Microwave, Ultrasound, and Mechanochemistry: Unconventional Tools that Are Used to Obtain “Smart” Catalysts for CO2 Hydrogenation



1
Department of Drug Science and Technology, NIS and INSTM reference Centres, Università degli Studi di Torino, Via Pietro Giuria 9, 10125 Torino, Italy
2
Department of Applied Science and Technology and INSTM-Unit of Torino Politecnico, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(7), 262; https://doi.org/10.3390/catal8070262
Submission received: 6 April 2018
/
Revised: 20 June 2018
/
Accepted: 21 June 2018
/
Published: 28 June 2018
(This article belongs to the Special Issue Catalytic, Photocatalytic and Electrocatalytic Processes for the Valorisation of CO2)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The most recent progress obtained through the precise use of enabling technologies, namely microwave, ultrasound, and mechanochemistry, described in the literature for obtaining improved performance catalysts (and photocatalysts) for CO2 hydrogenation, are reviewed. In particular, the main advantages (and drawbacks) found in using the proposed methodologies will be discussed and compared by focusing on catalyst design and optimization of clean and efficient (green) synthetic processes. The role of microwaves as a possible activation tool used to improve the reaction yield will also be considered.
1. Introduction
Atmospheric CO2 concentration is growing continuously: in 2017, a “record concentration” of 413 ppm was registered (according to https://www.co2.earth/daily-co2) and a concentration of ca. 570 ppm is expected to be reached by the end of this century [1].
The mitigation of CO2 (the most popular among atmospheric greenhouse gases) is currently a scientific, technological, social, and economic issue [1]. Several strategies have been proposed to limit/decrease CO2 concentration in the atmosphere: besides lowering CO2 emissions with the help of low-C fuels (and of appropriate policies from developed countries), there is a great spur of studies and technologies aimed at optimizing several processes of CO2 capture & storage [2,3,4,5,6,7,8,9,10] and reutilization [11,12]. The first two steps, currently fairly developed, are indeed functional to CO2 reutilization processes, which mostly need a concentrated and rather pure CO2 stream apt to further chemical transformations.
The chemical processes for CO2 reutilization are catalytic [13,14,15], photocatalytic [16,17,18,19,20], electrocatalytic [21,22,23,24,25], or photoelectrocatalytic [18,26]. As a whole, the chemical reduction of CO2 is a challenge from both the industrial and the catalytic point of view [27], but it is also a hot scientific topic, since CO2 is an ideal C1 feedstock for producing different types of chemicals and, ultimately, fuels. Unfortunately, its high thermodynamic stability limits the number of processes in which CO2 can be utilized as a chemical feedstock (e.g., the synthesis of urea), and usually electrocatalytic processes are needed [28].
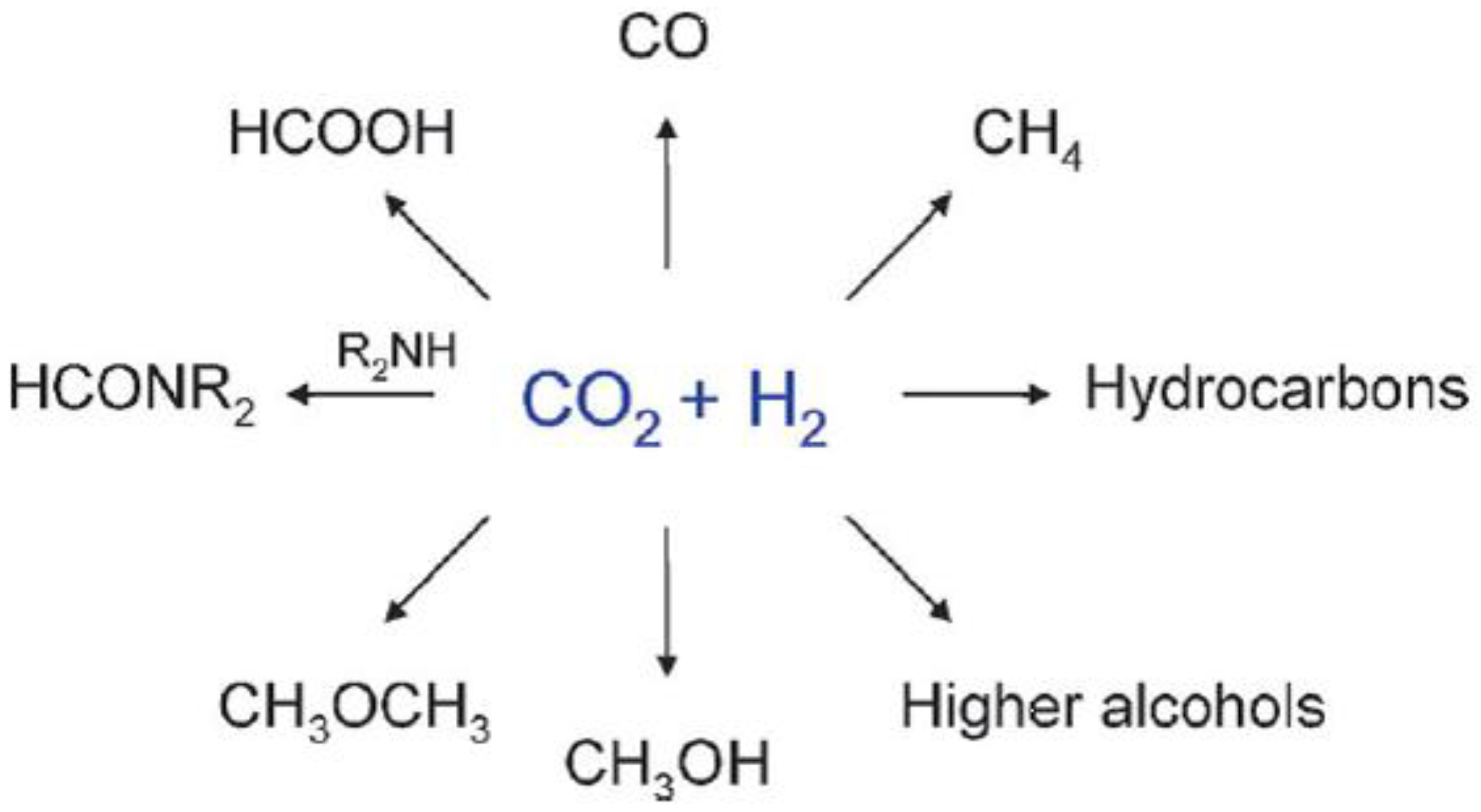
Most of the catalytic processes (like those reported in Scheme 1) imply the use of H2, which is per se a costly reagent due to its scarce natural abundance (from which arises the issue of its production), along with safety and transportation issues. All this notwithstanding, the products of CO2 reduction are ultimately fuels, raw materials, and/or chemical intermediates [1].
Though some homogenous catalysts are highly active and selective in several CO2 reduction processes [29], they are hardly applicable on an industrial scale, and therefore there is a great interest towards the development of efficient heterogeneous catalysts and/or the immobilization of homogeneous ones [30]. For the industrial application of CO2 hydrogenation processes, heterogeneous catalysts (e.g., Fe-, Cu-, and Ni-based solid catalysts [31]) offer some practical advantages compared to homogeneous ones: solids with large specific surface area, small particles of active phase, and fair metal dispersion are usually more active and selective, and also more stable and characterized by longer life. However, they often provide low yields and poor selectivity, mainly due to fast kinetics of C–H bond formation [1].
In order to allow industrial applications of heterogeneous catalysis, green processes should be developed, which often require efficient (nano)catalysts that are able to selectively transform CO2 into added value molecules [27]. Depending on the reaction adopted for CO2 reduction, different active phases (mostly Ni, but also more noble metals [31]) have to be supported on different oxides (γ-Al2O3 being the most used one [31]) that have to be stable at high temperatures, provide a certain amount of acid/base sites, and stabilize the catalytically active metal particles, etc.
Whatever the type of CO2 hydrogenation process, active, selective, and stable heterogeneous catalysts are preferable in view of an actual industrial application [1]. They should also be based on earth-abundant and non-toxic elements and obtained by green synthesis methods: in one word, they should be “smart”. Though the outputs of CO2 reduction are diverse [1], “smart” catalysts have to meet some requirements that can be obtained by optimizing their preparation strategy, for instance. The physico-chemical properties of a heterogeneous catalyst are determined and controlled by its preparation method: therefore, catalysts prepared by different means may have markedly different features and, thus, different catalytic performances [32,33].
In order to allow an actual development of feasible industrial CO2 hydrogenation processes, new catalysts should be obtained by methods that are able to control and tailor the physico-chemical properties in order to improve the catalyst performance [32]. Moreover, a deeper understanding of the reaction mechanisms with the help of both experiments and molecular simulations is needed [1], especially as far as the first step of CO2 reduction, i.e., its adsorption at the catalyst surface, is concerned. Notwithstanding the plethora of works concerning CO2 reduction, a thorough understanding of the mechanisms is still lacking and/or debated [18]. As far as heterogeneous catalysts are concerned, there is a general consensus about the idea that the active site is provided by the primary catalyst (e.g., the metal particle) acting synergistically with the support (and/or the promoter) [32]. Evidence of such synergy is provided in several works, but the actual mechanism is still unclear, even for the synthesis of methanoic acid (HCOOH), which is only the first step of CO2 hydrogenation [1].
From both industrial and scientific points of view, the great interest in heterogeneous catalysts based on abundant and low cost metals like Ni, Cu, and Fe is frustrated by their poor activity in mild reaction conditions, in that an extreme dispersion of metal particles and a strong interaction with the support should be attained [31].
As mentioned before, from an environmental point of view, catalysts should be obtained by green synthesis methods [27]. In our opinion, the use of microwaves, ultrasound, or mechanochemistry represents promising and simple approaches, which are alternatives to conventionally adopted synthetic protocols, to obtain novel catalysts with improved activity and selectivity, in contrast to conventional methods, which usually encompass multi-step processes, heating, and/or the addition of expensive and/or hazardous chemicals. The development of such techniques has increased significantly in recent years, with most reports published over the past 5–10 years (vide infra).
The aim of the present review is to supply a brief (but exhaustive) description of the most recent advances in the synthesis of solid catalysts (and photocatalysts) by microwave (MW), ultrasound (US), and mechanochemistry (MC) to be compared to conventional methods.
2. Microwaves as a Tool to Improve Catalytic CO2 Hydrogenation
2.1. General Principles of MW-Assisted Synthesis of (Nano)Materials
MW-assisted synthesis simply requires the presence of a MW reactor, simultaneously ensuring shorter reaction time, no contact between the radiation source and the reacting mixture, and a homogeneous heating of the latter [34,35].
Homogeneous heating is a crucial point, as thermal gradients affect both nucleation and growth steps, which finally determine the physico-chemical properties of the resulting nanomaterials. For this reason, MW-assisted synthesis was initially developed to prepare ceramic materials, for which it is crucial to avoid temperature gradients that may induce formation of fractures in the final product [36].
MW irradiation favors fast nucleation and crystalline growth, which can accelerate the formation of hierarchical structures, like the bismuth oxyiodide (BiOI) architectures shown in Figure 1 [37]. Hierarchical porous solids can be produced by MW-assisted synthesis in non-aqueous solvents, since organic solvents with high dielectric loss factor are required to ensure efficient MW absorption and rapid heating [37].
Additionally, MW heating can be easily coupled with several chemical methods including combustion synthesis (CS), hydrothermal (HT) synthesis, and sol-gel (SG). For instance, powders obtained by MW-assisted CS are more chemically and thermally stable than those obtained by “conventional” CS; moreover, smaller particles with a lower tendency to aggregation are obtained [36]. This occurs because MW irradiation uniformly heats the reagents mixture, whereas an electric oven heats a mixture from the exterior to the interior, finally creating a temperature gradient and leading to heterogeneous powders. Moreover, MW interacts with polar reagents, inducing dipole oscillations: the oscillating species collide more frequently and at higher energy, facilitating bond breakage and new bond formation. Consequently, the heat needed for the reaction is generated within the mixture with no need of external heat supply [34,35].
MW-assisted CS, for instance, allows obtaining sizeable amounts of phases (mainly oxides, nitrides, and carbides) that are stable at high temperatures and have nanometer size, highly specific surface area and defectivity, and mechanically stable pore structure [36]. MW is also known to favor the formation of given crystalline phases and can be efficiently used to drive the synthesis towards the desired products. All the above parameters are indeed important for catalytic applications.
2.2. MW-Assisted Syntheses of Heterogeneous Catalysts for CO2 Reduction
Cai et al. [38] supported some Cu-based catalysts on ZnO and on Ga2O3 and tested them in the CO2 hydrogenation to CH3OH:
CO2 + 3H2 ⇌ CH3OH + H2O
The Cu-based catalysts were prepared by co-precipitation and impregnation, and the effect of MW on both preparation methods was studied: as a whole, the catalysts prepared by MW-assisted synthesis showed better Cu dispersion and enhanced catalytic activity in terms of both CO2 conversion and CH3OH yield, especially when a single-step MW-assisted method was adopted [38]. The catalysts were characterized by X-Ray Diffraction (XRD), H2 temperature programmed reduction (TPR), N2 adsorption/desorption isotherms at −196 °C, CO2 temperature-programmed desorption (TPD), adsorption calorimetry, and X-ray Photoemission Spectroscopy (XPS). Since all the samples are calcined at 350 °C, no significant changes in their surface area were observed, although the sample obtained by a single-step, MW-assisted method showed slightly larger values for both surface area and pore volume. The XRD patterns of the same sample revealed the likely presence of small crystallites of Cu2O (whereas CuO mostly formed in the other samples) and the smallest size of ZnO particles. TPR showed the occurrence of a strong Cu/support interaction in the sample obtained by single-step, MW-assisted method, which, according to CO2 TPD and CO2 adsorption calorimetry, adsorbed a considerable amount of CO2 with the largest average adsorption heat value (ca. −20 kJ mol−1). This latter result showed that the use of MW seemed to improve the support basicity, a feature that helps CO2 adsorption, a crucial step during the catalytic process. The joint presence of well-dispersed Cu particles and strong basic sites at the support surface resulted in strong Cu/support synergy, leading to better catalytic performance. Besides strong Cu/support interactions, which took place with the catalysts prepared under MW irradiation, the authors observed that Cu particles were particularly active, probably because their surface was partially oxidized due to migration of Zn species from the support to the active metal (Cu) surface, which finally improved CO2 adsorption. On the contrary, such synergistic effects were not observed with same composition catalysts that were prepared by conventional methods.
Huang et al. [39] studied the effect of MW heating during HT synthesis of CuO-ZnO-ZrO2 catalysts for CO2 hydrogenation to CH3OH (Equation (1)), for which CuO-ZnO is usually considered the best catalyst and where ZrO2 has the role of imparting high thermal and chemical stability under redox conditions. This type of catalyst is usually prepared by HT method, requiring long reaction times: the (expected) effect of MW irradiation was indeed the shortening of preparation time. Different temperatures (i.e., 80, 100, 120, and 150 °C) were adopted during the MW-assisted HT method to obtain the catalysts named CZZ80, CZZ100, CZZ120, and CZZ150 [39].
