Whole-Cell Biocatalyst for Chemoenzymatic Total Synthesis of Rivastigmine
by
 , , ,
, , ,
Paola Vitale
1,2,* ,
,
Filippo Maria Perna
1,2,
Gennaro Agrimi
3,4,
Isabella Pisano
3,4,
Francesco Mirizzi
1,
Roberto Vito Capobianco
1 and
Vito Capriati
1,2,* 1
Dipartimento di Farmacia-Scienze del Farmaco, Università degli Studi di Bari «Aldo Moro», Via E. Orabona 4, I-70125 Bari, Italy
2
Consorzio C.I.N.M.P.I.S., Dipartimento di Farmacia-Scienze del Farmaco, Via E. Orabona 4, I-70125 Bari, Italy
3
Dipartimento di Bioscienze, Biotecnologie e Biofarmaceutica, Università degli Studi di Bari «Aldo Moro», Via E. Orabona 4, I-70125 Bari, Italy
4
Consorzio CIRCC, Via Celso Ulpiani 27, I-70126 Bari, Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2018, 8(2), 55; https://doi.org/10.3390/catal8020055
Submission received: 16 January 2018
/
Revised: 26 January 2018
/
Accepted: 29 January 2018
/
Published: 31 January 2018
Abstract
:Biocatalytic processes are increasingly playing a key role in the development of sustainable asymmetric syntheses, which are central to pharmaceutical companies for the production of chiral enantiopure drugs. This work describes a simple and economically viable chemoenzymatic process for the production of (S)-rivastigmine, which is an important drug for the treatment of mild to moderate dementia of the Alzheimer’s type. The described protocol involves the R-regioselective bioreduction of an aromatic ketone by Lactobacillus reuteri DSM 20016 whole cells in phosphate buffered saline (PBS) (37 °C, 24 h) as a key step. Biocatalytic performance of baker’s yeast whole cells in water and in aqueous eutectic mixtures have been evaluated and discussed as well. The route is scalable, environmentally friendly, and the target drug is obtained via four steps in overall 78% yield and 98% ee.
1. Introduction
Nowadays, the biocatalytic processes promoted by whole cells or isolated enzymes represent one of the most heavily used green methodologies in preparative chemistry to perform highly chemo-, regio-, and stereoselective organic transformations, as they are typically run under very mild reaction conditions (e.g., almost neutral pH buffer solutions, ambient temperature or mild heating, atmospheric pressure, etc.) [1,2]. Isolated enzymes are certainly specific catalysts for such transformations, with a history of high cost-effectiveness, especially on a preparative scale [3]. Whole-cell microorganisms, on the other hand, offer several positive features associated with their use as biocatalysts as they are more accessible, stable and easier to handle [4]. Moreover, the presence of efficient internal cofactor (e.g., NADH or NADPH in reduction reactions) regeneration systems allow for cheap biocatalytic processes to be carried out often using glucose as the sole carbon source [5]. Along with these properties, however, associated disadvantages are low yield processes, low reactant solubility in organic or aqueous media, low product recovery from reaction media, possible toxicity of substrates and products, and undesired side reactions because of the different enzymatic consortia usually present [4]. These drawbacks have prompted the isolation of new microorganisms and the development of new strains through metabolic and evolutionary engineering of known biocatalysts to obtain cheaper, faster, and more selective processes.
Among the known microorganisms, Saccharomyces cerevisiae (baker’s yeast) has been the most extensively utilized and studied whole-cell biocatalyst for the enantioselective reduction of prochiral ketones to enantiomerically enriched secondary alcohols, also on a preparative scale, because of its easy availability, economicity, and safety (GRAS status) [6]. Safe bacteria strains are also well-known and effective biocatalysts to perform bioreductions of carbonyl compounds with high performances [7]. Use of water is generally practical for carrying out bioreductions, although in many cases the additional employment of cosolvents (e.g., ethanol, 2-propanol, etc.) or different additives has proved to be necessary to overcome the scarce solubility of organic substrates, or to improve the selectivity and the recovery of products from the aqueous environment [8,9].
