Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series


Institut Lavoisier de Versailles, UVSQ, CNRS, Université Paris-Saclay, 78035 Versailles, France
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(4), 139; https://doi.org/10.3390/catal8040139
Submission received: 12 March 2018
/
Revised: 26 March 2018
/
Accepted: 29 March 2018
/
Published: 31 March 2018
(This article belongs to the Special Issue Catalyzed Mizoroki–Heck Reaction or C–H activation)
Abstract
:5,6-Dihydrobenzo[c]acridine belongs to the large aza-polycyclic compound family. Such molecules are not fully planar due to the presence of a partially hydrogenated ring. This paper describes the first Pd-catalyzed alkoxylation via C-H bond activation of variously substituted 5,6-dihydrobenzo[c]acridines. We determined suitable conditions to promote the selective formation of C-O bonds using 10% Pd(OAc)2, PhI(OAc)2 (2 eq.) and MeOH as the best combination of oxidant and solvent, respectively. Under these conditions, 5,6-dihydrobenzo[c]acridines bearing substituents at both rings A and D were successfully functionalized, giving access to polysubstitutited acridine motifs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Acridines and related derivatives represent an important class of aza-polycyclic compounds that have attracted considerable interest in the last century because of their broad range of properties and applications. For example, acridines are well-known as antibacterial, antimalarial, and anticancer agents [1,2,3], and have also been used in pigments, dyes, and sensor devices for decades [4]. More recently, acridine motifs have found additional applications such as cell imaging probes [5], catalysis [6], Organic Light-Emitting Diodes (OLEDs) [7] and organic semiconductors [8]. The modulation and/or enhancement of these properties drove the development of synthetic methodologies to (1) construct the acridine backbone; (2) selectively install substituents; (3) modulate the substitution pattern, fusing additional rings towards extended molecules; and (4) induce distortion from planarity by including a partially saturated fragment [9,10,11,12,13,14,15,16,17].
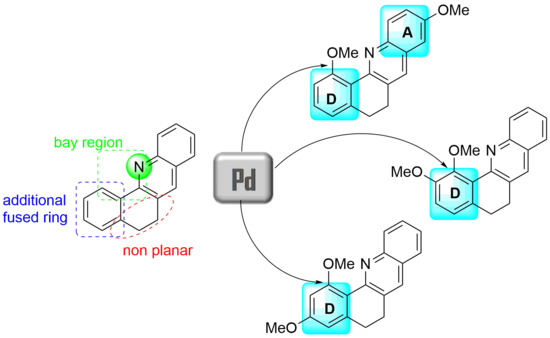
In this context, 5,6-dihydrobenzo[c]acridine is an intriguing member of the large aza-polycyclic compound family. Indeed, 5,6-dihydrobenzo[c]acridine is a tetracyclic molecule comprising four fused cycles including one pyridine ring and one partially hydrogenated cycle (Figure 1). In fact, this molecule represents a mix of bicyclic quinoline and tricyclic benzo[h]quinoline or acridine scaffolds. Within the structure of 5,6-dihydrobenzo[c]acridine, the presence of a nitrogen atom, an additional condensed ring, and a cyclic “dihydro” fragment provides its originality and interest by comparison with the parent and fully aromatic quinoline or (benzo)acridine skeletons. Moreover, the joint presence of a nitrogen atom and an peri-fused aromatic ring defines an aza-bay region and the presence of the non-planar ethylene bridge induces a deviation from the planarity compared with fully aromatic analogues. This deviation from planarity has been exploited recently in the preparation of helical-shaped molecules and the evaluation of photophysical and magnetic properties of helicate-like ligands [18,19,20].
