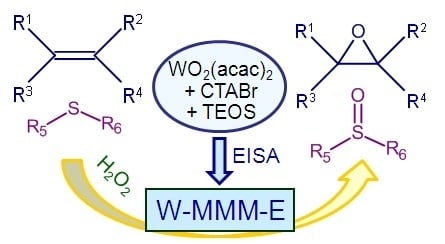
2.1. Catalyst Synthesis and Characterization
Acetylacetone has been widely employed as a hydrolysis-retarding dopant in sol-gel and template syntheses of mesoporous metal-silicates [
20,
52]. Use of this chelating agent in the synthesis of Ti-MMM-E catalysts by EISA not only prevented TiO
2 precipitation but also assisted in the generation of specific di(oligo)meric Ti centers within the silica matrix [
48]. The same acac chelating ligand was successfully employed for stabilization of Nb precursors in the synthesis of Nb-MMM-E materials using the same EISA methodology [
48]. The formation of oxo alkoxide/acac WO(OR)
x(acac)
y complexes by the interaction of W oxo alkoxides with acac was addressed in the literature [
53,
54]. All of these prompted us to use acac in the synthesis of W-MMM-E materials.
Method A (see
Section 3) was, therefore, similar to the procedure reported earlier for the synthesis of Ti-MMM-E [
47] and Nb-MMM-E [
48] materials. Tungsten(VI) ethoxide modified with acetylacetone was taken as the W precursor. The molar ratios of W/Si were 0.18/50 and 0.35/50 (samples
A-1 and
A-2, respectively).
In Methods B and C, we used WO
2(acac)
2 as the tungsten precursor, which was synthesized and characterized beforehand. In Method B, we also attempted to prevent hydrolysis and oligomerization of tungsten species by eliminating water content in the reaction mixture, i.e., using dry ethanol instead of 95% and excluding HCl. According to the EISA methodology, the introduction of tungsten into the final material was almost quantitative (except for samples
A due to poor solubility of tungsten ethoxide), as confirmed by the elemental analysis data (
Table 1).
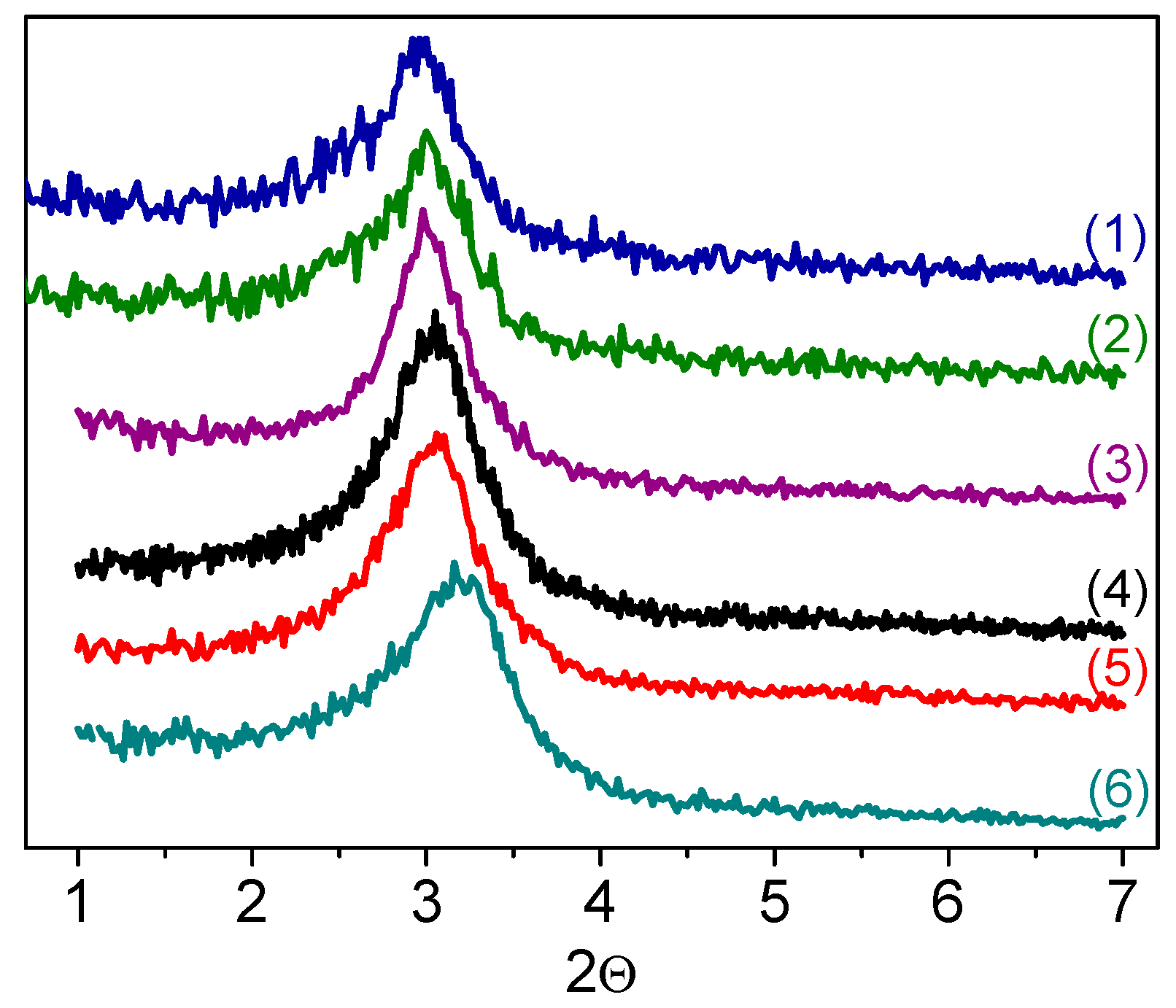
The small angle XRD patterns of W-MMM-E samples, shown in
Figure 1, exhibit a broad diffraction reflex around 3.0° (2
θ). This indicates a long-range structural order of cylindrical mesopores, which is typical for silicate materials prepared by EISA [
47,
48,
50]. The lattice constant, calculated using the XRD (10) reflection position for W-MMM-E, is close to that reported for Ti-MMM-E (5.0 vs. 5.2 nm) [
47]. Moreover, the lack of any additional reflexes at higher 2
θ values (
Figure S1), except for the broad one corresponding to amorphous silicate, indicates the absence of crystalline WO
3.
The textural properties of all W-MMM-E samples measured by low-temperature nitrogen adsorption are presented in
Table 1. All the solids possessed only mesopores and no micropores. Interestingly, the mean pore diameter for W-MMM-E is slightly less than that of Ti- and Nb-MMM-E materials (2.4–2.6 vs. 2.9–3.2 nm), while the silicate wall is oppositely thicker (2.5 vs. 2.2 nm). Texture parameters depend a little on the W precursor used. Both specific BET surface area and pore volume were slightly bigger for material A prepared via modification of tungsten ethoxide than for samples
B and
C synthesized using isolated WO
2(acac)
2 complex (1330 m
2/g and 0.57 cm
3/g for
A-1, versus 1170–1240 m
2/g and 0.46–0.51 cm
3/g for
C-1 and
B-1). Variations in the Si/W ratio produced very little effect on the texture parameter of the materials.