In recent years, deep eutectic solvents (DESs) have emerged as an interesting class of alternative reaction media for conducting organic reactions [10,11,12,13,14,15,16,17], including biotransformations [18,19,20,21]. Easily formed by mixing and gently heating naturally occurring hydrogen bond donors (e.g., polyols, carboxylic acids, amides, sugars, etc.) and hydrogen bond acceptors (e.g., choline chloride (ChCl), phosphonium salts, etc.), DESs are inexpensive, do not require further purification, are nontoxic and biodegradable. A multitude of factors (e.g., modification of cell membranes and permeability, interference with the cofactor-regeneration system, enzyme inhibition, etc.) seem to affect the catalytic performance of whole cells in DESs [18]. Moreover, in some cases, DES components have also been proved to be rapidly used by microorganisms in biocatalytic processes contributing to influence their metabolic equilibria [22]. As recently shown by other groups [23,24], and our own [25], a fascinating anti-Prelog baker’s yeast-promoted bioreduction of prochiral β-ketoesters and arylpropanones can be performed by simply changing the solvent from water to mixtures of water and DESs, thereby allowing the preparation of (R)-chiral alcohols as synthons for the manufacture of chemicals, modern pharmaceutical drugs, and flavors.
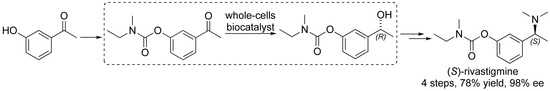
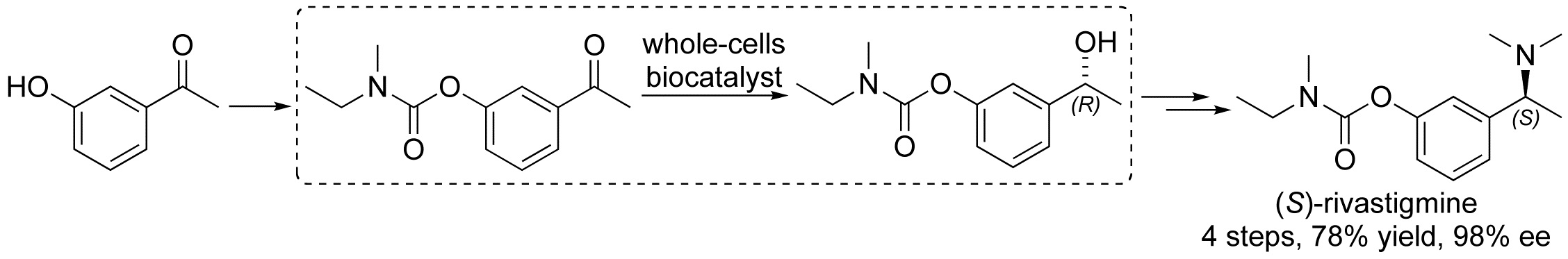
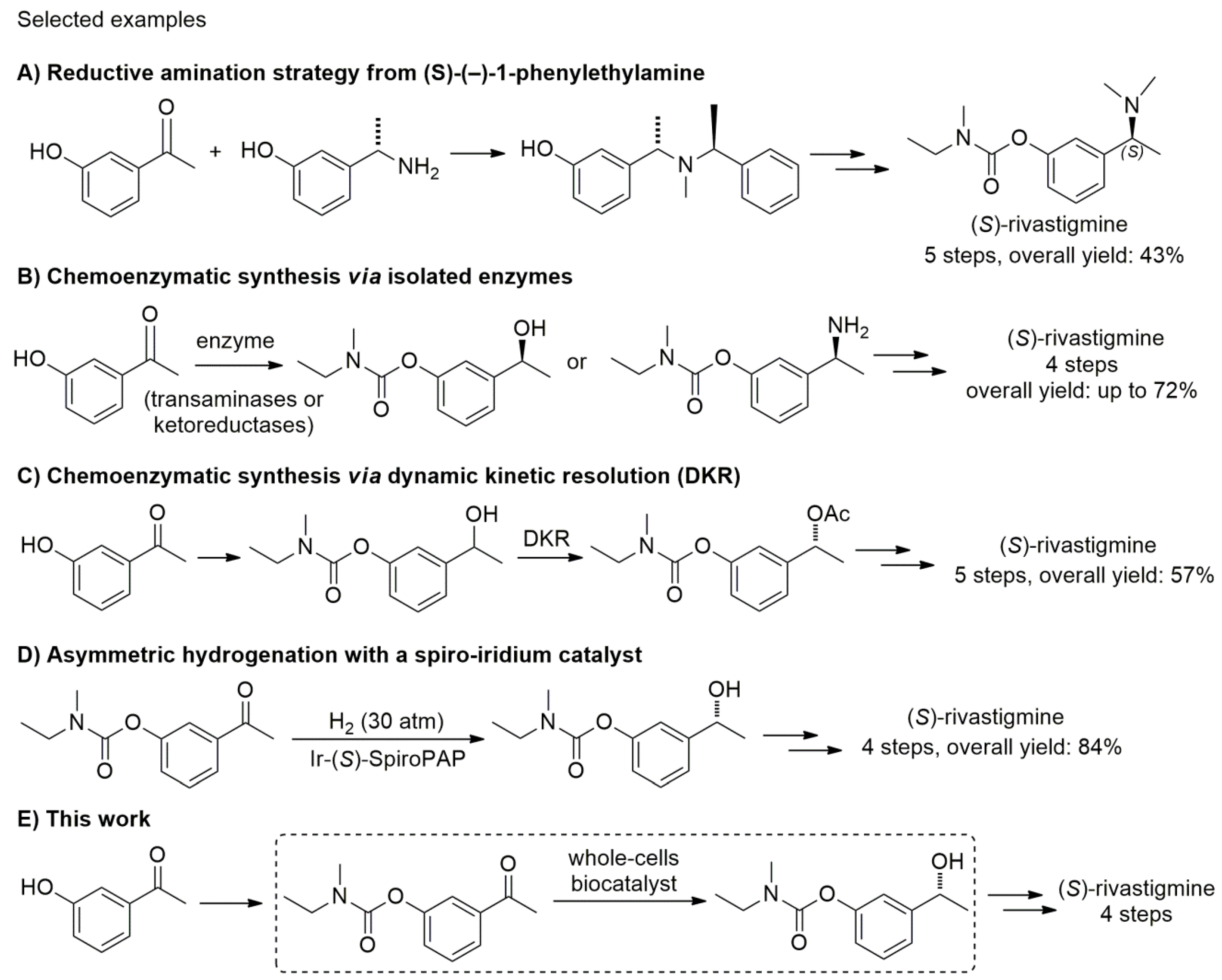
Building on our prior studies in using whole-cell biocatalysts (yeasts [26,27,28,29] and bacteria [30]) for the preparation of enantioenriched secondary alcohols by stereoselective bioreduction of prochiral ketones, we envisaged to apply such methodologies to the total synthesis of an important API (active pharmaceutical ingredient): (S)-rivastigmine (Scheme 1). This is a blockbuster drug extensively used for the treatment of mild to moderate dementia in patients with Alzheimer’s disease and Parkinson’s disease because of its dual inhibition of both acetylcholinesterase and butyrylcholinestrase [31,32]. Recent published methodologies of synthesis of (S)-rivastigmine focused on (i) multistep processes with chiral resolving agents or reductive amination strategies starting from enantiopure amines, however, with low overall yields (up to 43%) (Scheme 1A) [33,34,35,36]; (ii) enzyme-mediated reactions (lipases, transaminases, and ketoreductases) (Scheme 1B) [37,38,39,40,41,42], or dynamic kinetic resolution (DKR) using a polymer-bound ruthenium complex and a lipase as a key step (Scheme 1C) [43]; and (iii) the direct asymmetric reduction of carbonyl derivatives with a chiral spiro-iridium catalyst (Ir-SpiroPAP) (Scheme 1D) [44].
Herein we wish to report a particularly efficient four-step chemoenzymatic total synthesis of (S)-rivastigmine [overall yield: 78%; enantiomeric excess (ee): 98%] based on a highly stereoselective bioreduction of a key prochiral carbonyl derivative mediated by Lactobacillus reuteri DSM 20016 whole cells (Scheme 1E). The biocatalytic performance of such a microorganism, compared to the use of baker’s yeast in DESs as unconventional reaction media, is discussed as well.
2. Results and Discussion
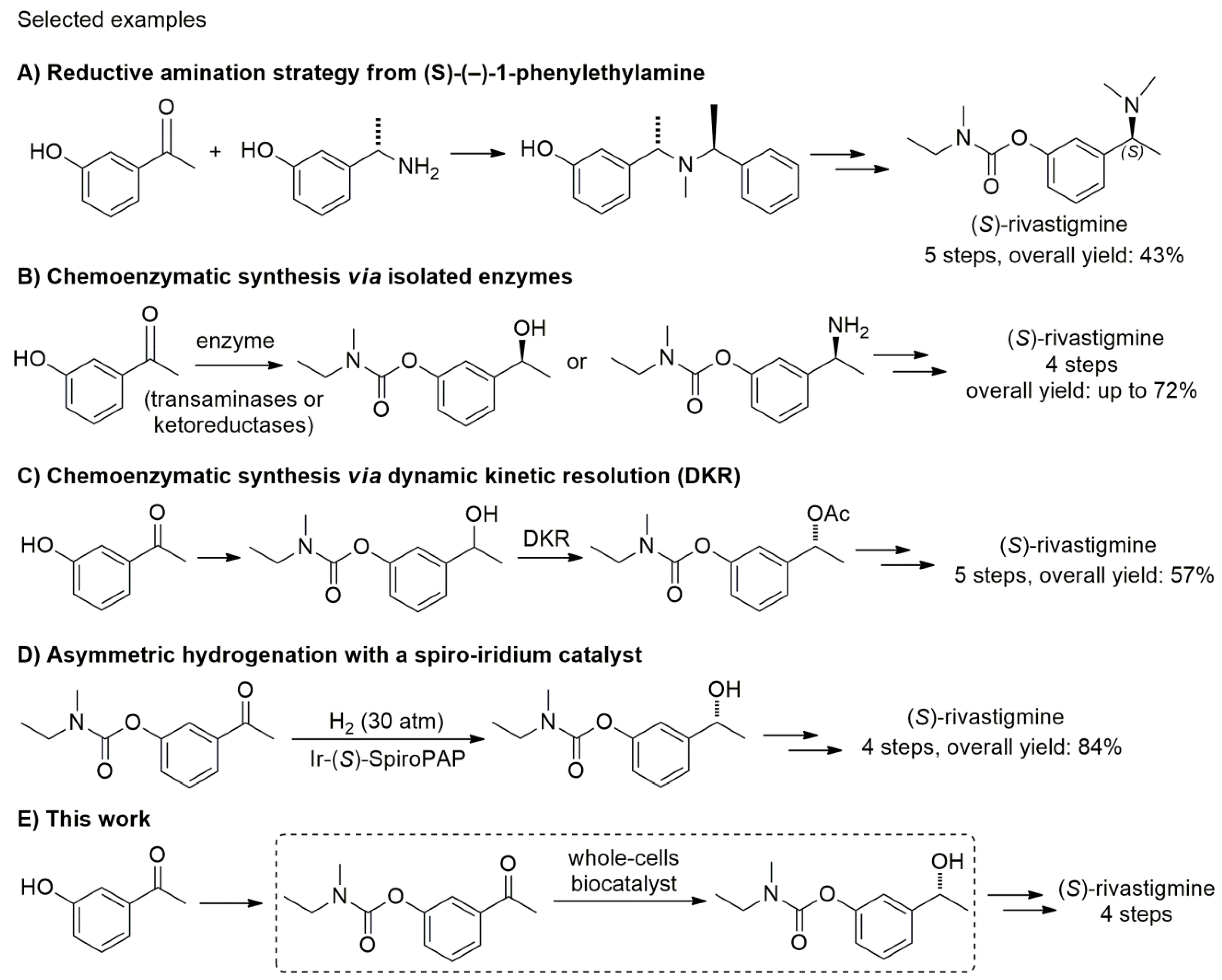
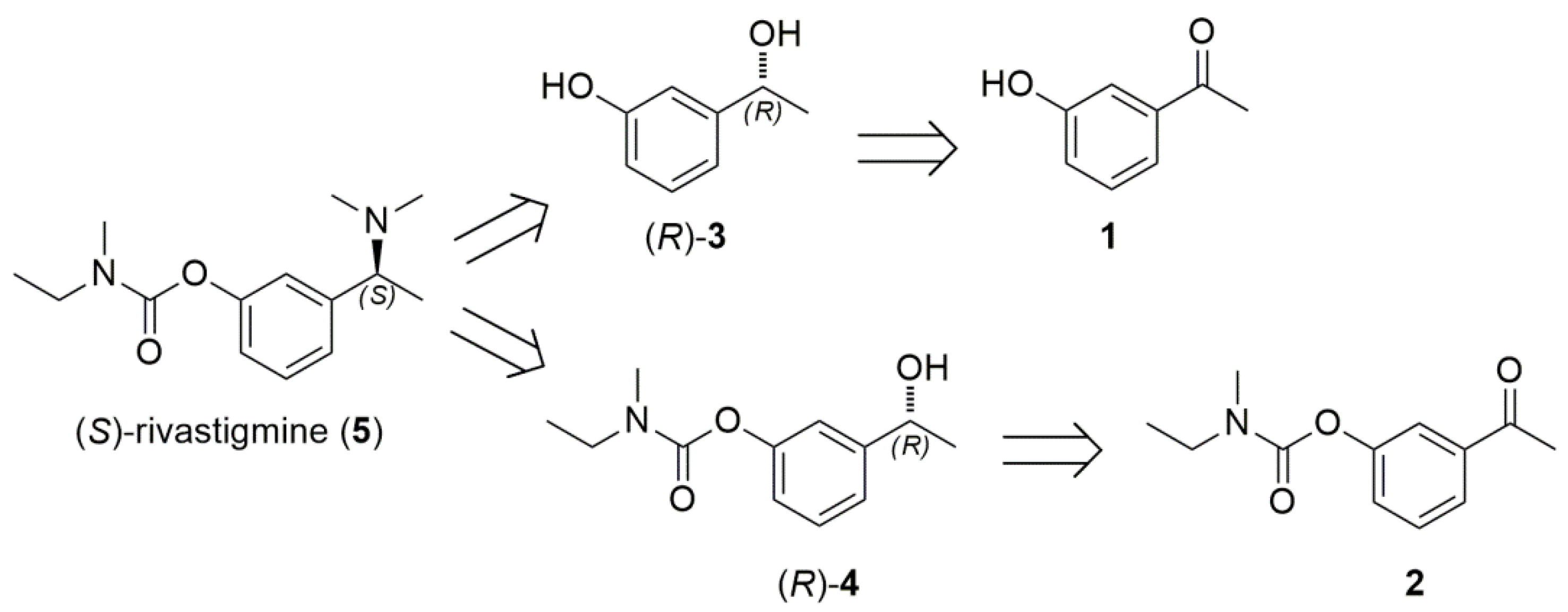
As different alcohol dehydrogenases (ADHs) with different stereopreference and substrate specificity can be expressed in various microorganisms, also by varying metabolic growth conditions and phase [26,27,28,29,30], we envisaged the whole-cell bioreduction of prochiral aryl ketone 1 or 2 as the key step in obtaining the appropriate precursor (R)-3 or (R)-4, useful for the synthesis of the target eutomer of (S)-rivastigmine (5) (Scheme 2). In order to establish the feasibility of this strategy, we explored different experimental conditions in water and in DES-water mixtures using baker’s yeast resting cells (RC) or Lactobacillus reuteri DSM 20016 whole cells. A set of representative results are summarized in Table 1 and Table 2.
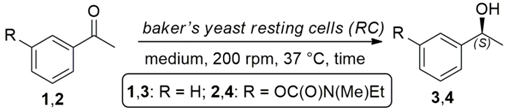
2.1. Bioreduction of Prochiral Ketones 1 and 2 with Baker’s Yeast RC
After incubating a mixture of ketone 1 and baker’s yeast RC (0.3 g/L) in tap water at 37 °C for 24 h, the expected alcohol 3 was obtained in a very low yield (less than 10%) (Table 1, entries 1,2). Under the same experimental conditions and by increasing the incubation time to up to 40 h, alcohol 3 could now be isolated in 25% yield, and with an enantiomeric excess (ee) >98% in favor of the S-enantiomer (Table 1, entry 3).