As already mentioned, installation of substitution at the 5,6-dihydrobenzo[c]acridine platform proved to be crucial to modulate both properties and shape of 3D-shaped molecular architectures. 5,6-Dihydrobenzo[h]acridine targets are usually obtained by two main routes using (1) Friedländer cyclisation between tetralone derivatives and o-aminoacetophenones [18] or (2) thermally-induced or acid-catalyzed cyclisation of 1-halovinyl-2-carboxaldehyde derivatives and anilines [14,15,20,21,22]. In both methodologies, substituents on rings A, C, and D usually arise from commercially available starting reactants. Post-functionalization of the acridine motif at both strategic sites (positions 1 and 11 in Figure 1) is more challenging. As already described, only the presence of bromide or iodide atoms at both positions allows metal-catalyzed installation of substituents. As examples, the formation of C-C and C-N bonds was achieved using Pd- and Cu-catalyzed strategies, respectively, from precursor bearing an iodide atom at position 11 [21] and the formation of homocoupling products was realized using the Cu-catalyzed Ullman reaction from precursor bearing a bromide atom at position 1 [18,19]. The development of methodologies that avoid the mandatory presence of halides represents a challenging alternative in the 5,6-dihydrobenzo[c]acridine series. In deep contrast to the fully aromatic benzo[h]quinoline derivatives where C-H activation and the formation of the corresponding metallacycles (Ru, Pd, Ir) at position 1 are well known and documented [23,24,25,26,27,28], C-H activation in the 5,6-dihydrobenzo[c]acridine series is scarcely reported [29]. In the dihydro analogues, the crucial point was whether the distortion from planarity due to the presence of the partially hydrogenated ring C would allow or hamper the transient palladacycle to form through C-H activation and the selective installation of substituents at position 1. In this communication, we disclose our preliminary results in the Pd-catalyzed alkoxylation via C-H bond activation within the 5,6-dihydrobenzo[c]acridine series (Figure 2).
2. Results and Discussion
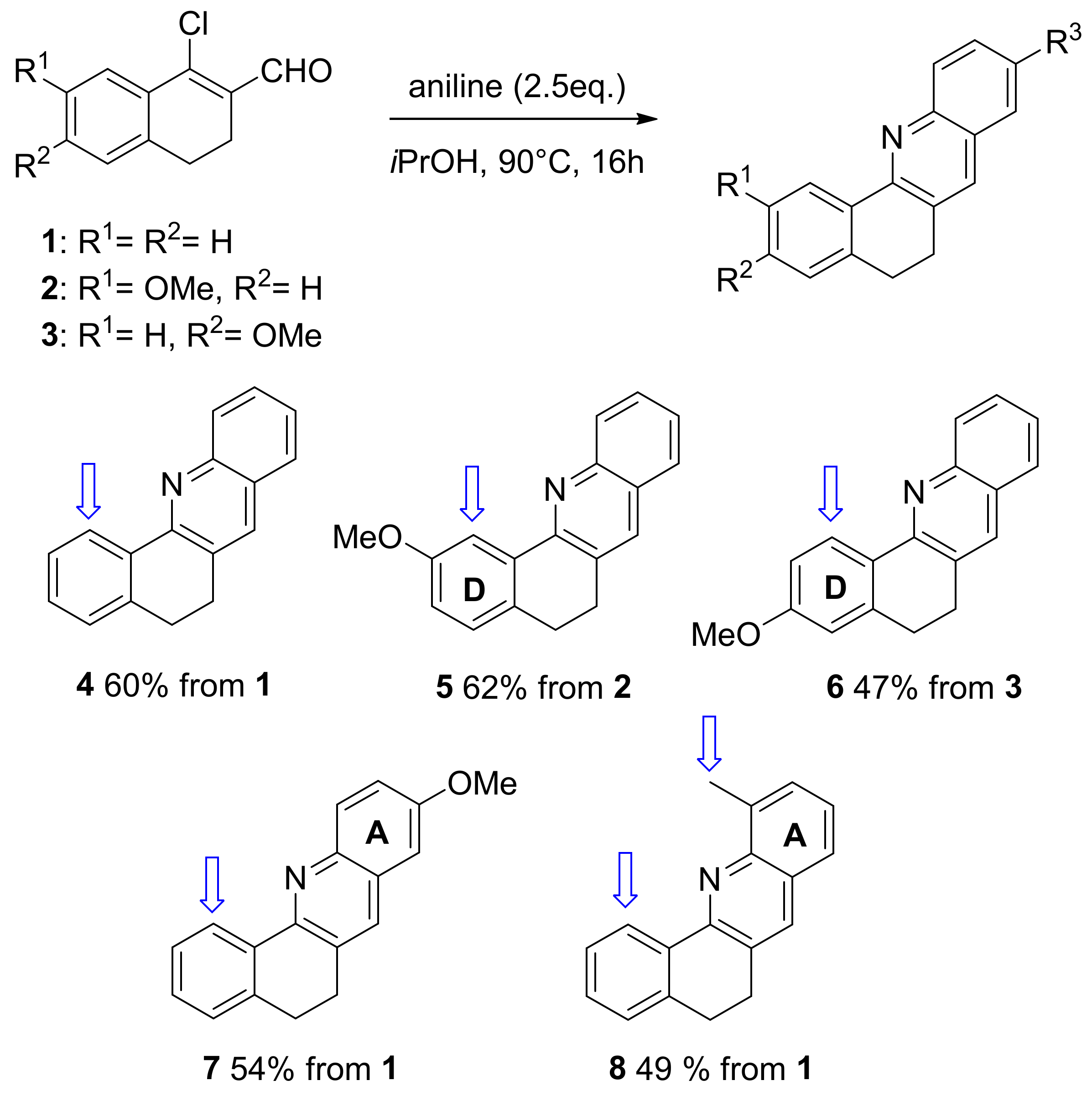
