The Deoxygenation Pathways of Palmitic Acid into Hydrocarbons on Silica-Supported Ni12P5 and Ni2P Catalysts

Abstract
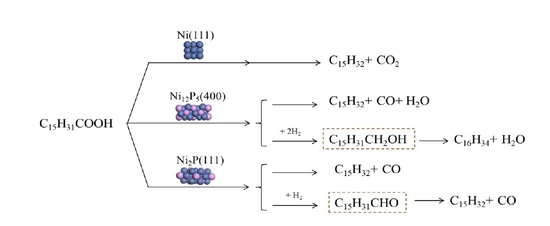
:
1. Introduction
2. Results and Discussion
2.1. Characterization of the Catalysts
2.1.1. Ni and P Elemental Analysis
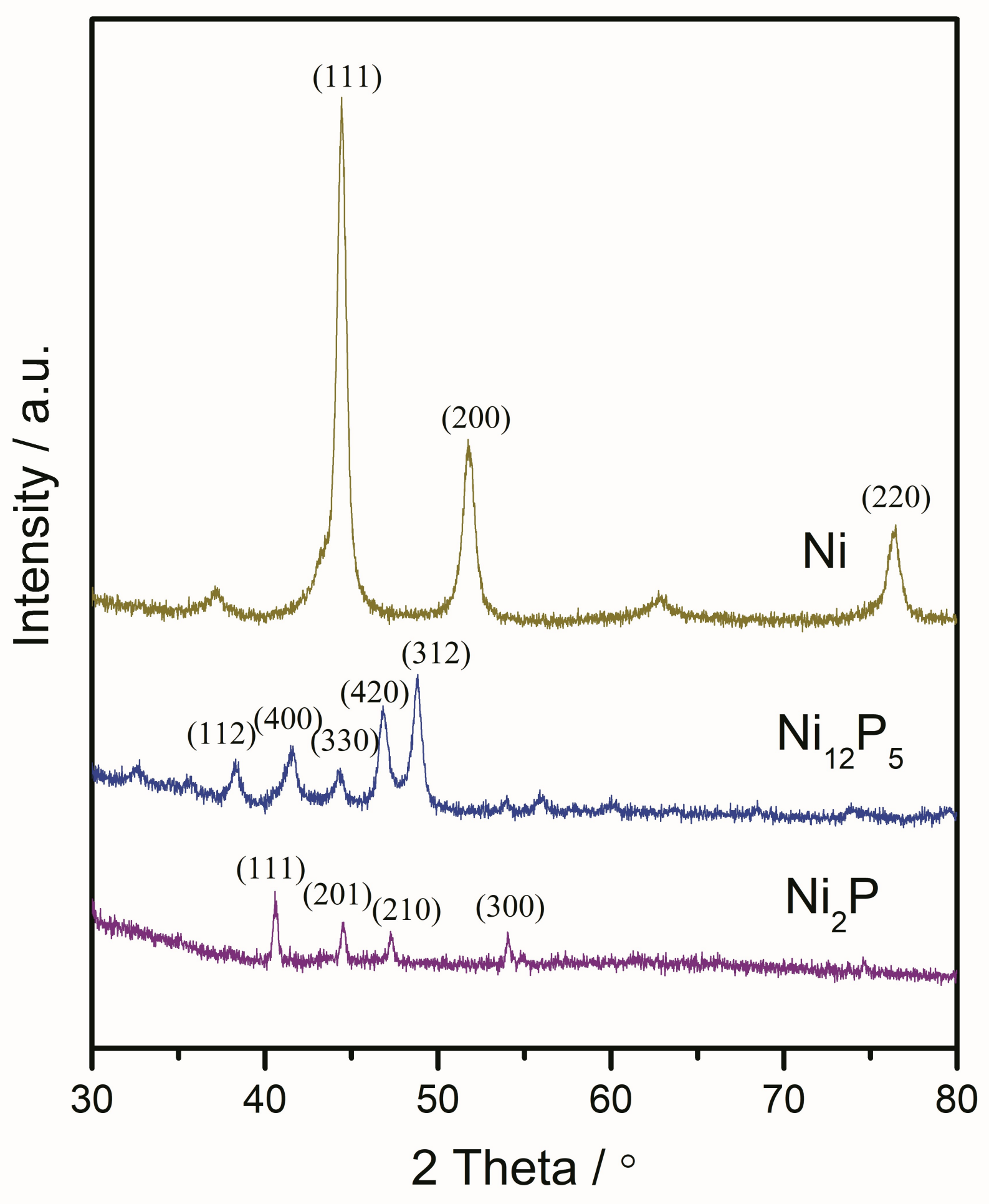
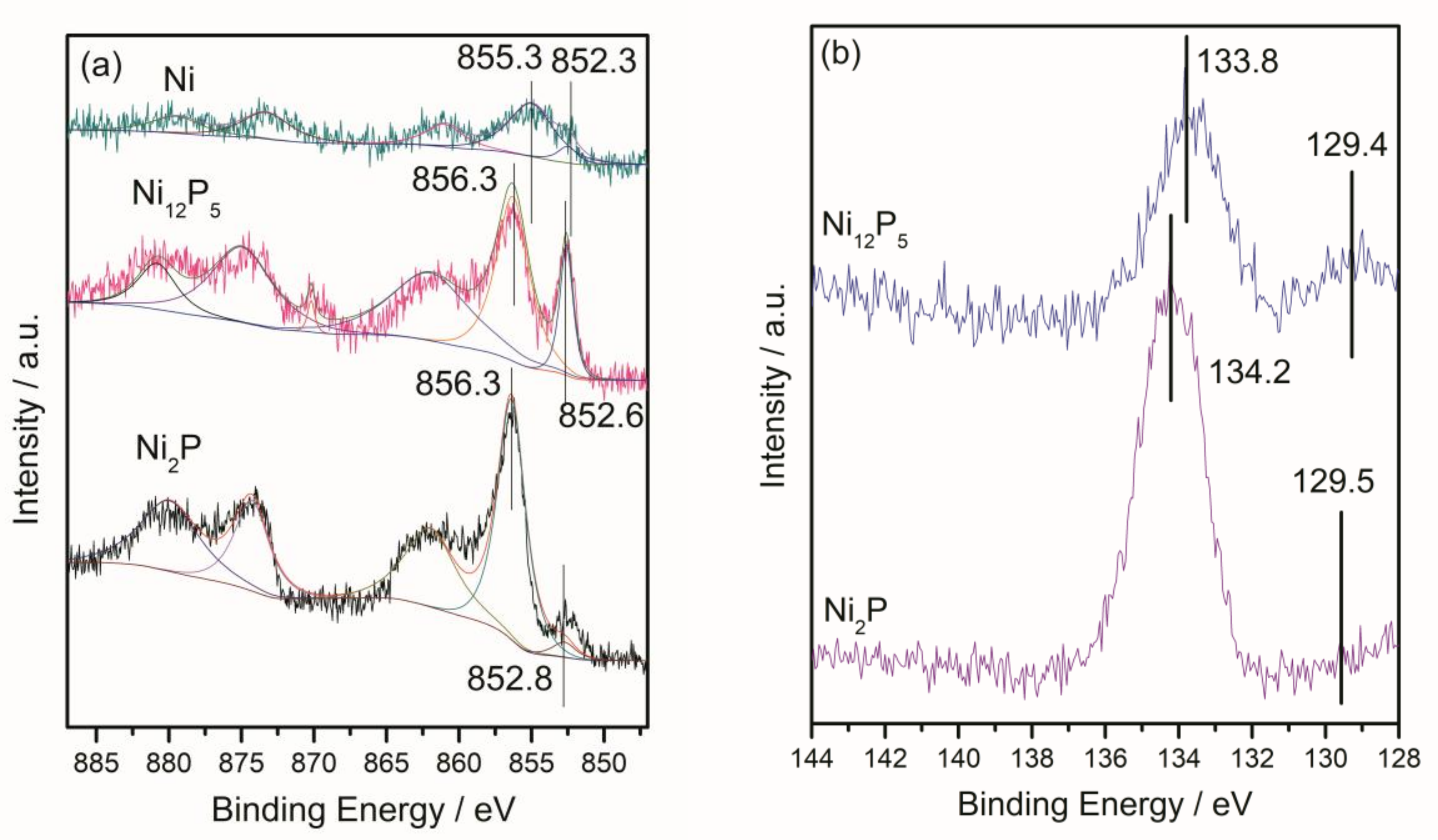
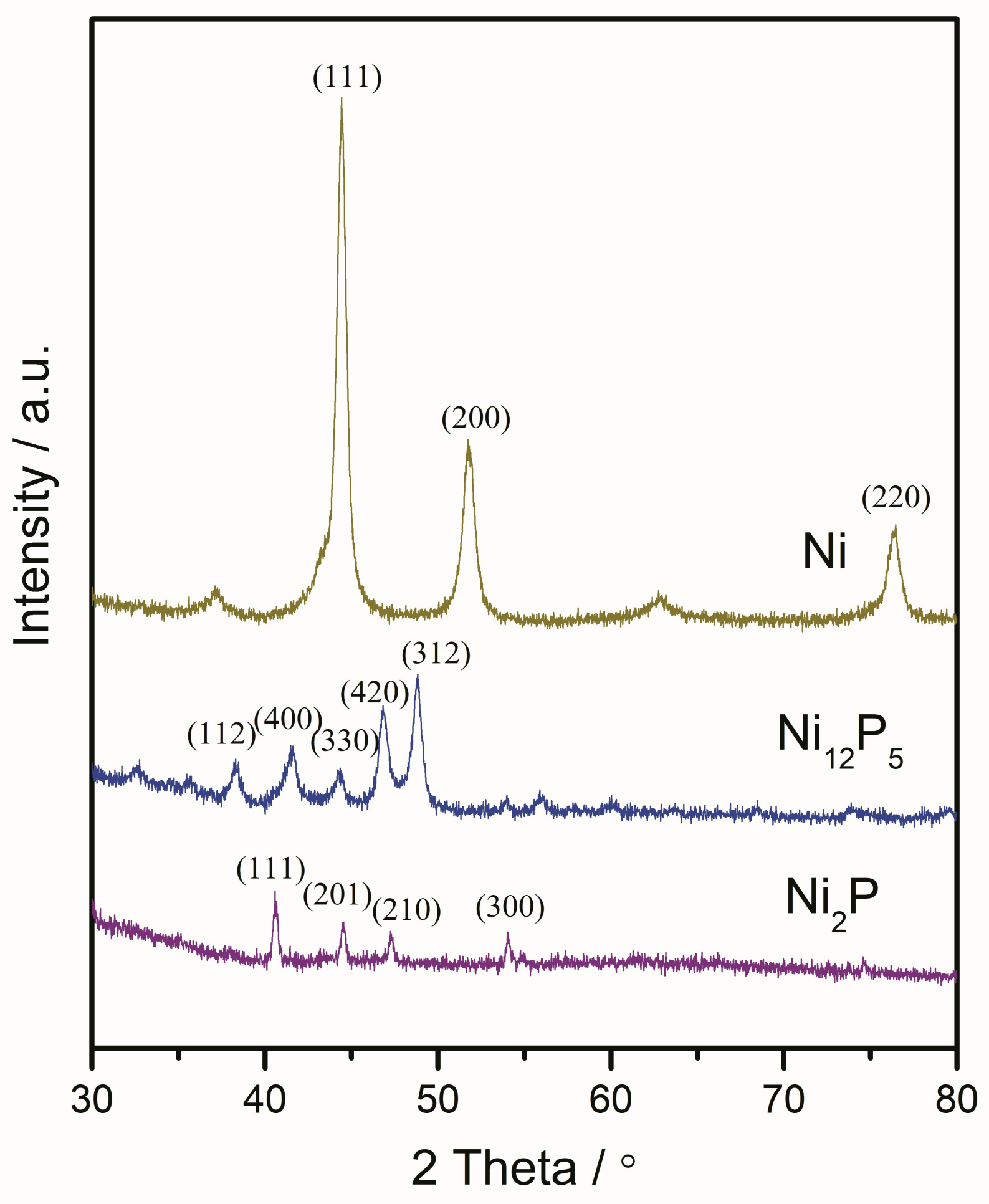
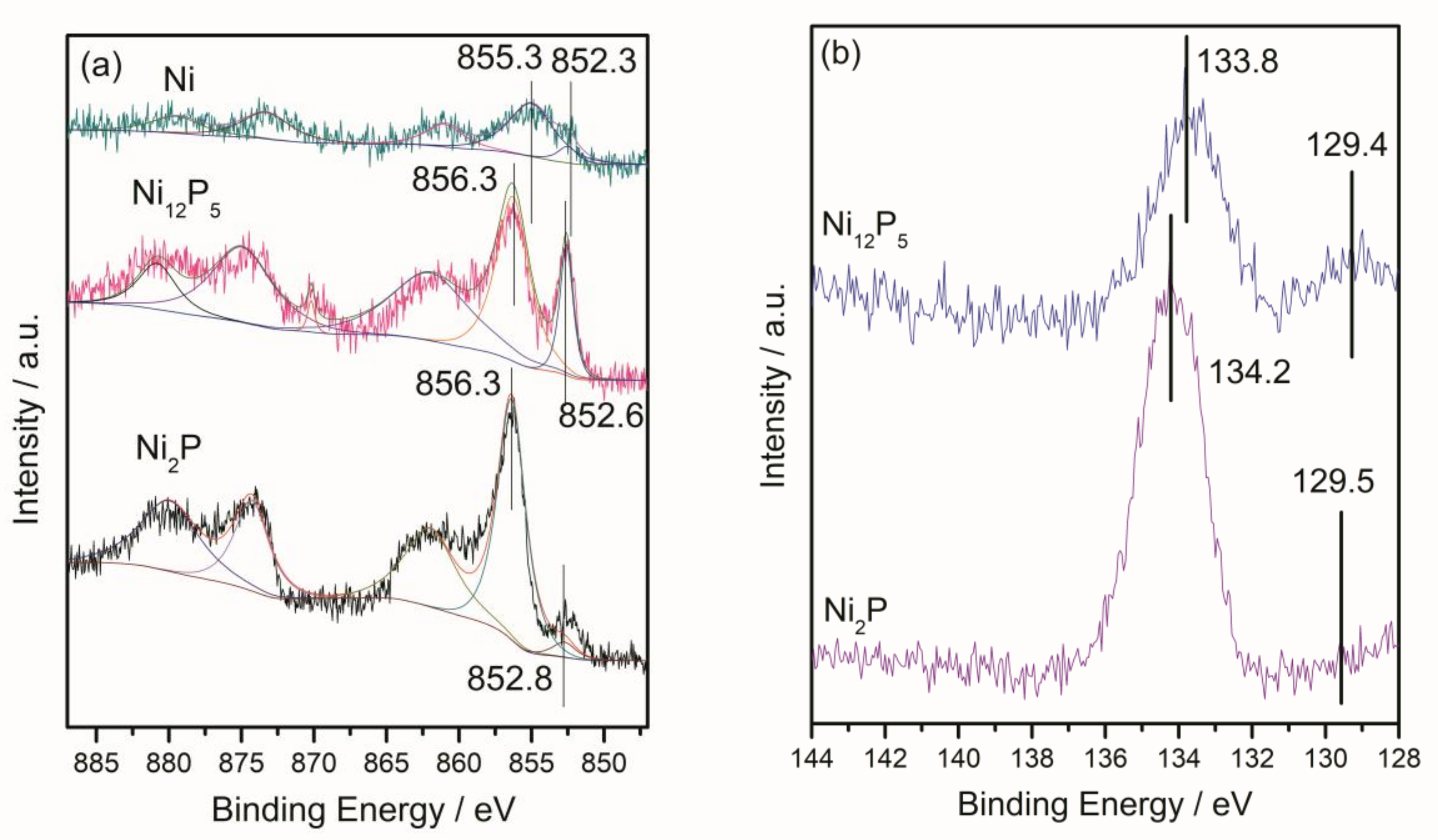
2.1.2. X-ray Diffraction (XRD) and X-ray Photoelectron Spectroscopy (XPS) Results
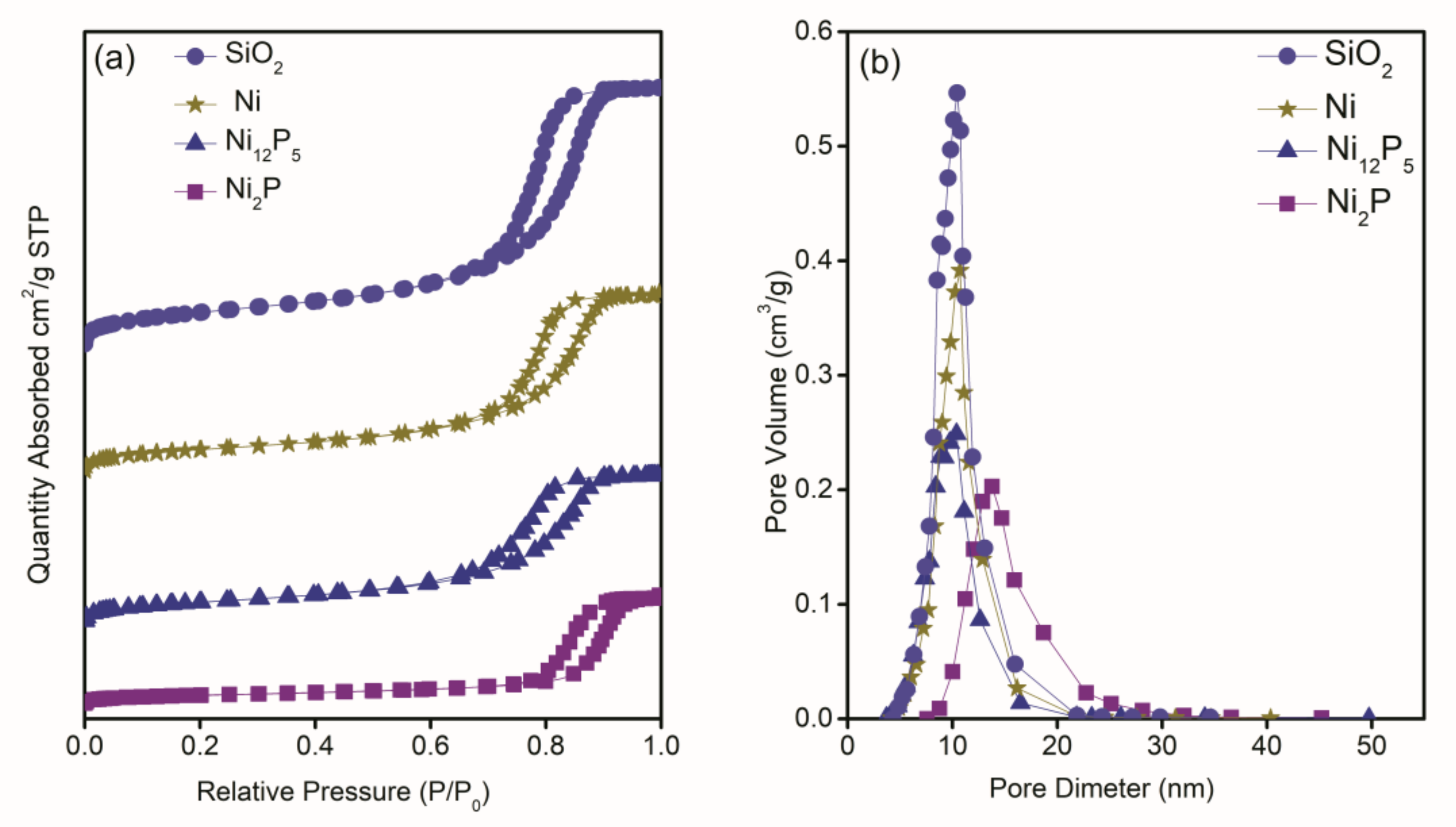
2.1.3. Textural Properties
2.1.4. H2-Chemisorption Measurements
2.1.5. Transmission Electron Microscopy (TEM)
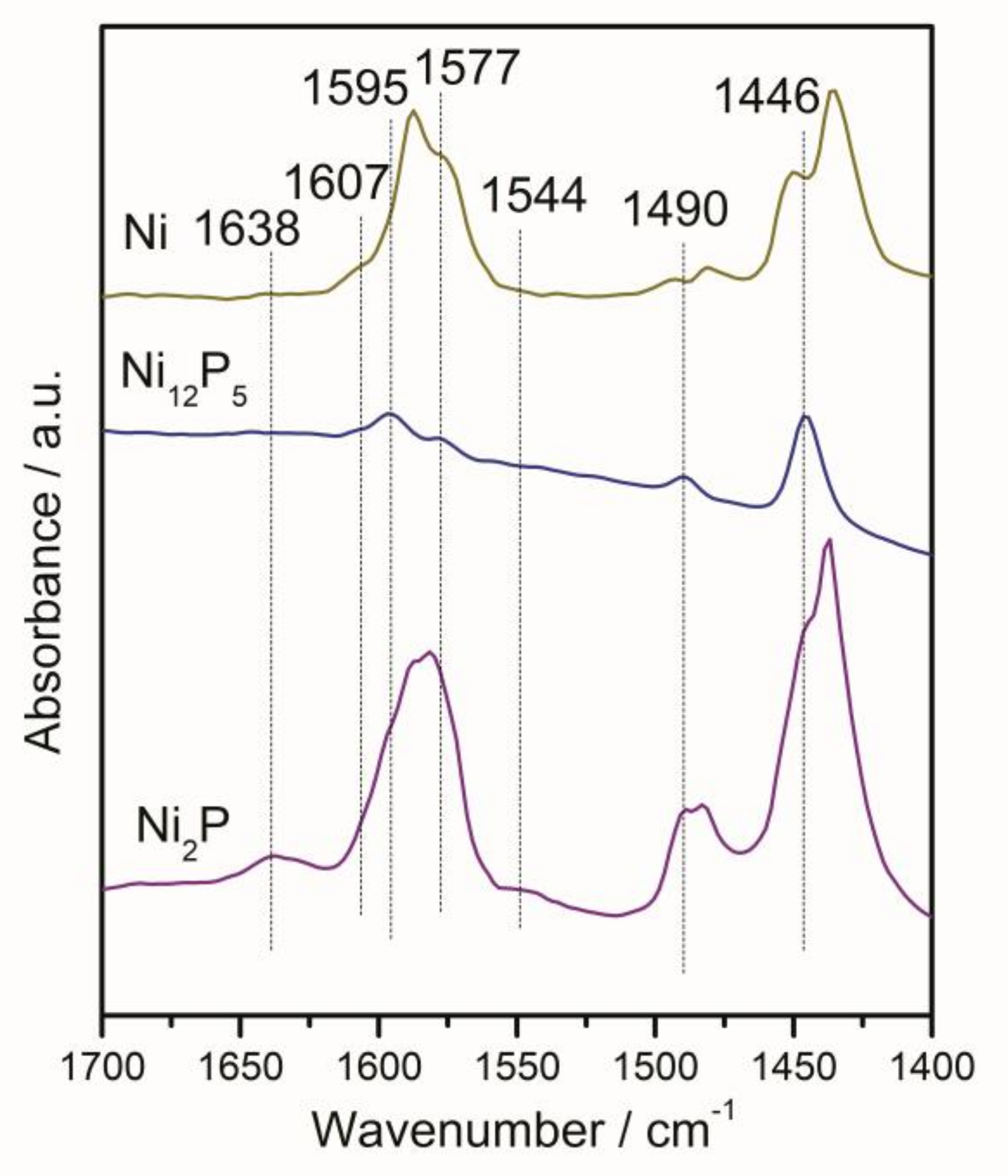
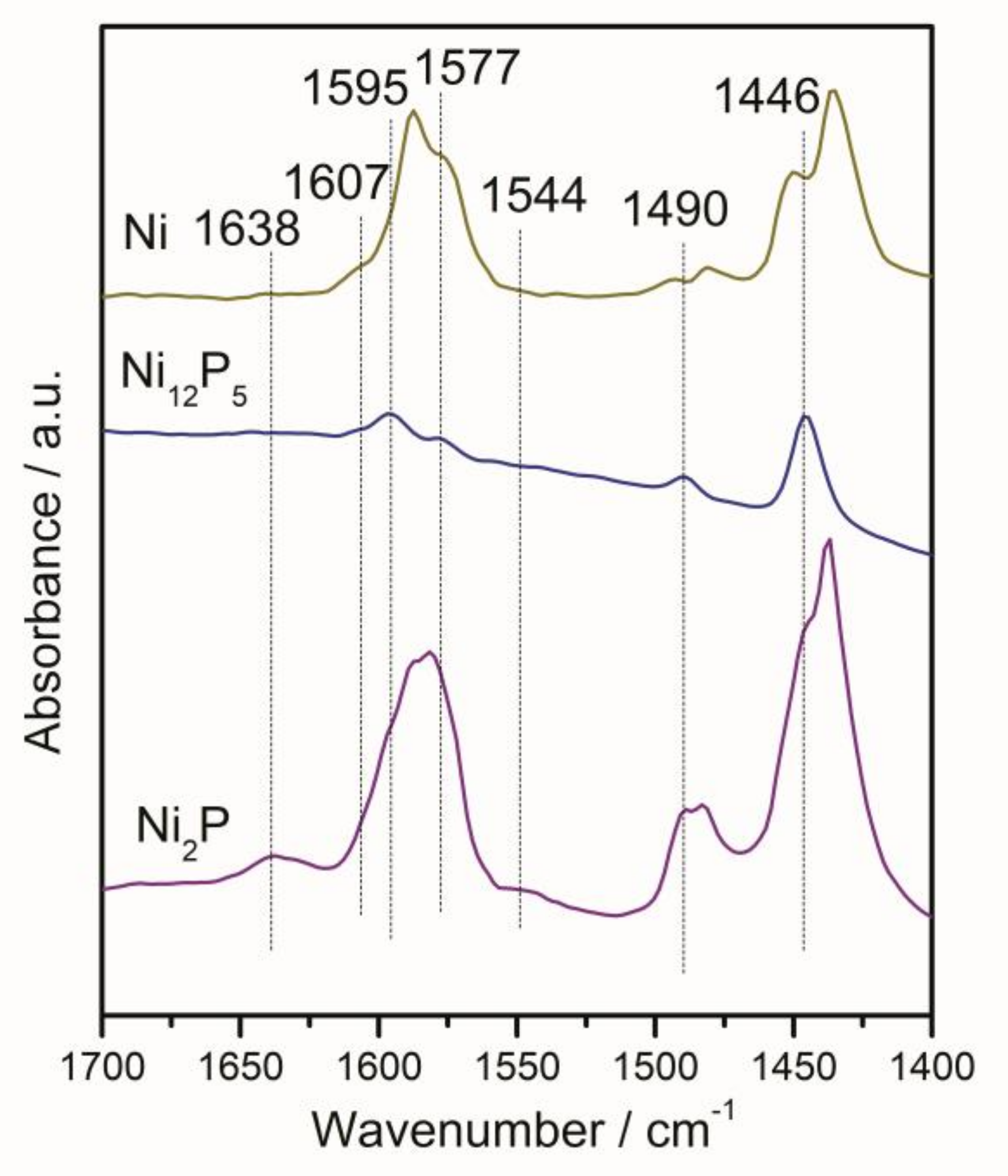
2.1.6. Infrared Spectroscopy of Pyridine Adsorption (Py-IR)
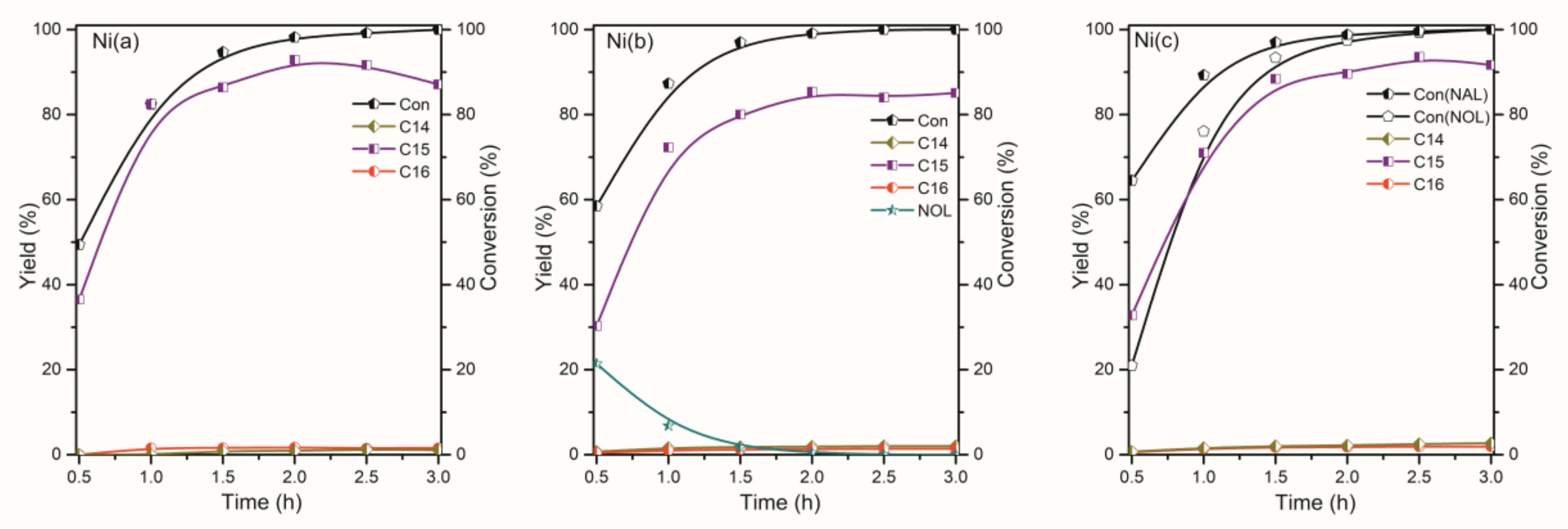
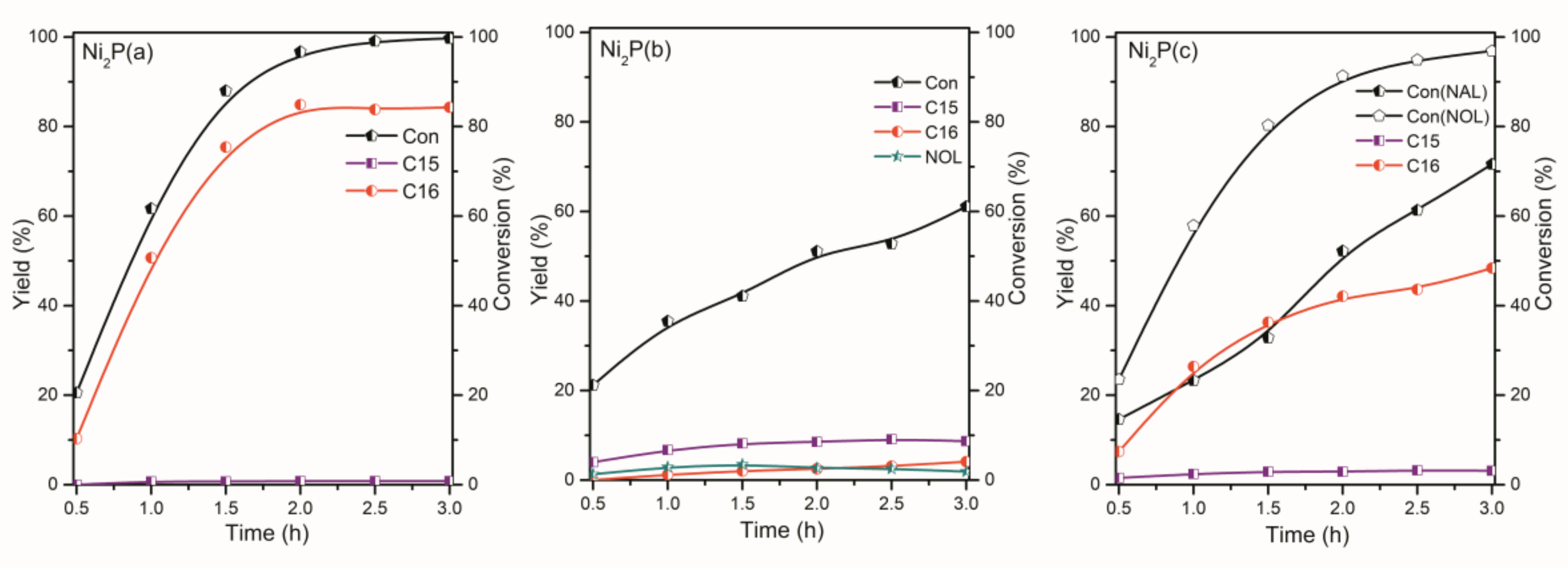
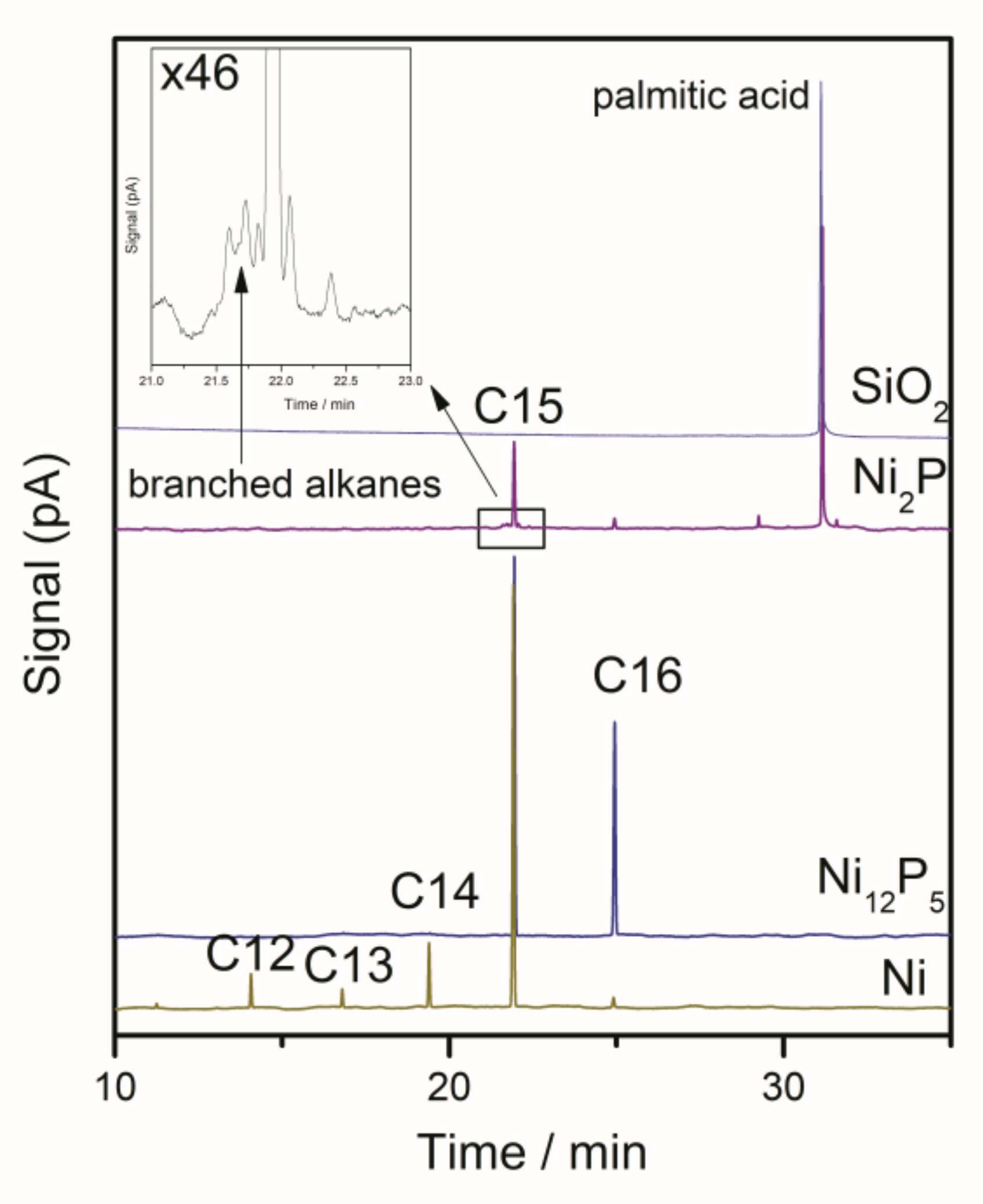
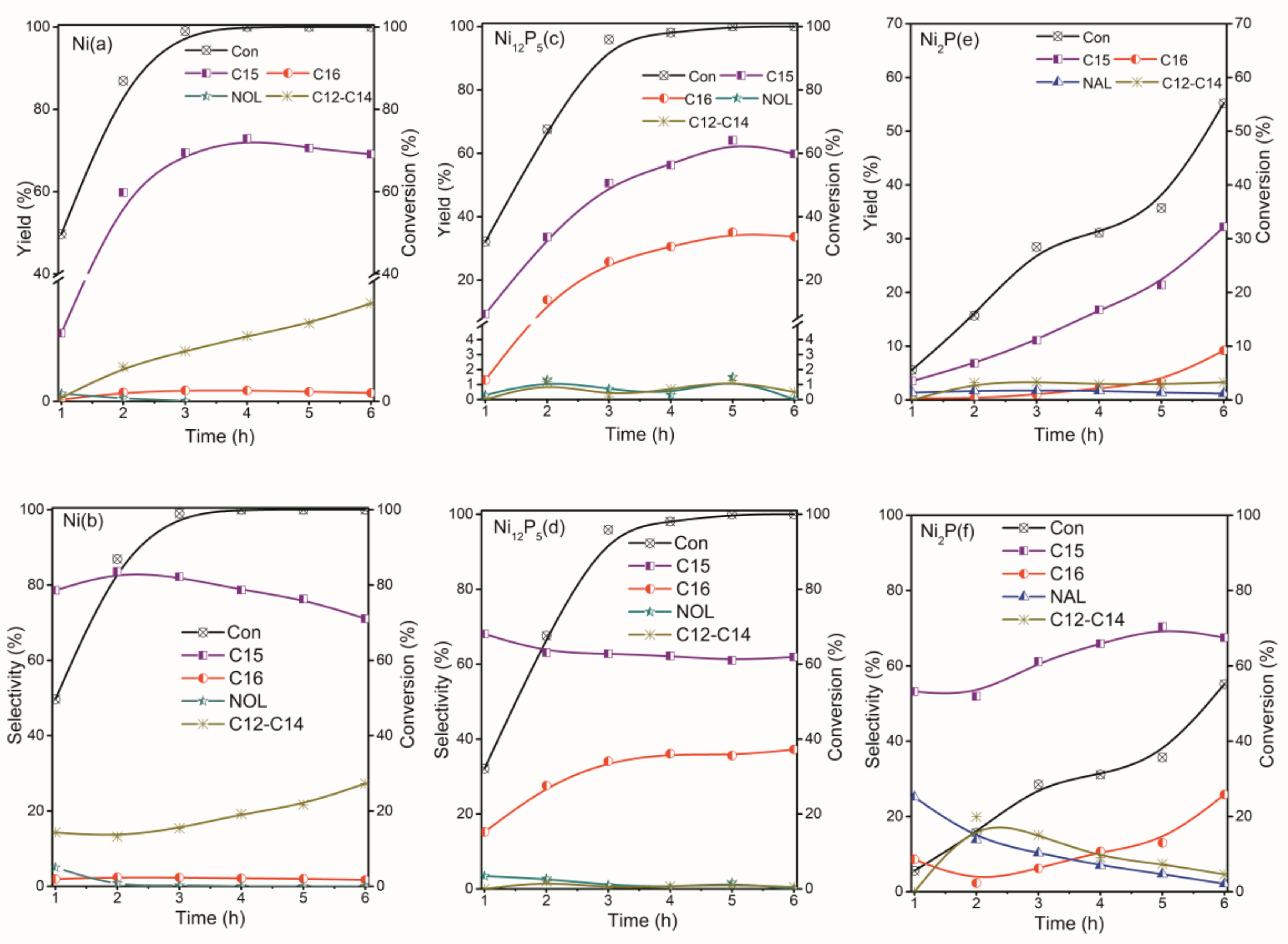
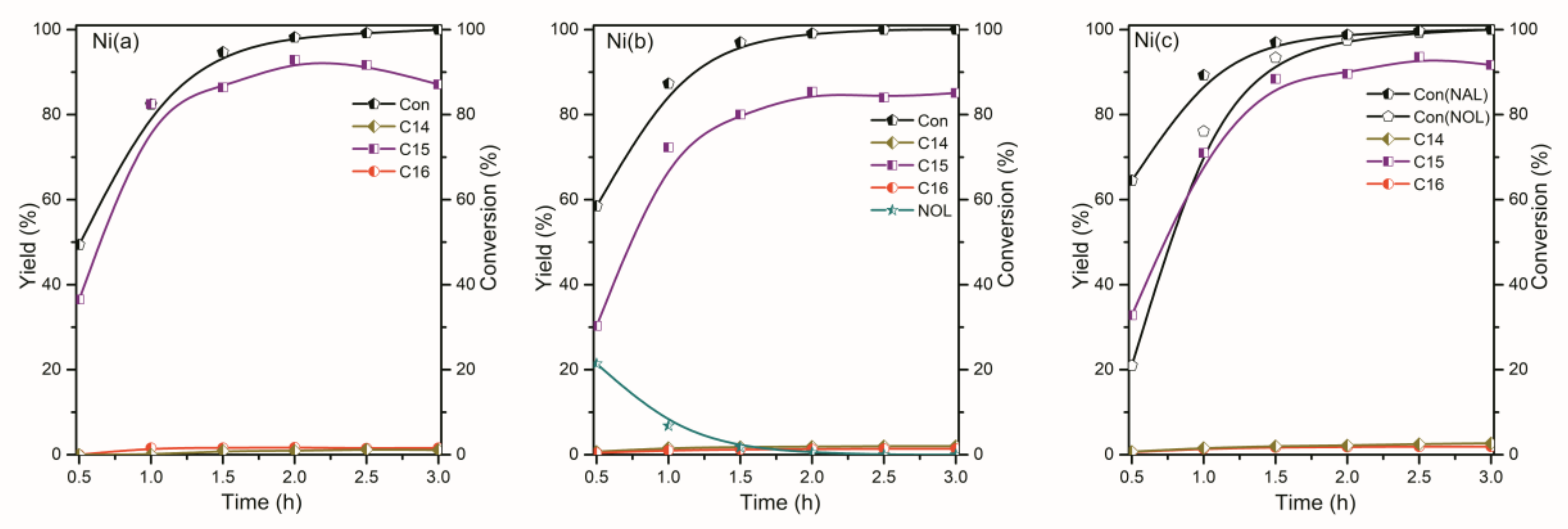
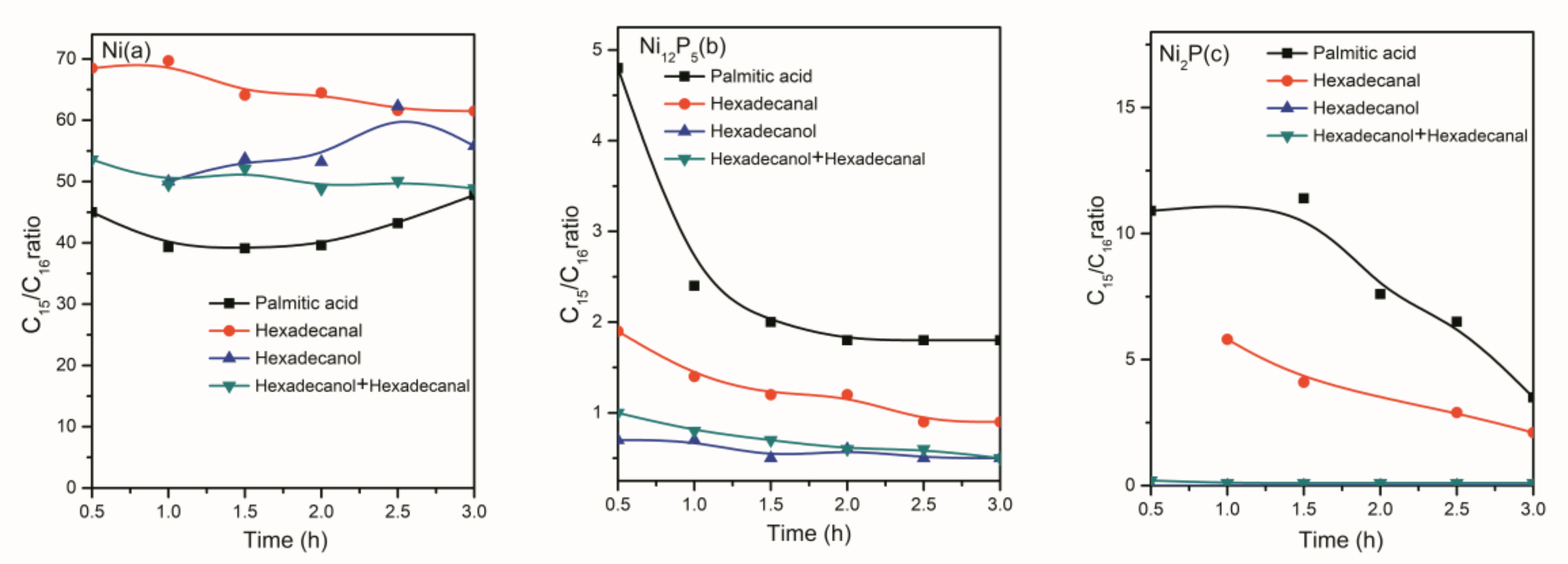
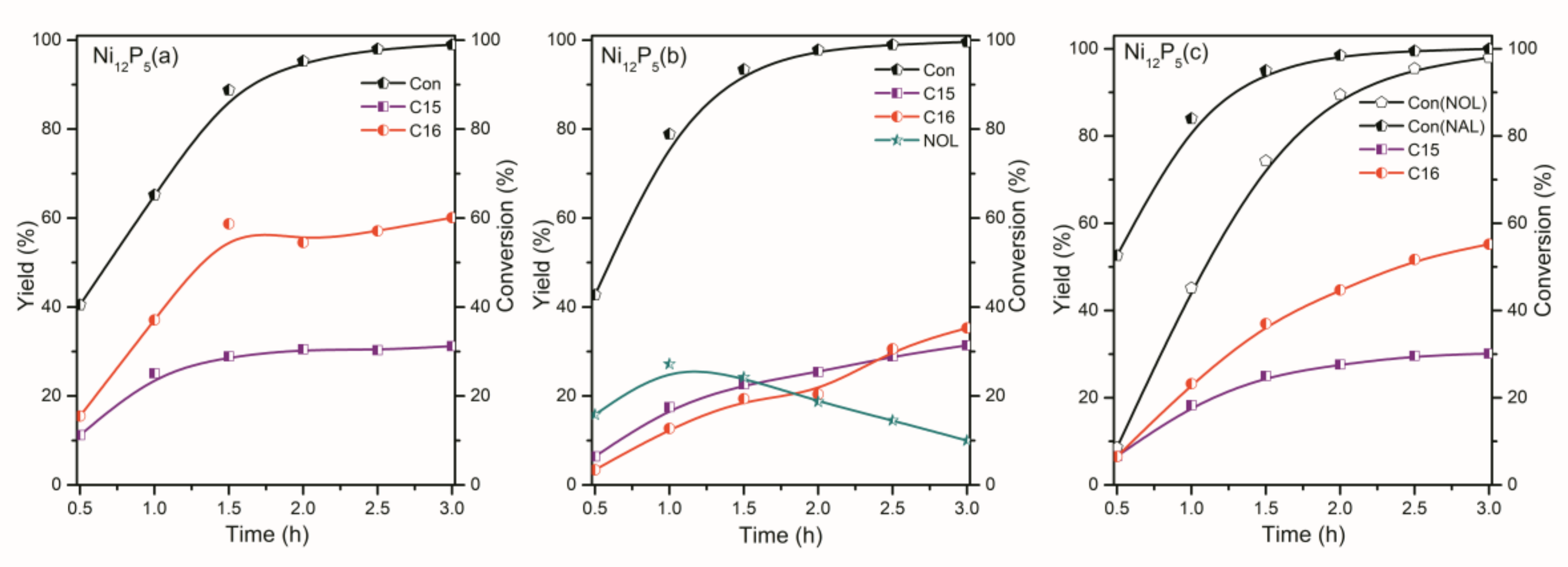
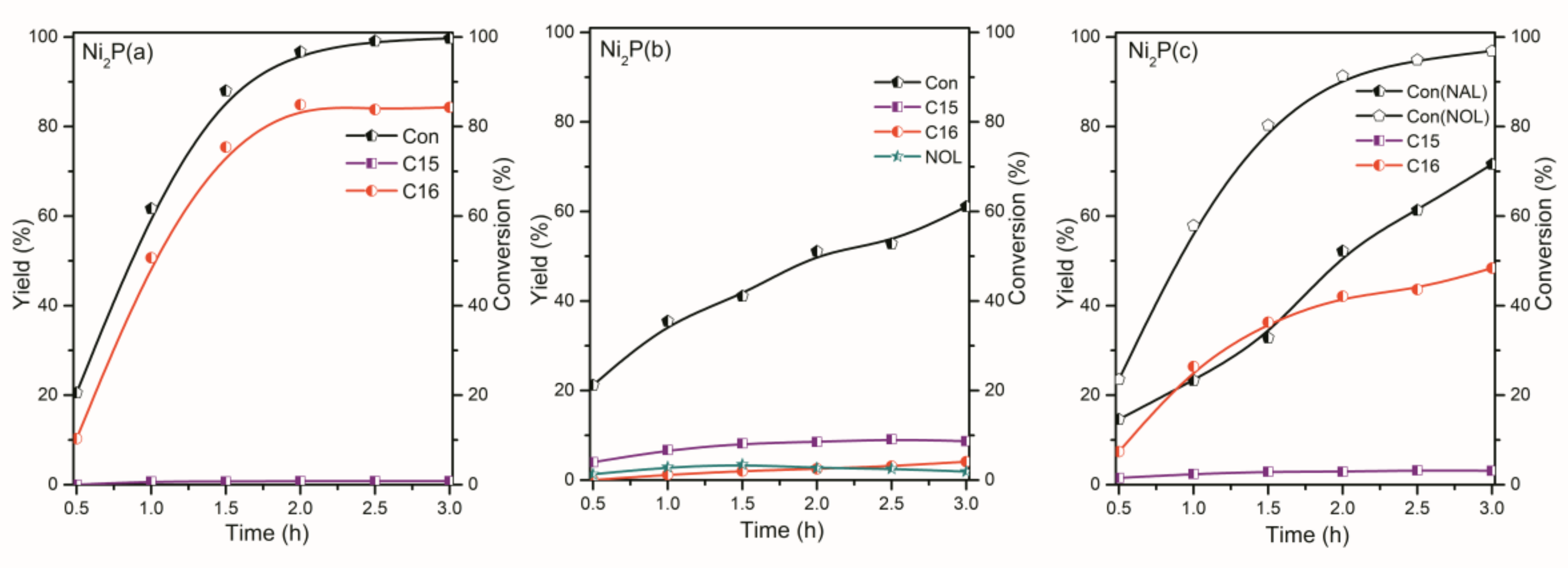
2.2. The Conversion of the Palmitic Acid on the Ni/SiO2, Ni12P5/SiO2 and Ni2P/SiO2 Catalysts
2.3. The Conversion of Intermediate Hexadecanol and Hexadecanal on the Ni/SiO2, Ni12P5/SiO2 and Ni2P/SiO2 Catalysts
2.4. The Kinetic Parameters
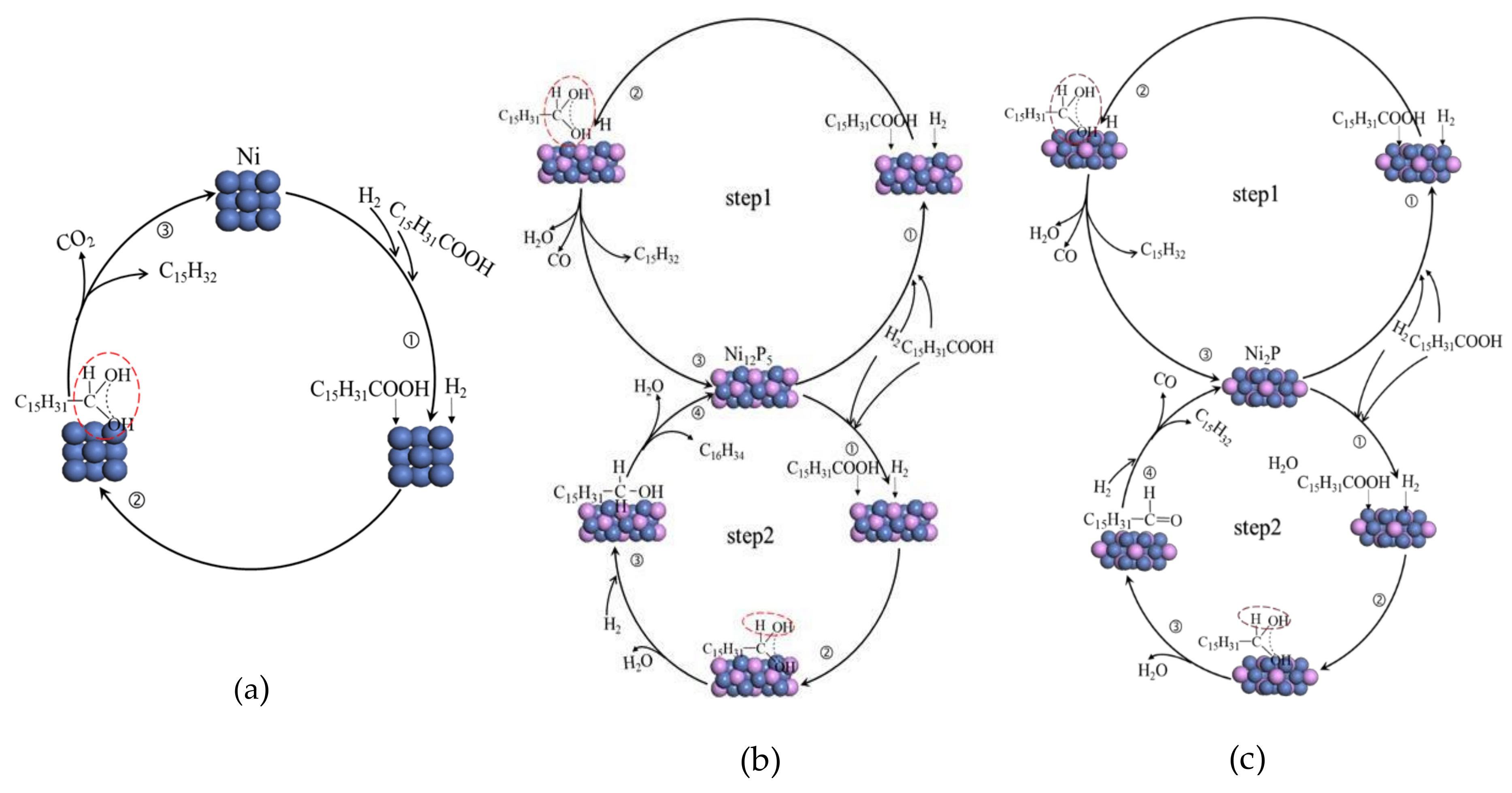
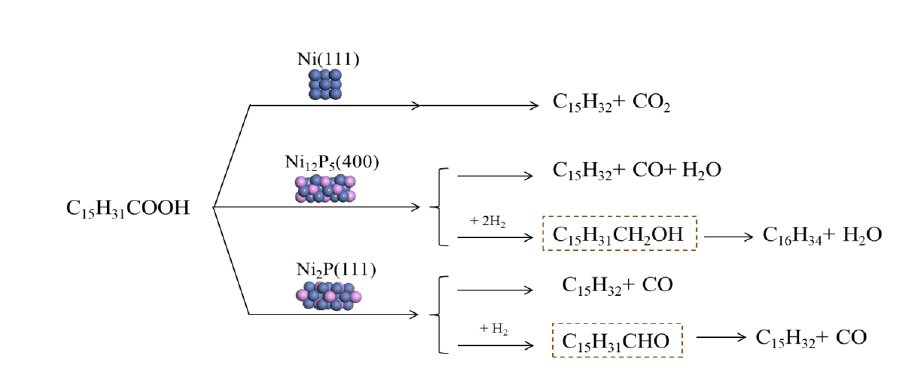
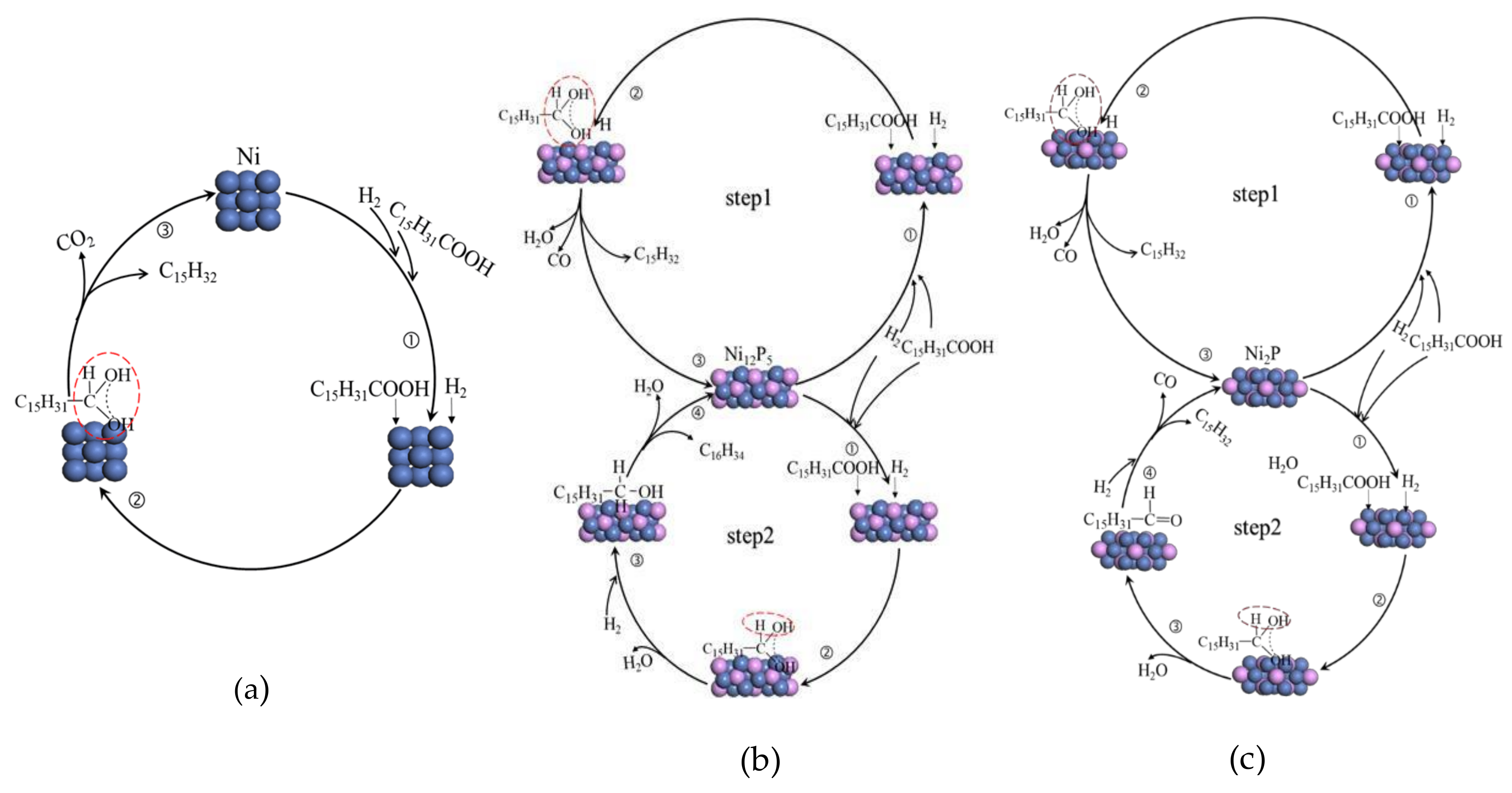
2.5. Summary of Reaction Pathway
3. Experiment
3.1. Catalyst Synthesis
3.2. Catalyst Characterization
3.3. Measurement of Catalytic Reaction
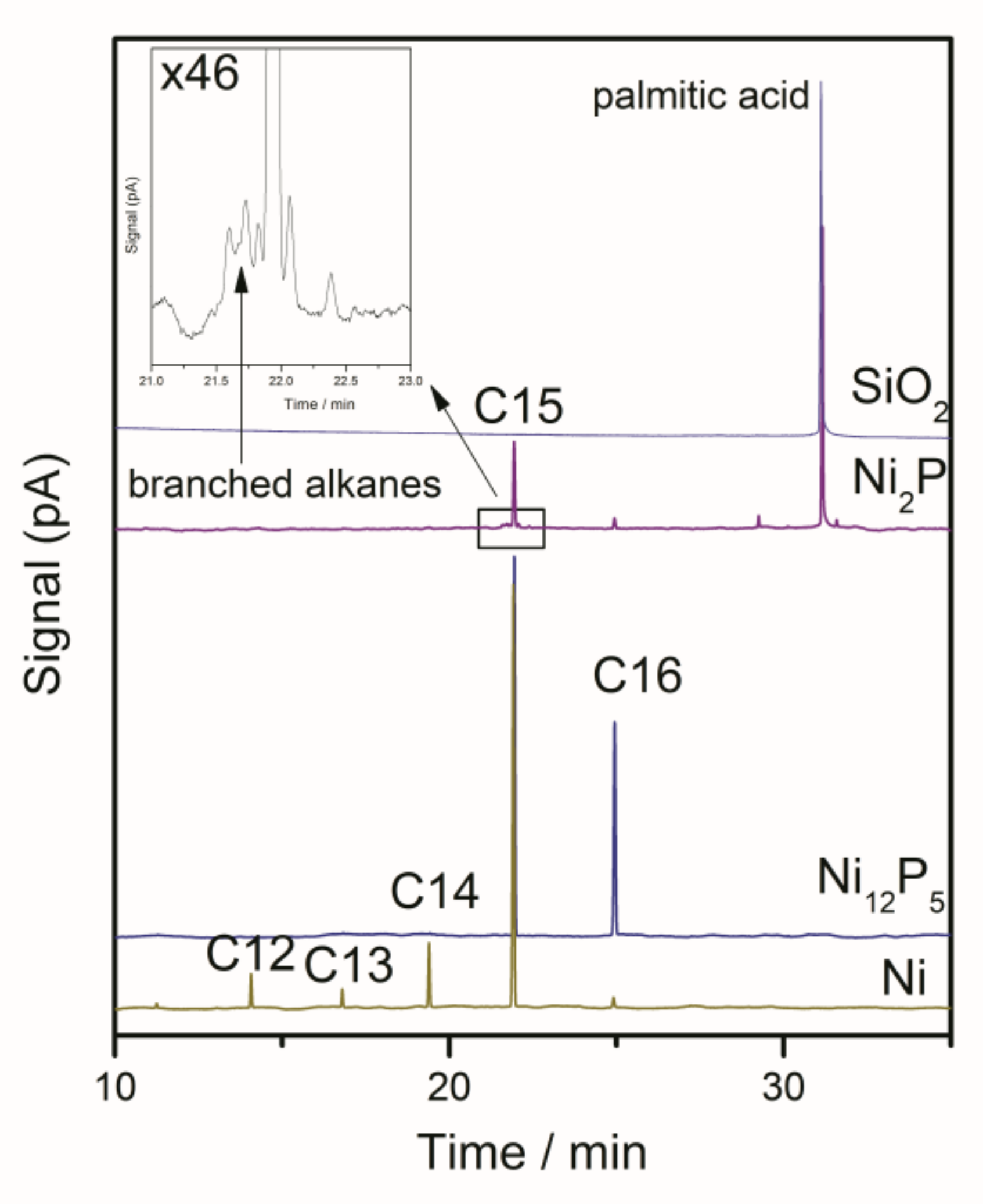
3.4. Product Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, B.; Zhao, C. High-grade diesel production by hydrodeoxygenation of palm oil over a hierarchically structured Ni/HBEA catalyst. Green Chem. 2015, 17, 1692–1701. [Google Scholar] [CrossRef]
- Koike, N.; Hosokai, S.; Takagaki, A.; Nishimura, S.; Kikuchi, R.; Ebitani, K.; Suzuki, Y.; Oyama, S.T. Upgrading of pyrolysis bio-oil using nickel phosphide catalysts. J. Catal. 2016, 333, 115–126. [Google Scholar] [CrossRef]