A prolonged reaction time to up to 120 h, however, did not affect the chemical yield of 3, while a slight erosion of the ee (98%) was detected (Table 1, entry 4). The replacement of water by neat ChCl/D-fructose (3:2 w/w), ChCl/glycerol (Gly) (1:2 mol/mol), and L-(+)-lactic acid/ChCl (2:1 mol/mol) eutectic mixtures did not lead to any conversion of 1 to up to 120 h of incubation (Table 1, entries 5–7). The same occurred after addition of up to 10 w% of water (Table 1, entries 8–10). On the other hand, the employment of a ChCl/Gly eutectic mixture with increasing percentages of water provided 3 with yields up to 32% and an ee >98%, however, still in favor of the S-enantiomer (Table 1, entries 11,12). Thus, an inversion of stereopreference promoted by baker’s yeast was not observed compared to the bioreduction carried out in pure water (Table 1, entry 4) [25]. As for ketone 2, by running its bioreduction in pure water, the expected alcohol 4 could be isolated in only 12% yield after 24 h incubation, although enantiomerically enriched (ee >98%) in favor of the S-enantiomer (Table 1, entry 13), whereas an erosion of its ee (95%) was detected after 120 h of incubation in a ChCl/Gly-based eutectic mixture with 20 w% of water (Table 1, entry 14).
Thus, the required R-enantioselectivity for alcohols 3 and 4 necessary for the synthesis of (S)-5 in the final steps, according to the retrosynthetic approach depicted in Scheme 2, was never observed, most probably because of a lower affinity of the starting prochiral ketones 1 and 2 towards R-ADHs, the bioreduction promoted by S-ADHs always taking place at a higher reaction rate in pure water.
2.2. Bioreduction of Ketones 1 and 2 with Lactobacillus reuteri DSM 20016 Whole Cells
We then investigated the stereochemical outcome of the bioreduction of ketones 1 and 2 using Lactobacillus reuteri DSM 20016 whole cells as biocatalyst (Table 2). Pleasingly, when 1.1 g cell dry weight (CDW)/L of Lactobacillus reuteri RC were incubated anaerobically for 24 h in a phosphate buffered saline (PBS) supplemented with 1% glucose at 37 °C in the presence of 1 g/L of ketone 2 in a shake flask with orbital shaking, alcohol 4 was isolated in 60% yield and with an ee of 98% in favor of the R-enantiomer (Table 2, entry 1). A less efficient conversion (47%) was instead obtained after 24 h by using growing cells (GC) of Lactobacillus reuteri DSM 20016 strain, inoculated at the initial density of 1.1 g CDW/L in MRS (De Man, Rogosa and Sharpe medium) medium [45] together with 1 g/L of ketone 2 (Table 2, entry 2).
By prolonging the incubation of ketone 2 in PBS supplemented with 1% glucose with Lactobacillus reuteri RC, alcohol 4 could now be smoothly recovered in 64 and 74% yield after 48 and 72 h, respectively, the ee always being 98% (Table 2, entries 3,4). Under the same experimental conditions, however, the bioreduction of ketone 1 provided alcohol (R)-3 only in 16% yield after 72 h although with an ee >98% (Table 2, entry 5). Further optimization of the reaction conditions focused on using a twin bioreactor system.
The biotransformation performance (yield and enantioselectivity) was experimentally verified on a gram scale, under controlled conditions (200 rpm, 37 °C, anaerobiosis, 1.1 g CDW/L), in three parallel reactors: a shake-flask in an orbital shaker (Table 2, entry 6) and two bioreactors (Table 2, entries 7 and 8), equipped with in situ pH and pO2 electrodes, temperature sensors and stirring speed control. In one vessel (Table 2, entry 7), pH was maintained at 7.0 by the addition of a NaOH solution (0.1 M), whereas the parallel experiment (Table 2, entry 8) was performed in the absence of an active pH control to evaluate the effect of the acidification induced by the metabolism of Lactobacillus reuteri cells on the conversion rate and on the ee of the product. After 72 h of incubation of ketone 2 with Lactobacillus reuteri DSM 20016 (RC), appreciable and very similar amounts of alcohol (R)-4 (63–71%) were detected in all the experiments, with an ee to up to 98%, regardless of the kind of shaking (orbital or mechanical) or pH control. This suggests that other factors, such as the toxicity of the substrate and the product, may limit the yield of conversion. In order to understand if the biotransformation performance could be improved by increasing the amount of the biocatalyst, we carried out the bioreduction of 2 with Lactobacillus reuteri DSM 20016 whole cells using a fourfold higher cell concentration (4.4 g CDW/L). Under these conditions, we reached the best conversion after only 24 h of incubation in anaerobiosis at 37 °C, in PBS and 1% glucose, in a shake flask with orbital shaking: enantiomerically enriched alcohol (R)-4 was isolated in 90% yield and in 98% ee by means of a clean extraction process (Table 2, entry 9). As for bioreductions with Lactobacillus reuteri DSM 20016 run in eutectic mixtures, preliminary experiments performed in carbohydrate-based DESs led to complete recovery of the acetophenone derivative as starting material. Glycerol, on the other hand, is known to be metabolized by the above microorganism into 1,3-propanediol as the final product [46].