First we focused on the preparation of variously substituted acridines (Scheme 1). Based on our previous reports [15,20,21], we envisioned the synthesis of the acridine platforms from 1-chlorovinylcarboxaldehydes 1–3 (see supplementary material). The latter are readily obtained from commercially available tetralones and Vilsmeier–Haack reagent in high to quantitative yields [14,15,30]. Compounds 1–3 were reacted with 2.5 eq. of aniline derivatives in iPrOH at 90 °C for 16 h to yield acridines 4 to 7 in yields ranging from 47 to 60%. We chose a combination of various anilines including aniline, p-anisidine, and o-toluidine, and substituted 1-chlorovinylcarboxaldehydes 1–3 in order to prepare acridines which display a different substitution pattern. Indeed, as shown in Scheme 1, acridines 5 and 6 are substituted at ring D in the 2 and 3 positions, respectively. In contrast, in acridines 7 and 8, substituents are located at ring A in the 9 and 11 positions, respectively.
With acridines 4 to 8 in hand, our next goal was to examine the C-H activation step. As represented in scheme 1, the acridine platform is expected to undergo cyclometallation at the 1 position in good agreement with previous reports [23,24,25,26,27,28] dealing with the fully aromatic benzo[h]quinoline analogues. Thus Pd-catalyzed alkoxylation should take place in the 1 position for substrates 4 to 7. In contrast, acridine 8 is a more challenging substrate which displays two potential reaction sites: the sp2 carbon atom located in the 1 position at ring D and the sp3 benzylic carbon atom located at ring A. Indeed, both C-H bond might afford a five membered cyclometallated adduct [31] and thus undergo subsequent alkoxylation.
Substrate 4 was selected for initial investigation because it presents one single bond C-H(1) for directed C-H activation. Various Pd-based conditions were tested in order to determine suitable catalytic combination for the alkoxylation reaction.
We found that Pd(OAc)2 (10%) was effective to obtain 1-methoxy-5,6-dihydrobenzo[c]acridine 9 in 82% yield (Scheme 2). Among several oxidants tested, PhI(OAc)2 (2 eq.) proved superior to I2 or oxone. The solvent was also a crucial parameter to ensure high conversion. Indeed, only the use of MeOH at 100 °C in a sealed tube gave the expected alkoxy acridine 9 in high yield. Decreasing the temperature even to 80 °C led to a severe decrease of conversion. Mixtures of solvents such as dioxane/MeOH similarly afforded poor conversion. The use of dichloroethane (DCE)/ MeOH as the solvent led to mixtures of 1-methoxy and 1-chloro derivatives 9 and 10. The formation of the C-Cl bond could be unambiguously evidenced when the reaction was realized in DCE without the presence of MeOH. In this case, compound 10 was isolated in 63% yield. Moving from MeOH to EtOH and iPrOH led to different issues. If the use of EtOH afforded the ethoxy analogue 11 in satisfactory 61% yield, iPrOH failed to react.