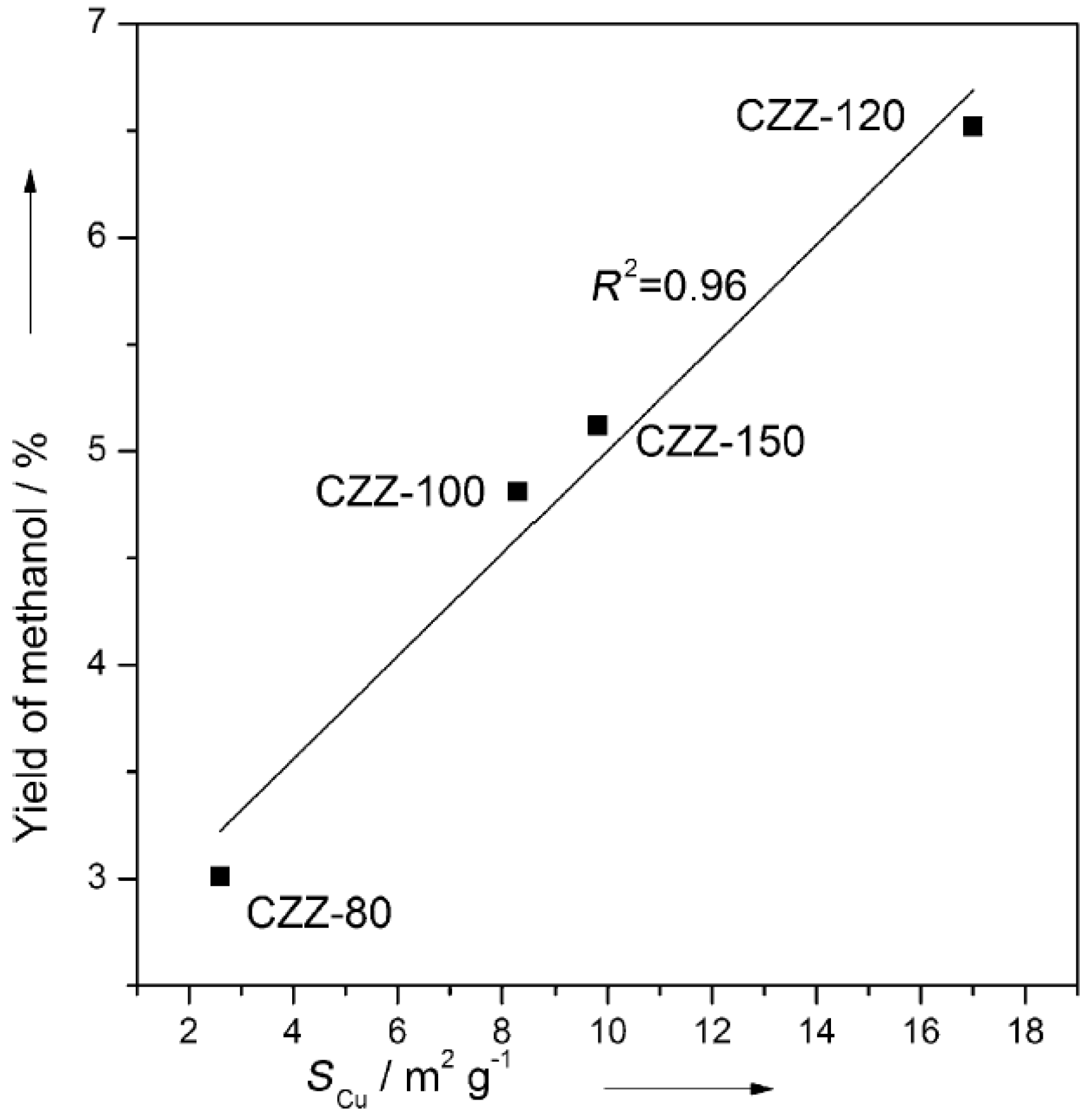
The catalyst obtained at 120 °C (CZZ120) showed the best catalytic properties in terms of both CO2 conversion and selectivity to CH3OH, becoming rather stable after 100 h reaction. XRD analysis allowed the calculation of the size of both CuO and Cu crystallites: the CZZ120 catalyst showed the smallest CuO and Cu crystallites before and after reduction, respectively, though Cu crystallites (ca. 18 nm) were larger than parent CuO crystallites (ca. 7.6 nm). The same catalyst was also characterized by the largest Cu area (SCu); the largest specific surface area and by a higher amount of CO2 molecules adsorbed by relatively weak basic sites. CuO crystallites in CZZ120 were easily reduced, as the samples showed the lowest reduction temperature during H2 TPR analysis. The CH3OH yield was directly related to the surface of Cu particles (SCu), as shown in Figure 2. On the contrary, XPS analysis did not show significant differences in the type of surface Cu species in catalysts obtained at different temperatures. As a whole, an optimum temperature existed for the synthesis of CuO-ZnO-ZrO2 catalysts [39]: Cu particle size, surface area, and basicity exhibited a volcano trend with a maximum at 120 °C.
CO2 methanation (Equation (2)) is a process that allows transformation of CO2 in a fuel through an exothermic reaction (∆H0 = −165 kJ mol−1), for which Ni/Al2O3 is the commercial catalyst [40]:
CO2 + 4H2 ⇌ CH4 + 2H2O
Besides Al2O3, other supports can be (mostly) SiO2 and TiO2, ZrO2 or CeO2. When other (more noble) metals like Ru, Pt, Pd, or Au are used, selectivity is lower with respect to Ni, as they do not produce exclusively CH4, but also CH3OH and CO (by Reversed Water Gas Shift, RWGS):
CO2 + H2 ⇌ CO + H2O
As a whole, Ni dispersion is crucial in the catalytic activity towards CO2 methanation [41,42,43], and therefore the catalyst synthesis method plays a prominent role [44,45,46]: according to Stangeland et al. [40], catalysts should be developed with improved activity at lower temperatures and high activity/stability up to ca. 500 °C. Conventional co-precipitation and impregnation methods may require several hours in order to obtain a fair Ni dispersion, whereas under MW irradiation the preparation time may decrease from hours to 15 min (this is one of the greatest advantages of using MW irradiation during co-precipitation and/or impregnation).
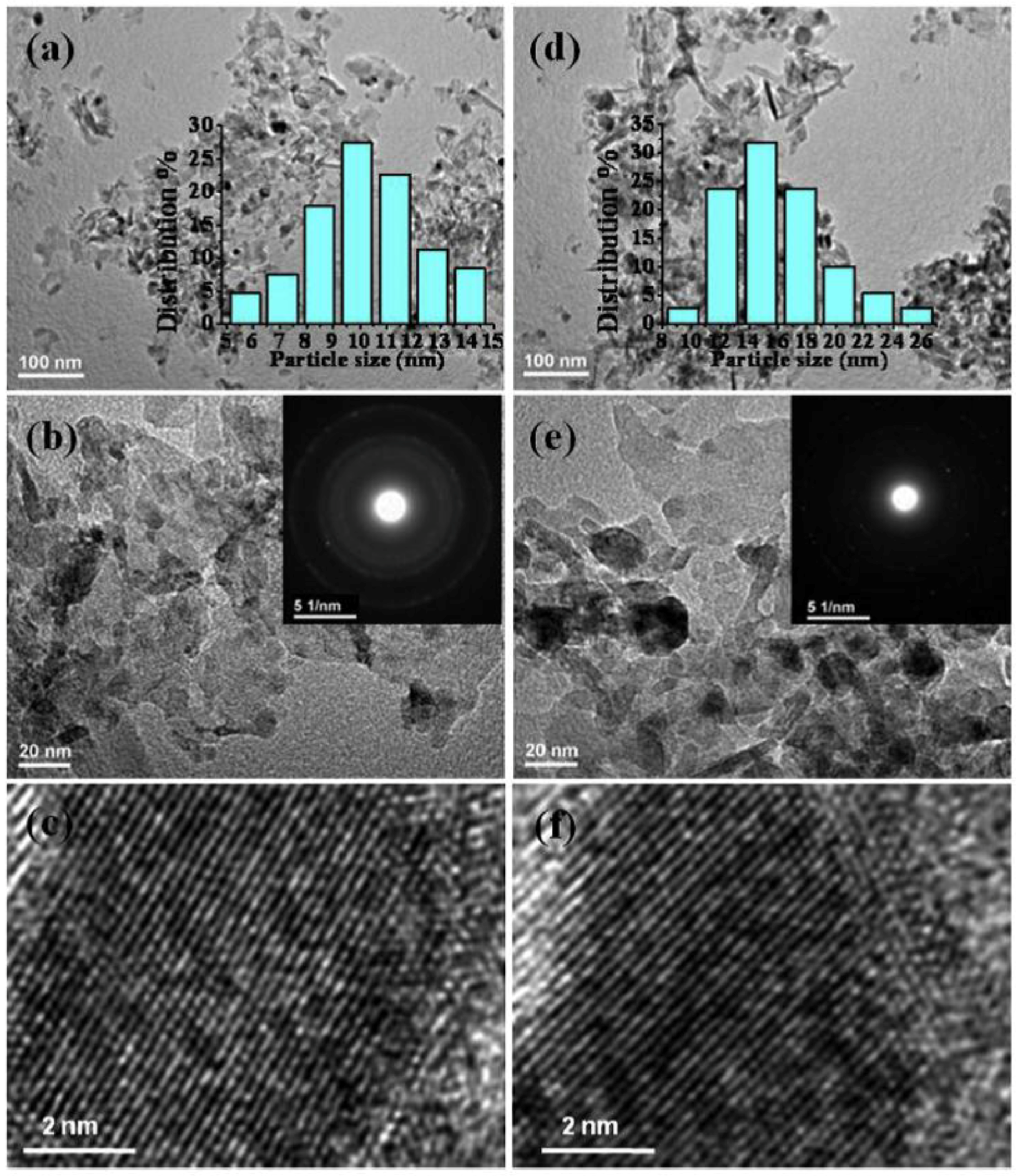
Moreover, MW-assisted synthesis of Ni/Al2O3 catalysts for CO2 methanation [47] resulted in higher Ni dispersion, as well as higher basicity of the support with respect to Ni/Al2O3 catalysts obtained by impregnation. Physico-chemical characterization of the catalysts showed some relevant differences: for instance, Raman spectroscopy revealed a higher amount of NiO oxide in the sample prepared by impregnation, which also showed lower surface area (≅143 m2 g−1 against 163 m2 g−1). The most important property was, however, the Ni dispersion: it was likely favored by the fact that MW helps the dispersion of inorganic salts on the support, as it can be clearly seen in Figure 3, showing the TEM analysis of two Ni/Al2O3 catalysts produced with and without MW assistance. The average size of Ni particles obtained by MW assisted synthesis was 10 nm and 16 nm in the sample prepared by impregnation. The sample obtained by MW-assisted synthesis also consumed ca. 25% more H2 during TPD analysis.
The better Ni dispersion obtained by MW joint with a stronger basicity of support (as determined by CO2 TPD) likely led to enhanced CO2 methanation activity at lower temperature. The corresponding CH4 selectivity was also maintained over the whole temperature range (200–400 °C), and the catalyst was rather stable under prolonged (72 h) catalytic tests without appreciable agglomeration of Ni species [47].
MW can be used during catalyst preparation in different manners: for instance, the effect of MW drying vs. conventional drying in the preparation of Ni-exchanged zeolites for CO2 methanation (2) has been recently studied [48]. The catalysts were prepared by incipient wetness impregnation of USY (Ultra Stabilized Y) zeolite with 5 wt % Ni. The effects of drying method after impregnation, calcination temperature, and pre-reduction temperature on the catalyst performance were evaluated. The drying method deeply affected the catalyst performance: in particular, MW-assisted drying induced remarkable changes on the type, location, and reducibility of Ni species within the zeolite structure and, simultaneously, led to effects on the support structural and textural properties and on the average size of Ni particles.
DR-UV-Vis spectroscopy showed that after conventional drying, most NiO species occurred at the outer surface of zeolite particles, whereas with MW-assisted drying, octahedral Ni2+ species formed if drying was performed in a closed container (when MW-assisted drying was performed in an open container, similar Ni species were obtained as they were during conventional drying). According to the authors, during MW heating in a close container, higher temperature and pressure led to formation of Ni2+ species in exchange position within the zeolite cavities, whereas if the container was left open during drying, NiO species mostly occurred at the outer surface of zeolite particles.
MW-assisted drying also deeply affected zeolite crystallinity: in a closed container, some amorphous phase formed (as detected by X-Ray powders diffraction), likely due to the high temperature reached within the container, whereas with MW-assisted drying in an open container, the zeolite crystallinity was preserved (as for conventional drying). Finally, MW-assisted drying led to less reducible Ni species, especially when octahedral Ni2+ species occurred. As observed with other Ni-based catalysts, MW-assisted drying led to formation of smaller Ni particles (in the 17–27 nm range). Unfortunately, the positive effects brought about by MW-assisted drying, like better Ni dispersion, were counterbalanced by the loss in zeolite crystallinity and, as a result, no appreciable improvement in the catalytic activity was obtained in terms of both CO2 conversion and selectivity to CH4. This was likely due to the fact that the support was a zeolite, for which the crystallinity of porous cavities is a crucial aspect [48].
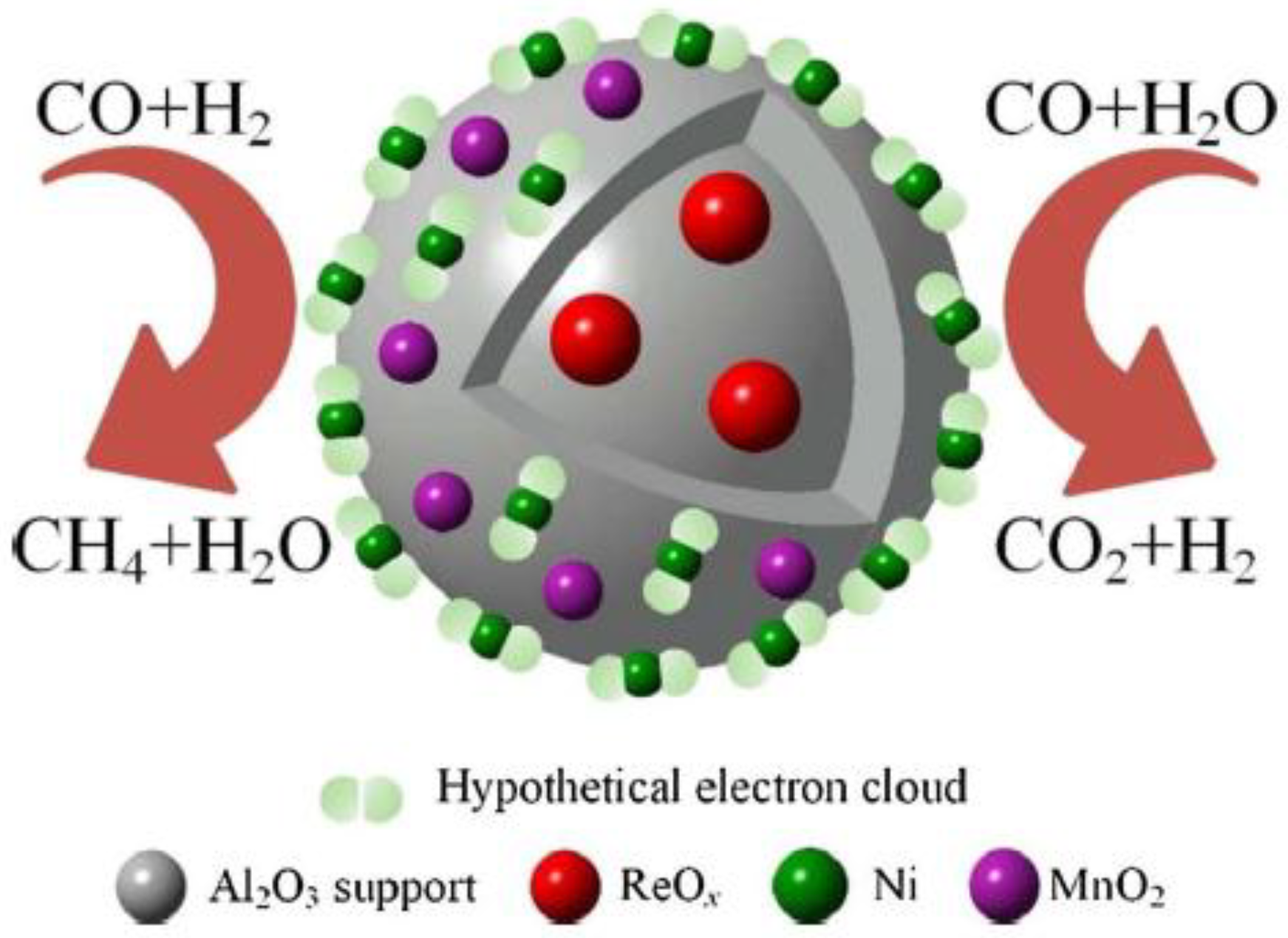
The material thermal response to MW heating is a crucial aspect, as shown by Dong et al. [49], with Re promoted Ni-Mn/Al2O3 catalysts for partial methanation (Scheme 2), a process in which the amount of (noxious) CO is reduced by coupling CO methanation (∆H0 = −206 kJ mol−1):
to water gas shift (WGS, ∆H0 = −41 kJ mol−1)
CO + 3H2 ⇌ CH4 + H2O
CO + H2O ⇌ CO2 + H2
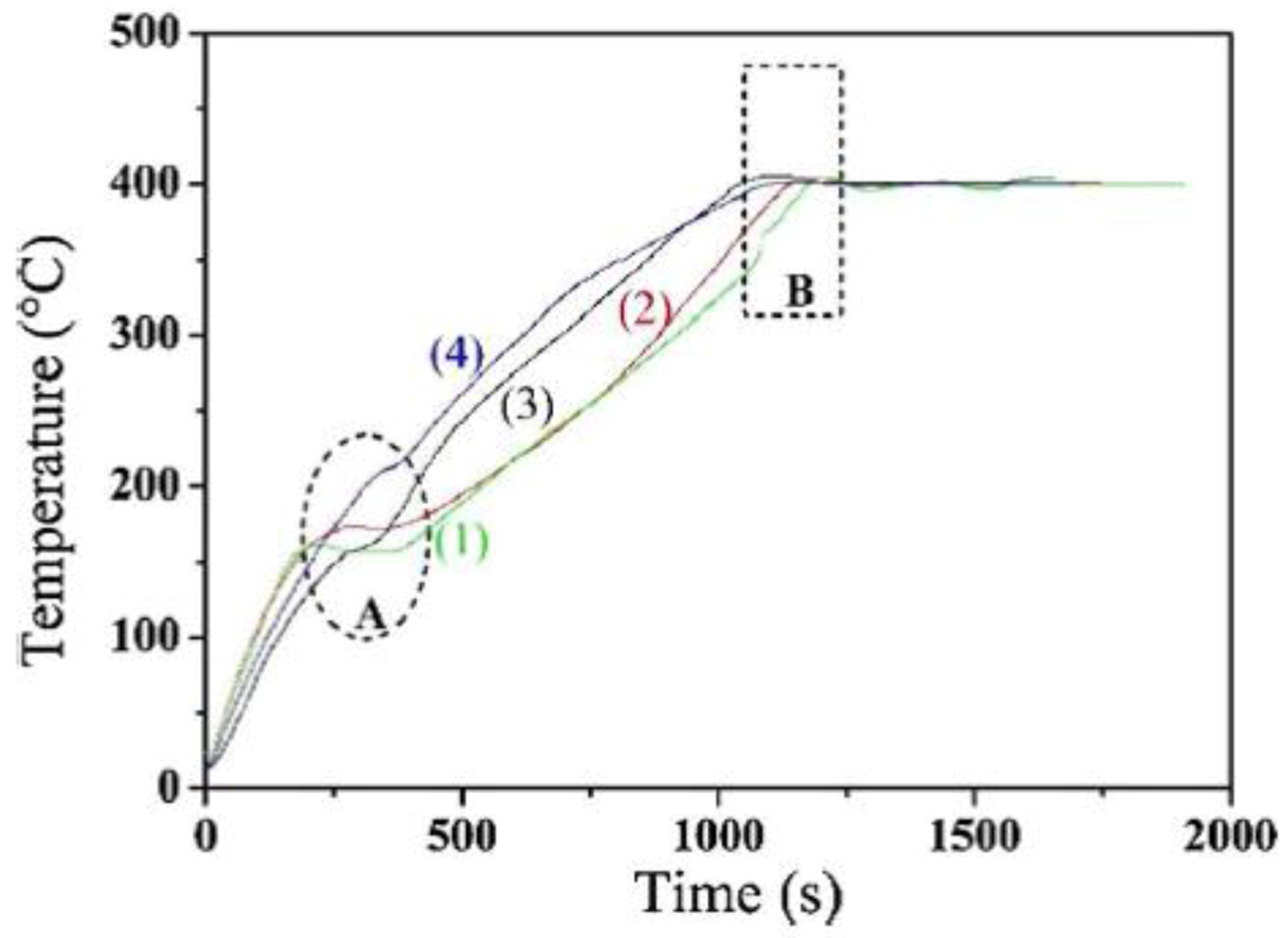
Figure 4 reports heating curves under MW irradiation (1.0 kW output power) of the different reagents used to produce the catalysts: Al2O3 is a poor MW absorber, but when Re was firstly incorporated in γ-Al2O3 (Re-Al2O3), the materials showed heating curves with higher slope (curves 3 and 4 in Figure 4). Due to the presence of Re, both the Re-Al2O3 and Ni-Mn/Re-Al2O3 materials absorbed more MW with respect to pure γ-Al2O3. This had a deep impact on the reaction time, in that the synthesis temperature (ca. 400 °C) was reached shortly when Re-Al2O3 was used for the deposition of the bimetallic Ni-Mn catalyst starting from the respective nitrates.