- Pattanaik, B.P.; Misra, R.D. Effect of reaction pathway and operating parameters on the deoxygenation of vegetable oils to produce diesel range hydrocarbon fuels: A review. Renew. Sustain. Energy Rev. 2017, 73, 545–557. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Morgan, T.; Lacny, J.; Mohapatra, S.; Crocker, M. Catalytic deoxygenation of triglycerides and fatty acids to hydrocarbons over carbon-supported nickel. Fuel 2013, 103, 1010–1017. [Google Scholar] [CrossRef]
- Kim, S.K.; Han, J.Y.; Lee, H.-S.; Yum, T.; Kim, Y.; Kim, J. Production of renewable diesel via catalytic deoxygenation of natural triglycerides: Comprehensive understanding of reaction intermediates and hydrocarbons. Appl. Energy 2014, 116, 199–205. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, L.; Xin, H.; Wang, G.; Li, D.; Hu, C. The production of diesel-like hydrocarbons from palmitic acid over HZSM-22 supported nickel phosphide catalysts. Appl. Catal. B 2015, 174, 504–514. [Google Scholar] [CrossRef]
- Moore, R.H.; Thornhill, K.L.; Weinzierl, B.; Sauer, D.; D’Ascoli, E.; Kim, J.; Lichtenstern, M.; Scheibe, M.; Beaton, B.; Beyersdorf, A.J.; et al. Biofuel blending reduces particle emissions from aircraft engines at cruise conditions. Nature 2017, 543, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Srifa, A.; Viriya-empikul, N.; Assabumrungrat, S.; Faungnawakij, K. Catalytic behaviors of Ni/γ-Al2O3 and Co/γ-Al2O3 during the hydrodeoxygenation of palm oil. Catal. Sci. Technol. 2015, 5, 3693–3705. [Google Scholar] [CrossRef]
- Chen, J.; Xu, Q. Hydrodeoxygenation of biodiesel-related fatty acid methyl esters to diesel-range alkanes over zeolite-supported ruthenium catalysts. Catal. Sci. Technol. 2016, 6, 7239–7251. [Google Scholar] [CrossRef]