2.3. Chemoenzymatic Total Synthesis of (S)-Rivastigmine
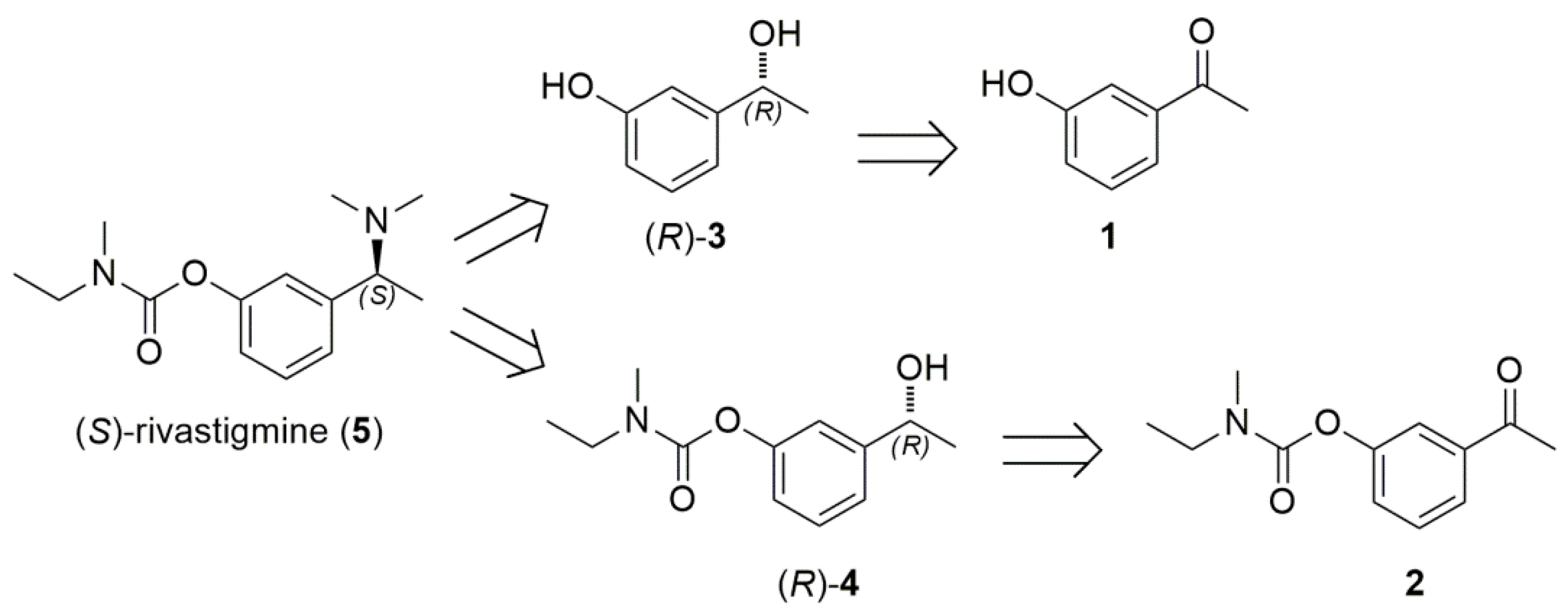
With the optimal reaction conditions in hand, the total chemoenzymatic synthesis of (S)-5 was then performed in four steps. Carbamate 2 was obtained from the commercially available 3-hydroxyacetophenone (1) in 91% yield according to reported procedures (Scheme 3) [43,44]. Then, it was subjected to an enantioselective biocatalytic reduction by Lactobacillus reuteri DSM 20016 whole cells into alcohol (R)-4, isolated in 90% yield and 98% ee. Finally, the transformation of (R)-4 into (S)-5 was carried out in a one-pot two-step process by first converting (R)-4 into the corresponding mesylate, and then by exploiting a stereospecific nucleophilic substitution with a solution of Me2NH in THF, which took place with inversion of configuration at the chirality center [43], thereby providing (S)-5 in 95% yield and 98% ee (Scheme 3).
3. Materials and Methods
3.1. General Methods
Melting points were taken on an electrothermal apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker 600 MHz spectrometer and chemical shifts are reported in parts per million (δ). Absolute values of the coupling constants are reported. FT-IR spectra were recorded on a Perkin-Elmer 681 spectrometer. GC analyses were performed on a HP 6890 model, Series II by using a HP1 column (methyl siloxane; 30 m × 0.32 mm × 0.25 μm film thickness). Analytical thin-layer chromatography (TLC) was carried out on pre-coated 0.25 mm thick plates of Kieselgel 60 F254; visualisation was accomplished by UV light (254 nm) or by spraying a solution of 5% (w/v) ammonium molybdate and 0.2% (w/v) cerium (III) sulfate in 100 mL 17.6% (w/v) aq. sulfuric acid and heating to 473 K until blue spots appeared (visualisation of alcohols 3 and 4). Chromatography was carried out by using silica gel 60 with a particle size distribution 40–63 μm and 230–400 ASTM. GC-MS analyses were performed on HP 5995C model and elemental analyses on an Elemental Analyzer 1106-Carlo Erba-instrument. MS-ESI analyses were performed on Agilent 1100 LC/MSD trap system VL. Optical rotation values were measured at 25 °C using a Perkin Elmer 341 polarimeter with a cell of 1 dm path length; the concentration (c) is expressed in g/100 mL. The enantiomeric excess (ee) was determined by HPLC analysis using a Phenomenex LUX Cellulose-1 [Cellulose tris(3,5-dimethylphenylcarbamate)] column (250 × 4.6 mm). Cell density was determinated by optical density at 620 nm (OD620) with a Genesys TM 20 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Gram-scale reaction were performed in a bioreactor (Sartorius Biostat B plus Twin System, Göttingen, Germany) equipped with in situ pH and pO2 electrodes, a temperature sensor, stirring speed and gas-flow ratio controllers.
Deep eutectic solvents ChCl/Gly (1:2 mol/mol), ChCl/D-fructose (3:2 w/w), and L-(+)-lactic acid/ChCl (2:1 mol/mol) were prepared by gently heating under stirring at 60–80 °C for 5 min the corresponding individual components until a clear solution was obtained [25]. All the chemicals and solvents were of commercial grade, further purified by distillation or crystallization prior to use. The progress of the reactions was monitored by TLC and/or GC. The racemic alcohols 3, 4 were prepared by NaBH4 reduction in EtOH in 91–95% yields [26,27,28,29,30]. The optically active alcohols 3 and 4, obtained from baker’s yeast or Lactobacillus reuteri bioreductions, had analytical and spectroscopic data identical to the previously synthesized or commercially available racemic compounds. All the reported reactions were repeated at least twice without any noticeable change in the results.
3-(1-Hydroxyethyl)phenol (3) [47]. (5–32% yield); Rf 0.30 (2:8 EtOAc:hexane); M.p. 118−119 °C; (S)-4 (from baker’s yeast): [α]D20 = −36.5 (c 1.0, CH3OH); er >99:1 (HPLC LUX Cellulose-1, hexane/IPA = 90:10, 1 mL/min, λ = 210 nm); (R)-isomer: 20.1 min, (S)-isomer: 22.2 min; νmax/cm−1 3382, 1617, 1590, 1481, 1407, 1372, 1294, 1269, 1168, 1085, 1070, 1009, 997, 939, 868, 787, 736, 699, 626. δH (600 MHz, CDCl3) 7.23–7.20 (m, 1H, ArH), 6.93–6.92 (m, 1H), 6.89–6.87 (m, 1H), 6.75–6.73 (m, 1H), 4.87 (q, J = 6.4 Hz, 1H), 1.63–1.58 (br s, 1H, OH, exchanges with D2O), 1.49 (d, J = 6.4 Hz, 3H). δC (150 MHz, CDCl3): 159.0, 149.8, 131.0, 118.4, 115.6, 113.9, 71.4, 26.2. HRMS (ESI−) calcd for [M – H, C8H9O2]−: 137.0603, Found 137.0608.