1H NMR analysis of crude products allows an easy identification of both reactants and products. Indeed, except for compound 5, all other acridines 4, 6–8 display characteristic chemical shifts for H(1) ranging from 8.55 to 8.70 ppm. 1H NMR of compound 4 shows two characteristic signals at 8.60 and 8.30 ppm, accounting for protons H(1) and H(11). As evidenced by Figure 3, alkoxylation or chlorination is selective at position 1 of the dihydrobenzo[c]acridine platform. Indeed, only H(11) remains unchanged in both cases.
Based on mechanistic studies reported by Sandford [31] on related 2-phenylpyridines and benzo[h]quinoline, a potential catalytic cycle is shown in Figure 4. The latter would involve successively a ligand-directed C-H activation to form a cyclometallated dimer, oxidation to generate a Pd(IV) species, and a release of the product after C-O bond-forming reductive elimination. The number and role of other ancillary ligands remain under investigation and are represented as sticks in Figure 4.
The last step might proceed either by intramolecular C-OR bond elimination from the metal center or by attack of an external nucleophile in an “SN2-like” reaction as suggested recently [23]. The in situ transformation of PhI(OAc)2 with alcoholic solvents to afford PhI(OR)2 is also suggested as a key step in alkoxylation reactions which account for the obtention C-O bonds [32].
Thus Pd(OAc)2, PhI(OAc)2, in methanol(or ethanol) at 100 °C afforded suitable conditions to promote alkoxylation in the dihydrobenzo[c]acridine series. With this conditions in hand, we next tried to install an additional methoxy group when ring D is already bearing a methoxy substituent, in order to prepare 1,2- and 1,3-bismethoxy acridine motifs (Scheme 3). Under the aforementioned conditions, acridines 12 and 13 were readily obtained in 70 and 59% isolated yield, respectively. Thus, the presence of a strong donating group at ring D does not hamper the C-H activation and the subsequent C-O bond formation.
Under similar catalytic conditions, acridine 7 afforded the expected bismethoxy derivative 14 in 81% yield (Scheme 4). The latter compound displays complementary substitution pattern by comparison with acridines 12 and 13. In this case both rings A and D are independently functionalized.
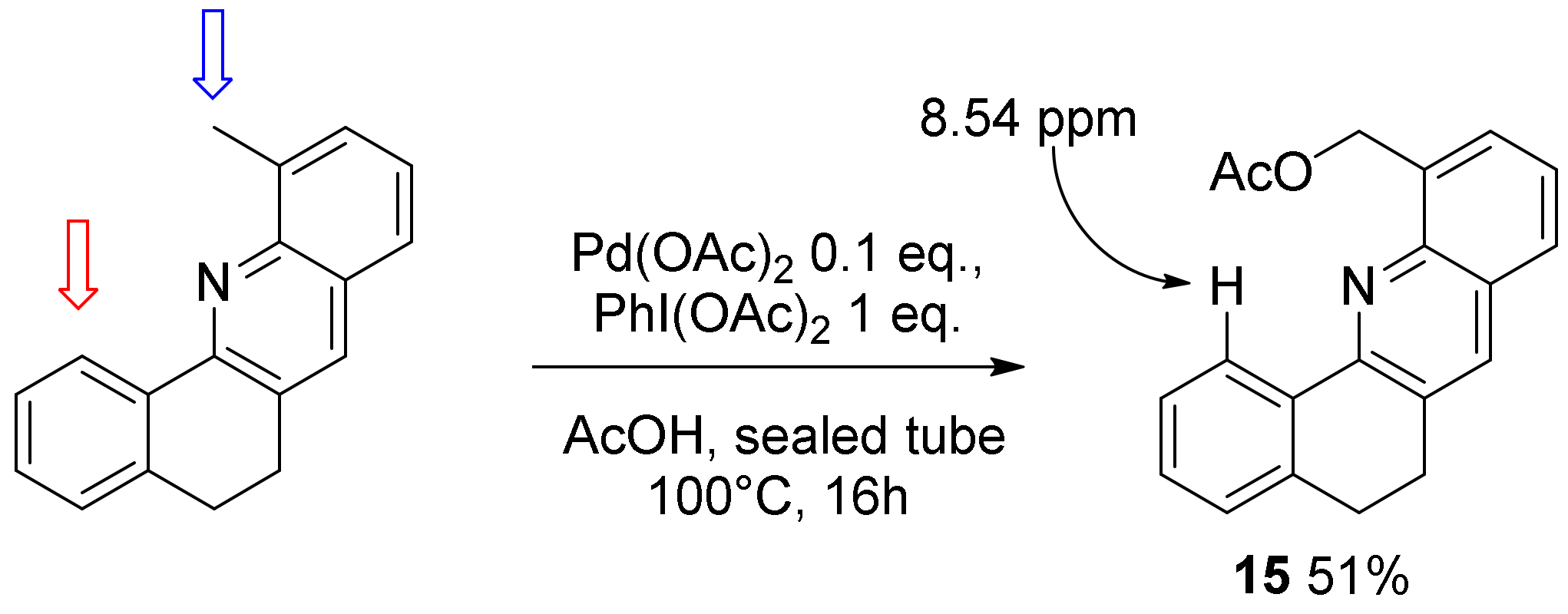
Finally, we decided to test our aforementioned C-H activation conditions in acridine 8. In contrast to acridine substrates 4–7, compound 8 displays two different C-H activation sites. Each of them might produce a transient five-membered palladacycle through C-H activation and might allow alkoxylation (Scheme 5). Unfortunately, under the aforementioned conditions in MeOH at 100 °C using one or two equivalents of oxidant, complex mixtures of alkoxylated products were obtained. In contrast, moving from MeOH to AcOH and using one equivalent of PhI(OAc)2 allowed to isolate acridine 15 as the major product in 51% isolated yield. 1HNMR spectra evidenced the presence of the characteristic signal of H(1), which resonates at 8.54 ppm.