- Xia, Q.; Zhuang, X.; Li, M.M.; Peng, Y.K.; Liu, G.; Wu, T.S.; Soo, Y.L.; Gong, X.Q.; Wang, Y.; Tsang, S.C. Cooperative catalysis for the direct hydrodeoxygenation of vegetable oils into diesel-range alkanes over Pd/NbOPO4. Chem. Commun. 2016, 52, 5160–5163. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Chen, P.; Duan, J.; Han, J.; Lou, H.; Zheng, X.; Hong, H. Carbon nanofibers supported molybdenum carbide catalysts for hydrodeoxygenation of vegetable oils. RSC Adv. 2013, 3, 17485. [Google Scholar] [CrossRef]
- Liu, J.; Liu, C.; Zhou, G.; Shen, S.; Rong, L. Hydrotreatment of jatropha oil over NiMoLa/Al2O3 catalyst. Green Chem. 2012, 14, 2499. [Google Scholar] [CrossRef]
- Han, J.; Duan, J.; Chen, P.; Lou, H.; Zheng, X.; Hong, H. Nanostructured molybdenum carbides supported on carbon nanotubes as efficient catalysts for one-step hydrodeoxygenation and isomerization of vegetable oils. Green Chem. 2011, 13, 2561. [Google Scholar] [CrossRef]
- Hermida, L.; Abdullah, A.Z.; Mohamed, A.R. Deoxygenation of fatty acid to produce diesel-like hydrocarbons: A review of process conditions, reaction kinetics and mechanism. Renew. Sustain. Energy Rev. 2015, 42, 1223–1233. [Google Scholar] [CrossRef]
- Kay Lup, A.N.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. A review on reaction mechanisms of metal-catalyzed deoxygenation process in bio-oil model compounds. Appl. Catal. A 2017, 541, 87–106. [Google Scholar] [CrossRef]
- Lestari, S.; Maki-Arvela, P.; Beltramini, J.; Lu, G.Q.; Murzin, D.Y. Transforming triglycerides and fatty acids into biofuels. ChemSusChem 2009, 2, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Gosselink, R.W.; Hollak, S.A.; Chang, S.W.; van Haveren, J.; de Jong, K.P.; Bitter, J.H.; van Es, D.S. Reaction pathways for the deoxygenation of vegetable oils and related model compounds. ChemSusChem 2013, 6, 1576–1594. [Google Scholar] [CrossRef] [PubMed]
- Wagenhofer, M.F.; Baráth, E.; Gutiérrez, O.Y.; Lercher, J.A. Carbon–carbon bond scission pathways in the deoxygenation of fatty acids on transition-metal sulfides. ACS Catal. 2017, 7, 1068–1076. [Google Scholar] [CrossRef]
- Snåre, M.; Kubicˇkovaʹ, I.; Ma1ki-Arvela, P.i.; Era1nen, K.; Murzin, D.Y. Heterogeneous catalytic deoxygenation of stearic acid for production of biodiesel. Ind. Eng. Chem. Res. 2006, 45, 5708–5715. [Google Scholar] [CrossRef]
