyclodextrin: Molecular Orbital Calculations with the Semiempirical AM1 Method and the Langevin Dipole Solvent Model. J. Phys. Chem. 1995, 99, 2312–2323. [Google Scholar] [CrossRef]
- Eto, M.; Kubota, S.; Nakagawa, H.; Yoshitake, Y.; Harano, K. Molecular Orbital Studies on Pericyclic Reactions of Cinnamyl Xanthates in beta-Cyclodextrin Cavities. Chem. Pharm. Bull. 2000, 48, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Granados, G.A.; de Rossi, R.H.; Barbiric, D.A.; Castro, E.A. Effect of β-cyclodextrin on the hydrolisis of N-phenylphtalamide and N-adamantylphtalamide—A two-sided semiempirical approach. J. Mol. Struct. THEOCHEM 2002, 619, 91–100. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, X.; Liu, N. The reactivity of phenancyl bromide under β-cyclodextrin as supramolecular catalyst: A computational survey. J. Mol. Model. 2015, 21, 131. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gilson, M.K.; Webb, S.P.; Potter, M.J. Modeling Protein-Ligand Binding by Mining Minima. J. Chem. Theory Comput. 2010, 6, 3540–3557. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, C.; Mao, J.; Yu, Y. A facile and practical approach to randomly methylated β-cyclodextrin. J. Chem. Technol. Biotechnol. 2010, 85, 248–251. [Google Scholar] [CrossRef]
- Musslimani, T.H.; Mettee, H. AM1 and PM3 semi-empirical study of the Diels–Alder reaction between N-, P-, O-and S-substituted aromatic heterocyclic five-membered rings with acrolein. J. Mol. Struct. 2004, 35–43. [Google Scholar] [CrossRef]
- Li, P.; Wong, B.M.; Zakharov, L.N.; Jasti, R. Investigating the Reactivity of 1,4-Anthracene-Incoporated Cycloparaphenylene. Org. Lett. 2016, 18, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Rowley, D.; Steiner, H. Kinetics of diene reactions at high temperatures. Discuss. Faraday Soc. 1951, 10, 198–213. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brown, F.K.; Houk, K.N. Transition structures of the Diels-Alder reaction of butadiene with acrolein. J. Org. Chem. 1989, 54, 1129–1134. [Google Scholar] [CrossRef]
- Daver, H.; Harvey, J.N.; Rebek, J., Jr.; Himo, F. Quantum Chemical Modeling of Cycloaddition Reaction in a Self-Assembled Capsule. J. Am. Chem. Soc. 2017, 139, 15494–15503. [Google Scholar] [CrossRef] [PubMed]
- Cézard, C.; Trivelli, X.; Aubry, F.; Djedaïni-Pilard, F.; Dupradeau, F.Y. Molecular dynamics studies of native and substituted cyclodextrins in different media: 1. Charge derivation and force field performances. Phys. Chem. Chem. Phys. 2011, 13, 15103–15121. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Chang, C.E.; Gilson, M.K. Tork: Conformational Analysis Method for Molecules and Complexes. J. Comput. Chem. 2003, 24, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Huang, J.; Gilson, M.K. Identification of Symmetries in Molecules and Complexes. J. Chem. Inf. Comput. Sci. 2004, 44, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.E.; Potter, M.J.; Gilson, M.K. Calculation of Molecular Configuration Integrals. J. Phys. Chem. B 2003, 107, 1048–1055. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham Iii, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Goetz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Doll, J.D.; Dion, D.R. Generalized langevin equation approach for atom-solid-surface scattering—Numerical techniques for gaussian generalized langevin dynamics. J. Chem. Phys. 1976, 65, 3762–3766. [Google Scholar] [CrossRef]
- Adelman, S.A. Generalized langevin theory for many-body problems in chemical-dynamics—General formulation and the equivalent harmonic chain representation. J. Chem. Phys. 1979, 71, 4471–4486. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L. A Smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-interaction of cartesian equations of motion of a system with constraints—Molecular-dynamics of N-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- GaussView; Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009.
- Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- McQuarrie, D.A.; Simon, J.D. Molecular Thermodynamics; University Science Books: Sausalito, CA, USA, 1999; ISBN 1-891389-05-X. [Google Scholar]
- Li, X.; Frisch, M.J. Energy-represented DIIS within a hybrid geometry optimization method. J. Chem. Theory Comput. 2006, 2, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods for Finding Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]

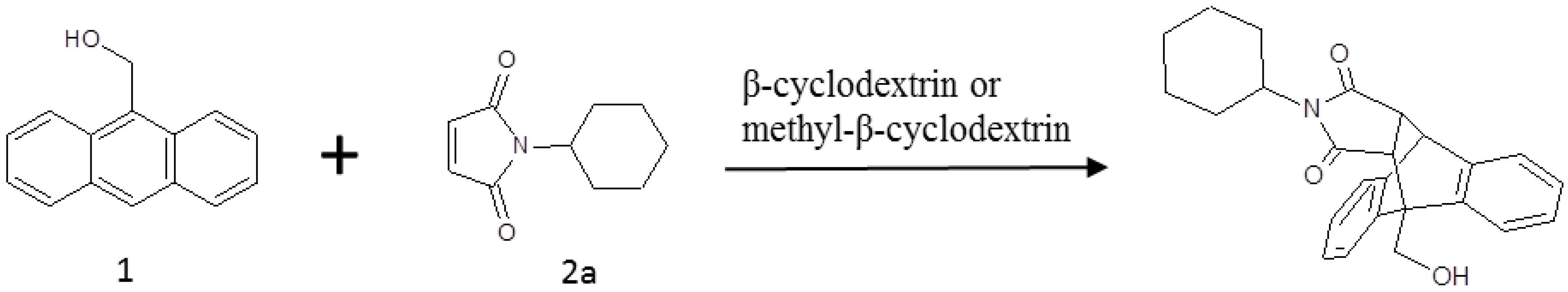


{kind=link}
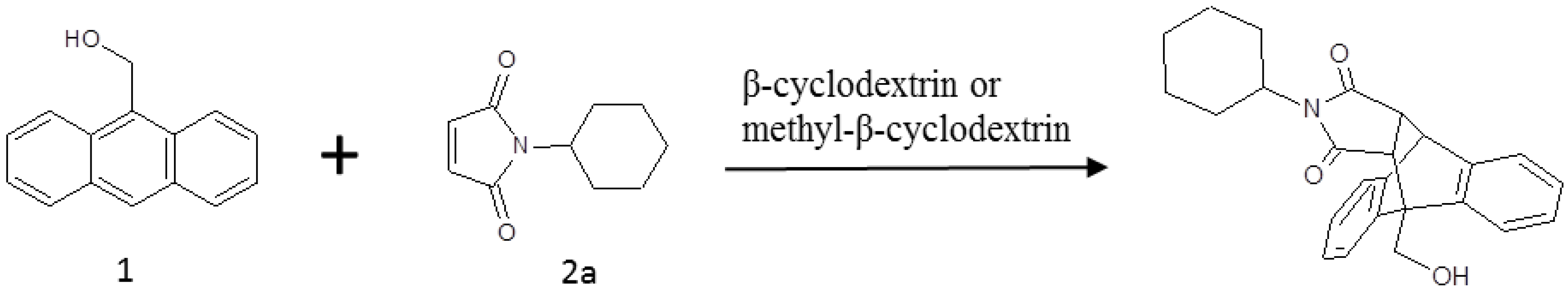{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔGcalc | ΔEValence | ΔECoulomb | ΔEPB | ΔENP | ΔEVDW | ΔH | -TΔS |
|---|---|---|---|---|---|---|---|---|
| β-CD and 1 | −2.71 | 1.96 | −8.04 | 15.69 | −2.86 | −25.27 | −18.52 | 15.81 |
| dimethyl-β-CD and 1 | −4.09 | 3.01 | −8.50 | 14.84 | −2.91 | −26.06 | −19.62 | 15.53 |
| β-CD and 2a | −6.58 | 0.39 | −5.98 | 13.23 | −2.76 | −25.12 | −20.23 | 13.65 |
| dimethyl-β-CD and 2a | −6.17 | 2.57 | −11.25 | 15.28 | −2.76 | −25.22 | −21.39 | 15.21 |
| Complex | ΔECoulomb | ΔEPB | ΔENP | ΔEVDW | ΔHMMPB/SA |
|---|---|---|---|---|---|
| β-CD with 2a | −7.24 | 17.23 | −2.19 | −23.07 | −15.27 |
| dimethyl-β-CD with 2a | −3.38 | 13.35 | −2.18 | −24.51 | −16.72 |
| β-CD with 1 and 2a | −16.00 | 27.79 | −2.66 | −25.88 | −16.75 |
| dimethyl-β-CD with 1 and 2a | −5.51 | 19.17 | −2.96 | −31.09 | −20.39 |

| Catalyst | MD | VM2 | |
|---|---|---|---|
| β-CD | d1 | 1.44 ± 0.70 | 0.38 ± 0.11 |
| d2 | −0.03 ± 0.87 | −1.39 ± 0.15 | |
| d3 | 3.35 ± 0.68 | 2.14 ± 0.11 | |
| dimethyl-β-CD | d1 | 2.05 ± 0.73 | 1.42 ± 0.13 |
| d2 | 0.67 ± 0.87 | −0.10 ± 0.17 | |
| d3 | 3.95 ± 0.70 | 3.37 ± 0.12 |
| No. | Bond C1–C3 | Bond C2–C4 | Improper C1–C2–H1–C5 | Improper C2–H2–C1–C6 | Improper C3–H3–C7–C9 | Improper C4–C8–C11–C10 |