3. Conclusions
In conclusion, we succeeded in the alkoxylation of 5,6-dihydrobenzo[h]acridine via Pd-catalyzed C-H activation. Alkoxylation occurs selectively in position 1 of the acridine platform using 10% Pd(OAc)2, PhI(OAc)2 and MeOH as the best combination of catalyst, oxidant and solvent, respectively. Several bismethoxy acridine derivatives bearing all substituents at ring D or at both ring D and A have been successfully obtained. Our strategy allowed a selective functionalization of sp3 carbon atom located at the benzylic position of ring A. Current studies are focused on further exploration of the substrate scope and the extension of this methodology to the selective formation of C-C bonds via C-H activation at 5,6-dihydrobenzo[h]acridine architectures.
4. Materials and Methods
4.1. General Information
All reagents and solvents were obtained from commercial sources and used without further purification. Reactions were routinely carried out under nitrogen and argon atmosphere with magnetic stirring. 1H and 13C NMR spectra were recorded on a Bruker AV1 300 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) working at 300 MHz, 75 MHz respectively for 1H and 13C, with chloroform-d as solvent. Chemicals shifts were reported in δ, parts per million (ppm), relative to chloroform (δ = 7.28 ppm) as international standards unless otherwise stated for proton nuclear magnetic resonance (1H NMR). Chemical shifts for carbon nuclear magnetic resonance (13C NMR) were reported in δ, parts per million (ppm), relative to the center line of the chloroform triplet (δ = 77.07 ppm). Coupling constants, J, were reported in Hertz (Hz) and refer to apparent peak multiplicities and not true coupling constants. The abbreviations s, d, dd, t, q, br and m stand for resonance multiplicities singlet, doublet, doublet of doublet, triplet, quartet, broad, and multiplet, respectively. Allylation diastereoselectivity was determined by 1H NMR integrations of the methylene signals in the crude products. High resolution mass spectrometry data were recorded with an accuracy within 5 ppm on a quadrupole-TOF mass spectrometer (Xevo Q-Tof, Waters, Guyancourt, France) using an electrospray ionization source operating in positive mode. Thin-layer chromatography (TLC) was carried out on aluminum sheets pre-coated with silica gel plates (Fluka Kiesel gel 60 F254, Merck, Bucharest, Romania) and visualized by a 254 nm UV lamp and potassium permanganate. Melting points (Mp) were determined on a System Kofler type WME apparatus (Fisher Scientific SAS, Illkirch, France).