- Lestari, S.; Simakova, I.; Tokarev, A.; Mäki-Arvela, P.; Eränen, K.; Murzin, D.Y. Synthesis of biodiesel via deoxygenation of stearic acid over supported Pd/C catalyst. Catal. Lett. 2008, 122, 247–251. [Google Scholar] [CrossRef]
- Snåre, M.; Kubičková, I.; Mäki-Arvela, P.; Chichova, D.; Eränen, K.; Murzin, D.Y. Catalytic deoxygenation of unsaturated renewable feedstocks for production of diesel fuel hydrocarbons. Fuel 2008, 87, 933–945. [Google Scholar] [CrossRef]
- Simakova, I.; Simakova, O.; Mäki-Arvela, P.; Simakov, A.; Estrada, M.; Murzin, D.Y. Deoxygenation of palmitic and stearic acid over supported Pd catalysts: Effect of metal dispersion. Appl. Catal. A 2009, 355, 100–108. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Snåre, M.; Eränen, K.; Myllyoja, J.; Murzin, D.Y. Continuous decarboxylation of lauric acid over Pd/C catalyst. Fuel 2008, 87, 3543–3549. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, C.; Kasakov, S.; Foraita, S.; Lercher, J.A. Manipulating catalytic pathways: Deoxygenation of palmitic acid on multifunctional catalysts. Chem. Eur. J. 2013, 19, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Kim, S.-H.; Lee, D.; Shim, S.E.; Baeck, S.-H.; Kim, B.S.; Chang, T.S. Hydrogenation of lactic acid to propylene glycol over a carbon-supported ruthenium catalyst. J. Mol. Catal. A Chem. 2013, 380, 57–60. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Zheng, H.; Zhang, C.; Zhang, B.; Li, Y. Aqueous-phase hydrodeoxygenation of carboxylic acids to alcohols or alkanes over supported Ru catalysts. J. Mol. Catal. A Chem. 2011, 351, 217–227. [Google Scholar] [CrossRef]
- Li, W.; Ye, L.; Chen, J.; Duan, X.; Lin, H.; Yuan, Y. Fe-SBA-15-supported ruthenium catalyst for the selective hydrogenolysis of carboxylic acids to alcoholic chemicals. Catal. Today 2015, 251, 53–59. [Google Scholar] [CrossRef]
- Lugo-José, Y.K.; Monnier, J.R.; Williams, C.T. Gas-phase, catalytic hydrodeoxygenation of propanoic acid, over supported group viii noble metals: Metal and support effects. Appl. Catal. A 2014, 469, 410–418. [Google Scholar] [CrossRef]