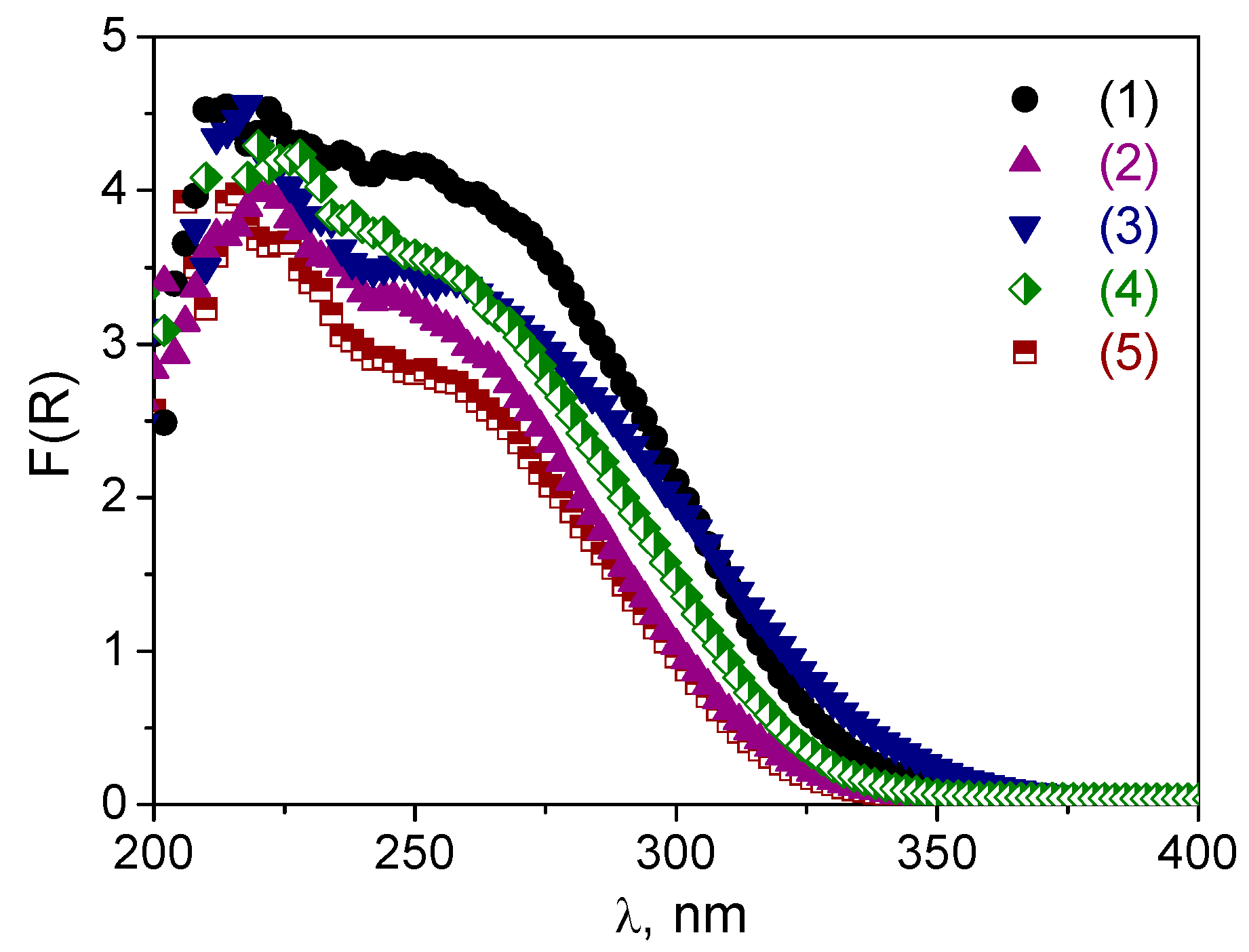
The DR UV-vis spectra of the W-MMM-E catalysts are shown in
Figure 2.
All materials exhibit two main absorptions—a sharp band with a maximum centered at ca. 220–225 and a broad one at 250–255 nm. The adsorption peak at 230 nm is attributed to isolated tetrahedral [WO
4]
2− species because the spectrum of Na
2WO
4 has a spinel structure [
21,
23]. Broad absorptions around 240–290 nm are typically assigned to octahedrally coordinated small oligomeric tungsten oxide species [
23,
31,
32,
41]. Thus, we may assume the presence of two types of tungsten oxo species in the W-MMM-E samples: isolated tetrahedrally coordinated and oligomeric octahedrally coordinated ones. The lower content of tungsten in the sample indicates a higher contribution of isolated species. A shoulder at 310–320 nm in the spectra of samples with a higher amount of W (
A-2 and
B-1.4) may indicate the presence of octahedrally coordinated polytungstate species or formation of WO
3-like nano-aggregates [
41]. Importantly, for all the elaborated W-MMM-E materials, no adsorptions were detected around 380–450 nm, indicating the absence of crystalline WO
3 on the catalyst surface [
21,
23,
25,
33,
39,
42,
43]. As opposed to other known tungsten–silicates with close W loading (Si/W ≥ 122), i.e., W-MCM-48 [
32,
33] or WO
3-MCF [
39], the EISA methodology enables better isolation of tungsten in the silicate matrix and prevents formation of WO
3 agglomerates.