|---|---|---|---|---|---|---|
| 1 | 2.203 | 2.391 | 21.498 | 18.172 | 16.042 | 11.803 |
| 2 | 2.112 | 2.603 | 25.175 | 13.678 | 18.587 | 9.557 |
| 3 | 2.171 | 2.473 | 22.346 | 17.424 | 16.720 | 11.938 |
| 4 | 2.230 | 2.398 | 20.148 | 18.362 | 14.907 | 11.434 |
| 5 | 2.264 | 2.342 | 19.919 | 21.709 | 13.786 | 11.243 |
| Reaction | ΔG | ΔH | -TΔS |
|---|---|---|---|
| non-catalyzed forward reaction | 16.89 | 0.86 | 16.03 |
| non-catalyzed reverse reaction | 48.06 | 48.66 | −0.60 |
| forward reaction catalyzed by β-CD, set 1 a | 22.75 | 21.29 | 1.47 |
| reverse reaction catalyzed by β-CD, set 1 a | 47.83 | 49.44 | −1.61 |
| forward reaction catalyzed by β-CD, set 2 b | 24.12 | 21.14 | 2.98 |
| reverse reaction catalyzed by β-CD, set 2 b | 48.67 | 49.92 | −1.25 |
| forward reaction catalyzed by dimethyl-β-CD, set 1 a | 13.73 | 10.05 | 3.68 |
| reverse reaction catalyzed by dimethyl-β-CD, set 1 a | 48.51 | 51.50 | −2.99 |
| forward reaction catalyzed by dimethyl-β-CD, set 2 b | 19.68 | 20.18 | −0.50 |
| reverse reaction catalyzed by dimethyl-β-CD, set 2 b | 49.48 | 51.25 | −1.77 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Sun, L.; Tang, Z.; Ali, Z.A.; Wong, B.M.; Chang, C.-e.A. An MM and QM Study of Biomimetic Catalysis of Diels-Alder Reactions Using Cyclodextrins. Catalysts 2018, 8, 51. https://doi.org/10.3390/catal8020051
Chen W, Sun L, Tang Z, Ali ZA, Wong BM, Chang C-eA. An MM and QM Study of Biomimetic Catalysis of Diels-Alder Reactions Using Cyclodextrins. Catalysts. 2018; 8(2):51. https://doi.org/10.3390/catal8020051
Chicago/Turabian StyleChen, Wei, Lipeng Sun, Zhiye Tang, Zulfikhar A. Ali, Bryan M. Wong, and Chia-en A. Chang. 2018. "An MM and QM Study of Biomimetic Catalysis of Diels-Alder Reactions Using Cyclodextrins" Catalysts 8, no. 2: 51. https://doi.org/10.3390/catal8020051