Other phenomena occurring during MW heating can be inferred by a close inspection of the curves in Figure 4: when Re is absent, the temperature plateau reached at ca. 150 °C is ascribed to thermal balance between (Ni and Mn) nitrates decomposition and MW irradiation. The phenomenon does not occur in the heating curves of Re-added samples, likely because of the enhancement of thermal decomposition of nitrates due to the presence of Re. Simultaneously, MW heating favors the formation of hot spots that induce an effective surface dispersion of the metal. A side-effect of fast MW heating was observed, i.e., formation of AlO(OH) phase (as detected by X-ray diffraction). MW heating, however, favored the dispersion of the NiO phase, which showed broader X-ray diffraction peaks with respect to the catalyst obtained by conventional heating. A better dispersion of the Ni active phase was also observed after reduction, confirming the overall positive effect of MW. At variance with what was observed with Ni-USY zeolite, MW heated samples were more easily reducible, as the onset of NiO reduction shifted to lower temperatures. Another important point observed by the authors was that MW-heated catalysts led to lower formation of carbon deposits, an undesired phenomenon that can lead to catalyst deactivation [49].
The presence of Re, however, also affected some important structural and electronic properties of the catalysts, as shown by the characterization of their physico-chemical properties. For instance, H2 TPR analysis showed a higher reducibility of β-type NiO (a type of NiO that has medium strong interaction with γ-Al2O3), indicating the facile formation of catalytically active Ni atoms. XPS analysis pointed out also a decrease of the binding energy of the Ni 2p line when Re was present, ascribed to the electron transfer from ReOx species to Ni atoms. The increased electron cloud density of Ni atoms could indeed favor the breakage of the C-O bond, positively affecting the catalytic activity.
CO2 reforming of methane (Equation (6)) is an endothermic reaction (∆H0 = +247 kJ mol−1, also known as dry reforming) to produce syngas mixtures with low H2:CO ratio that could be used for further Fischer-Tropsch synthesis (FTS) to produce light olefins:
CO2 + CH4 ⇌ 2CO + 2H2
The reaction has not yet been industrially implemented, since there are no commercial catalysts that can operate without undergoing deactivation due to carbon deposition [50]. An alternative to Ni-based catalysts (and to noble metals like Ru, Rh, Pd, Ir, and Pt) is provided by Ni-containing oxides, i.e., either Ni perovskites with defined structure or solid solutions with La, Al, or Mg (spinel, fluorite, and perovskite) [51]. Ni perovskites with defined structure are promising catalysts, with respect to noble metals, due to their low cost, thermodynamic stability at relatively high temperatures, and catalytic activity. Barros et al. [51] prepared La2NiO4/α-Al2O3 catalysts for CO2 reforming of CH4 by MW-assisted CS, by using glycine or urea as fuel and metal nitrates as oxidizers. For comparison, a catalyst with similar chemical composition was prepared by impregnation of α-Al2O3 prepared by MW.
XRD diffraction showed the formation of La3Ni2O6.92, La2NiO4, and LaAlxNix−1O3 crystalline structures independently of the preparation method. The sample prepared by impregnation was very active, but the crystalline structure was practically destroyed, with significant carbon deposition. The catalysts prepared by MW-assisted CS showed CH4 conversion above 80% at 700 °C, good stability, and a low coke formation after about 60 h on stream. The catalyst prepared by impregnation showed instead a decrease of CH4 conversion after 60 h, likely due to deactivation by coke deposition, according to TPO (Temperature Programmed Oxidation) analysis and TEM inspection, which showed the occurrence of carbon nanotubes, not observed when the catalysts were obtained by MW-assisted CS (Figure 5).
Moreover, the crystalline structure of the catalysts prepared by MW-assisted CS was preserved after CO2 reforming of CH4, whereas a prominent loss of crystallinity occurred in the catalyst obtained by impregnation (which was also deactivated) [51]; the authors ascribed the stability achieved by MW-assisted CS to the strong interaction of Ni active sites with the perovskite structure.
CO2 selective reduction to light olefins is a process that recently has progressed due to the urgent need of converting CO2 into building-block chemicals [13,14,52]. To raise selectivity towards light olefins, catalysts that are more effective are needed, since CO2 is very stable, and FTS usually leads to mixtures of products. K-promoted Fe catalyst is very selective towards CO2 hydrogenation to light olefins; additionally to K, Zn is also used as a promoter that accelerates FTS, increases catalyst stability, enhances light olefins selectivity, improves CO2 adsorption, promotes H2 dissociation, and is highly active in both WGS and RWGS reactions [52]. The effect of MW was studied on the synthesis of HT precipitation followed by incipient wet impregnation (to add K) of a series of K-promoted Fe-Zn catalysts with different Fe/Zn molar ratios. Physico-chemical characterization of the catalysts showed they were characterized by rather uniform particle size (ca. 100 nm). XRD analysis showed the Zn addition to the K-promoted Fe2O3 matrix led to formation of ZnFe2O4 spinel phase and ZnO phase, and also brought about an increase of the surface area. Moreover, the Fe-Zn interaction affected the adsorption and reduction of CO2 adsorption.
The catalysts exhibited excellent activity for CO2 hydrogenation and high selectivity toward C2–C4 olefins owing to the promoting effect of Zn, due to the fact that particles with uniform shape were obtained, and a fair dispersion of active components occurred with respect to conventional co-precipitation. Additionally, the formed ZnFe2O4 phase imparted stability to the catalyst, although lowering its activity due to segregation of a fraction of Fe (the active phase).
2.3. MW-Assisted Synthesis of Photocatalysts for CO2 Reduction
Photocatalytic CO2 reduction processes (i.e., biomimetic artificial photosynthesis) are very challenging due to intrinsic limitations of the used semiconductors, like poor absorption in the visible range and fast electron/holes recombination rate (e.g., for TiO2) and stability (e.g., for CdS). “Plasmonic” photocatalysis is a plasmonic metal-enhanced photocatalytic process occurring when noble metal nanoparticles (NPs) decorate the surface of a semiconductor (e.g., TiO2) to improve its photocatalytic efficiency through the localized surface plasmon resonance (LSPR) effect of NPs [53].
Most of the studies on the subject concern either Au or Ag NPs supported on TiO2, but their actual use is hampered by the cost and the limited abundance of the noble metals. Recently, it has been shown that also Cu NPs can have LSPR effect, indicating that Cu could replace noble metals.
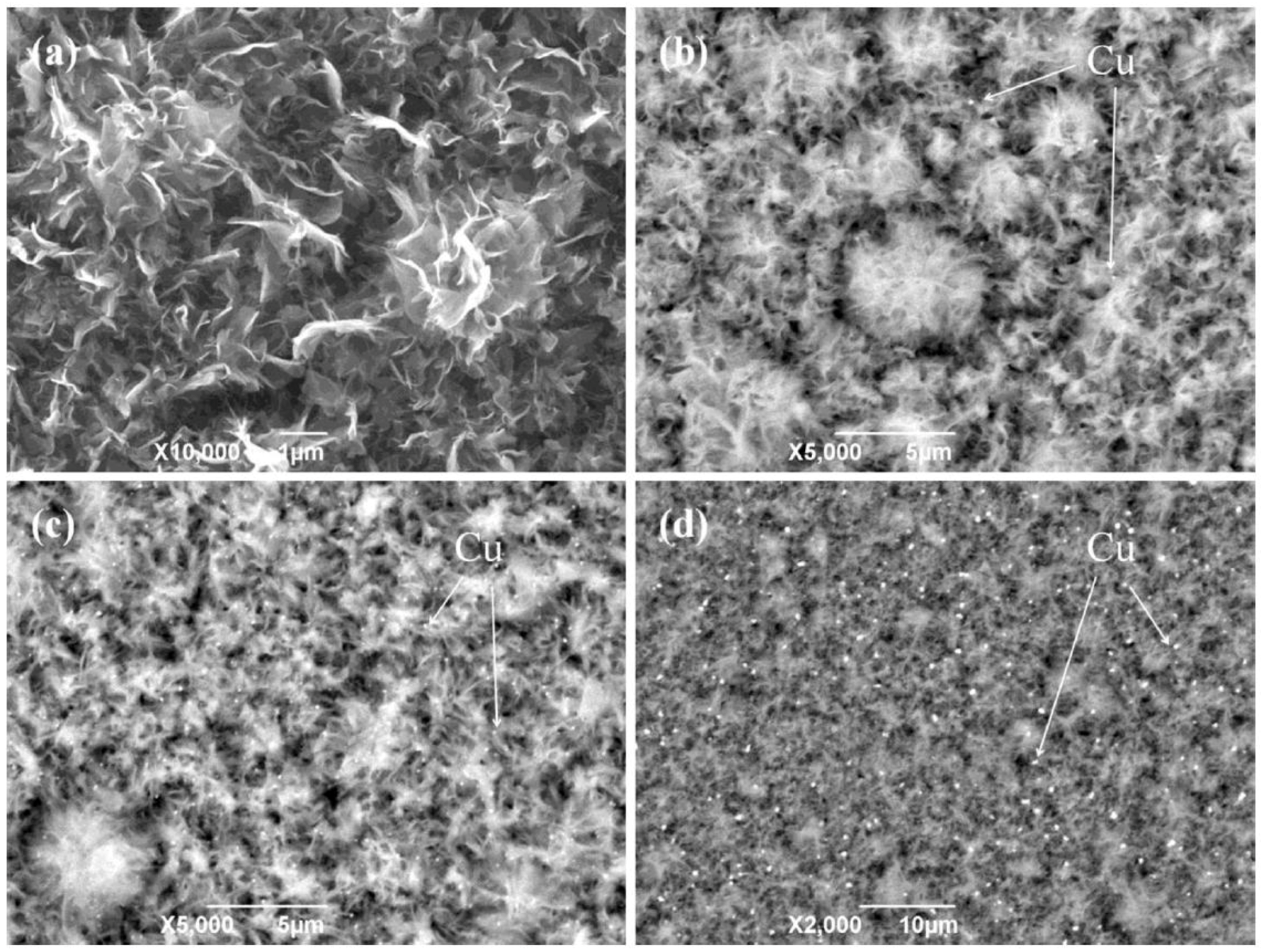
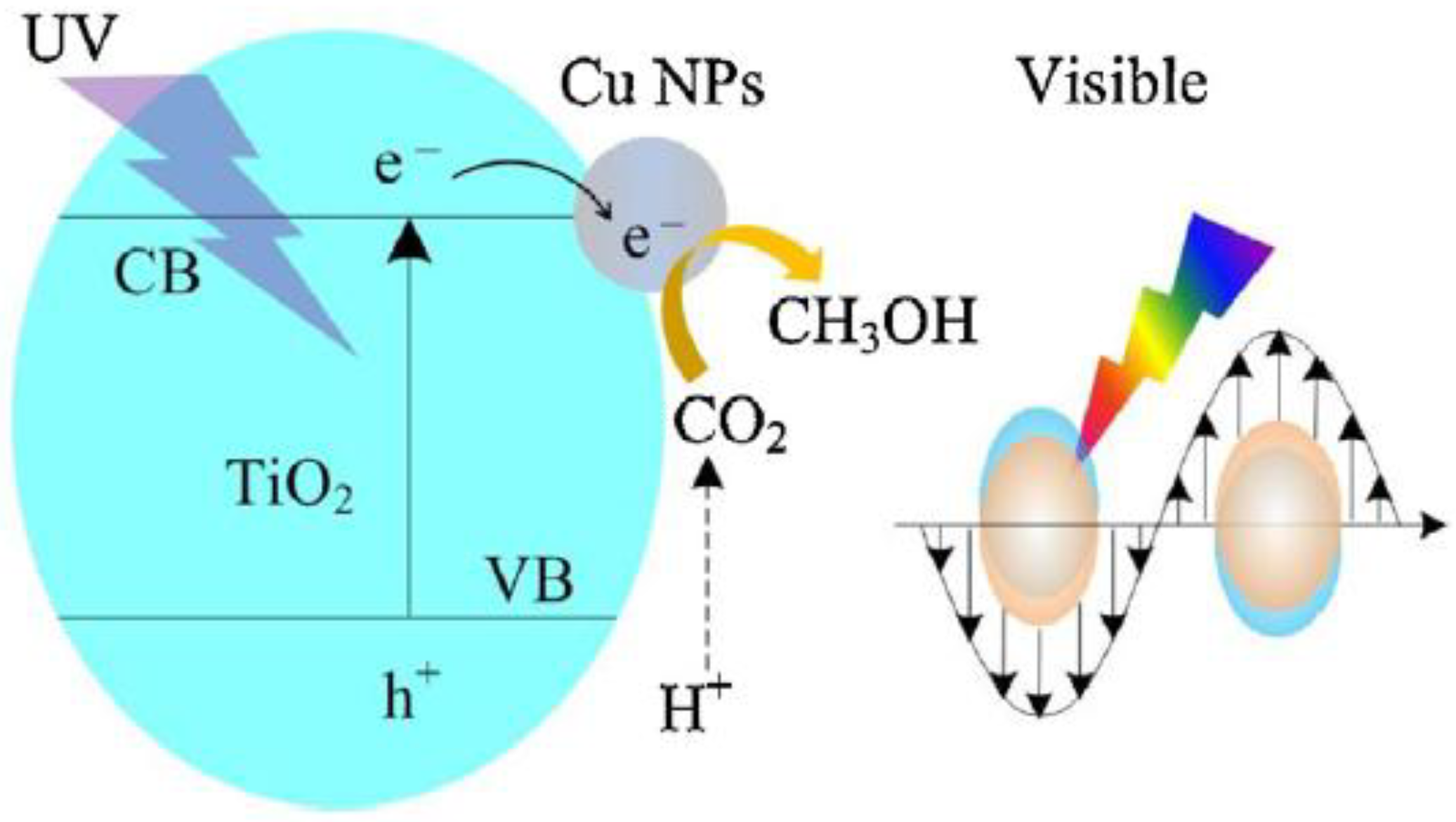
Cu NPs-decorated TiO2 nanoflower films (Cu/TiO2 NFFs) were prepared using both HT method and MW-assisted reduction process. Polyvinylpyrrolidone was used as capping agent and NaH2PO2*H2O as reducing agent in diethylglycole solution under MW irradiation. The catalysts were tested in the photocatalytic reduction of CO2 to CH3OH [54].