N-Ethyl-N-methyl-O-[3-(1-hydroxyethyl)phenyl]carbamate (4) [44,48]. (5–90% yield); Rf 0.30 (4:6 EtOAc:hexane); (R)-4 from Lactobacillus reuteri: [α]D20 = +23.8 (c 1.1, CHCl3); ee = 98% (HPLC LUX Cellulose-1, hexane/IPA = 90:10, 1 mL/min, λ = 210 nm); (S)-isomer: 11.4 min, (R)-isomer: 12.6 min; νmax/cm−1 3422, 2971, 2927, 2874, 1706, 1475, 1432, 1400, 1303, 1286, 1261, 1229, 1169, 1086, 956, 872, 784, 757, 697. δH (600 MHz, CDCl3) 7.33–7.26 (m, 1H, ArH), 7.18–7.16 (m, 1H, ArH), 7.15–7.11 (m, 1H, ArH), 7.03–6.98 (m, 1H), 4.87 (q, J = 6.4 Hz, 1H), 3.51–3.44 (m, 1H, NCH2 of one rotamer), 3.43–3.36 (m, 1H, NCH2 of one rotamer), 3.06 (s, 1.5H, NCH3 of one rotamer), 2.98 (s, 1.5H, NCH3 of one rotamer), 1.47 (d, J = 6.4 Hz, 3H), 1.25–1.23 (m, 1.5H, NCH3 of one rotamer), 1.19–1.17 (m, 1.5H, NCH3 of one rotamer). δC (150 MHz, CDCl3): 154.6 (rotamer), 154.4 (rotamer), 151.6, 147.5, 147.4, 129.2, 122.1, 120.6, 118.8, 69.9, 44.0, 34.2 (rotamer), 33.8 (rotamer), 25.0, 13.2 (rotamer), 12.4 (rotamer). GC-MS (70 eV) m/z (rel. int.): 223 (M+, 12), 86 (100), 58 (45). HRMS (ESI−) calcd for [M – H, C12H16NO3]−: 222.1130, Found 222.1134.
3-[1-(Dimethylamino)ethyl]phenyl-N-ethyl-N-methylcarbamate, (S)-5 (rivastigmine) [43,44]. (95% yield); Rf 0.35 (1:1 EtOAc:hexane); (S)-5 [α]D20 = −34.5 (c 1.0, CHCl3); ee = 98% (HPLC LUX Cellulose-1, hexane/IPA = 99:1, 0.7 mL/min, λ = 210 nm); (R)-isomer: 30.2 min, (S)-isomer: 31.8 min. δH (600 MHz, CDCl3) 7.29–7.26 (m, 1H, ArH), 7.12–7.10 (m, 1H, ArH), 7.07–7.05 (m, 1H), 7.01–6.97 (m, 1H, ArH), 3.48–3.43 (m, 1H, NCH2 of one rotamer), 3.42–3.36 (m, 1H, NCH2 of one rotamer), 3.24 (q, J = 6.4 Hz, 1H, NCHCH3), 3.04 (s, 1.5H, NCH3 of one rotamer), 2.97 (s, 1.5H, NCH3 of one rotamer), 2.19 (s, 6H), 1.35 (d, J = 6.7 Hz, 3H), 1.23–1.21 (m, 1.5H, NCH3 of one rotamer), 1.18–1.16 (m, 1.5H, NCH3 of one rotamer). δC (150 MHz, CDCl3): 154.4, 151.5, 145.5, 128. 9, 124.3, 120.8, 120.3, 65.6, 44.0, 43.1, 34.2, 33.8, 20.0, 13.2, 12.5.
3.2. Microorganisms and Media
Lactobacillus reuteri DSM 20016 was obtained from DSMZ culture collection (Braunschweig, Germany). Cells were maintained at −80 °C in culture broth supplemented with 25% (w/v) glycerol. Cultures were carried out in MRS medium [45] containing: 20g/L glucose, 10 g/L peptone, 8 g/L meat extract, 4 g/yeast extract, 1 g/L Tween 80, 2 g/L di-potassium hydrogen phosphate, 5 g/L sodium cetate∙3H2O, 2 g/L tri-ammonium citrate, 0.2 g/L of magnesium sulfate∙7H2O, 0.05 g/L manganese sulfate∙2H2O. Cells were incubated at 37 °C for 24 h, statically. Cell density was monitored using optical density at 620 nm (OD620) with a Genesys TM 20 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Baker’s yeast was commercialized by LESAFFRE Italia Spa as fresh yeast in cubes (Lievital, LESAFFRE ITALIA S.p.A, Sissa Trecasali (PR), Italy) [49].
3.3. Blank Experiments
Ketone 1 or 2 (40 mg) was added to a 100 mL flask containing 40 mL of the culture medium, and stirred at 37 °C on an orbital shaker at 200 rpm. The content of the flask was extracted with Et2O and analyzed. After 24 h, no reaction took place, as resulted by NMR analysis.
3.4. Bioreduction of Acetophenones 1,2 by Using Baker’s Yeat Cells in Acqueous DESs Mixtures (the General Procedure Described Refers to a ChCl/Gly (1:2 mol/mol) + 10 w% Water Mixture)
The eutectic mixture was prepared by gently heating and stirring at 60 °C for 5 min the corresponding individual components (28.5 g of Gly and 21.5 g of ChCl kept in an Erlenmeyer flask) until a clear solution was obtained. Then, water (5 mL (10% w/w)) was added to the DES kept at 37 °C, and the baker’s yeast [25] (12.5 g) was dispersed to give a smooth paste in the mixture. Ketone 1 or 2 (50 mg) was added, and the mixture was stirred at 37 °C in an orbital shaker (250 rpm), monitoring the reaction progress by 1H NMR. After a fixed time (Table 1), the reaction was stopped by the addition of EtOAc followed by centrifugation, decantation, and extraction with EtOAc. The extracts were dried over anhydrous Na2SO4, and the solvent evaporated under reduced pressure. The residue was purified by silica gel column chromatography using hexane/EtOAc (10:1) as the eluent to yield the desired alcohol 3 or 4. Absolute configurations of secondary alcohols 3 and 4 obtained from the bioprocess were determined by comparison of their optical specific rotation signs and/or retention times with known data (see Section 3.1).
3.5. Bioreduction of Ketone 2 by Using Lactobacillus reuteri Growing Cells (Table 2, Entry 2)
Lactobacillus reuteri DSM 20016 was grown overnight in MRS medium [45], as described in Section 3.2. The cells were inoculated at OD620 = 0.05 in flasks containing 50 mL of MRS to which the ketone 2 was added at the final concentration of 1 g/L. To ensure the anaerobic conditions, flasks were degassed with N2 for 5 min. The reaction mixture was incubated at 37 °C, 200 rpm for 24 h. Then, the suspension was centrifuged (2800× g, 10 min, 4 °C), and the aqueous phase was extracted with EtOAc (3 × 15 mL). The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure. The product was isolated by silica gel column chromatography using hexane and EtOAc (10:1) as the eluents to yield the desired alcohol 4 (Table 2, entry 2).
3.6. Bioreduction of Acetophenones 1,2 by Using Lactobacillus reuteri Resting Cells: General Procedure (Table 2)
Lactobacillus reuteri DSM 20016 was grown overnight in MRS medium. This pre-culture was inoculated in 50 mL of MRS and incubated for 24 h (37 °C). Cells were harvested by centrifugation (2800× g, 10 min) and washed twice with phosphate buffered saline pH 7.4 (PBS, Sigma-Aldrich, St. Louis, MO, USA). Then, the cells were suspended in the same buffer and adjusted for cell density (1.1 or 4.4 g CDW/L). To this cell suspension, 1% glucose and the desired concentration of arylketone 1 or 2 were added (total volume 50 mL). To ensure the anaerobic conditions, flasks were degassed with a N2 flux for 3 min. The reaction mixture was incubated at 37 °C, 200 rpm. After the appropriate conversion time (Table 2), the suspension was centrifuged (2800× g, 10 min, 4 °C), and the aqueous phase was extracted with EtOAc (3 × 15 mL). The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure. The product was isolated by silica gel column chromatography using hexane and EtOAc (10:1) as the eluents to yield the desired alcohol 3 or 4 (Table 2). Absolute configurations of alcohols 3 and 4 obtained from the bioprocess were determined by comparison of their optical specific rotation signs and/or retention times with known data (see Section 3.1).