4.2. General Procedure for Pd-Catalyzed Alkoxylation
The acridine derivative (1 eq.), PhI(OAc)2 (2 eq.), and Pd(OAc)2 (0.1 eq.) were place in screw-capped tube. MeOH (3 mL) was next added and the reaction mixture was stirred for 15 min. The tube was sealed and the suspension was heated with stirring to 100 °C for 16 h. The crude mixture was filtered through Celite and the solvent evaporated. The solid residue was extracted between ethyl acetate and successively water and brine. The organic layers were dried over sodium sulfate and the solvent was removed under vacuum. In all cases the residue was purified by flash column chromatography on silica gel (petroleum ether/dichloromethane, 6:4) to afford the expected alkoxyacridine derivative.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/8/4/139/s1, experimental procedure for the alkoxylation reaction couplings as well as analytical data for new compounds.
Acknowledgments
University of Versailles St Quentin, MENRT-France, and LabEx CHARMMMAT (ANR-11-LABEX-0039) are gratefully acknowledged for financial supports and grant (BL).
Author Contributions
Benjamin Large and Damien Prim performed the experiments, Anne Gaucher, Aurélie Damond and Flavien Bourdreux analyzed the data. Damien Prim wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhou, Y.-J.; Chen, D.-S.; Li, Y.-L.; Liu, Y.; Wang, X.-S. Combinatorial synthesis of pyrrolo[3,2-f]quinoline and pyrrolo[3,2-a]acridine derivatives via a three-component reaction under catalyst-free conditions. ACS Comb. Sci. 2013, 15, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-M.; Ramiandrasoa, F.; Guetzoyan, L.; Pradines, B.; Quintino, E.; Gadelle, D.; Forterre, P.; Cresteil, T.; Mahy, J.-M.; Pethe, S. Synthesis and biological evaluation of acridine derivatives as antimalarial agents. ChemMedChem 2012, 7, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Suryadi, J.; Bierbach, U. Cellular recognition and repair of monofunctional-interactive platinum-DNA adducts. Chem. Res. Toxicol. 2015, 28, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Geddes, C.D. Optical thin film polymeric sensors for the determination of aqueous chloride, bromide and iodide ions at high pH, based on the quenching of fluorescence of two acridinium dyes. Dyes Pigment. 2000, 45, 243–251. [Google Scholar] [CrossRef]
- Warther, D.; Bolze, F.; Leonard, J.; Gug, S.; Specht, A.; Puliti, D.; Sun, X.-H.; Kessler, P.; Lutz, Y.; Vonesch, J.-L.; et al. Live-cell one- and two-photon uncaging of a far-red Emitting acridinone fluorophore. J. Am. Chem. Soc. 2009, 132, 2585–2590. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Yoo, C.; Lee, Y. Direct CO2 addition to a Ni(0)–CO species allows the selective generation of a Nickel(II) carboxylate with expulsion of CO. J. Am. Chem. Soc. 2018, 140, 2179–2185. [Google Scholar] [CrossRef]
- Dos Santos, P.L.; Ward, J.S.; Bryce, M.R.; Monkman, A.P. Using guest–host interactions to optimize the efficiency of TADF Oleds. J. Phys. Chem. Lett. 2016, 7, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Kumar, V.; Singh, S.P.; Sharma, A.; Prakash, S.; Singh, C.; Anand, R.S. Non-aggregating solvatochromic bipolar benzo[f]quinolines and benzo[a]acridines for organic electronics. J. Mater. Chem. 2012, 22, 14880–14888. [Google Scholar] [CrossRef]