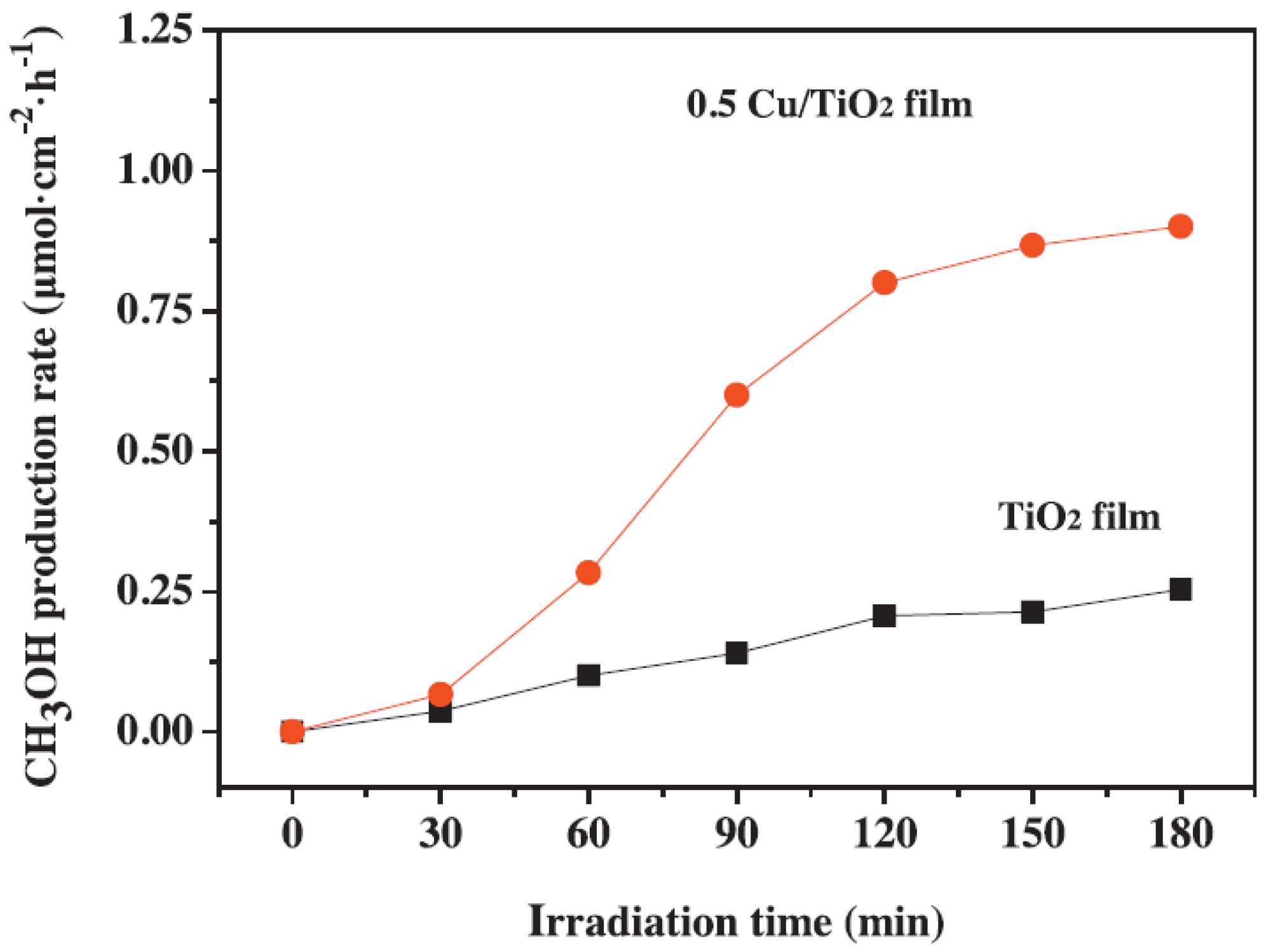
Physico-chemical characterization of the films showed that both Cu NPs and TiO2 films were characterized by a fair dispersion of Cu NPs on the TiO2 NFFs (Figure 6) and by visible light harvesting properties, due to LSPR of Cu NPs and the peculiar nanostructure of the TiO2 film. Moreover, the presence of Cu NPs allowed fluorescence quenching due to the suppression of charge carriers. The CH3OH production rate reached 1.8 mmol cm−2 h−1 (with an energy efficiency of 0.8%) with a 0.5 wt % Cu/TiO2 NFFs under UV and visible light irradiation: with Cu NPs, the production rate was ca. 6.0 times higher than with pure TiO2 film (Figure 7).
The positive photocatalytic performance towards CO2 reduction to CH3OH observed when Cu NPs were present (Figure 7) was ascribed to a synergistic mechanism of Cu NPs at the surface of TiO2 (Scheme 3): Cu NPs obtained by MW-assisted reduction method likely suppressed the recombination of charge carriers and improved the charge transfer efficiency.
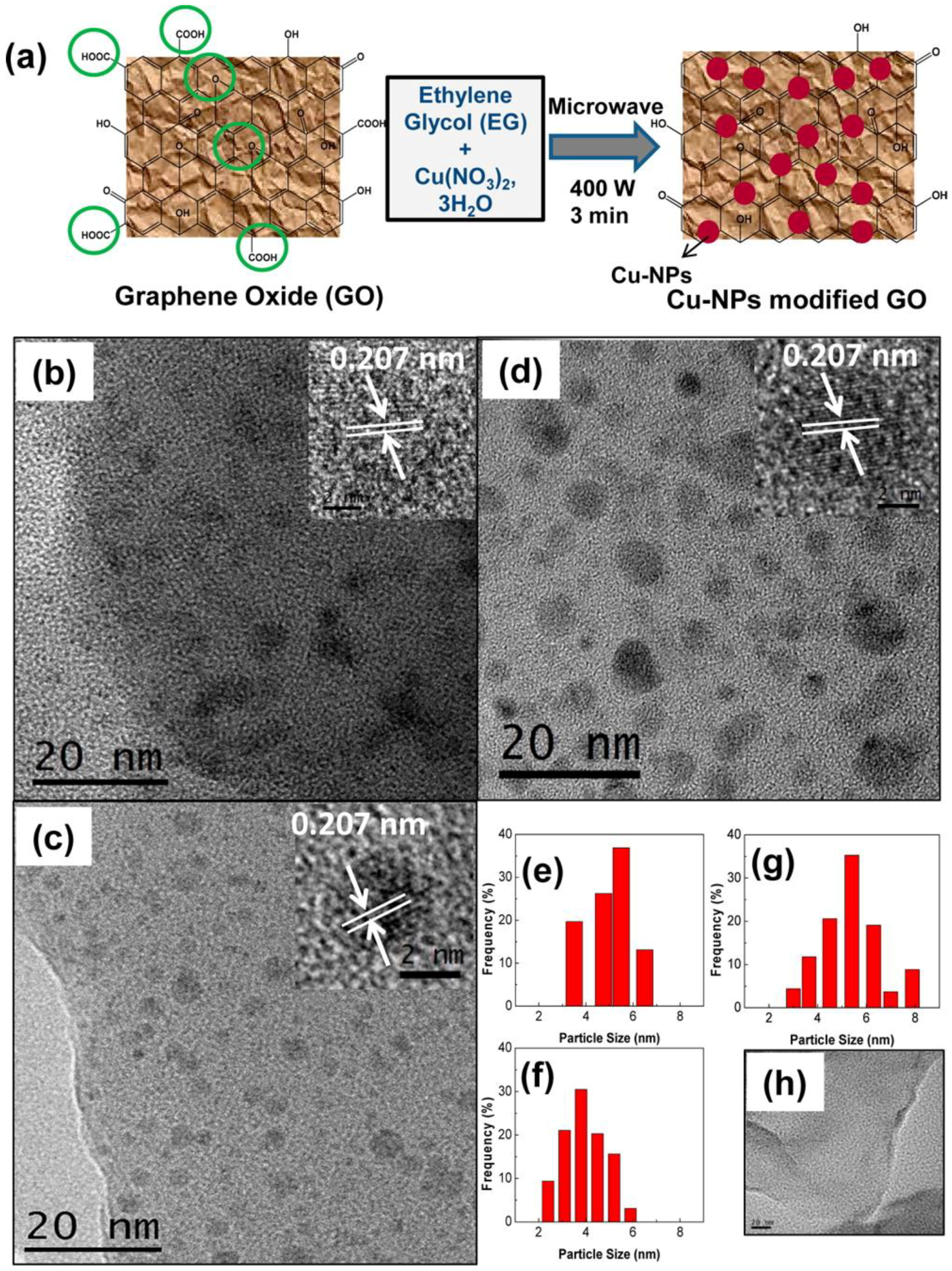
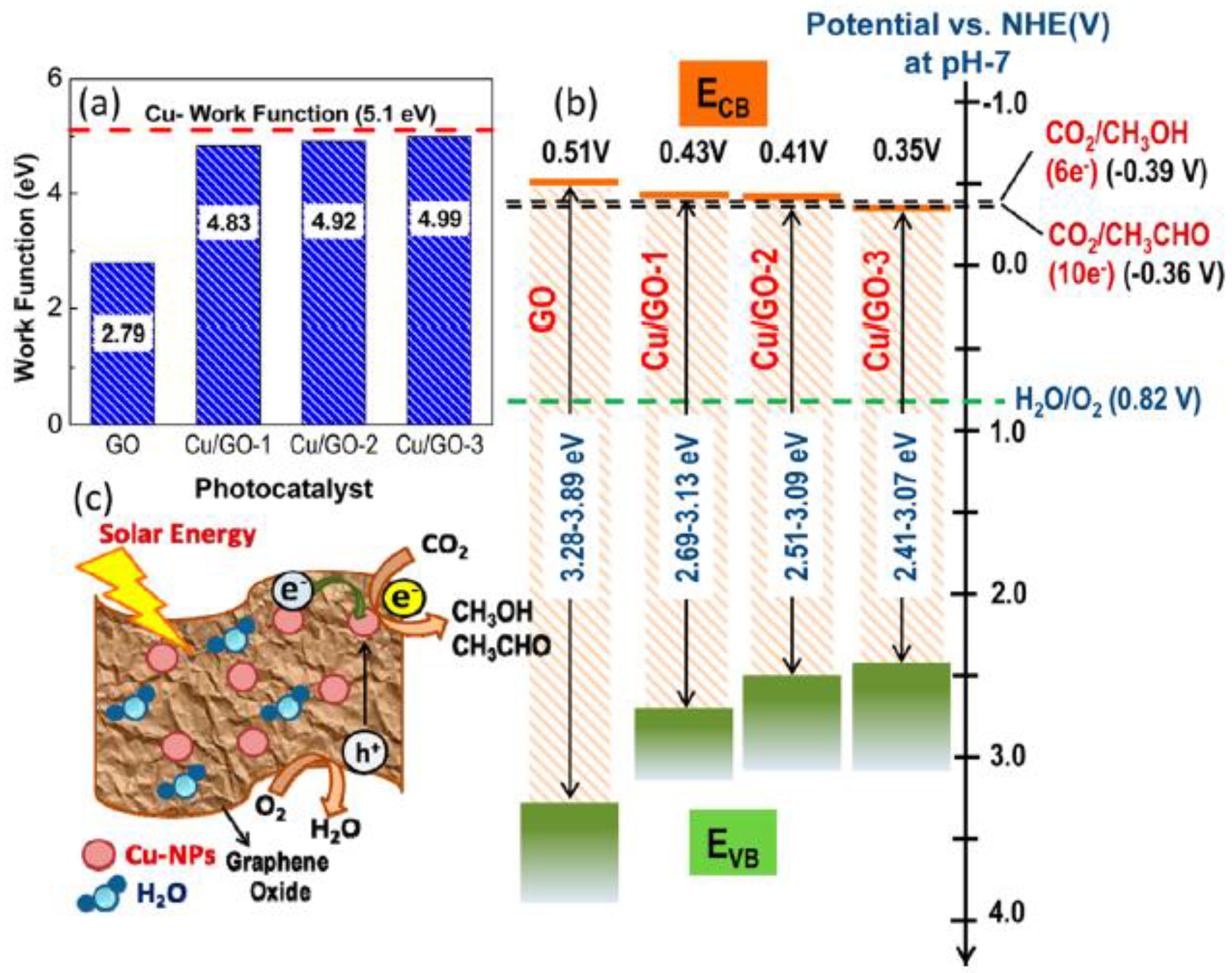
In another study, Cu-Graphene Oxide (Cu/GO) composite photocatalysts were obtained using a simple and rapid one-pot MW process [55], shown in Figure 8a.
By using Cu(NO3)2 as metal precursor, short MW heating (ca. 3 min at 400 W) was performed in ethylene glycol solution (Figure 8a), leading to a fair dispersion of crystalline Cu NPs in the GO.
The obtained Cu NPs (with a size of ca. 4–5 nm, Figure 8) were strongly interacting with GO: the obtained composite was particularly active under solar irradiation in the production of both CH3OH and acetaldehyde. Cu NPs were shown to enhance the GO photocatalytic activity of GO, essentially by suppressing electron-hole recombination, and also by reducing the GO bandgap and by modifying its work function (Scheme 4). The presence of Cu NPs greatly enhanced absorption of GO in the visible range, though the shift in the band gap by effect of Cu NPs was not well understood. XPS analysis also showed that oxygenated groups of GO (i.e., -COOH and C-O-C groups) were almost completely removed by the MW treatment. No other Cu oxides were detected, indicating that the MW heating led to the reduction of all the Cu ions.
XPS analysis [55] showed the likely occurrence of a charge transfer from GO to Cu NPs, and a correlation was found between the Cu content and the rate of formation and selectivity of the products. The Cu/GO catalyst with 10 wt % Cu was more than 60 times more active than pure GO and ca. 240 times more active than commercial TiO2 (Degussa P25) photocatalyst under visible light.
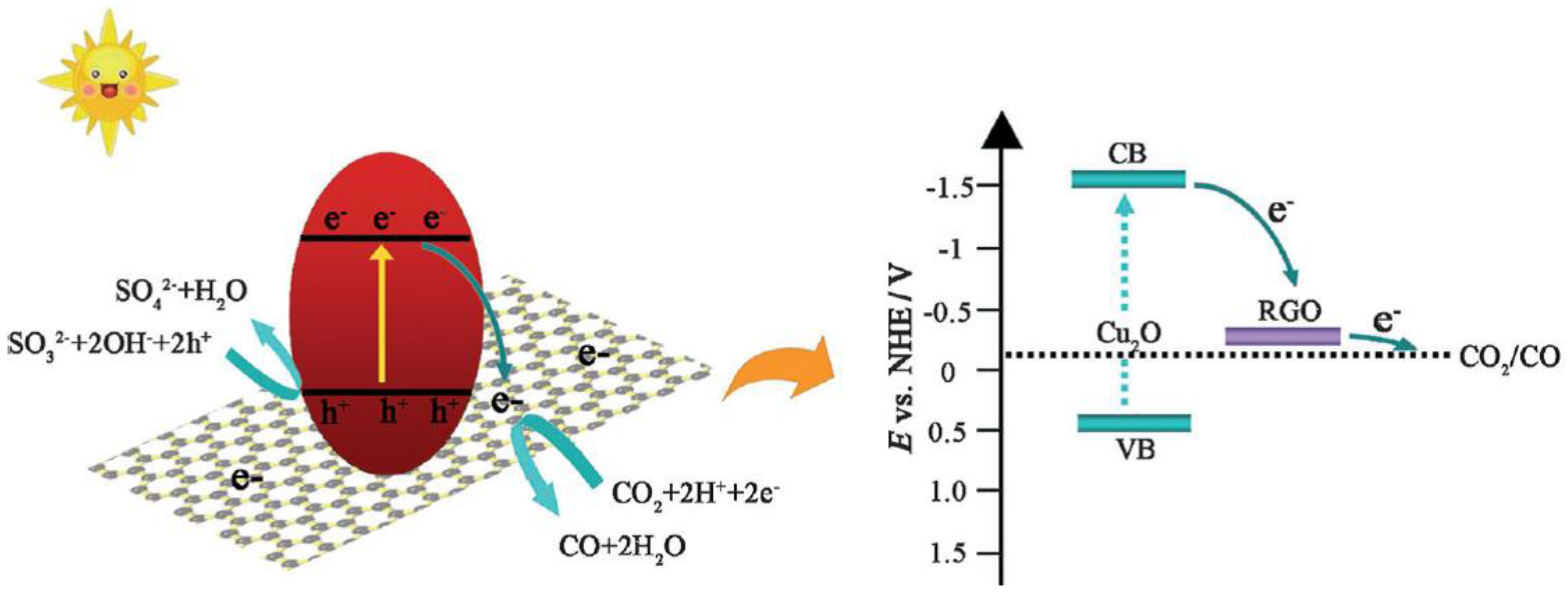
Also, Cu2O can be used for CO2 photoconversion: An et al. [56] produced Cu2O/RGO (Reduced Graphene Oxide) composites using a facile, one-step, MW-assisted HT synthesis. X-ray diffraction showed that the Cu2O was likely the only Cu-containing phase in the composites, and MW-assisted synthesis allowed the formation of composites where intimate contact between RGO sheets and Cu2O microspheres (a result of the strong affinity between the metal oxide and the abundant functional groups of GO) was enhanced. Such strong interaction between Cu2O and RGO was attributed to the adopted MW-assisted synthesis: such interaction was very effective in that RGO positively affected the photocatalytic activity, improving both activity and stability of Cu2O, due to an efficient charge separation and transfer to RGO (Scheme 5).