3.7. Bioreduction of Acetophenones 1,2 by Using Lactobacillus reuteri Resting Cells in Bioreactor: General Batch Procedure (Table 2, Entries 7,8)
Lactobacillus reuteri DSM 20016 was grown overnight in MRS medium. 50 mL of this pre-culture were inoculated into 1.4 L of MRS and incubated for 24 h at 37 °C. The cell suspension was centrifuged at 4000 rpm for 10 min and the harvested cells were re-suspended in a 2 L stirred tank bioreactor (Sartorius Biostat B plus Twin System, Germany) containing 0.725 L of sterile PBS supplemented with 1% glucose, at an initial optical density of 2.5 O.D. (corresponding to 1.1 g CDW). To this cell suspension, arylketone 1 or 2 was added, monitoring the reaction progress by 1H NMR. The bioreactors were equipped with in situ pH and pO2 electrodes, a temperature sensor, stirring speed and gas-flow ratio controllers. Cells were cultured under the following conditions: temperature was maintained at 37 °C; pH 7.5; when requested, pH was automatically controlled by additions of NaOH 0.1 M (entry 7); stirring speed was maintained at 200 rpm; anaerobic conditions were maintained by continuous injection of a stream of pure sterile N2 at 50 mL/min through a sparger placed at the bottom of the bioreactor. At the end of biotrasformation, bacterial cultures were centrifuged at 4000 rpm for 10 min, and the aqueous solution was extracted with EtOAc (5 × 100 mL). The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure. The product was isolated by silica gel column chromatography using hexane and EtOAc (10:1) as the eluents to yield alcohols 3 or 4 (Table 2).
4. Conclusions
In summary, a novel, whole-cell-based chemoenzymatic total synthesis of (S)-rivastigmine (5) (overall yield: 78%, ee: 98%) has been developed. The key step is the biocatalytic reduction of 3-acetylphenyl-N-ethyl-N-methylcarbamate (2) (obtained in 91% yield by commercially available 3-hydroxyacetophenone (1)) by Lactobacillus reuteri DSM 20016 whole cells to the corresponding secondary alcohol (R)-4 (90% yield; 98% ee; PBS as the reaction medium, 24 h, 37 °C). The latter is finally converted into the target drug (S)-5 following a straightforward stereospecific two-step mesylation-amine nucleophilic substitution route. This simple, cheap, environmentally friendly and easy-to-make procedure is highly competitive with recently reported multistep approaches based on chiral resolving agents, dynamic resolution processes, or asymmetric hydrogenation reactions which make use of expensive chiral catalysts. It is worth underlying that the described procedure has also been performed on a gram scale, and is valuable for setting up a large-scale process production of (S)-5, as the preparation of Lactobacillus reuteri DSM 20016 whole cells is very simple and inexpensive. Moreover, these cells require as a reducing equivalents source just glucose or cheaper alternatives such as spruce lignocelluloses hydrolysates or cheese whey [30,46]. Baker’s yeast (RC) has also been investigated as an alternative whole-cell biocatalyst. The bioredution of key carbonyl derivatives 1 and 2, run both in water and in different aqueous eutectic mixtures, did indeed provide the corresponding secondary alcohols 3 and 4 with ee >98% (conversion up to 32%), but with stereochemistry opposite (that is S) to that necessary for the preparation of the optically pure eutomer of rivastigmine. Biocatalytic processes have become increasingly demanding in preparative chemistry in recent years as green methodologies. They, indeed, allow highly chemo-, regio-, and stereoselective transformations to be carried out under very mild reaction conditions, and are thus a valid alternative to all those approaches in asymmetric synthesis relying on expensive (organo)catalysts, chiral ligands and transition metal complexes, and that still make use of hazardous chemicals and toxic, volatile organic compounds.
Acknowledgments
This work was financially supported by the University of Bari within the framework of the Project “Sviluppo di nuove metodologie di sintesi mediante l’impiego di biocatalizzatori e solventi a basso impatto ambientale” (code: Perna01333214Ricat), and by both CIRCC and C.I.N.M.P.I.S. (project code: COCM8470P0) consortia. This work was partially supported also by the “Reti di Laboratori—Produzione Integrata di Energia da Fonti Rinnovabili nel Sistema Agroindustriale Regionale” program funded by the “Apulia Region Project Code 01”. (Intervento cofinanziato dall’Accordo di Programma Quadro in materia di Ricerca Scientifica—II Atto Integrativo—PO FESR 2007–2013, Asse I, Linea 1.2-PO FSE 2007–2013 Asse IV “Investiamo nel vostro futuro”.
Author Contributions
Paola Vitale and Gennaro Agrimi conceived and designed the experiments; Francesco Mirizzi, Roberto Vito Capobianco and Isabella Pisano performed the experiments; Paola Vitale, Filippo Maria Perna, Gennaro Agrimi and Isabella Pisano analyzed the data; Vito Capriati, Filippo Maria Perna and Gennaro Agrimi contributed reagents/materials/analysis tools; Vito Capriati and Paola Vitale wrote the paper.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Faber, K. Biotransformations. In Organic Chemistry: A Textbook, 6th ed.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Simon, R.C.; Mutti, F.G.; Kroutil, W. Biocatalytic synthesis of enantiopure building blocks for pharmaceuticals. Drug Discov. Today Technol. 2013, 10, e37–e44. [Google Scholar] [CrossRef] [PubMed]
- Rozzell, D. Tabular Survey of Available Enzymes. In Enzyme Catalysis in Organic Synthesis, 3rd ed.; Drauz, K., Gröger, H., May, O., Eds.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2012; pp. 1847–1938. [Google Scholar]
- Pscheidt, B.; Glieder, A. Yeast cell factories for fine chemical and API production. Microb. Cell Fact. 2008, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.A.R.; Moran, P.J.S.; Conceiçao, G.J.A.; Fardelone, L.C. Recent Advances in the Biocatalytic Asymmetric Reduction of Acetophenones and α,β-Unsaturated Carbonyl Compounds. Food Technol. Biotechnol. 2004, 42, 295–303. [Google Scholar]
- Csuk, R.; Glaenzer, B.I. Baker’s yeast mediated transformations in organic chemistry. Chem. Rev. 1991, 91, 49–97. [Google Scholar] [CrossRef]
- Lamed, R.J.; Keinan, E.; Zeikus, J.G. Potential applications of an alcohol-aldehyde/ketone oxidoreductase from thermophilic bacteria. Enzym. Microb. Technol. 1981, 3, 144–148. [Google Scholar] [CrossRef]
- Pàmies, O.; Bäckvall, J.-E. Combination of Enzymes and Metal Catalysts. A Powerful Approach in Asymmetric Catalysis. Chem. Rev. 2003, 103, 3247–3262. [Google Scholar] [CrossRef] [PubMed]
- García-Urdiales, E.; Rebolledo, F.; Gotor, V. Study of the enantioselectivity of the CAL-B-catalysed transesterification of α-substituted α-propylmethanols and α-substituted benzyl alcohols. Tetrahedron Asymmetry 2001, 12, 3047–3052. [Google Scholar] [CrossRef]
- Francisco, M.; van den Bruinhorst, A.; Kroon, M.C. Low-transition-temperature mixtures (LTTMs): A new generation of designer solvents. Angew. Chem. Int. Ed. 2013, 52, 3074–3085. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Hao, J.-W.; Mo, L.-P.; Zhang, Z.-H. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F.M. Stereoselective organocatalysed reactions in deep eutectic solvents: Highly tunable and biorenewable reaction media for sustainable organic synthesis. Green Chem. 2016, 18, 792–797. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Hevia, E.; Capriati, V. Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities. Eur. J. Org. Chem. 2015, 6779–6799. [Google Scholar] [CrossRef] [Green Version]