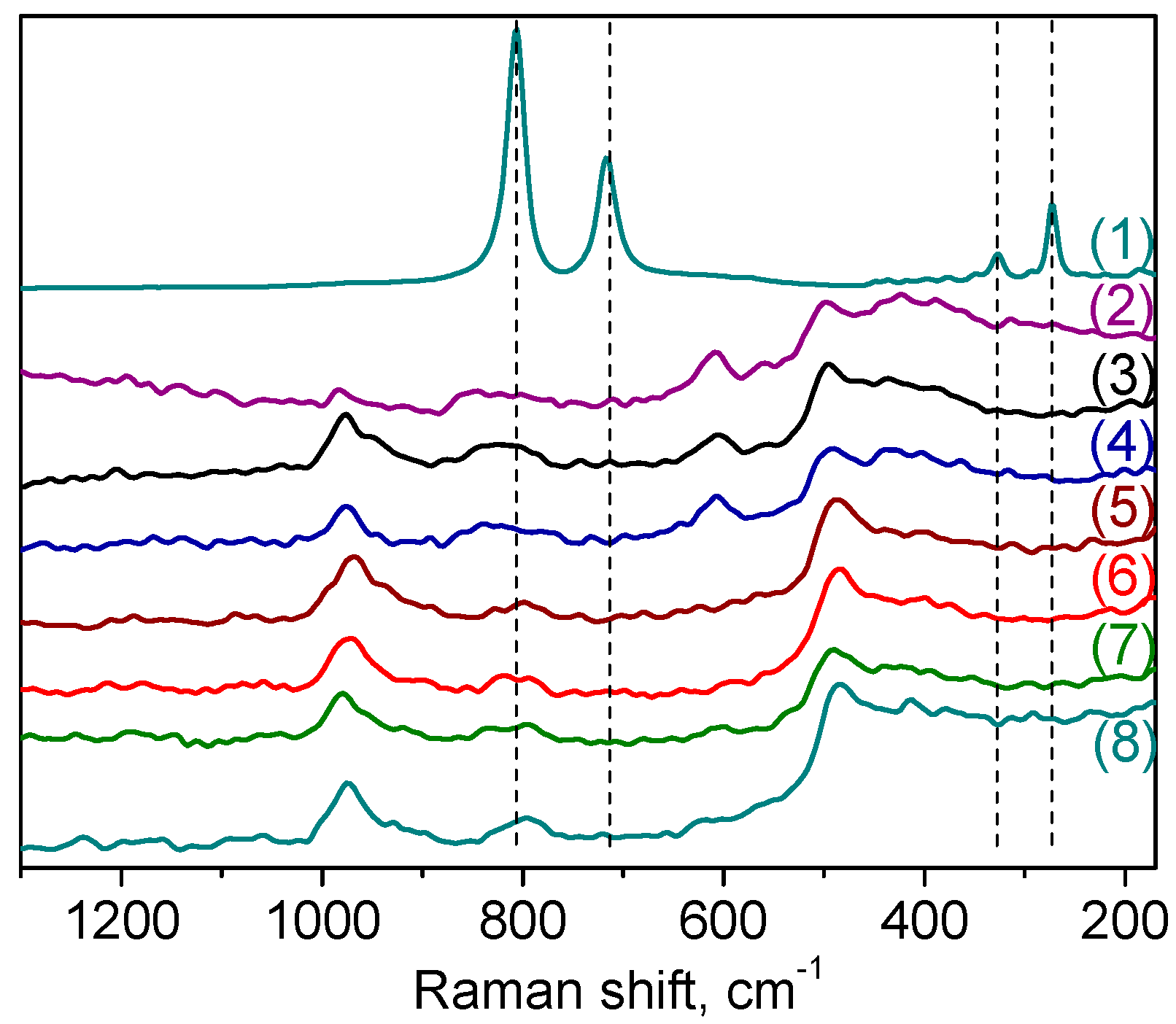
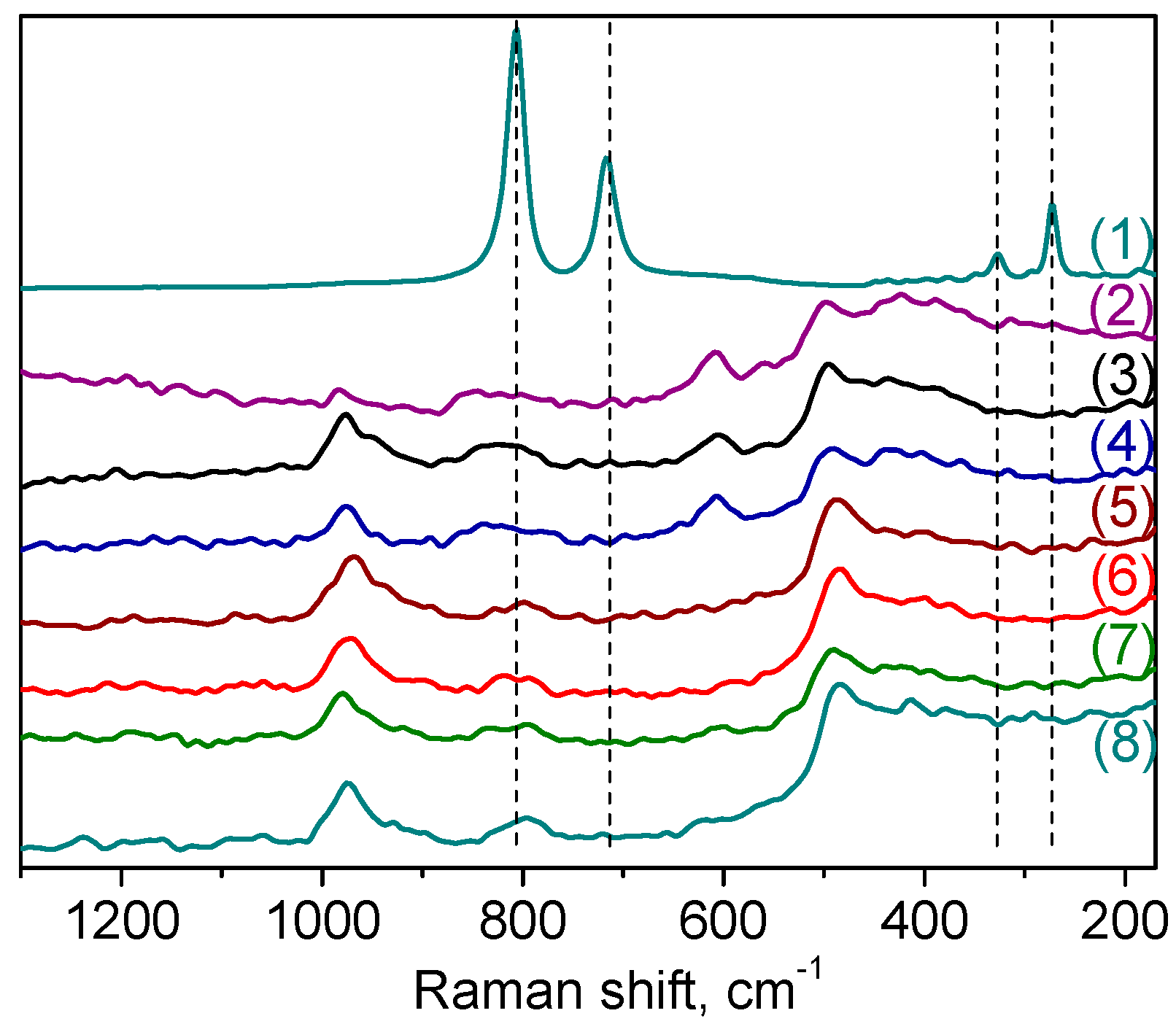
Raman spectroscopy was also employed to explore further the nature of the W species formed on the surface of the catalyst. The spectra of samples
A-1 and
A-2 are shown in
Figure 3, in comparison with the spectra of bulk WO
3 and tungsten-free silicate matrix. In spite of the distinction in the DRS-UV spectra, the Raman spectra of these samples were quite similar. The Raman spectrum of crystalline WO
3 is characterized by strong bands at 808 and 720 cm
−1 along with peaks at 324 and 272 cm
−1 (
Figure 3, curve (1)) [
33,
43,
55,
56]. Significantly, these peaks are absent in the Raman spectra of the W-MMM-E catalysts, corroborating the absence of WO
3 crystallites and incorporation of well dispersed tungsten oxide species into the silica matrix. The spectra of W-MMM-E samples (
Figure 3, curves (3) and (4)) are very similar to those of the pure EISA silica (
Figure 3, curve (2)) except for the intense band at 975 cm
−1 which may be attributed to vibrations of the terminal W=O bonds [
55,
57,
58]. Raman features between 1000 and 900 cm
−1 are typically observed in mono- and polytungstate species [
22,
41,
57,
58].
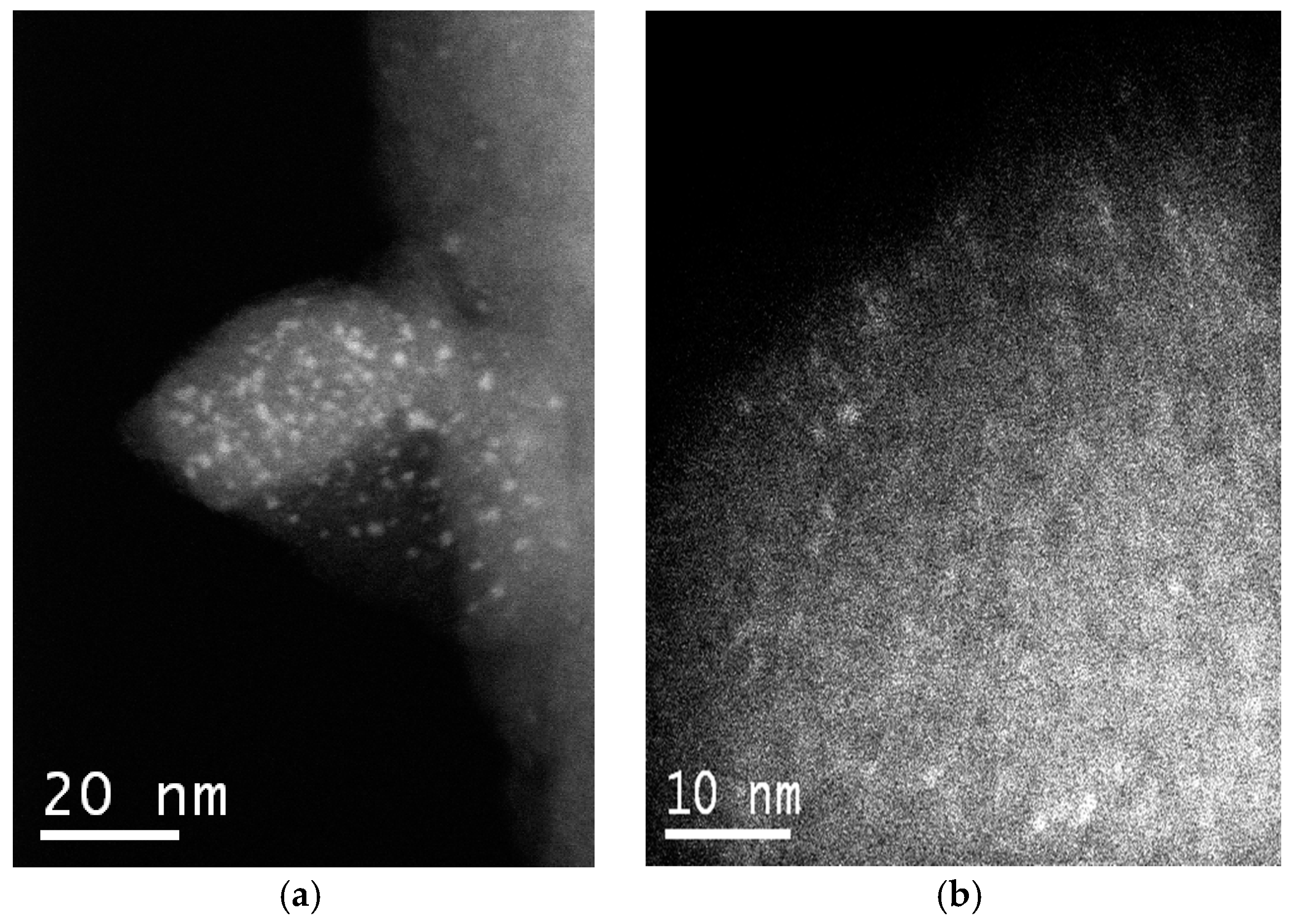
STEM-HAADF (high angle annular dark field in scanning-TEM mode) images of the W-MMM-E (sample
A-1) exhibit nanoscale inclusions of tungsten associates, unevenly distributed over the silica surface. Two representative images are given in
Figure 4.