- Chen, J.; Shi, H.; Li, L.; Li, K. Deoxygenation of methyl laurate as a model compound to hydrocarbons on transition metal phosphide catalysts. Appl. Catal. B 2014, 144, 870–884. [Google Scholar] [CrossRef]
- Bui, P.; Cecilia, J.A.; Oyama, S.T.; Takagaki, A.; Infantes-Molina, A.; Zhao, H.; Li, D.; Rodríguez-Castellón, E.; Jiménez López, A. Studies of the synthesis of transition metal phosphides and their activity in the hydrodeoxygenation of a biofuel model compound. J. Catal. 2012, 294, 184–198. [Google Scholar] [CrossRef]
- Li, K.; Wang, R.; Chen, J. Hydrodeoxygenation of anisole over silica-supported Ni2P, MoP, and NiMoP catalysts. Energy Fuels 2011, 25, 854–863. [Google Scholar] [CrossRef]
- Bowker, R.H.; Smith, M.C.; Pease, M.L.; Slenkamp, K.M.; Kovarik, L.; Bussell, M.E. Synthesis and hydrodeoxygenation properties of ruthenium phosphide catalysts. ACS Catal. 2011, 1, 917–922. [Google Scholar] [CrossRef]
- Kubička, D.; Kaluža, L. Deoxygenation of vegetable oils over sulfided Ni, Mo and NiMo catalysts. Appl. Catal. A 2010, 372, 199–208. [Google Scholar] [CrossRef]
- Peng, B.; Yuan, X.; Zhao, C.; Lercher, J.A. Stabilizing catalytic pathways via redundancy: Selective reduction of microalgae oil to alkanes. J. Am. Chem. Soc. 2012, 134, 9400–9405. [Google Scholar] [CrossRef] [PubMed]
- Brillouet, S.; Baltag, E.; Brunet, S.; Richard, F. Deoxygenation of decanoic acid and its main intermediates over unpromoted and promoted sulfided catalysts. Appl. Catal. B 2014, 148, 201–211. [Google Scholar] [CrossRef]
- Ruinart de Brimont, M.; Dupont, C.; Daudin, A.; Geantet, C.; Raybaud, P. Deoxygenation mechanisms on Ni-promoted MoS2 bulk catalysts: A combined experimental and theoretical study. J. Catal. 2012, 286, 153–164. [Google Scholar] [CrossRef]
- Toba, M.; Abe, Y.; Kuramochi, H.; Osako, M.; Mochizuki, T.; Yoshimura, Y. Hydrodeoxygenation of waste vegetable oil over sulfide catalysts. Catal. Today 2011, 164, 533–537. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Ryymin, E.M.; Viljava, T.R.; Krause, A.O.I. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts. J. Mol. Catal. A Chem. 2007, 277, 107–112. [Google Scholar] [CrossRef]