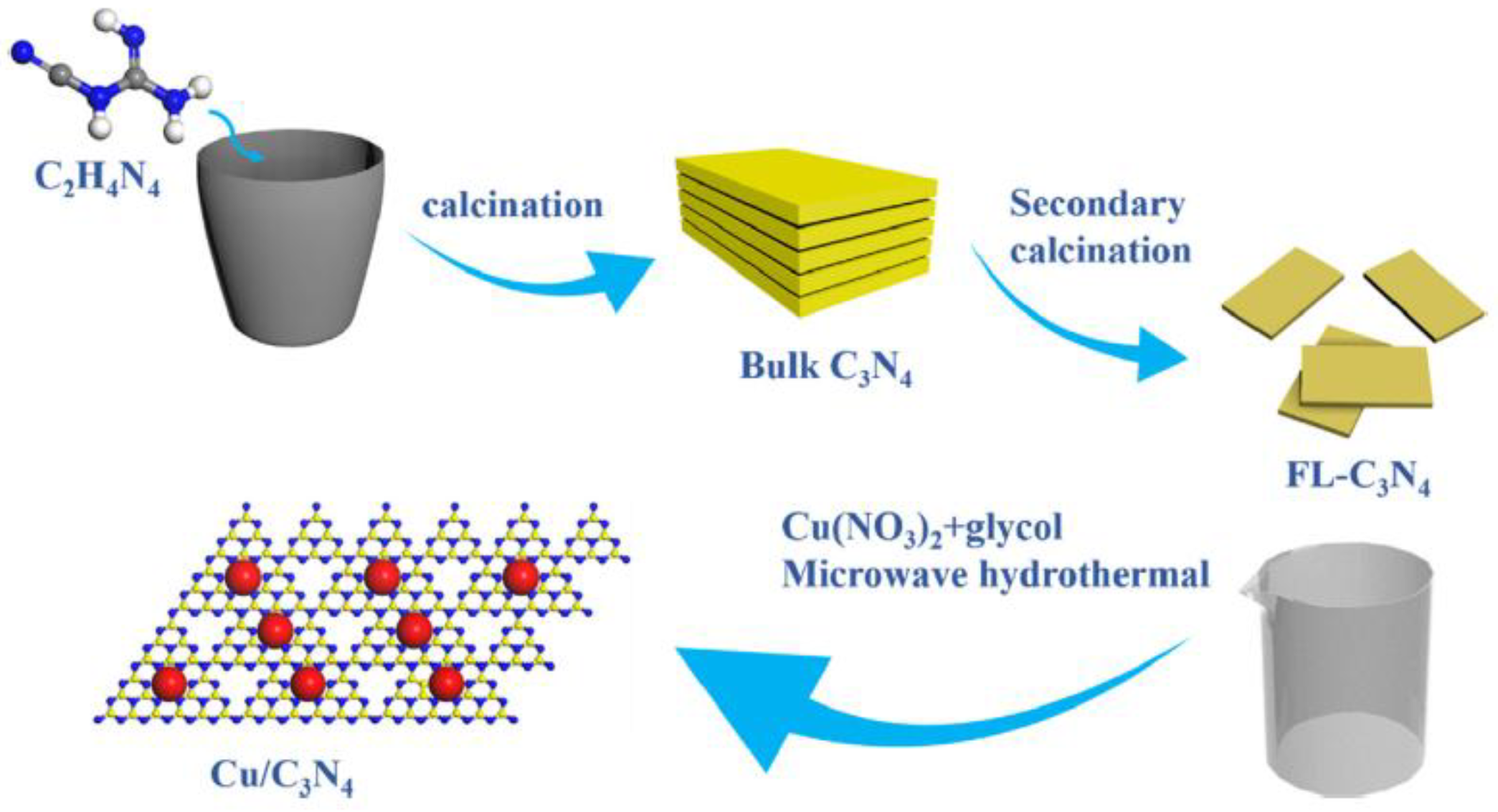
Cu-decorated graphitic carbon nitride (g-C3N4) nanosheets were obtained by a procedure depicted in Scheme 6, in which the final step involved the MW-assisted HT synthesis of Cu NPs [57].
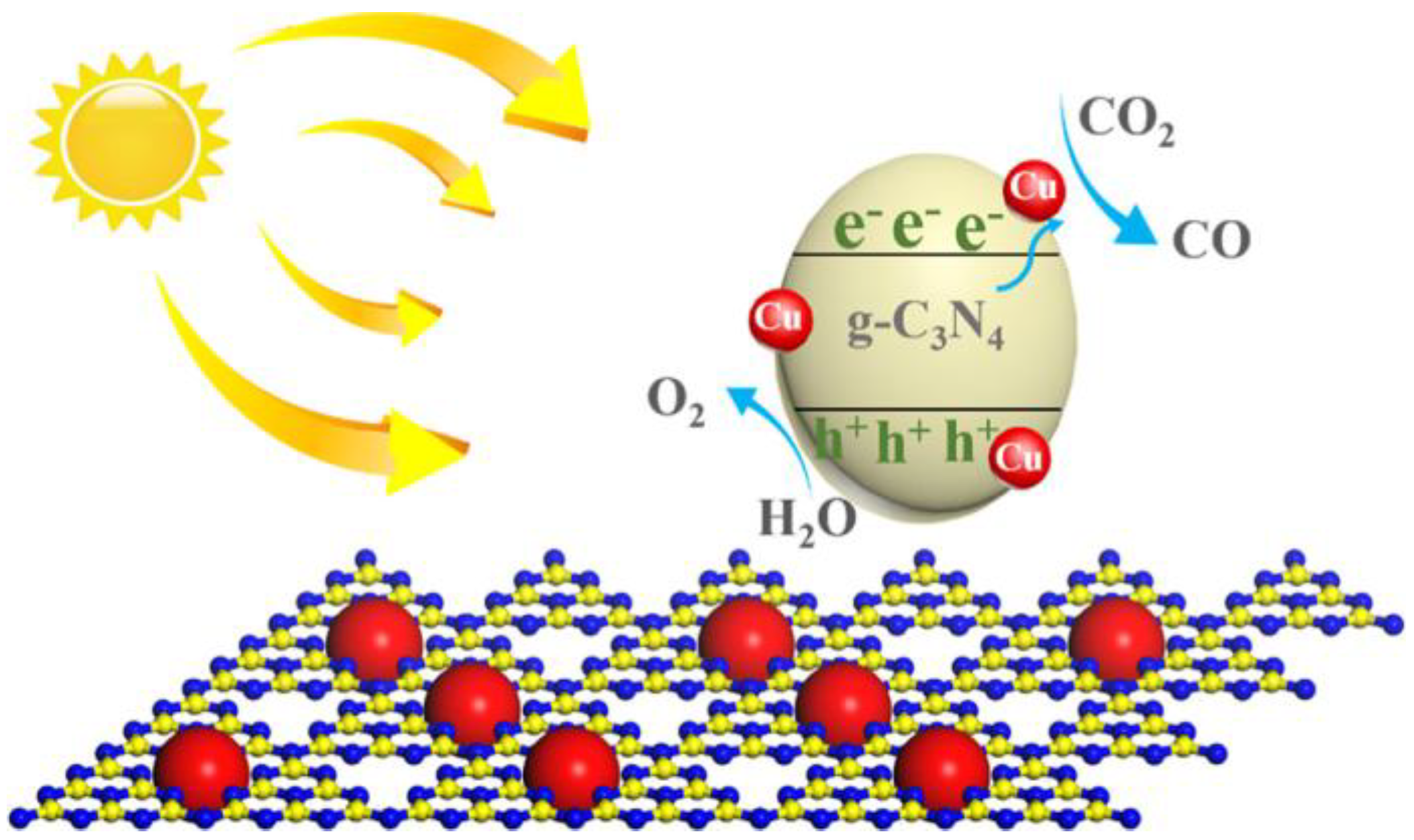
The composites were tested for the photocatalytic reduction of CO2 to CO. During MW-assisted HT synthesis, crystalline Cu NPs were obtained: some of them were deposited at the outer surface of nanosheets, and other were wrapped within them, as shown by TEM pictures in Figure 9. Such an intimate contact between the two phases likely enhanced the transfer of electrons and positively affected the photocatalytic activity.
The presence of Cu NPs also affected the ability of absorbing visible light, as shown by DR UV- Vis spectroscopy: Cu NPs likely facilitate the light harvesting process; moreover, the absorption edge of the composites shifted towards longer wavelength, indicating that a higher number of electrons/holes could be generated.
Photocatalytic tests showed that Cu had a positive effect of the CO yield. An optimal amount of Cu was found, corresponding to 6 wt % Cu. That content was a kind of tradeoff between 3 wt % (a too low Cu content) and 9 wt %, in which a higher number of Cu NPs can became centers for electrons/holes recombination. Scheme 7 reports the proposed mechanism, in which photogenerated electrons excited in g-C3N4 migrate to the surface of Cu NPs, acting both as trapping centers and catalytic sites, for the photocatalytic CO2 reduction to CO. Simultaneously, photogenerated holes migrated to the surface of the photocatalyst for water oxidation to O2.
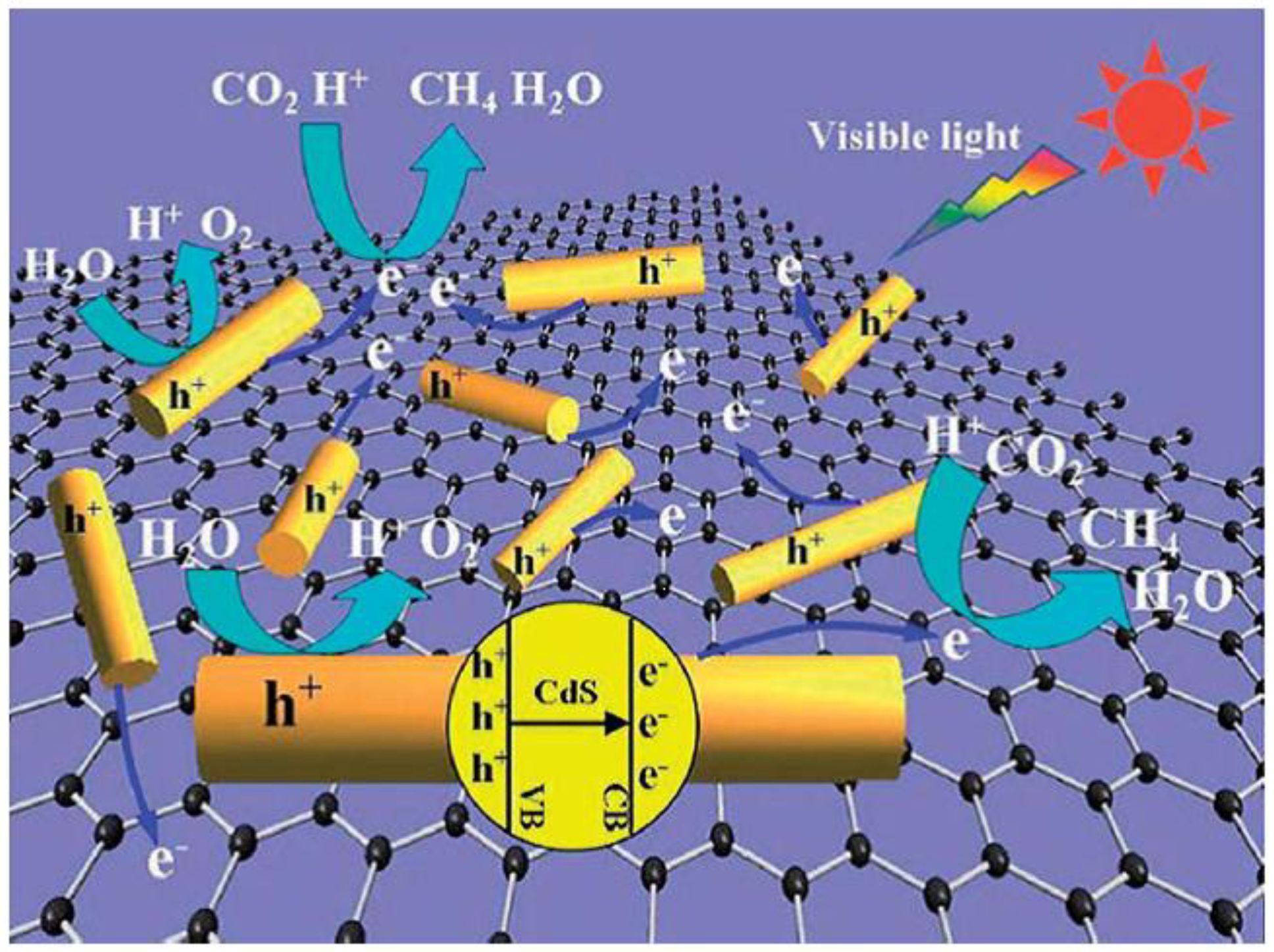
CdS is one of the most promising candidate semiconductors as photocatalysts active in the Vis range: Yu et al. [58] prepared RGO–CdS nanorods composites in ethanolamine–water solution using a one-step MW-assisted HT method. The composites exhibited a high activity in the photocatalytic reduction of CO2 to CH4, even without noble metal catalyst (i.e., Pt, which is usually adopted as co-catalyst). MW-assisted HT synthesis requires shorter time and less energy than conventional HT synthesis. Furthermore, particles with uniform size are usually obtained. Raman and XPS spectroscopies confirmed the reduction of parent GO using the MW-assisted method. TEM analysis (Figure 10) showed an intimate connection between the two materials.
An optimal RGO content of 0.5 wt % was found, leading to the highest CH4 yield. Higher RGO content led to lower CH4 yields; the authors ascribed this result to RGO shielding light absorption by CdS nanorods. RGO acted as co-catalyst, enhancing the photocatalytic reduction of CO2 to CH4, with the best performance attained with a RGO content of 0.5 wt %; the corresponding CH4 production rate was 2.51 mmol h−1 g−1, which exceeded that of pure CdS nanorods by more than 10 times. According to the authors, RGO was able to adsorb more CO2 molecules and to activate them very efficiently, simultaneously generating a higher number of photogenerated electrons on its surface, finally leading to an enhancement of CO2 reduction performance of the RGO–CdS composite (Scheme 8).
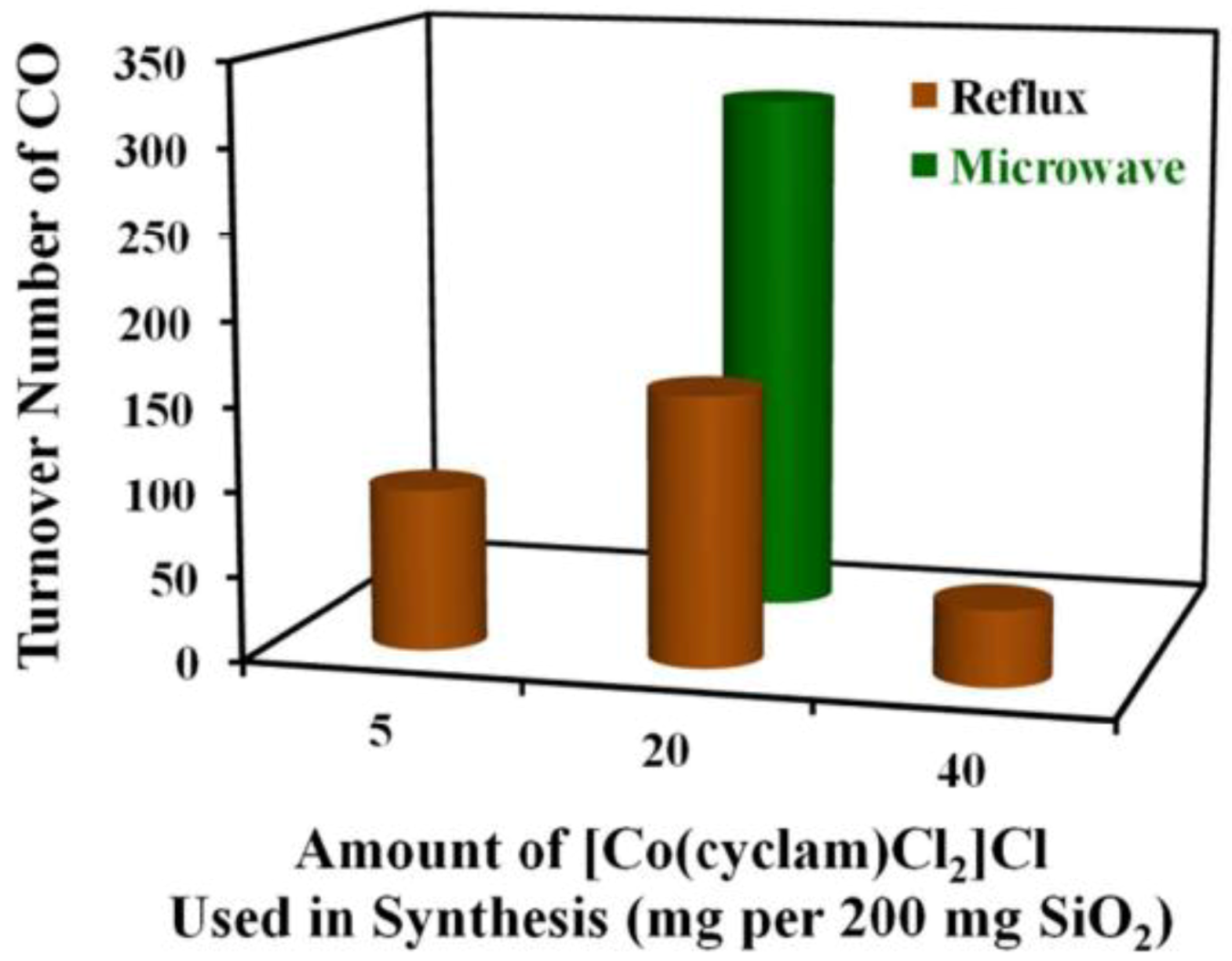
In another work, MW were found to positively affect the immobilization of homogeneous Co(III) cyclam complex (cyclam is 1,4,8,11-tetraazacyclotetradecane, namely: [Co(cyclam)Cl2]Cl) at the surface of SBA-15 silica. The photocatalytic activity towards CO2 reduction to CO was studied in the presence of p-terphenyl as a photosensitizer [59]. Usually, Co(III) cyclam immobilization is performed by reflux method, but MW-assisted immobilization of the Co(III) cyclam complex occurred more rapidly and led to more uniform Co(III) surface species. Moreover, a smaller amount of Co(III) complex was needed with respect to reflux method. This latter aspect led to well-defined, molecularly dispersed Co(III) sites within SBA-15 mesopores. IR spectroscopy showed that the complex interacts with the silica surface mainly by chemisorption on silanol (≡SiOH) groups. The photocatalyst prepared by MW-assisted synthesis was more active and selective than that prepared by reflux method, in the presence of a molecular photosensitizer and sacrificial electron donors (Figure 11).