Figure 4a shows a piece of the catalyst with W-containing particles of 1–2 nm dispersed on the surface. However, such large particles are much less common in other parts of the catalyst. Some ordering of the mesopores with a periodicity of ~2 nm can be noticed in
Figure 4b. One can also distinguish here bright clusters with a size less than 1 nm and spots of particles with an elongated shape, which correspond to polytungstate species. These spots have a lower contrast, most likely because they are located inside the mesopores and not on the external surface of the catalyst particles.
2.2. Hydrothermal Stability and Stability towards H2O2
Hydrothermal stability is essential for the catalysts applied for oxidation using aqueous hydrogen peroxide. However, as has already been mentioned, tungsten-containing mesoporous silicates usually suffer from active metal leaching under turnover conditions during liquid-phase oxidation.
The stability of all the elaborated W-MMM-E samples was examined by treatments: (i) with boiling water for 6 h and (ii) with 0.11 M solution of H
2O
2 in CH
3CN for 1 h at room temperature. Similar tests were previously employed for Ti-MMM-E [
47] and Nb-MMM-E [
48] materials.
Figure 1 (curves (5) and (6)) shows the XRD patterns of sample
A-2 after the treatments. Both the position and width of the diffraction reflexes are very close, which suggests fairly good structural stability in the W-MMM-E solids synthesized by the EISA technique. Raman spectra also did not show changes after treatment with H
2O
2 (
Figure 3, compare curves (3), (4) and (5), (6)).
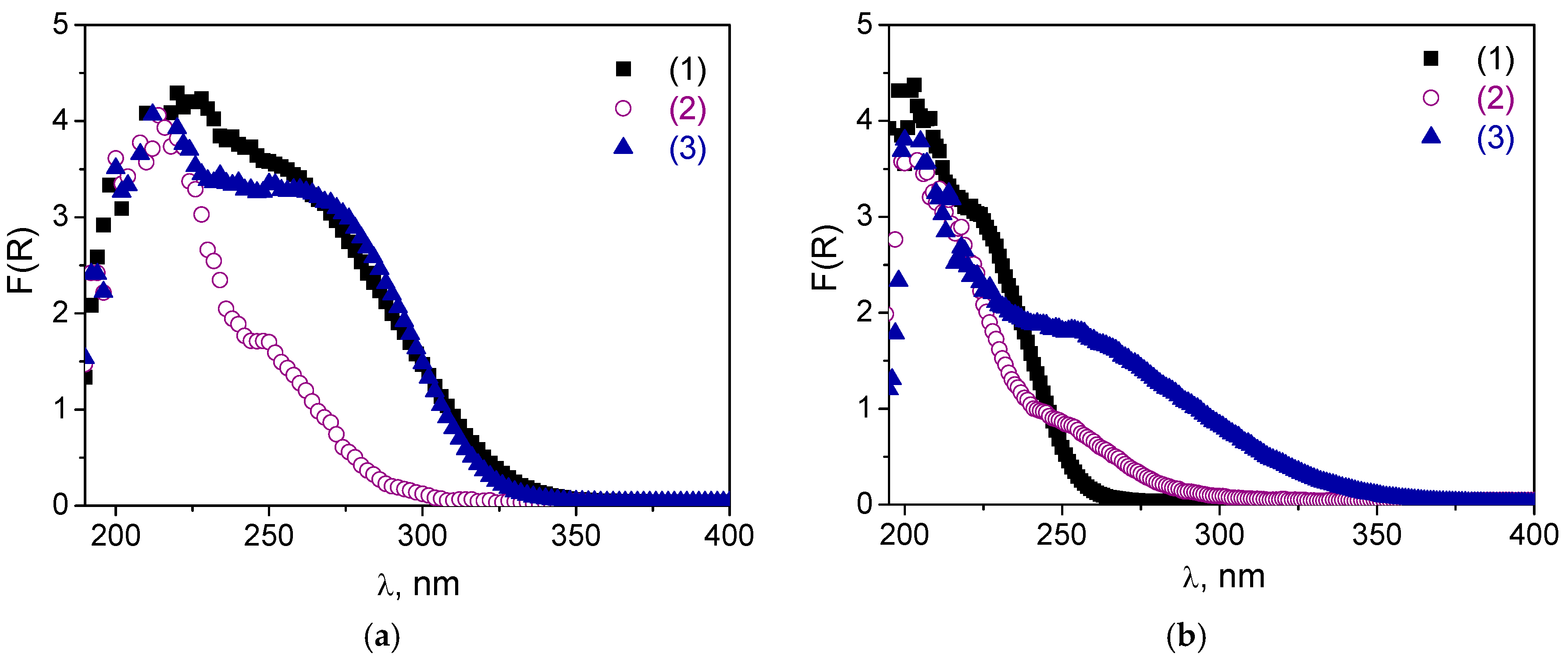
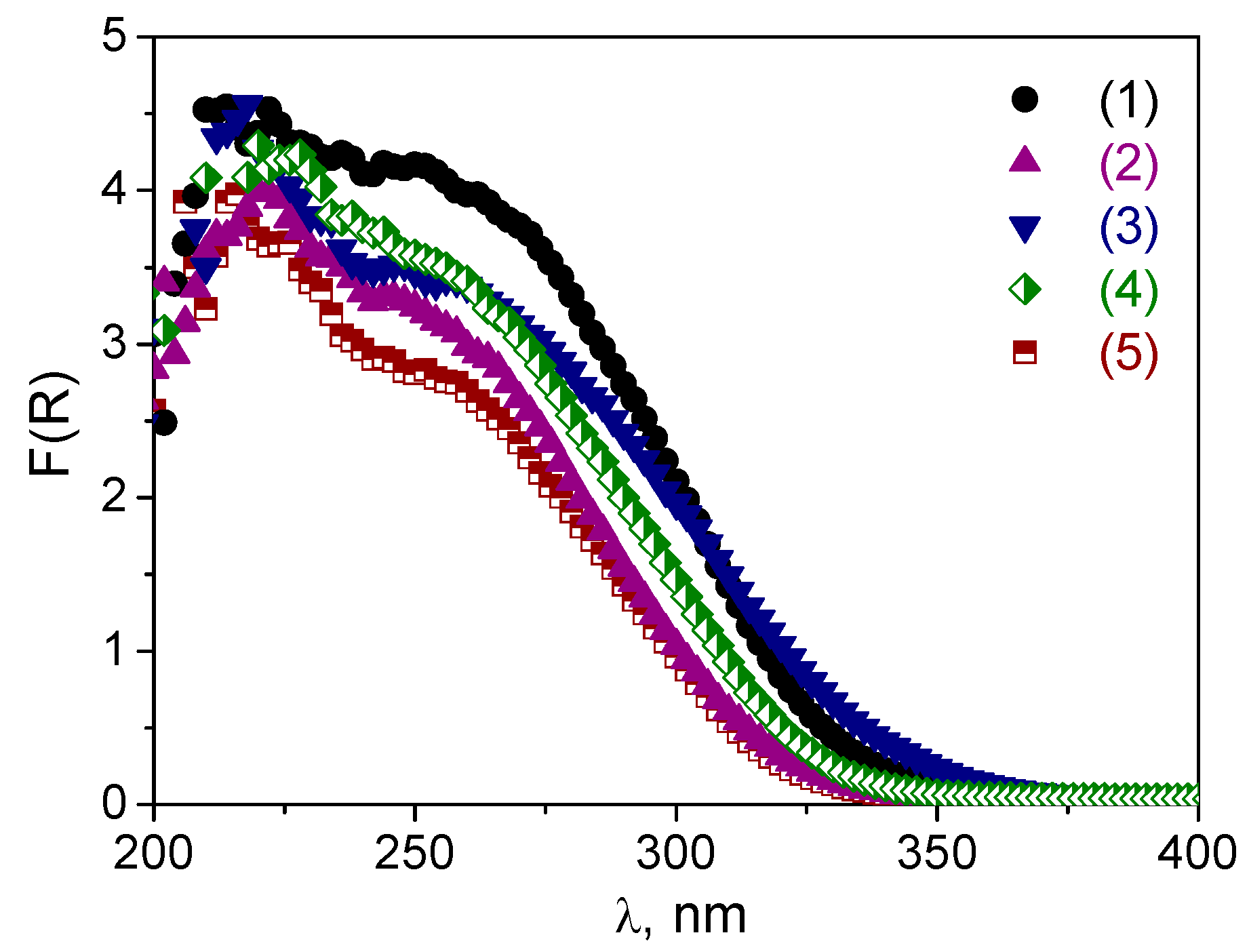
The DR UV-vis spectra after the treatments are given in
Figure 5 along with
Figures S2 and S3 in Supplementary Materials. One can see that, after the boiling procedure, the shoulder assigned to oligomeric tungsten oxo species (240–290 nm) decreased dramatically, and treated samples contained mostly isolated W. This effect was most pronounced for the A and B samples. Obviously, leaching of oligomeric W species occurred during the treatment, which was also confirmed by elemental analysis (
Table S1): 19–107 ppm of leached W was detected in the reaction mixture after the catalyst was separated by filtration. The A materials were also not stable during the treatment with H
2O
2.