- Vidal, C.; García-Álvarez, J.; Hernán-Gómez, A.; Kennedy, A.R.; Hevia, E. Exploiting Deep Eutectic Solvents and Organolithium Reagent Partnerships: Chemoselective Ultrafast Addition to Imines and Quinolines Under Aerobic Ambient Temperature Conditions. Angew. Chem. Int. Ed. 2016, 55, 16145–16148. [Google Scholar] [CrossRef] [PubMed]
- Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F.M.; Salomone, A.; Florio, S.; Capriati, V. Water opens the door to organolithiums and Grignard reagents: Exploring and comparing the reactivity of highly polar organometallic compounds in unconventional reaction media towards the synthesis of tetrahydrofurans. Chem. Sci. 2016, 7, 1192–1199. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Deep Eutectic Solvents: The Organic Reaction Medium of the Century. Eur. J. Org. Chem. 2016, 612–632. [Google Scholar] [CrossRef]
- Xu, P.; Zheng, G.W.; Zong, M.H.; Li, N.; Lou, W.Y. Recent progress on deep eutectic solvents in biocatalysis. Bioresour. Bioprocess. 2017, 4. [Google Scholar] [CrossRef]
- Lan, D.; Wang, X.; Zhou, P.; Hollmann, F.; Wang, Y. Deep eutectic solvents as performance additives in biphasic reactions. RSC Adv. 2017, 7, 40367–40370. [Google Scholar] [CrossRef]
- Sheldon, R.A. Biocatalysis and Biomass Conversion in Alternative Reaction Media. Chem. Eur. J. 2016, 22, 12984–12999. [Google Scholar] [CrossRef] [PubMed]
- Durand, E.; Lecomte, J.; Villeneuve, P. Deep eutectic solvents: Synthesis, application, and focus on lipase-catalyzed reactions. Eur. J. Lipid Technol. 2013, 115, 379–385. [Google Scholar] [CrossRef]
- Mbous, Y.P.; Hayyan, M.; Wong, W.F.; Looi, C.Y.; Hashim, M.A. Unraveling the cytotoxicity and metabolic pathways of binary natural deep eutectic solvent systems. Sci. Rep. 2017, 7, 41257. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, Z.; Domínguez de María, P. Whole-Cell Biocatalysis in Deep-Eutectic-Solvents/Aqueous Mixtures. ChemCatChem 2014, 6, 1535–1537. [Google Scholar] [CrossRef]
- Bubalo, M.C.; Mazur, M.; Radosevic, K.; Redovnikovic, I.R. Baker’s yeast-mediated asymmetric reduction of ethyl 3-oxobutanoate in deep eutectic solvents. Process Biochem. 2015, 50, 1788–1792. [Google Scholar] [CrossRef]
- Vitale, P.; Abbinante, V.M.; Perna, F.M.; Salomone, A.; Cardellicchio, C.; Capriati, V. Unveiling the Hidden Performance of Whole Cells in the Asymmetric Bioreduction of Aryl-containing Ketones in Aqueous Deep Eutectic Solvents. Adv. Synth. Catal. 2017, 359, 1049–1057. [Google Scholar] [CrossRef]
- Vitale, P.; Perna, F.M.; Perrone, M.G.; Scilimati, A. Screening on the use of Kluyveromyces marxianus CBS 6556 growing cells as enantioselective biocatalysts for ketone reductions. Tetrahedron: Asymmetry 2011, 22, 1985–1993. [Google Scholar] [CrossRef]
- Vitale, P.; D’Introno, C.; Perna, F.M.; Perrone, M.G.; Scilimati, A. Kluyveromyces marxianus CBS 6556 growing cells as a new biocatalyst in the asymmetric reduction of substituted acetophenones. Tetrahedron Asymmetry 2013, 24, 389–394. [Google Scholar] [CrossRef]
- Vitale, P.; Perna, F.M.; Agrimi, G.; Scilimati, A.; Salomone, A.; Cardellicchio, C.; Capriati, V. Asymmetric chemoenzymatic synthesis of 1,3-diols and 2,4-disubstituted aryloxetanes by using whole cell biocatalysts. Org. Biomol. Chem. 2016, 14, 11438–11445. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Digeo, A.; Perna, F.M.; Agrimi, G.; Salomone, A.; Scilimati, A.; Cardellicchio, C.; Capriati, V. Stereoselective Chemoenzymatic Synthesis of Optically Active Aryl-Substituted Oxygen-Containing Heterocycles. Catalysts 2017, 7, 37. [Google Scholar] [CrossRef]
- Perna, F.M.; Ricci, M.A.; Scilimati, A.; Mena, M.C.; Pisano, I.; Palmieri, L.; Agrimi, G.; Vitale, P. Cheap and environmentally sustainable stereoselective arylketones reduction by Lactobacillus reuteri whole cells. J. Mol. Catal. B Enzym. 2016, 124, 29–37. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Bhanja, C.; Jena, S. Synthesis Design of ‘Rivastigmine’—A Potent Therapeutic Agent for Alzheimer’s disease using Retrosynthetic Analysis. J. Chem. Pharm. Res. 2012, 4, 3171–3183. [Google Scholar]
- Hu, M.; Zhang, F.-L.; Xie, M.-H. Novel Convenient Synthesis of Rivastigmine. Synth. Commun. 2009, 39, 1527–1533. [Google Scholar] [CrossRef]
- Hana, S.; Josef, H.; Stanislav, S. A Method of Production of (–)-(S)-3-[1-(Dimethylamino)ethyl]phenyl-N-ethyl-N-methylcarbamate. Patent Application No. WO2004037771 A1, 6 May 2004. [Google Scholar]
- Rao, R.; Shewalkar, M.P.; Nandipati, R.; Yadav, J.S.; Khagga, M.; Shinde, D.B. General Strategy for Large-Scale Synthesis of (+)-Rivastigmine and (+)-NPS R-568. Synth. Commun. 2012, 42, 589–598. [Google Scholar] [CrossRef]
- Reddy, V.V.; Rao, M.V.N.B.; Ganesh, V.; Kumar, A.V.; Praveen, C.; Mukkanti, K.; Reddy, G.M.; Reddy, G.M. An Improved Process for the Production of Rivastigmine Tartrate, a Cholinesterase Inhibitor. Lett. Org. Chem. 2010, 7, 149–154. [Google Scholar] [CrossRef]
- Fuchs, M.; Koszelewski, D.; Tauber, K.; Kroutil, W.; Faber, K. Chemoenzymatic asymmetric total synthesis of (S)-Rivastigmine using ϖ-transaminases. Chem. Commun. 2010, 46, 5500–5502. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Koszelewski, D.; Tauber, K.; Sattler, J.; Banko, W.; Holzer, A.K.; Pickl, M.; Kroutil, W.; Faber, K. Improved chemoenzymatic asymmetric synthesis of (S)-Rivastigmine. Tetrahedron 2012, 68, 7691–7694. [Google Scholar] [CrossRef]
- Mangas-Sánchez, J.; Rodríguez-Mata, M.; Busto, E.; Gotor-Fernández, V.; Gotor, V. Chemoenzymatic Synthesis of Rivastigmine Based on Lipase-Catalyzed Processes. J. Org. Chem. 2009, 74, 5304–5310. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.K.; Bhandya, S.R.; Maddur, N.; Shukla, R.; Kumar, A.; Mittapalli, V.S.N.J. Asymmetric synthesis of an enantiomerically pure rivastigmine. Tetrahedron Asymmetry 2013, 24, 374–379. [Google Scholar] [CrossRef]
- Sethi, M.K.; Bhandya, S.R.; Kumar, A.; Maddur, N.; Shukla, R.; Mittapalli, V.S.N.J. Chemo-enzymatic synthesis of optically pure rivastigmine intermediate using alcohol dehydrogenase from baker’s yeast. J. Mol. Catal. B Enzym. 2013, 91, 87–92. [Google Scholar] [CrossRef]
- Nagai, T.; Sakurai, S.; Natori, N.; Hataoka, M.; Kinoshita, T.; Inoue, H.; Hanaya, K.; Shoji, M.; Sugai, T. Synthesis of enantiomerically enriched drug precursors and an insect pheromone via reduction of ketones using commercially available carbonyl reductase screening kit “Chiralscreen® OH”. Bioorg. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Park, J.; Kim, M.J. Chemoenzymatic Synthesis of Rivastigmine via Dynamic Kynetic Resolution as a Key Step. J. Org. Chem. 2010, 75, 3105–3108. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.-C.; Zhu, G.-L.; Xie, J.-H.; Zhang, X.-D.; Zhou, Q.-L.; Li, Y.-Q.; Shen, W.-H.; Che, D.-Q. Industrial Scal-Up of Enantioselective Hydrogenation for the Asymmetric Synthesis of Rivastigmine. Org. Proc. Dev. 2013, 17, 307–312. [Google Scholar] [CrossRef]
- De Man, J.C.; Rogosa, M.; Sharpe, M.E. A Medium for the Cultivation of Lactobacilli. J. Appl. Bacteriol. 1960, 23, 130–135. [Google Scholar] [CrossRef]
- Ricci, M.A.; Russo, A.; Pisano, I.; Palmieri, L.; de Angelis, M.; Agrimi, G. Improved 1,3-propanediol synthesis from glycerol by the robust Lactobacillus reuteri strain DSM 20016. J. Microbiol. Biotechnol. 2015, 6, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Kišic, A.; Stephan, M.; Mohar, B. ansa-Ruthenium(II) Complexes of R2NSO2DPEN-(CH2)n(η6-Aryl) Conjugate Ligands for Asymmetric Transfer Hydrogenation of Aryl Ketones. Adv. Synth. Catal. 2015, 357, 2540–2546. [Google Scholar] [CrossRef]
- Nasário, F.D.; Cazetta, T.; Moran, P.J.S.; Rodrigues, J.A.R. Deracemization of 1-phenylethanol via tandem biocatalytic oxidation and reduction. Tetrahedron Asymmetry 2016, 27, 404–409. [Google Scholar] [CrossRef]
- Baker’s Yeast LIEVITAL (25gx2—Cod. 100001181). Available online: https://lesaffre.ovh/ (accessed on 16 January 2018).