3. Ultrasound as Unconventional Activation Method
3.1. General Principles of US-Assisted Synthesis of (Nano)Materials
US is essentially a mechanical longitudinal wave with frequency equal to or higher than 20 kHz, which is the upper limit of human hearing range. US propagation gives sinusoidal variation in the static pressure in the liquid medium inducing cavitation, a phenomenon controlled by external parameters, i.e., US frequency and intensity and type solvent. Cavitation is based on radical and mechanical effects. The radical effect occurs either at the bubble interface or in the interior cavity and is ascribed to the occurrence of the sonolysis of molecules. The mechanical effect, occurring after the cavity collapse, arises from shear forces, microjets, and shock waves outside the bubble, therefore producing severe physical changes in the presence of solids or metals. The transient implosive collapse of the bubble is an adiabatic phenomenon, during which inside the bubble very high temperature and pressure occur (~4600 °C and ~50 MPa). The phenomenon affects the reaction system [60] by generating both an intense local convection in the reaction medium (finally enhancing the mass transfer processes) and highly reactive radical species within the bubbles, due to high local temperature and pressure. Upon fragmentation of the bubble, the radicals generated therein get released into the medium, where they can induce/accelerate chemical reactions [61].
On the one hand, US has emerged as a potential green technique for the intensification of different physical, chemical, and biological processes. Compared with conventional heating, the use of US irradiation reduced the reaction time from hours to minutes. On the other hand, US can be effectively employed in the synthesis of NPs with a high production yield, without using expensive instruments and/or extreme conditions [62,63], as, for example, carbon encapsulated Fe NPs with superior catalytic activity and reusability for hydrogenation reaction in the liquid phase [64]. US allow obtaining of highly reactive surfaces, enhancing mixing and increasing mass transport. Therefore, such unconventional method can be particularly helpful for preparing and activating catalytic materials [65].
3.2. US-Assisted Synthesis of Cu–ZnO Catalysts and of Their Improved Formulations
Under typical industrial conditions, CH3OH is formed mostly via CO2 hydrogenation, with CO acting as CO2 source and as oxygen scavenger from water, which inhibits the active metal sites [66]. A synergism between Cu0 and Cu+ sites is the most adopted explanation for the activity of Cu/ZnO-promoted catalysts [38,61,67,68]; however, Cu0 sites [69,70] and/or Cu–Zn alloy [71,72,73] have been proposed to have a catalytic role as well.
Many unconventional preparation routes have been adopted to obtain Cu–ZnO catalysts with improved activity, selectivity, resistance to poisons, and enhanced lifetime [38,74,75,76,77,78]. In this frame, the SG method was largely adopted in order to increase the metal surface area along with the total surface area [79,80]. Moreover, the excellent activity of oxalate co-precipitated catalysts was also demonstrated [81], and Cu–Zn catalysts prepared by “spark-erosion” showed superior performance in terms of selectivity [82], with noticeable TOF in the CO2 hydrogenation reaction that was ascribed to a close contact among the phases in Cu/ZnO/ZrO2 catalyst produced by combustion.
To further improve the physico-chemical properties of a Cu–ZnO/ZrO2 catalyst (Znat/Cuat, 0–3; ZrO2, 42–44 wt %), a new synthetic procedure consisting in the US-assisted reverse co-precipitation was proposed [83]. The US irradiation guaranteed highly increased total surface area (BET, 120–180 m2 g−1), although the Zn/Cu atomic ratio strongly affected either the specific surface area or the volume of the pores. Indeed, the materials with low (≤0.1) and high (>0.7) Zn/Cu ratios have lower surface area than those with a Zn/Cu ratio of 0.3–0.4, corresponding to a maximum of 175 m2/g. An analogous volcano trend was observed for the pore volume and pore diameter, in which the catalyst with Zn/Cu ratio equal to 0.4 contains mainly mesopores with diameter in the 70–120 Å range and, as a consequence, an enhanced cumulative volume (0.6 cm3 g−1). Moreover, the authors found that the US irradiation led to very high dispersion (3–58%) and exposure of Cu (SCu = 9–63 m2 g−1). The catalytic activity in the CH3OH production by CO2 hydrogenation was explored in a range of temperatures (160–260 °C) and pressures (1.0–3.0 MPa), and compared with a commercial Cu–ZnO/Al2O3 catalyst with optimum results ascribed to the presence of highly dispersed Cu particles.
The direct synthesis of dimethyl ether starting from syngas involves two consecutive reactions, i.e., CH3OH synthesis and CH3OH dehydration [84]. Alternatively, dimethyl ether can be directly obtained from syngas or CO2 hydrogenation.
The use of novel bi-functional catalysts based on metal oxides (namely, CuO, ZnO, Al2O3, and possibly some precious metal oxides, such as Cr2O3) to synthesize CH3OH and on acid solids (namely, HZSM-5 and HY zeolites or SAPOs) to convert it into dimethyl ether can significantly enhance the yield with respect to the two-step process, because CH3OH dehydration can interfere with the equilibrium of the synthesis reaction [85].
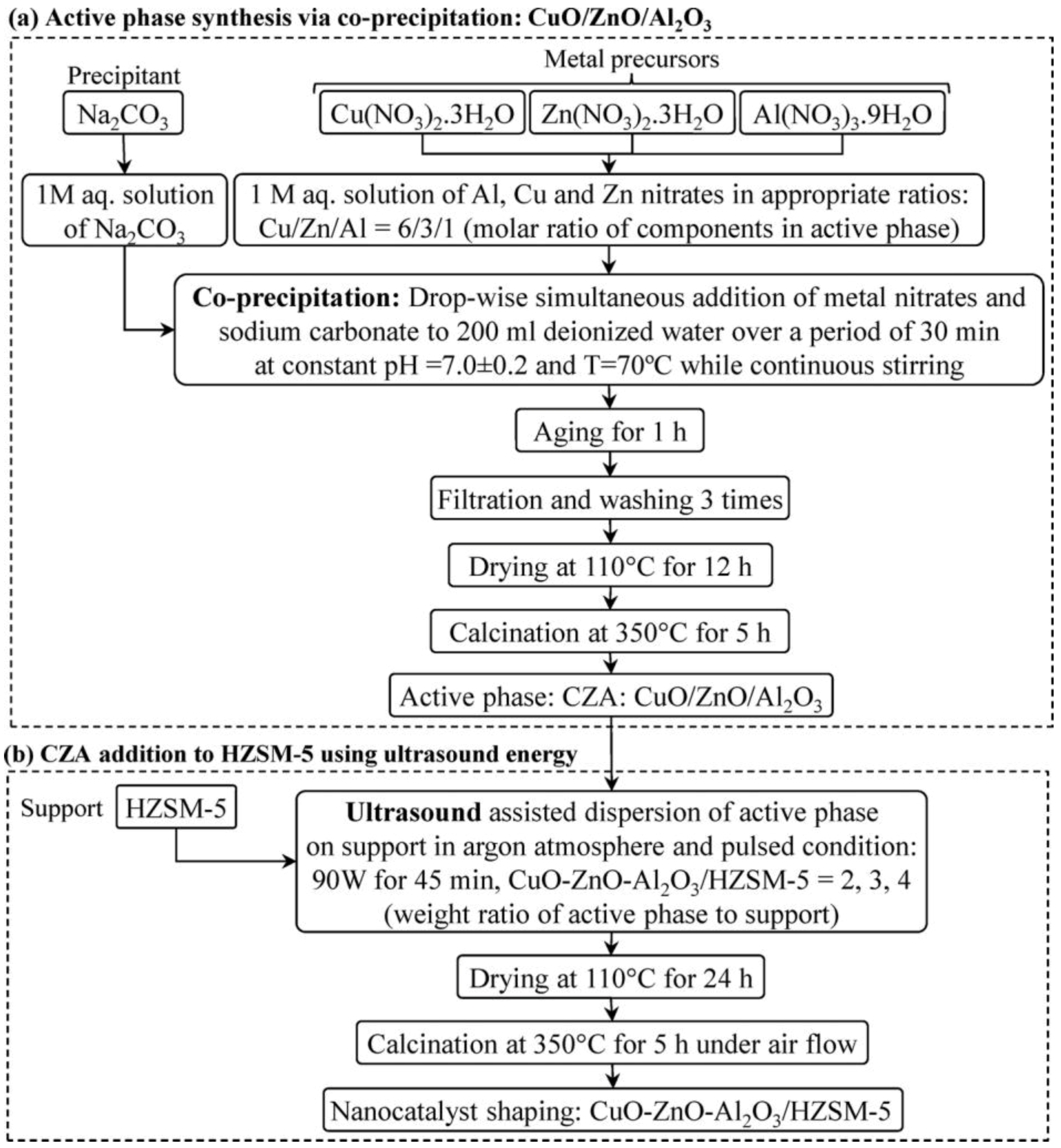
In this contest, Cu-ZnO-Al2O3, as well as Al2O3 and HZSM-5 catalysts, have been widely investigated [86,87,88]. In particular, a previous study reported on the preparation of CuO-ZnO-Al2O3/HZSM-5 hybrid catalysts (CZA/HZSM-5) using different techniques, such as impregnation, co-precipitation-physical mixing method, and a US-assisted co-precipitation procedure [89]. It was observed that the CZA/HZSM-5 catalysts prepared by US-assisted co-precipitation displayed higher activity than the systems obtained by conventional physical mixing and impregnation. Further, various catalysts with different CZA and HZSM-5 wt. ratio (namely, 2, 3, and 4) were synthesized by the US-assisted co-precipitation procedure summarized in Figure 12 in order to establish a correlation between the catalyst composition and the displayed activity [90]. The experimental conditions for US irradiation in Ar atmosphere were 90 W for 45 min, with 10 min current pulse time and 0.5 min rest time, by means of the experimental apparatus shown in Figure 13.
It is worth noting that the zeolite preserved its crystal structure during the preparation. Moreover, the crystallinity was significantly influenced by varying of the CZA content. XRD peaks related to monoclinic CuO, and hexagonal ZnO phases were observed. FESEM analyses indicated the presence of a uniform coating of the HZSM-5 surface by CZA upon US irradiation during the synthesis. In addition, the catalysts consisted of many small particles with almost spherical shape. At variance with reducibility, the surface area was not dependent on the CZA content. The HZSM-5 structure was not damaged upon loading CZA, as demonstrated by FTIR spectroscopy results. The optimal composition of the CZA/HZSM-5 catalysts for the conversion of syngas to dimethylether was 4:1 at 275 °C and 40 bar.
Interestingly, it was reported that the sonication time significantly affected both morphology and structure of the CZA/HZSM-5 catalyst [91]. In particular, larger CuO crystals formed at longer irradiation times, and at the same time the particle aggregates changed into smaller size and more uniform shape, resulting in an increase of the surface area. The catalyst synthesized under long US irradiation showed very good activity during time on stream test, whilst the non-sonicated sample lost its activity.
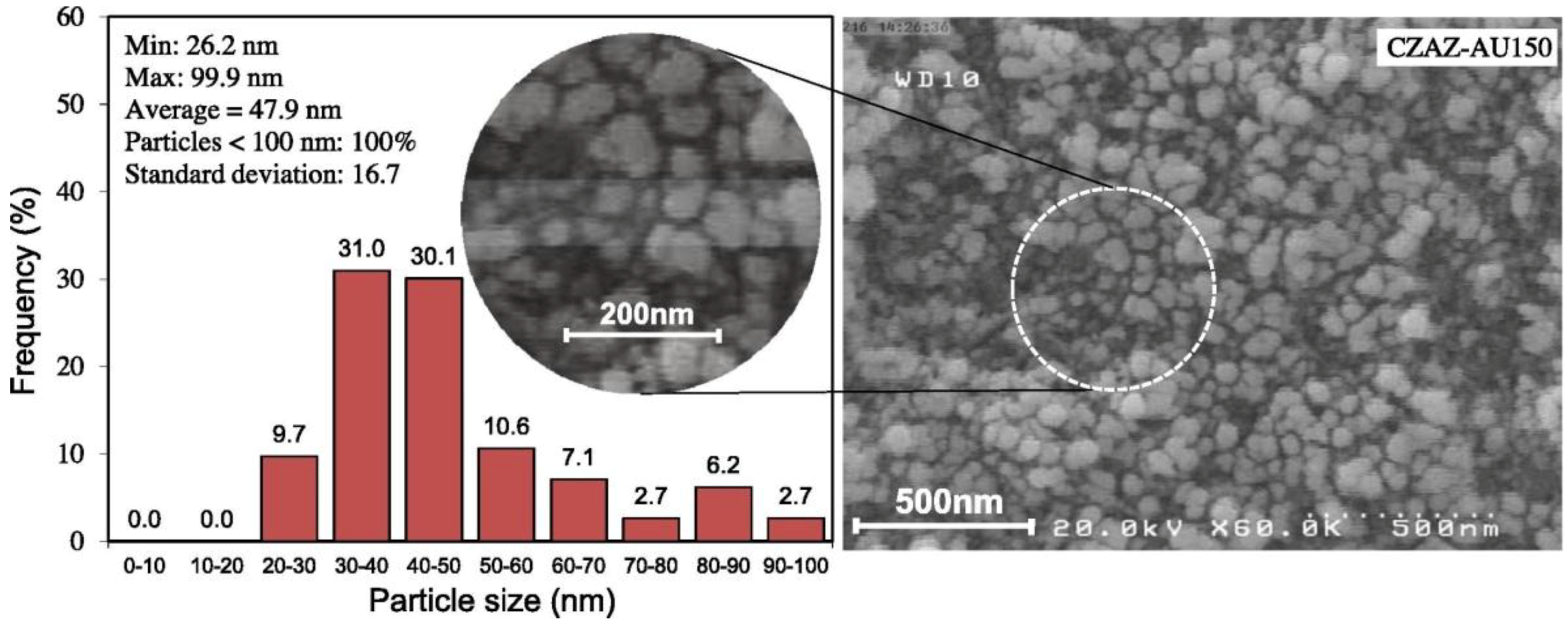
The effect of US irradiation power was evaluated on different CZA/HZSM-5 catalysts prepared by US-assisted co-precipitation method [92]. The results of a thorough physico-chemical characterization evidenced that the nature of the precursors and the time of irradiation strongly affected the morphology, the surface area, the functional groups, and the structure of the final materials. In particular, as shown in Figure 14, highly irradiated acetate-based catalysts had uniform morphology, as well as smaller CuO particles with higher surface area, due to high nucleation rate and high cooling rates, resulting in a stronger interaction among the components in comparison to the catalysts prepared by starting from nitrate precursors.
It is worth noting that an increase of the irradiation power gave rise to smaller particles, resulting in an improved dispersion and surface area of the catalysts. Moreover, CuO particles with lower crystallinity were generated at high power, resulting in an enhanced interaction among the metal oxide particles. In addition, such ultrasound-assisted co-precipitation method allowed to produce CZA/HZSM-5 catalysts that were more stable with respect to those prepared by conventional precipitation, for which 18% CO conversion and 58% dimethyl ether yield decreases in 24 h time on stream test were observed.