- Martins, A.P.; Frizzo, C.P.; Moreira, D.N.; Buriol, L.; Machado, P. Solvent-free heterocyclic synthesis. Chem. Rev. 2009, 109, 4140–4182. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fan, Q.; Jiang, X. Nitrogen-iodine exchange of dirayliodonium salts: Access to acridine and carbazole. Org. Lett. 2018, 20, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, P.; He, M.; Wu, Q.; Ye, L.; Mu, Y. Facile synthesis of acridine derivatives by ZnCl2-promoted intramolecular cyclization of o-arylaminophenyl Schiff bases. Org. Lett. 2014, 16, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-J.; Chen, W.-W.; Li, Y.; Xu, M.-H. Facile synthesis of acridines via Pd(0)-diphosphine complex-catalyzed tandem coupling/cyclization protocol. Org. Biomol. Chem. 2015, 13, 6580–6586. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Kindelin, P.J.; Klumpp, D.A. Charge migration in dicationic electrophiles and its application to the synthesis of aza-polycyclic aromatic compounds. Org. Lett. 2006, 8, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, P.; Meena Rani, A.; Saiganesh, R.; Balasubramanian, K.K.; Kabilan, S. Synthesis of 5,6-dihydrobenz[c]acridines: A comparative study. Tetrahedron 2009, 65, 811–821. [Google Scholar] [CrossRef]
- Souibgui, A.; Gaucher, A.; Marrot, J.; Bourdreux, F.; Aloui, F.; Ben Hassine, B.; Prim, D. New series of acridines and phenanthrolines: Synthesis and characterization. Tetrahedron 2014, 70, 3042–3048. [Google Scholar] [CrossRef]
- Gogoi, S.; Shekarrao, K.; Duarah, A.; Bora, T.C.; Gogoi, S.; Boruah, R.C. A microwave promoted solvent-free approach to steroidal quinolines and their in vitro evaluation for antimicrobial activities. Steroids 2012, 77, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Mishra, S.; Kakde, B.N.; Dey, D.; Bisai, A. Expeditious approach to pyrrolophenanthridones, phenanthridines, and benzo[c]phenanthridines via organocatalytic direct biaryl-coupling promoted by potassium tert-butoxide. J. Org. Chem. 2013, 78, 7823–7844. [Google Scholar] [CrossRef] [PubMed]
- Jierry, L.; Harthong, S.; Aronica, C.; Mulatier, J.-C.; Guy, L.; Guy, S. Efficient dibenzo[c]acridine helicene-like synthesis and resolution: Scale up, structural control, and high chiroptical properties. Org. Lett. 2012, 14, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Speed, S.; Pointillart, F.; Mulatier, J.-C.; Guy, L.; Golhen, S.; Cador, O.; Le Guennic, B.; Riobé, F.; Maury, O.; Ouahab, L. Photophysical and magnetic properties in complexes containing 3d/4f elements and chiral phenanthroline-based helicate-like ligands. Eur. J. Inorg. Chem. 2017, 14, 2100–2111. [Google Scholar] [CrossRef]
- Souibgui, A.; Gaucher, A.; Marrot, J.; Aloui, F.; Mahuteau-Betzer, F.; Ben Hassine, B.; Prim, D. A Flexible strategy towards thienyl-, oxazolyl- and pyridyl-fused fluorenones. Eur. J. Org. Chem. 2013, 21, 4515–4522. [Google Scholar] [CrossRef]
- Solmont, K.; Boufroura, H.; Souibgui, A.; Fornarelli, P.; Gaucher, A.; Mahuteau-Betzer, F.; Ben Hassine, B.; Prim, D. Divergent strategy in the synthesis of original dihydro benzo- and dihydronaphtho-acridines. Org. Biomol. Chem. 2015, 13, 6269–6277. [Google Scholar] [CrossRef] [PubMed]
- Some, S.; Ray, J.K. Chemoselective arylamination of β-bromovinylaldehydes followed by acid catalyzed cyclization: A general method for polycyclic quinolines. Tetrahedron Lett. 2007, 48, 5013–5016. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A highly selective catalytic method for the oxidative functionalization of C-H bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Seki, B. Arylation using ruthenium catalyst. In Catalytic Transformations via C-H Activation; Yu, J.-Q., Ed.; Georg thieme Verlag KG: Stuttgart, Germany, 2016; Volume 1, pp. 119–153. ISBN 978-3-13-171141-0. [Google Scholar]