Figure S2 showed a decrease in the shoulder at 240–290 nm in DR-UV spectra for both
A-1 and
A-2 samples, but it occurred less dramatically than in the case of the boiling procedure. On the other hand, the spectra of both
B-1 and
B-1.4 samples almost did not change (
Figures S2 and S3, and
Figure 5a). For sample
C-1, both treatments caused a long-wave shift in the absorption band that was manifested to a greater extent in the case of the boiling procedure than following treatment with H
2O
2 (
Figure 5b). This shift may indicate some further agglomeration of the tungsten species. Thus, one may conclude that materials
B and
C, synthesized using the WO
2(acac)
2 precursor, are more stable during treatment with aqueous H
2O
2 than material A, obtained through one-pot modification of tungsten ethoxide with acetylacetone. According to the elemental analysis data, tungsten leaching was less under the following conditions: (i) when treated with H
2O
2 rather than with boiling water and (ii) when initial W content was minimal, i.e., for
B-1 and
A-1 samples in contrast to
B-1.4 and
A-2, respectively (
Table S1).
2.3. Catalytic Studies
The catalytic properties of the tungsten-based silicates prepared by the EISA methodology was examined during the oxidation of cyclooctene (CyOct) as a model substrate with 1 equivalent of hydrogen peroxide using CH
3CN as the solvent. The main results are presented in
Table 2.
All the elaborated W-MMM-E materials exhibited excellent selectivity during CyOct epoxidation with aqueous H
2O
2, regardless of the synthesis procedure, the content or the state of W (isolated or oligomeric). Catalysts
B and
C were somewhat more active than material
A (turnover frequency (TOF) values 58–66 vs. 53,
Table 2). Importantly, tungsten-free silica matrix was almost inactive in this reaction, indicating that the observed catalysis is due to the presence of W (
Table 2, entry 14). Bulk tungsten oxide also had very low activity (
Table 2, entry 13), which is consistent with the literature [
22,
23,
24,
43]. It was reported previously that only nanoparticulate WO
3 demonstrates catalytic activity [
22,
43]. The acetylacetonate complex, WO
2(acac)
2, was more active than heterogeneous catalysts obtained using this precursor (compare entry 12 and entries 6, 8, and 10 in
Table 2); however, the latter can be used repeatedly (see
Section 2.4) and thus, allows higher total turnover number (TON) values to be reached (226–237 for four consecutive runs using
B-1 or
C-1 vs. 85 for one run with homogeneous WO
2(acac)
2). Among other known tungsten–silicates, a superior CyOct epoxide yield was reported for W-MCM-41 (Si/W = 31) synthesized via the peroxide route: 100% selectivity was reached at 98% substrate conversion after 24 h at room temperature using a five-fold excess of anhydrous H
2O
2 (solution in
t-BuOH) [
21].
As has already been mentioned, the main drawback of tungsten-containing mesoporous silicates is active metal leaching under turnover conditions during liquid-phase oxidation. Unfortunately, the amount of leached tungsten using W-MMM-E catalysts in CyOct oxidation with 30% aqueous H
2O
2 was considerable (50–58 ppm, see
Table 2). However, the leaching degree could be reduced to 17–20 ppm by using 50% hydrogen peroxide instead of 30%, or by using catalysts with a lower tungsten content (
Table 2), which is in agreement with the literature [
39] and our results for the catalyst’s stability to aqueous H
2O
2 and boiled water (see
Section 2.2). It is quite typical of mesoporous metal–silicates that metal leaching is caused by cooperative action of H
2O
2 and water (a combination of hydrolysis and complexing processes) [
20]. Thus, it can be reduced by decreasing the water content in the reaction mixture, i.e., by using more concentrated H
2O
2.
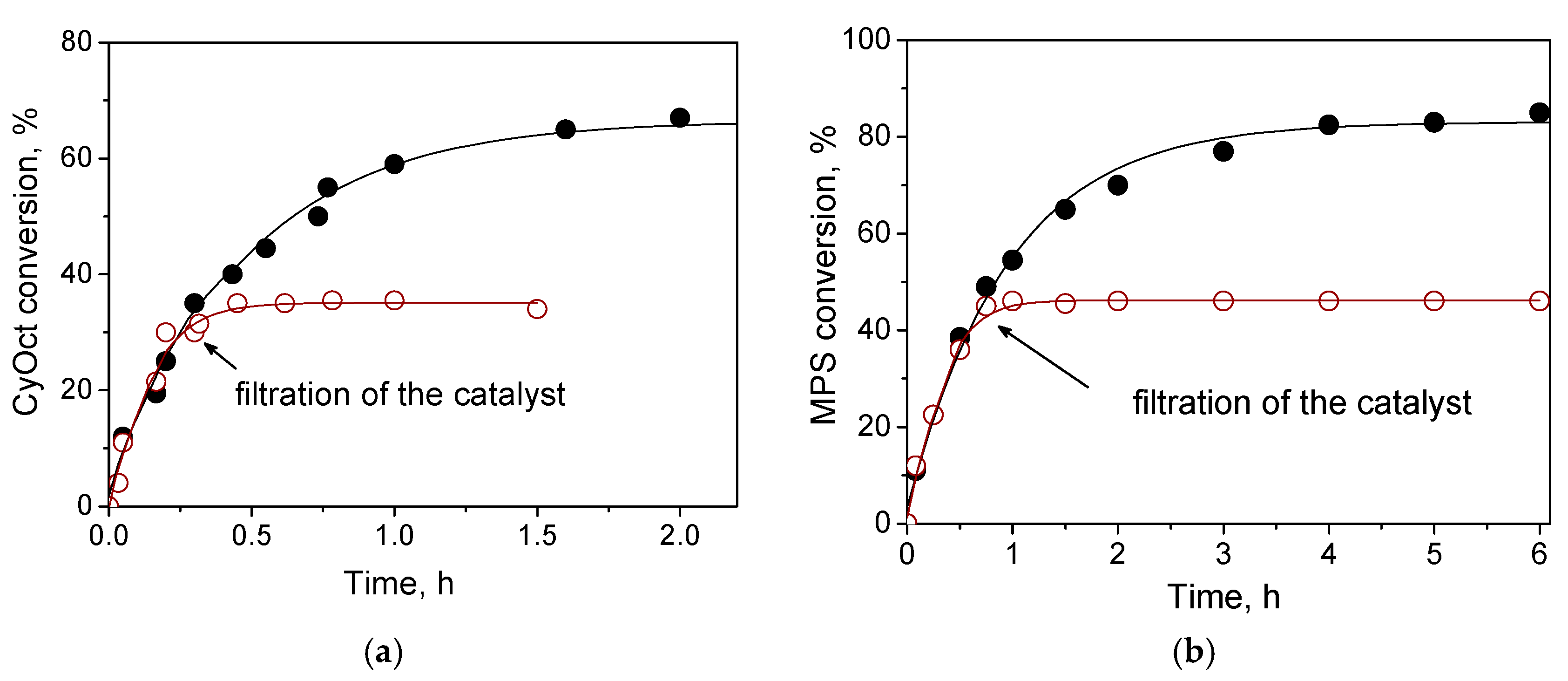
Despite appreciable leaching of the active metal under turnover conditions of CyOct oxidation with 30% H
2O
2, hot filtration tests exhibited neither substrate conversion nor epoxide formation in the filtrate after removal of the solid catalyst (
Figure 5a and
Figure S3), implying that inactive metal species leached out and corroborated the heterogeneous nature of the catalysis over W-MMM-E. These results are not surprising considering that bulk WO
3 was almost inactive during CyOct oxidation. The same behavior was reported for the W-SBA-16 and W-KIT-6 materials [
23,
24]. It was suggested that the epoxidation activity is mainly due to framework-incorporated W species and not the extra-framework metal oxide species [
23,
24].