- Oyama, S.T.; Onkawa, T.; Takagaki, A.; Kikuchi, R.; Hosokai, S.; Suzuki, Y.; Bando, K.K. Production of phenol and cresol from guaiacol on nickel phosphide catalysts supported on acidic supports. Top. Catal. 2015, 58, 201–210. [Google Scholar] [CrossRef]
- Iino, A.; Cho, A.; Takagaki, A.; Kikuchi, R.; Oyama, S.T. Kinetic studies of hydrodeoxygenation of 2-methyltetrahydrofuran on a Ni2P/SiO2 catalyst at medium pressure. J. Catal. 2014, 311, 17–27. [Google Scholar] [CrossRef]
- Sawhill, S.; Layman, K.; Vanwyk, D.; Engelhard, M.; Wang, C.; Bussell, M. Thiophene hydrodesulfurization over nickel phosphide catalysts: Effect of the precursor composition and support. J. Catal. 2005, 231, 300–313. [Google Scholar] [CrossRef]
- Franke, R.; Chassé, T.; Streubel, P.; Meisel, A. Auger parameters and relaxation energies of phosphorus in solid compounds. J. Electron. Spectrosc. Relat. Phenom. 1991, 56, 381–388. [Google Scholar] [CrossRef]
- Landau, M.V.; Herskowitz, M.; Hoffman, T.; Fuks, D.; Liverts, E.; Vingurt, D.; Froumin, N. Ultradeep hydrodesulfurization and adsorptive desulfurization of diesel fuel on metal-rich nickel phosphides. Ind. Eng. Chem. Res. 2009, 48, 5239–5249. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A.; Oyama, S.T. Oxygen-removal of dibenzofuran as a model compound in biomass derived bio-oil on nickel phosphide catalysts: Role of phosphorus. Appl. Catal. B 2013, 136–137, 140–149. [Google Scholar] [CrossRef]
- Koranyi, T.; Vit, Z.; Poduval, D.; Ryoo, R.; Kim, H.; Hensen, E. SBA-15-supported nickel phosphide hydrotreating catalysts. J. Catal. 2008, 253, 119–131. [Google Scholar] [CrossRef]
- Oyama, S.T.; Wang, X.; Lee, Y.-K.; Bando, K.; Requejo, F.G. Effect of phosphorus content in nickel phosphide catalysts studied by XAFS and other techniques. J. Catal. 2002, 210, 207–217. [Google Scholar] [CrossRef]