- Powers, D.C.; Benitez, D.; Tkatchouk, E.; Goddard, W.A., III; Ritter, T. Bimetallic reductive elimination from dinuclear Pd(III)complexes. J. Am. Chem. Soc. 2010, 132, 14092–14103. [Google Scholar] [CrossRef] [PubMed]
- Aiello, I.; Crispini, A.; Ghedini, M.; La Deda, M.; Barigelletti, F. Synthesis and characterization of a homologous series of mononuclear palladium complexes containing different cyclometalated ligands. Inorg. Chim. Acta 2000, 308, 121–128. [Google Scholar] [CrossRef]
- Selbin, J.; Gutierrez, M.A. Cyclometallation IV. Palladium(II) compounds with benzo[h]quinoline and substituted 2,6-diraylpyridines. J. Organomet. Chem. 1983, 246, 95–104. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Yuan, J.; Guo, S. Palladacycles incorporating a carboxylate-functionalized phosphine ligand: Syntheses, characterization and their catalytic applications toward Suzuki couplings in water. Transit. Met. Chem. 2017, 42, 727–738. [Google Scholar] [CrossRef]
- Li, C.; Sun, P.; Yan, L.; Pan, Y.; Cheng, C.-H. Synthesis and electroluminescent properties of Ir complexes with benzo[c]acridine or 5,6-dihydro-benzo[c]acridine ligands. Thin Solid Films 2008, 516, 6186–6190. [Google Scholar] [CrossRef]
- Sako, M.; Takeuchi, Y.; Tsujihara, T.; Kodera, J.; Kawano, T.; Takizawa, S.; Sasai, H. Efficient enantioselective synthesis of oxahelicenes using redox/acid cooperative catalysts. J. Am. Chem. Soc. 2016, 138, 11481–11484. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Schardt, B.C.; Hill, C.L. Preparation of iodobenzene dimethoxide. A new synthesis of [18O]iodosylbenzene and a reexamination of its infrared spectrum. Inorg. Chem. 1983, 22, 1563–1565. [Google Scholar] [CrossRef]
Figure 1.
Molecular structure, characteristic structural features, and strategic site of 5,6-dihydrobenzo[c]acridine.
Figure 1.
Molecular structure, characteristic structural features, and strategic site of 5,6-dihydrobenzo[c]acridine.

Figure 2.
Polysubstituted 5,6-dihydrobenzo[c]acridines via C-H bond activation.

Scheme 1.
General route towards variously substituted acridines 4–8. In blue C-H activation sites.

Scheme 2.
Determination of best experimental conditions for C-H activation of acridine 4.

Figure 3.
1H NMR characteristic signals for H(1) –red arrow and H(11) – blue arrow acridine derivatives 4, 9, and 10.
Figure 3.
1H NMR characteristic signals for H(1) –red arrow and H(11) – blue arrow acridine derivatives 4, 9, and 10.

Figure 4.
Possible mechanism for palladium-catalyzed regioselective alkoxylation.

Scheme 3.
Preparation of 1,2- and 1,3-bismethoxy acridine motifs 12 and 13.

Scheme 4.
Substitution at rings A and D of the acridine platform.

Scheme 5.
Selective C-H activation at the benzylic site.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Large, B.; Bourdreux, F.; Damond, A.; Gaucher, A.; Prim, D. Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series. Catalysts 2018, 8, 139. https://doi.org/10.3390/catal8040139
AMA Style
Large B, Bourdreux F, Damond A, Gaucher A, Prim D. Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series. Catalysts. 2018; 8(4):139. https://doi.org/10.3390/catal8040139
Chicago/Turabian StyleLarge, Benjamin, Flavien Bourdreux, Aurélie Damond, Anne Gaucher, and Damien Prim. 2018. "Palladium-Catalyzed Regioselective Alkoxylation via C-H Bond Activation in the Dihydrobenzo[c]acridine Series" Catalysts 8, no. 4: 139. https://doi.org/10.3390/catal8040139
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.