A range of alkenes, including bulky terpenes (3-carene and caryophyllene), can be epoxidized effectively producing epoxides with up to 99% selectivity (
Table 3).
Styrene and cyclohexene, the substrates which are difficult to epoxidize, showed moderate epoxide selectivity (
Table 3, entries 5, 6 and 9, 10). During styrene oxidation, the main byproduct was benzaldehyde (39–41%). Note that the oxidative C=C cleavage producing benzaldehyde is a typical side reaction for styrene epoxidation, specifically with H
2O
2, in the presence of W-based catalysts [
60,
61]. Cyclohexene also produced allylic oxidation products—cyclohexenyl hydroperoxide, 2-cyclohexene-1-ol, and 2-cyclohexene-1-one (totally 15–17%)—as well as 1,2-trans-cyclohexane diol and its overoxidation product, 2-hydroxycyclohexane-1-one. However, compared to Ti- and Nb-containing MMM-E (
Table 3, entries 7, 8 and 11, 12), W-MMM-E materials were more selective during epoxidation of cyclohexene and styrene [
59]. As with the other known tungsten–silicates, W-HMS gave only 2-cyclohexenol and 2-cyclohexenone during cyclohexene oxidation with a three-fold excess of 37% H
2O
2 [
38], while the reaction over W-SBA-16 yielded a 56% epoxidation selectivity with 36% conversion (1 equivalent of 30% H
2O
2, 50 °C, 6 h) [
23].
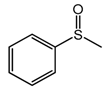
We also tested the catalytic activity of the elaborated W-MMM-E materials during the oxidation of methyl phenyl sulfide (MPS) to methyl phenyl sulfoxide (MPSO) at room temperature. The results are presented in
Table 4. All the samples demonstrated a similar catalytic performance during the reaction: 82–89% selectivity to MPSO at 95–98% MPS conversion.
C-1 was a bit more selective to the sulfoxide (89%), but, at the same time, the oxidant utilization efficiency in its presence was smaller in comparison to another catalyst: 89% vs. 97% for
B-1.4 and
B-1. It is noteworthy that methyl phenylsulfone (MPSO
2) was also formed as the concomitant product. As in the case of CyOct epoxidation, tungsten-free silica matrix was almost inactive (
Table 4, entry 6). Homogeneous WO
2(acac)
2 was as active as heterogeneous ones but gave more sulfone product (compare entry 5 and entries 2–4 in
Table 4).
All further studies were performed in the presence of
C-1. The decrease in the H
2O
2/MPS molar ratio, from 1.2 to 1 mol/mol, led to a decrease in substrate conversion, from 96 to 91%, but did not affect the selectivity of MPSO (
Table 5, entries 1 and 2). The efficiency of oxidant utilization also improved from 89 to 99%. Importantly, tungsten leaching in these reactions did not exceed 3 ppm.
Table 6 shows a comparison of the catalytic performance of W-MMM-E in MPS sulfoxidation with Ti- and Nb-MMM-E prepared by the same methodology [
47,
48]. One can see that tungsten-silicate
C-1 demonstrated superior catalytic properties in terms of activity (TOF 120 vs. 45 and 90 h
−1 for Ti- and Nb-MMM-E, respectively), total MPS conversion (91% vs. 82–83%), and sulfoxidation selectivity (91% vs. 81–82%) at similar values of H
2O
2 utilization efficiency (99% vs. 97–99%).
To evaluate the substrate scope, we studied the catalytic performance of
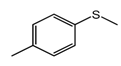
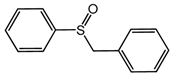
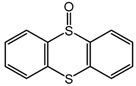
C-1 during the oxidation of a range of organic sulfides (
Table 5, entries 3–5). The results of the oxidation of methyl p-tolyl sulfide and benzyl phenyl sulfide were close to those for MPS oxidation: the sulfide conversions attained were 84% and 85% and the selectivities to corresponding sulfoxides were88 and 84%, respectively. During thianthrene oxidation, products of electrophilic oxidation, namely thiantherene-5-oxide and thiantherene-5,10-dioxide, were mainly formed (
Table 5, entry 5).
C-1 also catalyzed the oxidation of MPS with another oxidant,
tert-butylhydroperoxide (TBHP) (
Table 5, entry 6). In this case, the reaction was more selective to sulfoxide: selectivity reached 95% at 86% substrate conversion, but required a higher reaction temperature, 60 °C.
2.4. Catalyst Recyclability and Stability under Reaction Conditions
The hot catalyst filtration test for MPS oxidation indicated the heterogeneous nature of the catalysis over W-MMM-E (
Figure 6b), which was anticipated given that the tungsten leaching was negligible (2.5–3 ppm) under the turnover conditions of MPS oxidation, i.e., at room temperature instead of the 50 °C used for CyOct oxidation.
Although the question on the catalyst stability under turnover conditions is crucial for heterogeneous catalysis during liquid-phase processes [
62], only a few examples on the reusability of tungsten-based mesoporous silicates in the oxidations with aqueous H
2O
2 have been so far reported [
22,
23,
32,
37,
39]. Good recyclability within 3–5 consecutive runs was shown for (i) W-MCM-48 with a 38.5 Si/W molar ratio during cyclopentene oxidation with a two-fold excess of 50% H
2O
2 (35 °C, 35 h) [
32]; (ii) WO
3-MCF washed with ammonium acetate (Si/W = 941.7) during cycloocta-1,5-diene epoxidation with a three-fold excess of 50% H
2O
2 (60 °C, 24 h) [
39]; and (iii) nanoparticulate WO
3 prepared by flame spray pyrolysis during oxidation of cyclooctene with 1 equiv. H
2O
2 (80 °C, 4 h) [
22]. W-HMS (Si/W = 30) [
37] and W-SBA-16 (Si/W = 22) [
23] demonstrated deterioration of catalytic properties upon reuse.