- Xin, H.; Guo, K.; Li, D.; Yang, H.; Hu, C. Production of high-grade diesel from palmitic acid over activated carbon-supported nickel phosphide catalysts. Appl. Catal. B 2016, 187, 375–385. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Zhou, J.; Zhang, S.; Rao, D.; He, S.; Wei, M.; Evans, D.G.; Duan, X. Metal phosphides derived from hydrotalcite precursors toward the selective hydrogenation of phenylacetylene. ACS Catal. 2015, 5, 5756–5765. [Google Scholar] [CrossRef]
- Guan, Q.; Han, F.; Li, W. Catalytic performance and deoxygenation path of methyl palmitate on Ni2P/SiO2 synthesized using the thermal decomposition of nickel hypophosphite. RSC Adv. 2016, 6, 31308–31315. [Google Scholar] [CrossRef]
- Oyama, S.T.; Lee, Y.-K. Bifunctional nature of a SiO2-supported Ni2P catalyst for hydrotreating: EXAFS and FTIR studies. J. Catal. 2006, 239, 376–389. [Google Scholar] [CrossRef]
- Basila, M.I.; Kantner, T.R.; Rhee, K.H. Infrared spectroscopic studies of trimethylamine and pyridine chemisorption. Nat. Acidic Sites Silica-Alumina 1964, 68, 3197–3207. [Google Scholar]
- Wawrzetz, A.; Peng, B.; Hrabar, A.; Jentys, A.; Lemonidou, A.A.; Lercher, J.A. Towards understanding the bifunctional hydrodeoxygenation and aqueous phase reforming of glycerol. J. Catal. 2010, 269, 411–420. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, C.; Mejía-Centeno, I.; Fuentes, G.A.; Jentys, A.; Lercher, J.A. Comparison of kinetics and reaction pathways for hydrodeoxygenation of C3 alcohols on Pt/Al2O3. Catal. Today 2012, 183, 3–9. [Google Scholar] [CrossRef]
- Fu, J.; Lu, X.; Savage, P.E. Catalytic hydrothermal deoxygenation of palmitic acid. Energy Environ. Sci. 2010, 3, 311–317. [Google Scholar] [CrossRef]
- Li, W.; Gao, Y.; Yao, S.; Ma, D.; Yan, N. Effective deoxygenation of fatty acids over Ni(OAC)2 in the absence of H2 and solvent. Green Chem. 2015, 17, 4198–4205. [Google Scholar] [CrossRef]
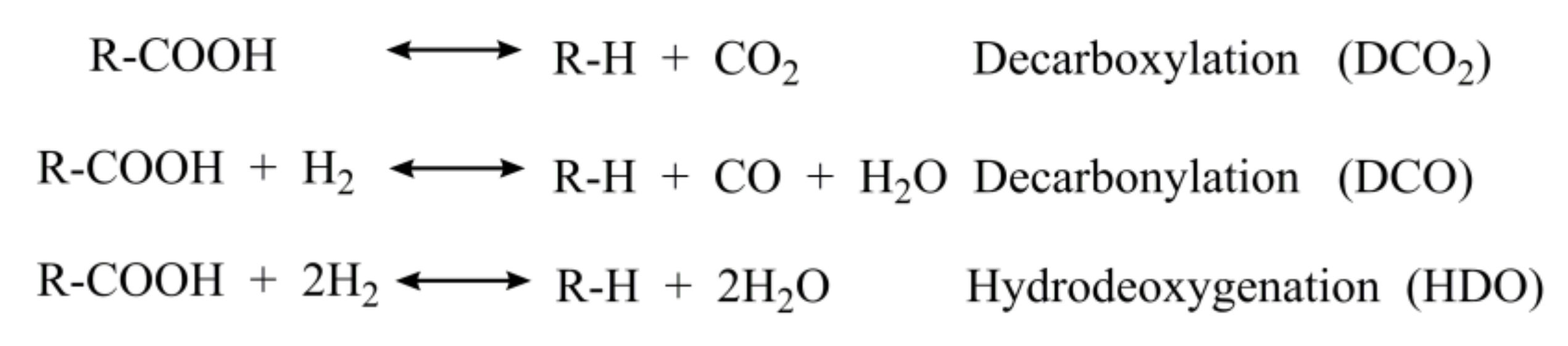






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
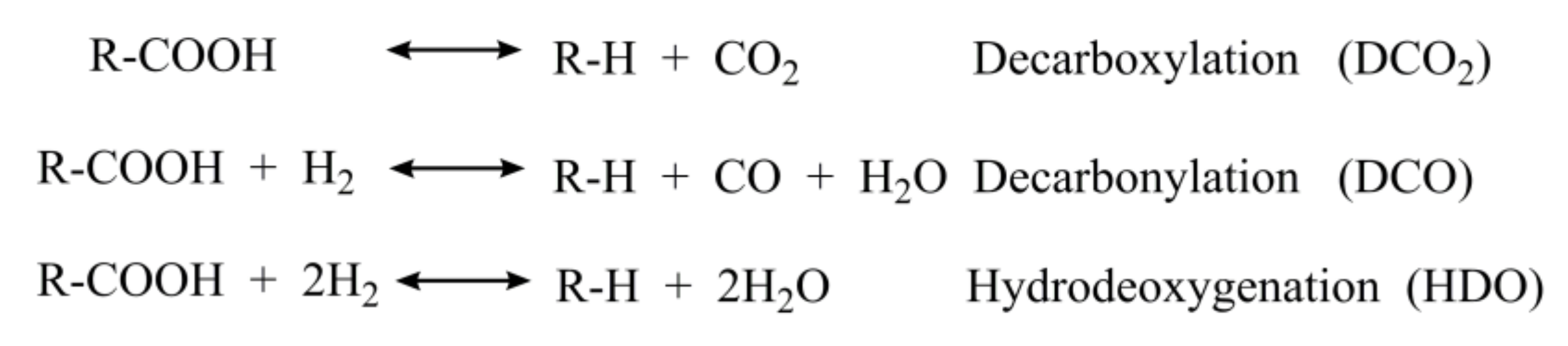{kind=link}
{kind=link}
| Element | Ni | Ni12P5 | Ni2P |
|---|---|---|---|
| Ni | 24.20 | 18.84 | 10.19 |
| P | - | 5.91 | 10.53 |