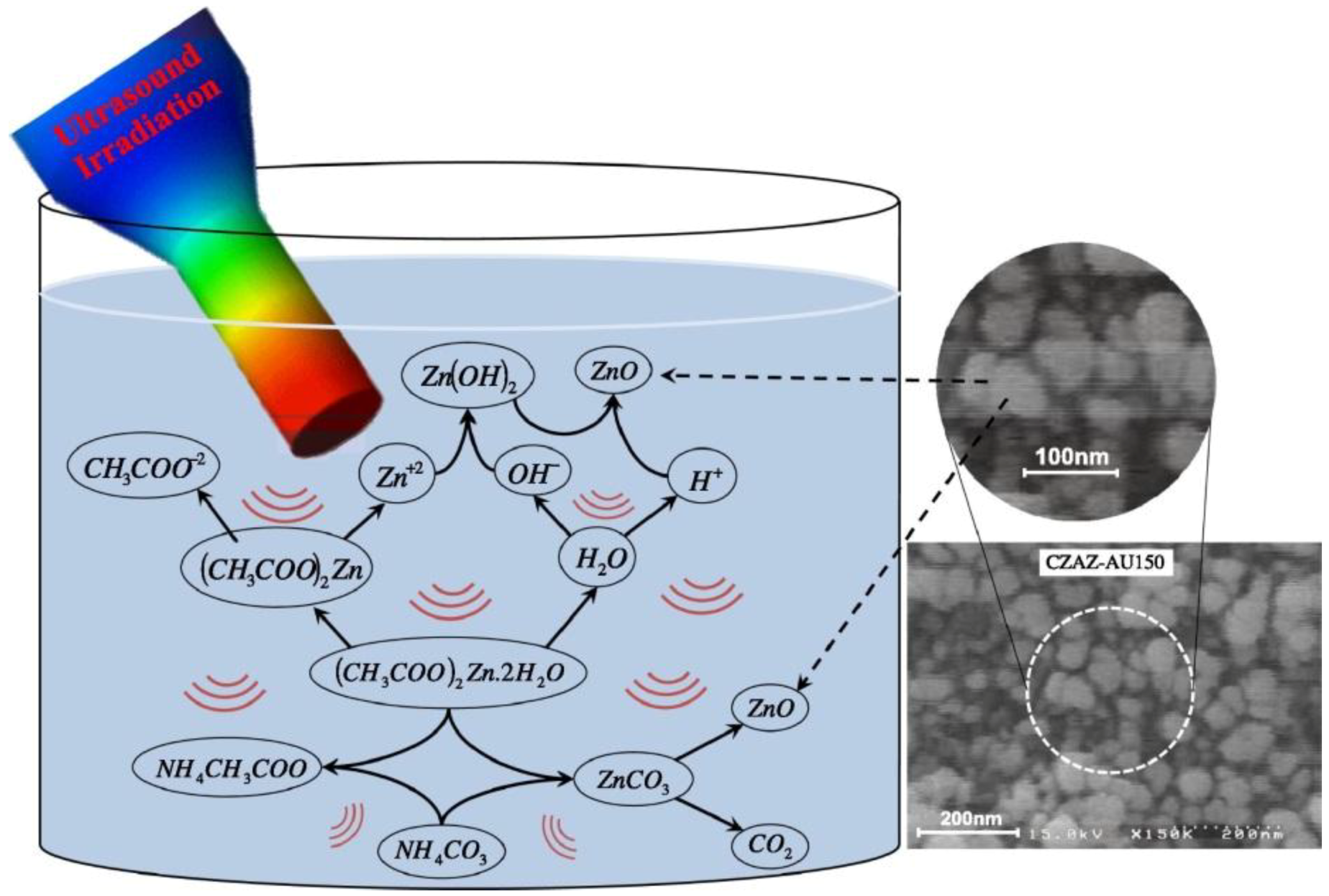
The authors proposed a pathway for the ZnO synthesis under ultrasound irradiation in CZA that is illustrated in Figure 15. Such pathway can be extended to other precursors, as well as to CuO and Al2O3. More specifically, the metallic precursors underwent decomposition and hydratation upon ultrasound irradiation, and H· and OH· radicals formed by water sonolysis [93]. The free OH· radicals formed in the cavities or at interface can react with the metal cations forming metal hydroxides that are converted to the corresponding metal oxides upon thermal treatment. Simultaneously, by co-precipitation of the metal precursor, the metal carbonates (that will then be converted into the metal oxides) are produced. Therefore, a strong synergistic action of US during co-precipitation was advised. In particular, the OH· radicals coming from the water sonolysis are able to convert a fraction of the metal salt to the corresponding metal oxide without any help from the precipitant. Moreover, a significant increase of the nucleation rate under peculiar temperature and pressure conditions upon cavity collapse gives rise to the formation of small and well-dispersed NPs. In particular, the irradiation cycles impede the growth of the nucleated particles, resulting in the production of small catalyst particles. Finally, particle agglomeration is prevented, indeed, the populations of OH· radicals and of the nucleation sites are enhanced at high US power, and the conversion to metal oxides is therefore improved.
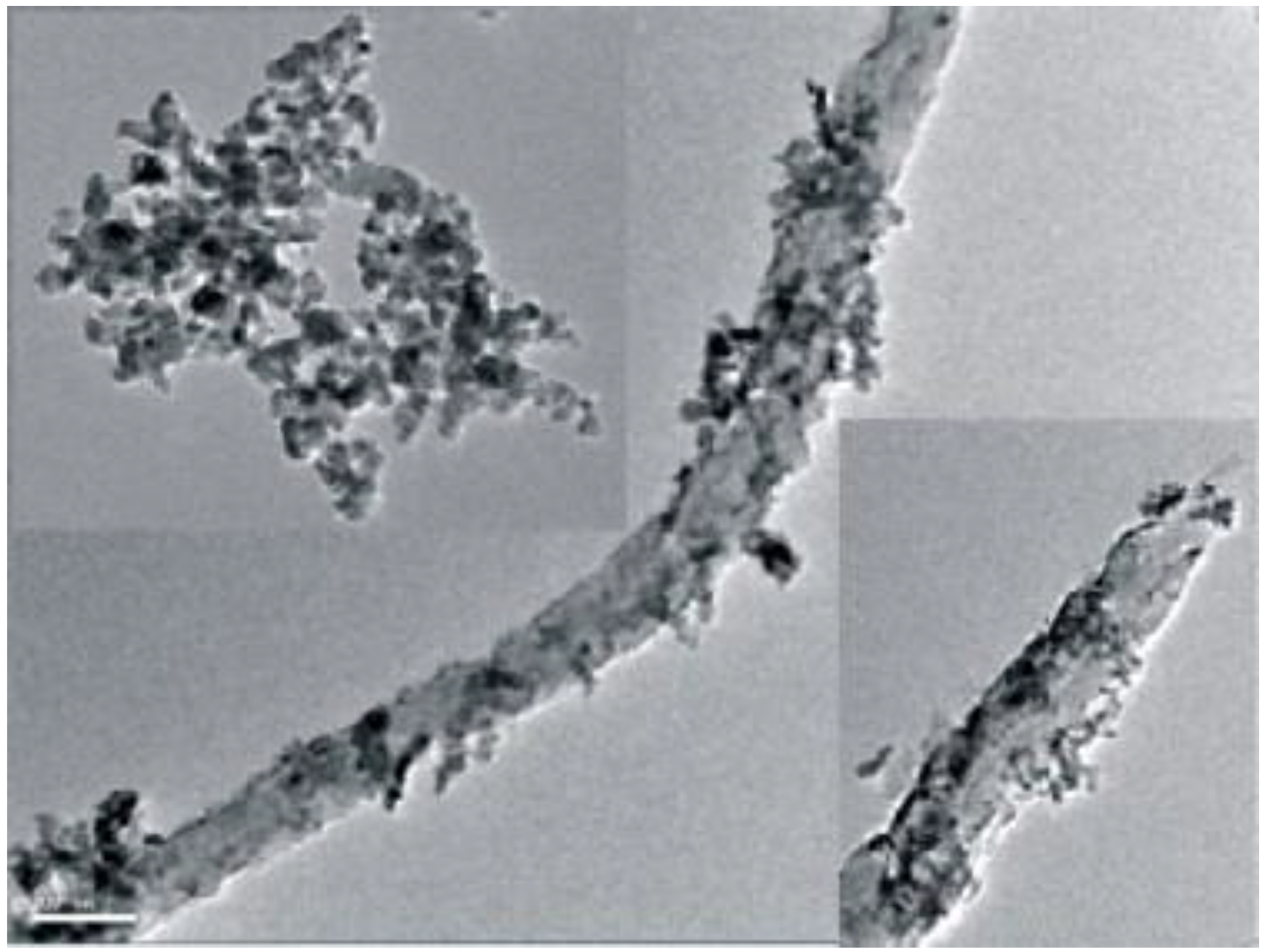
An US-assisted co-precipitation method was employed for the preparation of CZA/HZSM-5 catalysts supported on multi-walled carbon nanotubes pretreated with H2SO4/HNO3 mixture [94], in which the acid pretreatment had the aim of activating and functionalizing the surface of the nanotubes. The acid-pretreated carbon nanotubes were dispersed in deionized water by sonication more easily than the pristine ones, because the presence of the surface functional groups increased the capability to form hydrogen bonds, converting the surface of the multi-walled carbon nanotubes from hydrophobic to hydrophilic, therefore resulting in improved dispersion and suspension stability. The CZA NPs of about 10–20 nm were quite homogeneously distributed and densely deposited at the surface of the acid-pretreated, multi-walled carbon nanotubes (as shown in Figure 16).
The catalysts were tested in the hydrogenation of CO2 to produce dimethyl ether in a fixed-bed reactor. It was shown that the presence of the multi-walled carbon nanotubes has a promoting effect on the catalytic activity and on the dimethyl ether. The CO2 conversion was 46.2%, with dimethyl ether yield and selectivity of 20.9% and 45.2%, respectively, at 262 °C, 3.0 MPa, volume ratio H2/CO2 = 3, and space velocity = 1800 mL gcat−1h−1.
3.3. US-Assisted Synthesis of Catalysts with Ferromagnetic Properties
The synthesis of nanostructured materials based on ferromagnetic elements is of great interest due to the possibility to tune their magnetic properties [95,96,97], as well as to promote physico-chemical surface processes [98,99] by controlling the shape and size of the obtained NPs. In addition, magnetic catalysts can be easily positioned and recovered by using an external magnetic field.
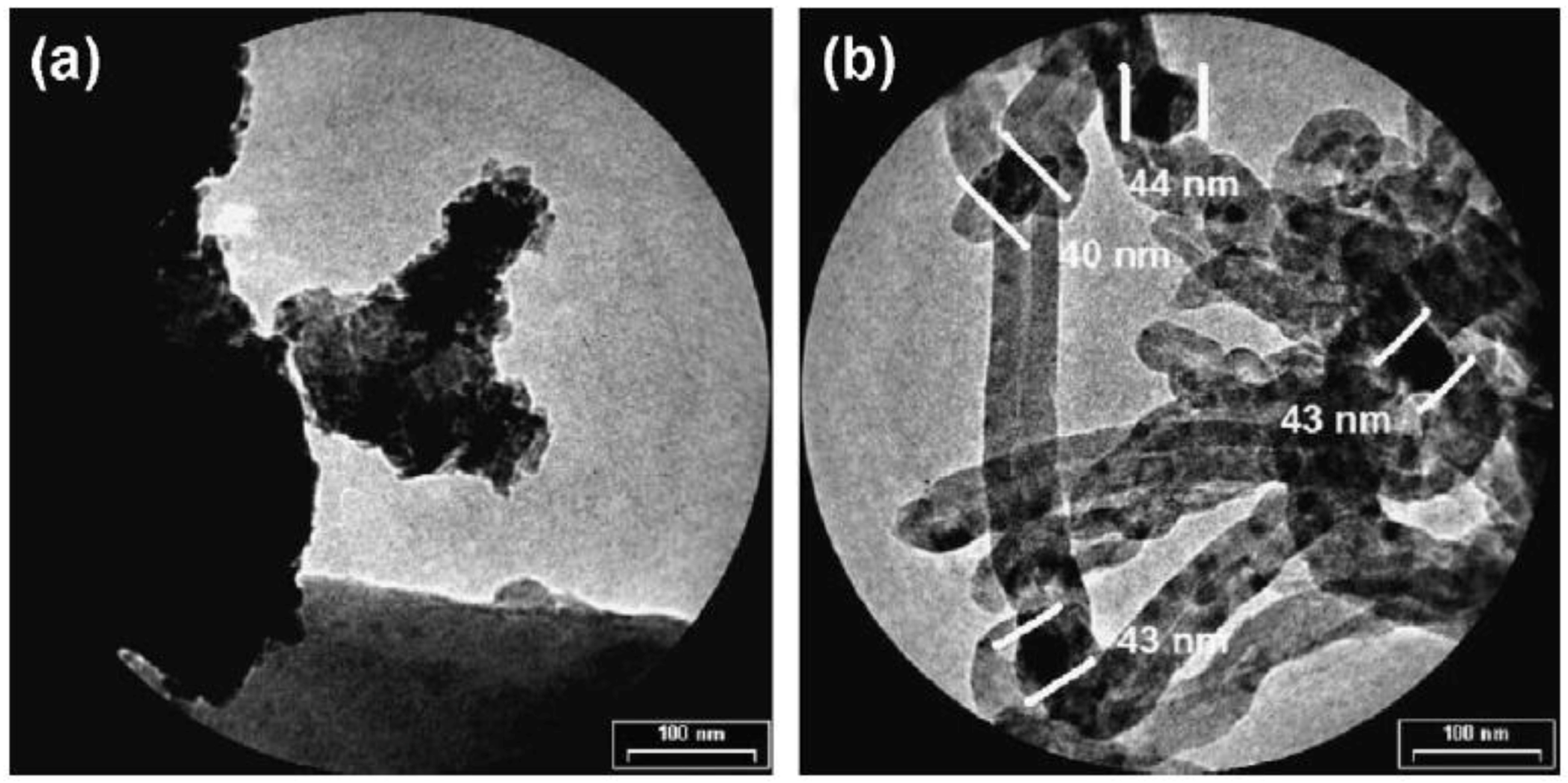
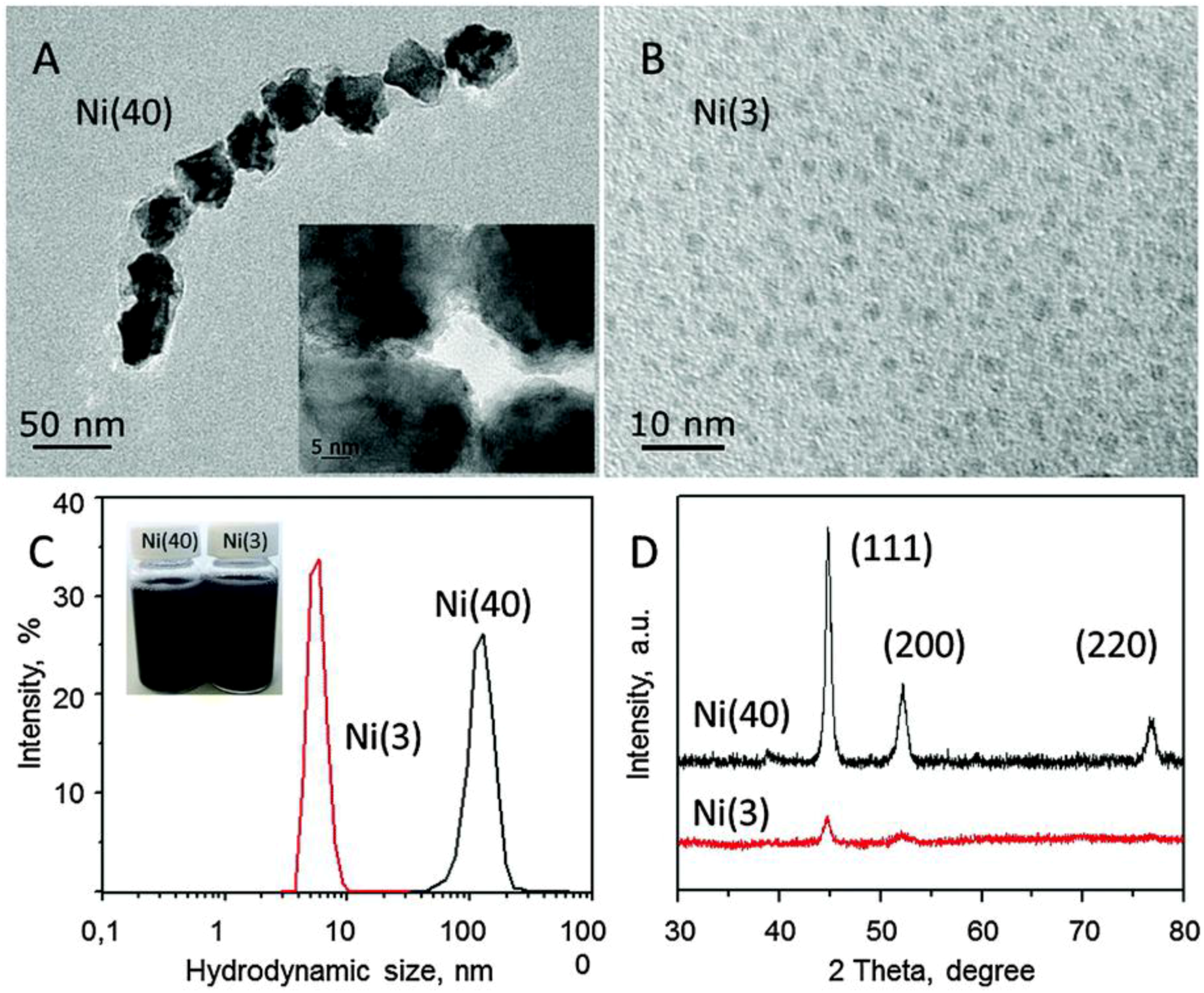
In a recent paper, Vargas et al. reported a facile two-step UP procedure to synthesize Ni NPs and then support them on m-ZrO2 to obtain low-cost and active catalysts for CO2 methanation (Equation (2)) [100]. More in detail, Ni NPs were firstly produced by chemical reduction of a metal precursor (nickel chloride) with either hydrazine hydrate (N2H4·H2O) or sodium borohydride (NaBH4) in water, in the presence of citric acid and sodium citrate as protecting agents. The chemical reduction was carried out under US irradiation (40 kHz, 300 W) without any additional stirring sources. Then, the black colored solution, indicating the presence of Ni0 NPs, was added to other aqueous suspensions of commercial monoclinic ZrO2. In order to obtain homogenous materials, the obtained mixtures were maintained overnight in a conventional ultrasonic bath at ambient conditions. The two samples were centrifuged, washed several times with a mixture of deionized water and ethanol, and dried under vacuum. The catalysts had the same metal loading (1 wt % Ni), but different metal particle sizes. It was found that the final particle size was strongly related to the employed reducing agent. Particularly, homogeneous Ni particles with globular shape and average size of 40 ± 1 nm formed in the presence of hydrazine, as shown in Figure 17A.