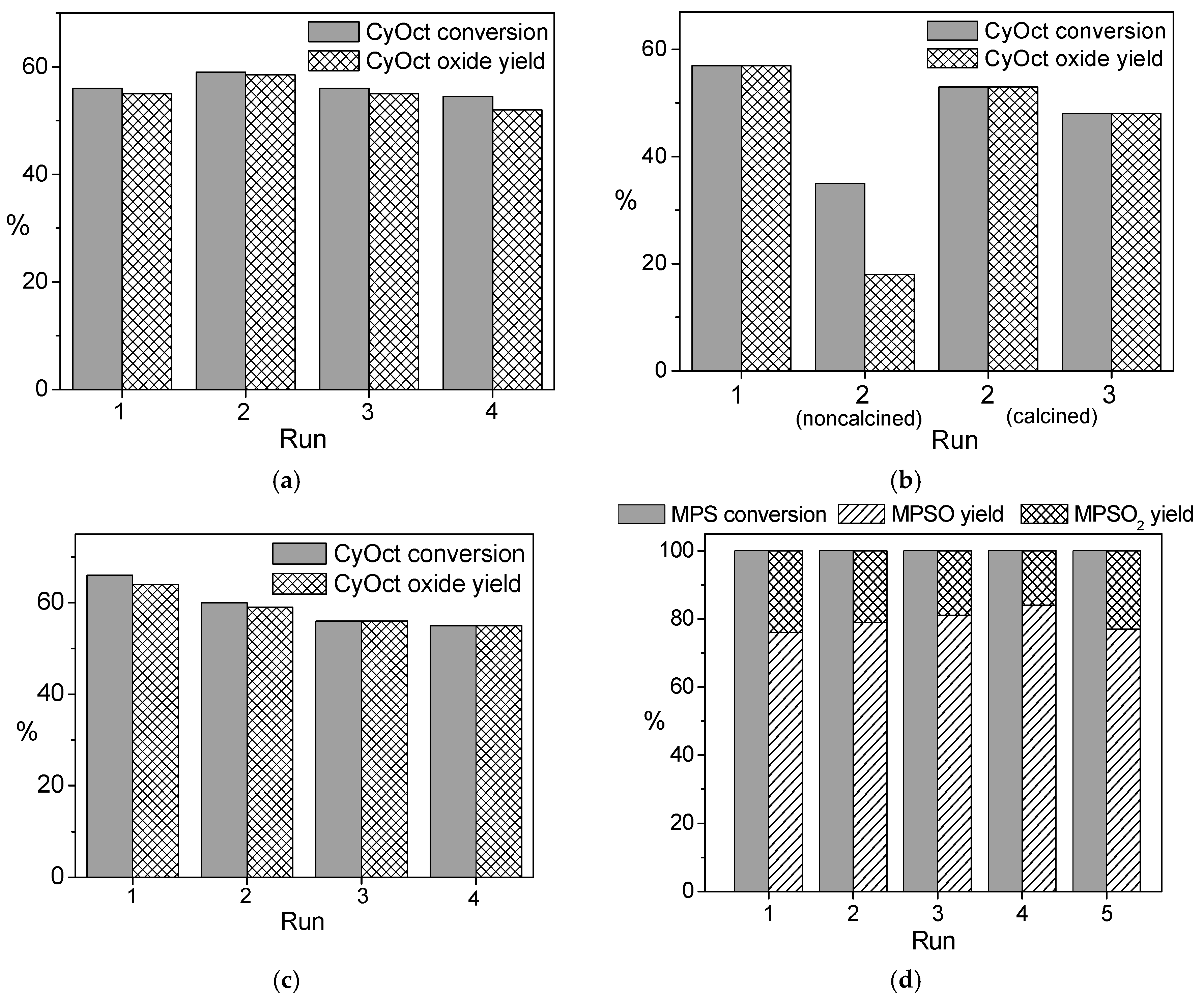
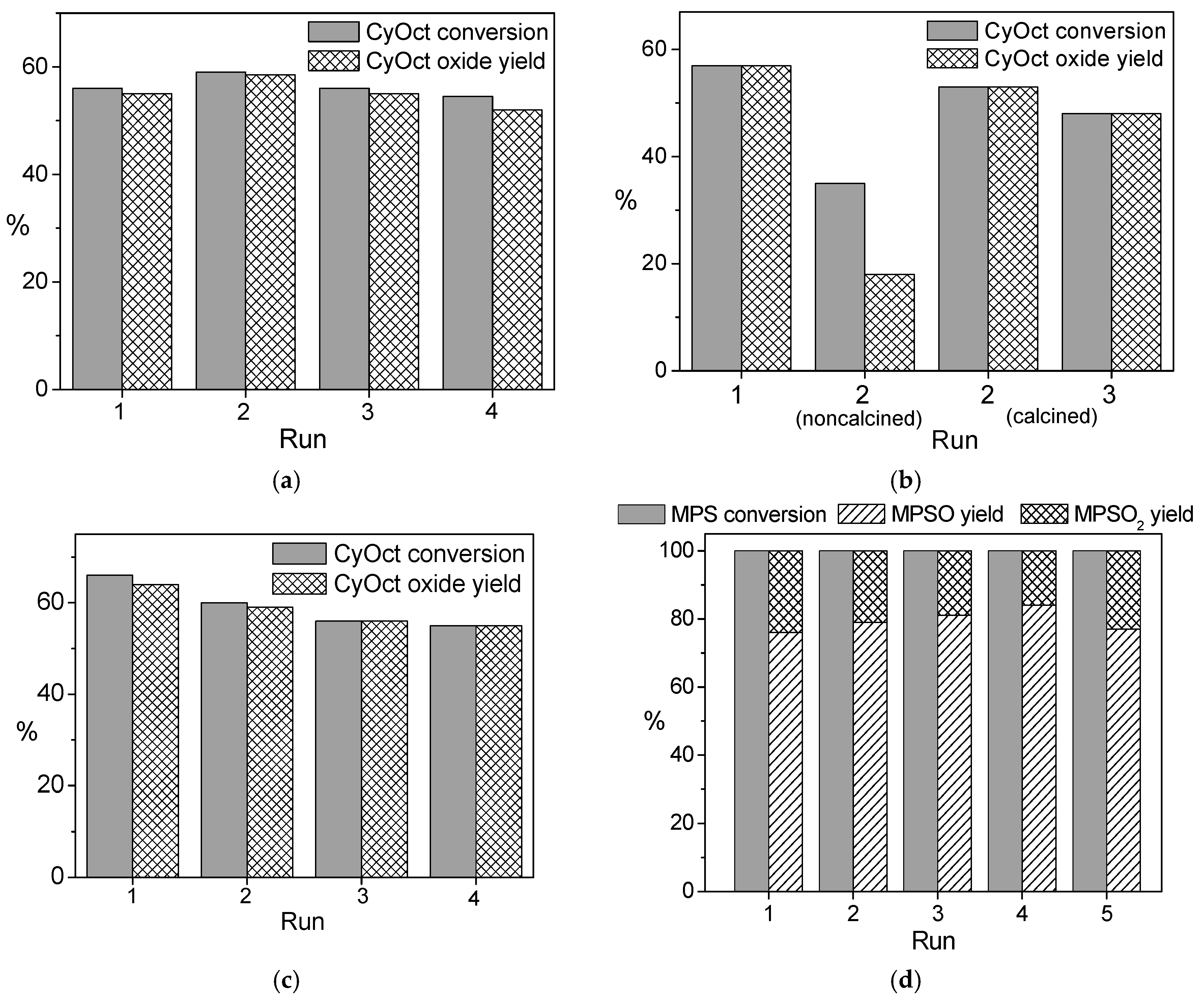
The recycling behavior of all the elaborated W-MMM-E catalysts was studied in three to four consecutive runs of the oxidation of CyOct with 30 or 50% H
2O
2 (
Figures S5–S7 and
Figure 7a–c). In contrast to Nb-MMM-E catalysts, calcination was necessary to regenerate the catalyst (
Figure S7 and
Figure 7b). In good agreement with the leaching tests, better recycling behavior was observed when using 50% H
2O
2 instead of 30%. One can see that A catalysts exhibited a gradual decrease in the maximal achievable substrate conversion during recycling at stably high epoxidation selectivity levels when using 30% aqueous H
2O
2 (
Figures S5a and S6). The
A-2 catalyst showed stability of catalytic properties after the first reuse with 50% H
2O
2 as the oxidant and then activity smoothly reduced (CyOct conversion decreased from 61% to 55% after the second reuse;
Figure S4b), while
A-1 lost neither activity nor selectivity during recycling using 50% H
2O
2 (
Figure 7a). The
B-1.4 sample turned out to be slightly less stable than
A-2: the value of CyOct conversion reduced from 61% to 42% after the second reuse using 50% H
2O
2 (
Figure S7). Both
B-1 and
C-1 catalysts showed good recycling behavior using 50% H
2O
2 (
Figure 7b,c). Thus, one may conclude that there is no necessity for eliminating water content in the reaction mixture during W-MMM-E synthesis or using isolated WO
2(acac)
2 as the tungsten precursor, since all
A-1,
B-1 and
C-1 samples showed good stability of catalytic properties during reuse (compare
Figure 7a–c). Under the milder conditions of MPS oxidation (25 °C), catalyst
C-1 did not lose its catalytic properties during at least five consecutive reuses using 30% aqueous H
2O
2 (
Figure 7d). Tungsten leaching was 17–24 and 2.5 ppm after the first run of CyOct (see
Table 2) and MPS oxidation, respectively, over the fresh catalysts. With the reused
C-1 catalyst, the degree of W leaching reduced to 7.5 (for CyOct) and <1 ppm (for MPS).