| Catalysts | Brunauer–Emmett–Teller (BET) Surface Area (m2·g−1) | Pore Volume (cm3·g−1) | Pore Size (nm) | Crystal Size (nm) | Metal Exposed (%) | Active Site (mmol·g−1) | TOF (h−1) |
|---|---|---|---|---|---|---|---|
| SiO2 | 342 | 0.95 | 11.0 | - | - | - | - |
| Ni/SiO2 | 237 | 0.66 | 11.0 | 15.6 | 0.34 | 0.03 | 468 |
| Ni12P5/SiO2 | 218 | 0.55 | 10.0 | 17.3 | 0.45 | 0.03 | 828 |
| Ni2P/SiO2 | 88 | 0.38 | 18.6 | 36.7 | 0.26 | 0.02 | 36 |
| Element | Ni | Ni12P5 | Ni2P |
|---|---|---|---|
| Ni | 2.82 | 5.22 | 4.02 |
| P | - | 2.06 | 5.27 |
| O | 54.93 | 56.90 | 57.12 |
| Si | 42.25 | 35.82 | 33.59 |
| Catalyst | Palmitic Acid | Hexadecanal | Hexadecanol |
|---|---|---|---|
| Ni | 4.0 | 6.2 | 4.5 |
| Ni12P5 | 2.6 | 4.1 | 3.7 |
| Ni2P | 0.2 | 1.9 | 1.9 |
| Catalyst | k (h−1) | Ea (kJ/mol) | A (h−1) | ||
|---|---|---|---|---|---|
| Palmitic Acid | Hexadecanal | Hexadecanol | Palmitic Acid | ||
| Ni/SiO2 | 1.50 | 1.02 | 1.02 | 85 | 4.5 × 108 |
| Ni12P5/SiO2 | 1.08 | 0.81 | 0.80 | 93 | 8.4 × 108 |
| Ni2P/SiO2 | 0.11 | 0.14 | 1.03 | 51 | 3.0 × 103 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, W.; Xin, H.; Yang, H.; Du, X.; Yang, R.; Li, D.; Hu, C. The Deoxygenation Pathways of Palmitic Acid into Hydrocarbons on Silica-Supported Ni12P5 and Ni2P Catalysts. Catalysts 2018, 8, 153. https://doi.org/10.3390/catal8040153
Zhou W, Xin H, Yang H, Du X, Yang R, Li D, Hu C. The Deoxygenation Pathways of Palmitic Acid into Hydrocarbons on Silica-Supported Ni12P5 and Ni2P Catalysts. Catalysts. 2018; 8(4):153. https://doi.org/10.3390/catal8040153
Chicago/Turabian StyleZhou, Wenjun, Hui Xin, Huiru Yang, Xiangze Du, Rui Yang, Dan Li, and Changwei Hu. 2018. "The Deoxygenation Pathways of Palmitic Acid into Hydrocarbons on Silica-Supported Ni12P5 and Ni2P Catalysts" Catalysts 8, no. 4: 153. https://doi.org/10.3390/catal8040153
APA StyleZhou, W., Xin, H., Yang, H., Du, X., Yang, R., Li, D., & Hu, C. (2018). The Deoxygenation Pathways of Palmitic Acid into Hydrocarbons on Silica-Supported Ni12P5 and Ni2P Catalysts. Catalysts, 8(4), 153. https://doi.org/10.3390/catal8040153