Scheme 1.
Previous syntheses of (S)-rivastigmine, and this work reporting a whole-cell biocatalytic reduction approach as a key step.
Scheme 1.
Previous syntheses of (S)-rivastigmine, and this work reporting a whole-cell biocatalytic reduction approach as a key step.
Scheme 2.
Retrosynthetic analysis of (S)-rivastigmine (5) starting from 3-hydroxyacetophenone (1) or 3-acetylphenyl-N-ethyl-N-methylcarbamate (2) via chiral secondary alcohols (R)-3 or (R)-4.
Scheme 2.
Retrosynthetic analysis of (S)-rivastigmine (5) starting from 3-hydroxyacetophenone (1) or 3-acetylphenyl-N-ethyl-N-methylcarbamate (2) via chiral secondary alcohols (R)-3 or (R)-4.
Scheme 3.
Chemoenzymatic total sythesis of (S)-rivastigmine (5) in four steps.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Stereoselective bioreduction of ketones 1 and 2 with baker’s yeast RC in different reaction media. 1
Table 1.
Stereoselective bioreduction of ketones 1 and 2 with baker’s yeast RC in different reaction media. 1
| Entry | Ketone | t (h) | Reaction Medium | Product (Yield %) 2 | ee (%) 3 | Absolute Configuration 4 |
|---|---|---|---|---|---|---|
| 1 | 1 | 15 | H2O | 3 (5) 5 | N.D. | N.D. |
| 2 | 1 | 24 | H2O | 3 (8) 5 | N.D. | N.D. |
| 3 | 1 | 40 | H2O | 3 (25) | >98 | S |
| 4 | 1 | 120 | H2O | 3 (27) | 98 | S |
| 5 | 1 | 120 | ChCl/Gly 2:1 | N.R. | N.D. | N.D. |
| 6 | 1 | 120 | L-(+)-lactic acid/ChCl 2:1 | N.R. | N.D. | N.D. |
| 7 | 1 | 120 | ChCl/D-fructose 2:1 | N.R. | N.D. | N.D. |
| 8 | 1 | 120 | ChCl/Gly 2:1 + 10 w% H2O | N.R. | N.D. | N.D. |
| 9 | 1 | 120 | L-(+)-lactic acid/ChCl 2:1 + 10 w% H2O | N.R. | N.D. | N.D. |
| 10 | 1 | 120 | ChCl/D-fructose 2:1 + 10 w% H2O | N.R. | N.D. | N.D. |
| 11 | 1 | 120 | ChCl/Gly 2:1 + 20 w% H2O | 3 (8) | >98 | S |
| 12 | 1 | 120 | ChCl/Gly 2:1 + 40 w% H2O | 3 (32) | >98 | S |
| 13 | 2 | 24 | H2O | 4 (12) | >98 | S |
| 14 | 2 | 120 | ChCl/Gly 2:1 + 20 w% H2O | 4 (5) | 95 | S |
1 Reaction conditions: ketone (1.5 mM), baker’s yeast (300 mg mL−1); deep eutectic solvent (DES) amount: 50 g. 2 Isolated yield by column chromatography with no other products formed. 3 Enantiomeric excess (ee) determined by HPLC. 4 Absolute configuration of the major enantiomer. 5 Calculated by 1H NMR with reference to diagnostic signals of the unreacted ketone in the crude; no other products were detected. N.R. = no reaction. N.D. = not determined.
Table 2.
Stereoselective reduction of ketones 1 and 2 with Lactobacillus reuteri DSM 20016 whole cells 1. CDW: cell dry weight.
Table 2.
Stereoselective reduction of ketones 1 and 2 with Lactobacillus reuteri DSM 20016 whole cells 1. CDW: cell dry weight.

| Entry | Ketone | t (h) | Reaction Medium | Cells Concentration(g CDW/L) | Product (Yield %) 2 | ee (%) 3 | Absolute Configuration 4 |
|---|---|---|---|---|---|---|---|
| 1 | 2 | 24 | PBS | 1.1 (RC) | 4 (60) | 98 | R |
| 2 | 2 | 24 | MRS | 1.1 (GC) 1 | 4 (47) | 98 | R |
| 3 | 2 | 48 | PBS | 1.1 (RC) | 4 (64) | 98 | R |
| 4 | 2 | 72 | PBS | 1.1 (RC) | 4 (74) | 98 | R |
| 5 | 1 | 72 | PBS | 1.1 (RC) | 3 (16) | >98 | R |
| 6 | 2 | 72 | PBS | 1.1 (RC) 5 | 4 (68) | 98 | R |
| 7 | 2 | 72 | PBS | 1.1 (RC) 6 | 4 (71) | 98 | R |
| 8 | 2 | 72 | PBS | 1.1 (RC) 7 | 4 (63) | 98 | R |
| 9 | 2 | 24 | PBS | 4.4 (RC) | 4 (90) | 98 | R |
1 Typical reaction conditions: resting cells (RC) or growing cells (GC); (see Experimental). 2 Isolated yield by column chromatography with no other products formed. 3 Enantiomeric excess (ee) determined by HPLC. 4 Absolute configuration of the major enantiomer. 5 Reaction with 5.83 mmol of ketone 2 performed in a shake flask (orbital shaking at 200 rpm, see Experimental). 6 Reaction with 5.83 mmol of ketone 2 performed in a bioreactor with pH stabilization (7.0). 7 Reaction with 5.83 mmol of ketone 2 performed in a bioreactor without pH stabilization (see Experimental).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vitale, P.; Perna, F.M.; Agrimi, G.; Pisano, I.; Mirizzi, F.; Capobianco, R.V.; Capriati, V. Whole-Cell Biocatalyst for Chemoenzymatic Total Synthesis of Rivastigmine. Catalysts 2018, 8, 55. https://doi.org/10.3390/catal8020055
AMA Style
Vitale P, Perna FM, Agrimi G, Pisano I, Mirizzi F, Capobianco RV, Capriati V. Whole-Cell Biocatalyst for Chemoenzymatic Total Synthesis of Rivastigmine. Catalysts. 2018; 8(2):55. https://doi.org/10.3390/catal8020055
Chicago/Turabian StyleVitale, Paola, Filippo Maria Perna, Gennaro Agrimi, Isabella Pisano, Francesco Mirizzi, Roberto Vito Capobianco, and Vito Capriati. 2018. "Whole-Cell Biocatalyst for Chemoenzymatic Total Synthesis of Rivastigmine" Catalysts 8, no. 2: 55. https://doi.org/10.3390/catal8020055
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.