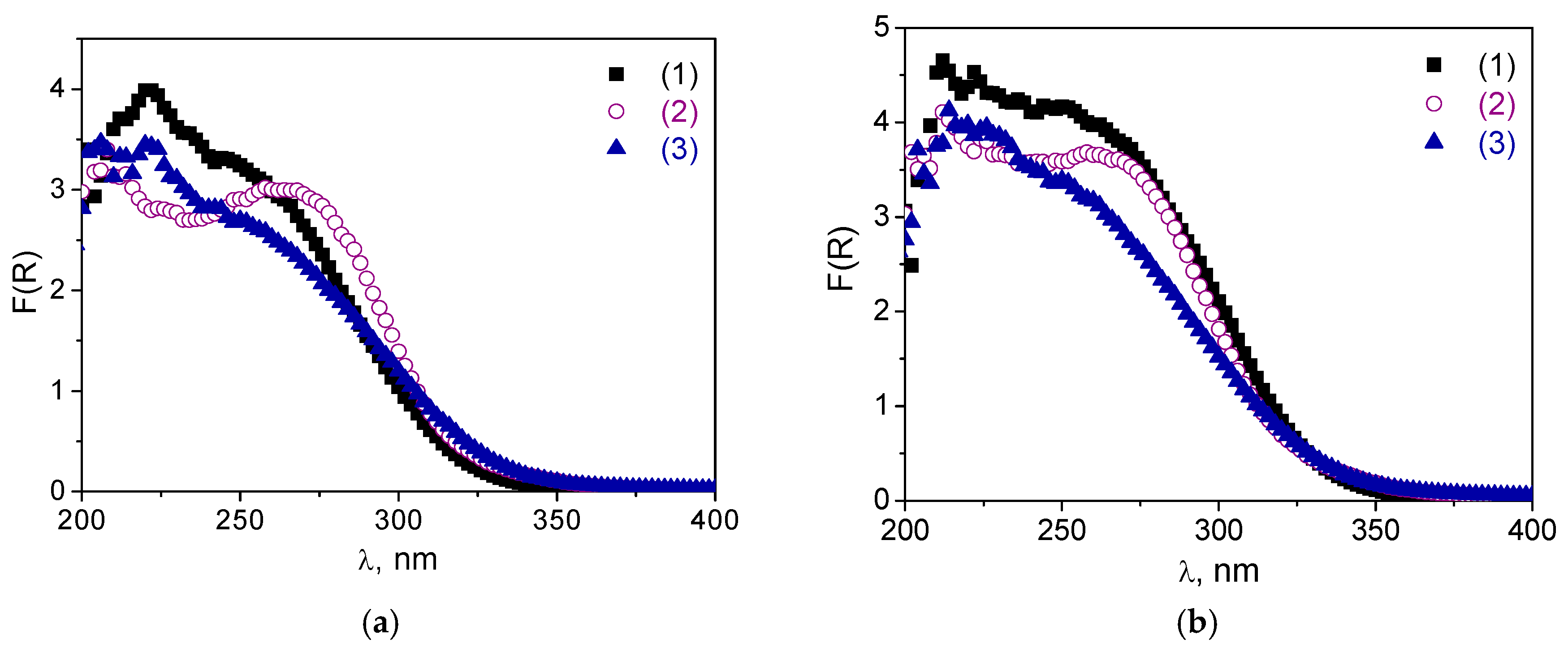
Raman spectra of catalysts
A-1 and
A-2 revealed no changes concerning the band attributed to ν(W=O) at 975 cm
−1 after CyOct catalytic oxidation
Figure 3, curves (7) and (8)). However, the low-intensity band at ca. 600 cm
−1, that refers to the spectrum of the tungsten-free silicate matrix decreased. Supposedly, this may be caused by some restructuring of the silica wall in the mesoporous material as a result of wetting and repeated calcinations after catalytic reactions. However, this does not affect the long-range ordering of the mesoporous material, since the textural properties of the
A-1 sample changed insignificantly after the catalytic reaction of CyOct oxidation (see
Table 1). The observed long–wave shift in the DR UV-vis spectra (
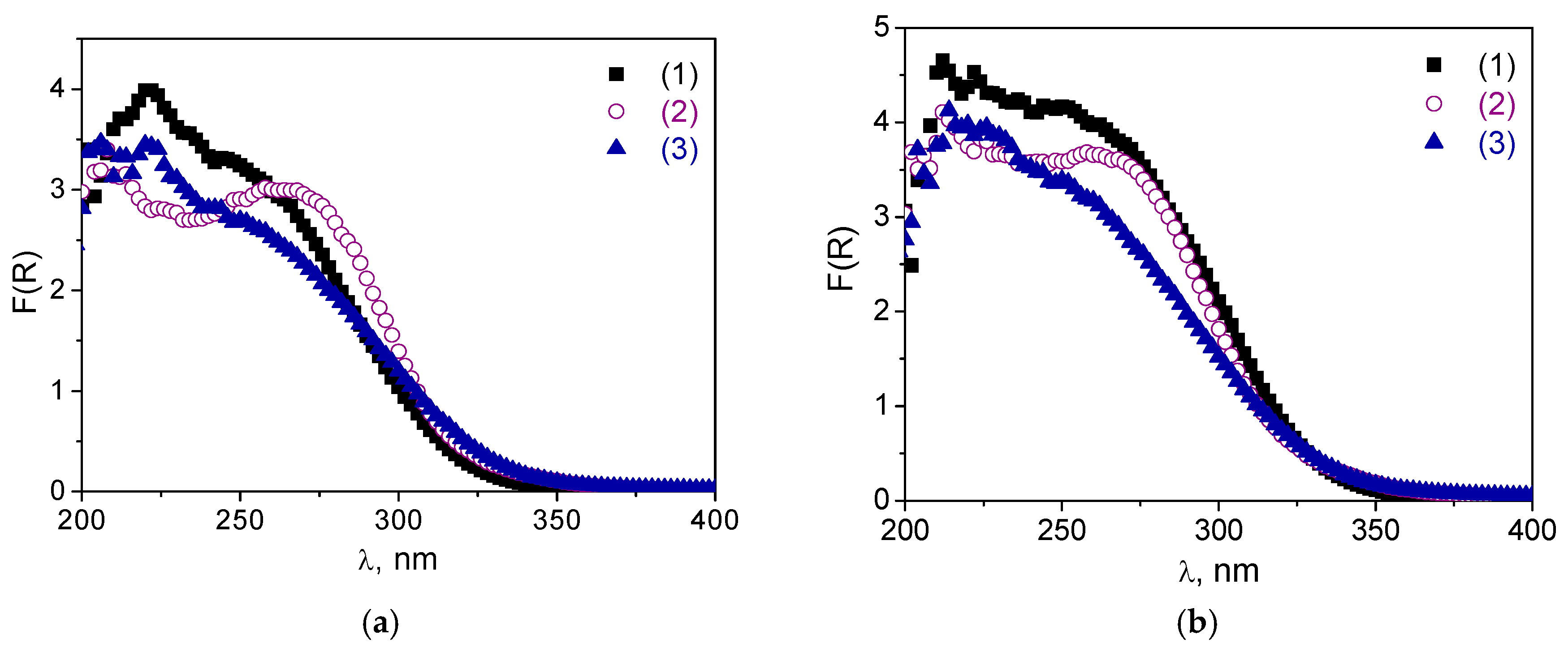
Figure 8a,b) of non-calcined catalysts
A-1 and
A-2 after the oxidation reaction may be due to adsorption of water or some organic molecules. Indeed, the DR UV-vis spectra after subsequent calcination revealed almost no changes compared to the spectrum of initial materials,
A-1 and
A-2 (
Figure 8a,b).

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}