On the contrary, when NaBH4 was employed, smaller and well isolated Ni NPs with average diameter equal to 2.7 ± 0.4 nm were observed (Figure 17B). Such differences can be explained by considering that in the case of N2H4*H2O, the formation of the [Ni(N2H4)3]2+ complex implied a slower kinetics, hence inhibiting the formation of nuclei and favoring particle growth. Instead, the use of sodium borohydride gave rise to a larger number of Ni nuclei and, consequently, to smaller particles. Hydrodynamic diameters larger than those measured by TEM were detected in both samples (Figure 17C) in agreement with the presence of citrate at the surface of NPs and of the occurrence of dynamics dipolar magnetic interactions. Peaks at 44.7°, 52.1°, and 76.7° 2 Theta degrees were observed in the diffraction pattern of the catalyst with large Ni particles (Ni(40) in Figure 18c), assigned to the (111), (200), and (220) reflections of the cubic Ni phase (Figure 17D). As for the catalyst with small NPs, (Ni(3) in Figure 18c), only the (111) peak was detected. The catalysts were tested in the CO2 methanation reaction, revealing high activity (70%) and excellent selectivity to CH4 (90%) at 720 K.
Ni/ZrO2 catalysts modified with rare earth elements, in particular Ni/(Zr-Sm)Ox catalysts, showed improved activity in the CO2 methanation (Equation (2)) [101,102]. Conventional wet impregnation method or preparation starting from amorphous Ni-Sm alloys were the preparation procedures usually adopted for these catalysts. In particular, the activity of these systems was correlated with the presence of (Zr–Sm)Ox units with the tetragonal ZrO2 structure. Moreover, the catalytic performance was also influenced by preparation method, surface area, and pore size distribution of the catalysts [101,103].
A Ni-based (40 at % Ni) catalyst supported on mesoporous yttria-stabilized-zirconia composite was prepared by a US method using sodium dodecyl sulfate as a templating agent to obtain high surface area (193 m2/g) [104]. The application of the US method resulted in short reaction time (6 h) and no requirement for the glycolation of nickel, yttrium, and zirconium ions. In another paper by the same research group, a simple one-step US-assisted synthesis (in which the corresponding inorganic salts were used as precursors) was applied to prepare 20–40 at % Ni supported on mesoporous ZrO2 [105]. In these catalysts, Ce and Sm rare earth ions were introduced in the support to further enhance its activity towards CO2 methanation. The obtained catalysts possessed in situ generated mesopores, and the Ni oxide (or hydroxide) that were about 10 nm were incorporated into tetragonal ZrO2 modified by the rare earth metals. In particular, the catalyst containing 30 at % Ni loading had maximum porous volume and size, and at the same time displayed the highest activity, which was further enhanced upon proper oxidation–reduction activation treatment, during which additional active centers were produced.
CO2 methanation was also performed over Ni/Al2O3 catalysts prepared by a US-assisted co-precipitation [106]. The authors observed that an increase in the Ni loading from 5 wt % up to 25 wt % produced a surface area enhancement, resulting in better catalytic performances, and simultaneously decreased the crystallinity and improved the catalyst reducibility. A further increase of the metal loading had a negative effect on the surface area, as well as on the catalytic activity, due to the drop of the Ni dispersion. The 25 wt % Ni/Al2O3 catalyst attained high CO2 conversion (74%) and almost complete CH4 selectivity (99%) at 350 °C. The catalyst demonstrated also high stability at the same temperature after 10 h time-on-stream. The catalytic performance was observed to decrease at higher Gas Hourly Space Velocity, due to both reduced contact time between the reactants and the catalyst surface, as well as the amount of adsorbed reactants. Conversely, a positive effect on the catalyst activity was observed when higher H2/CO molar ratio was employed. Furthermore, the performance of catalysts calcined at higher temperature was observed to decrease.
In another recent study, Co/SBA-15 and Co/SBA-16 catalysts with 20 wt % Co loading were prepared by conventional incipient wetness impregnation method [107]. Additional vacuum and US treatments were performed after the synthesis. N2 physisorption, small angle XRD (SAXRD), wide angle XRD, SEM and energy dispersive X-ray spectroscopy (SEM/EDX), TEM, and TPR were employed to characterize the catalysts. It was observed that Co dispersion on the SBA-15 support was enhanced upon such post-synthesis treatments, due to the decrease of the metal crystallite size. The presence of Co3O4 and of different various CoxOy species was detected. Both post-impregnation treatments affected the metal-support interaction, increasing the activity in the CO2 hydrogenation. It was observed that the vacuum treatment gave rise to a material in which crystalline cobalt oxide was located in the inner pores, whilst cobalt oxide agglomeration at the outer surface took place upon the US treatment.
A series of 0.5–5.0% Ru catalysts supported on Al2O3 for the selective methanation of CO in H2-containing streams were investigated with the aim to clarify the influence of process parameters, such as the metal loading, the temperature of calcination, the chlorine ions content, and the space velocity, on the catalytic performances [108]. Careful characterization by XRD, Thermal Gravim- etry/Differential Thermal Gravimetry (TG/DTG), and SEM was performed. A number of laboratory experiments were performed to clarify the influence of process parameters (Ru loading, calcinations temperature, space velocity, and chlorine ions content) on the activities of catalysts. It was observed that by carrying out the synthesis of the catalysts by US-assisted impregnation, the catalytic activity was significantly improved, and the impregnation time was significantly reduced. In particular, agglomeration accompanied by non-homogeneous distribution of Ru deposits on Al2O3 were observed when the catalysts were prepared by wetness impregnation method. Conversely, US impregnation strongly decreased the particle size and improved the distribution of Ru during the impregnation. The best results were achieved by the catalyst containing 2 wt % Ru on Al2O3 calcined at 400 °C. Long-term tests demonstrated high activity and stability under realistic reaction conditions.
4. The Role of Mechanochemical Activation in the Synthesis of Nanostructured Catalysts
4.1. General Principles of MC
MC induces chemical transformations by mechanical action (compression, shear or friction) that allows breaking intramolecular bonds and further chemical reactions. More in detail, radical species are mechanochemically produced due to the breaking of weak bonds and to the extreme surface plasma conditions generated by the mechanical impact. Mechanochemical activation processes have a long history, and their importance does not decrease with time, because many solid-phase reactions (with stoichiometric amount of reactants) can be quantitatively and rapidly promoted under solvent-free conditions [109]. Therefore, the mechanochemical mixing process can be an environmentally advantageous alternative to conventional chemical syntheses, in which solvents often play a key role in energy dispersion and chemicals solvation and transportation [110].
The preparation of nanomaterials for advanced applications by mechanochemical synthesis is among the most suitable alternative routes because of its simplicity and high reproducibility. Indeed, such methodology is also environmentally advantageous in comparison to conventional preparation procedures, since it is often solvent-free, and it is carried out under mild reaction conditions and involving short reaction times.
4.2. MC as a Tool for the Synthesis of Catalysts
The MC synthesis of heterogeneous catalysts strongly affects their catalytic properties. Indeed, the excess of potential energy produced during grinding, together with shear and friction forces, can deeply modify the materials by producing a large variety of defects, hence improving or modifying the reactivity [109,111].
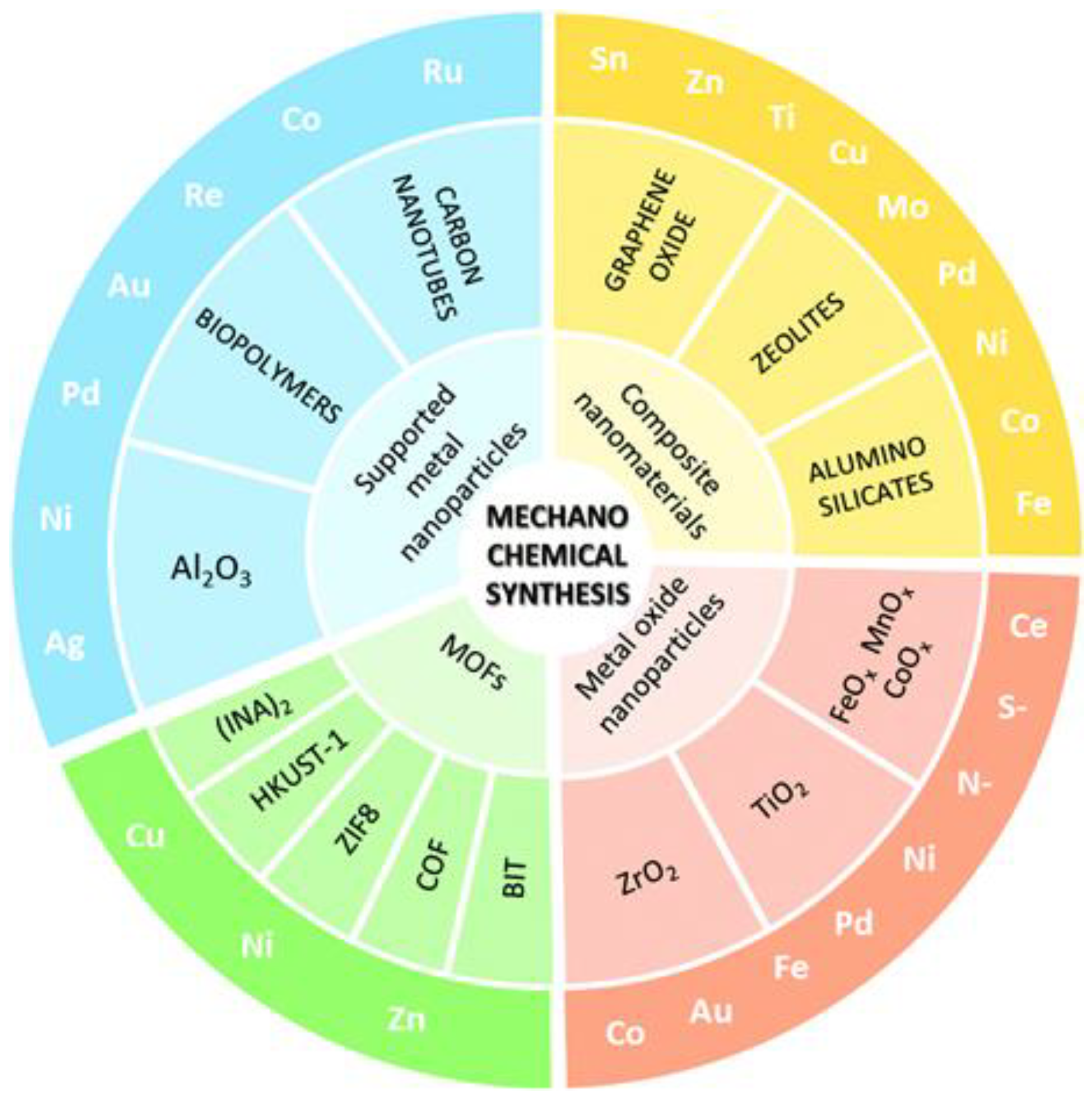
A variety of heterogeneous catalysts, ranging from supported NPs to nanocomposites has been obtained by using MC synthesis, as summarised in Scheme 9 [109].
MC catalyst activation has been successfully applied for many years. Mori et al. [112] investigated the influence of the mechanical milling of MgO-mixed catalysts of Ru, Ni, Fe, or their combinations at temperatures ranging from 80 to 150 °C under initial pressures of 100 Torr CO2 and 500 Torr H2 on the hydrogenation of CO2 to CH4. They found that the CH4 yield was related to the nature of the catalyst (Ru/MgO gave the best yield, whilst Ni/Fe showed the worst activity). Interestingly, the MC activation produced catalysts with improved CH4 production rate and significantly lowered the reaction activation energy. Indeed, an apparent activation energy of 39 kJ mol−1 and kJ mol−1 was accomplished for Ni-Fe-MgO and Ru-MgO catalysts, respectively, upon the milling activation. Such values were almost halved with respect to the same systems produced without mechanical activation. Interestingly, the authors found that the catalysts submitted to further milling activation pre-treatment did not shown any improvement in the activity, suggesting that the milling during reaction played the major role. SEM revealed an increased number of larger particles at increasing milling time, due to small particle agglomeration during milling. However, no significant increase of surface area was observed. Electron probe microanalysis (EPMA) results revealed uniform distribution and aggregation of Ni, Fe, and MgO particles during milling, indicating that the activity had to be ascribed to changes in the surface structure rather than to increased surface area.
In the same years, another study reported on the influence of the MC activation of several traditional catalysts such as Ni, Zr, Zr-Ni-containing materials, Zr hydride, and different Zr-Ni hydrides employed in the hydrogenation of CO and of CO2 [113]. Inactive NiO and ZrO2 were also investigated for comparison purposes. In particular, the study underlined the difference between the traditional catalytic hydrogenation and the mechanically induced hydrogenation to CH4. The study was carried out by employing a flow-milling vial as a catalytic reactor. The authors found that Zr and Zr-Ni hydrides were the most active in CH4 production, despite the fact the hydrogen present in the mixture had a negative effect, since it overcame the CH4 formation. On the contrary, Ni-based catalysts were unable to produce CH4, but were active in CO disproportionation. Zirconium hydride was the only one that showed activity for CO2 hydrogenation. Unexpectedly, the production of CH4 was observed on NiO and ZrO2. The differences in activity were ascribed to the structural transformations of the metal and hydride catalysts under milling, i.e., to the presence of low-coordinate centers upon MC treatment that were able to efficiently activate CO.
MC synthesis was also proven to be effective for obtaining nanophase carbides by facile ball milling of elemental iron and carbon powders at room temperature. Such catalysts were tested in the hydrogenation of CO2 and showed superior activity and selectivity with respect to Fe/C mixtures and coarse-grained conventionally synthesized carbides [114]. It was proposed that nanocrystalline iron carbides with sizes ranging between 8 and 16 nm were the catalytically active species. The presence of defects and/or highly active sites, strained regions, and grain boundaries formed during the grinding guaranteed effective hydrocarbons chain propagation. Interestingly, also a commercial cementite powder displayed some activity upon the ball-milling activation.
Re–Co/Al2O3 catalysts were synthesized via ball milling by using tungsten carbide balls in a tungsten carbide container [115]. In contrast to the preparation of catalysts containing noble metal NPs, in which an oxide precursor or a metal salt are usually employed, the preparation of catalysts with transition metal involves the direct milling of the metal precursors. The obtained bimetallic Re–Co-containing catalysts were more active in the CH4 conversion than in the CO hydrogenation, whilst the activity of the catalysts prepared by incipient wetness impregnation was exactly the opposite. Such difference was explained by assuming that crystallinity in MC prepared material was introduced upon the activation heat treatment at 650 °C. Indeed, X-ray powder diffraction indicated the presence of disordered Re–Co phases before the heat treatment.
Similar systems, such as Co–Fe and Ni catalysts supported on ZrO2 and TiO2, were also prepared by ball milling and showed enhanced catalytic activity in CO hydrogenation [116]. It is worth noting that upon mechanical treatment of TiO2 in the anatase form, the authors observed the transition to brookite and rutile already at room temperature. The activity was ascribed to Co50Fe50 and Ni domains with average size of about 30 and 50 nm, distributed on the supports. XRD measurements confirmed that the mechanochemical dispersion of the Co50Fe50 and Ni phases on TiO2 and ZrO2 prevented the Co, Fe, and Ni oxidation by reaction with the supports. Oxidation of such species usually takes place during calcination during preparation of the catalysts.
Among the most effective and economic materials able to hydrogenate CO2 to CH3OH, Cu–Zn-based catalysts have been extensively studied [117,118,119,120,121]. Usually, commercial Cu–Zn catalysts are prepared by co-precipitation on Al2O3 support: such method implies several steps (precipitation, aging, filtration, drying, calcination, and reduction). Along with specific surface area and CuO and ZnO distribution, such steps have a strong influence on the activity of the final catalyst [122,123,124,125].
In particular, the distribution of CuO and ZnO has pivotal relevance during the aging, because the ion exchange between Cu2+ in malachite (Cu2CO3(OH)2) and Zn2+ in hydrozincite (Zn5(OH)6(CO3)2) occurs, and the formation of zincian malachite ((Cu,Zn)2CO3(OH)2) and/or aurichalcite ((Cu,Zn)5(OH)6(CO3)2) hydroxy carbonate precursors are extremely sensitive to pH, duration, temperature, and stirring [126,127,128].
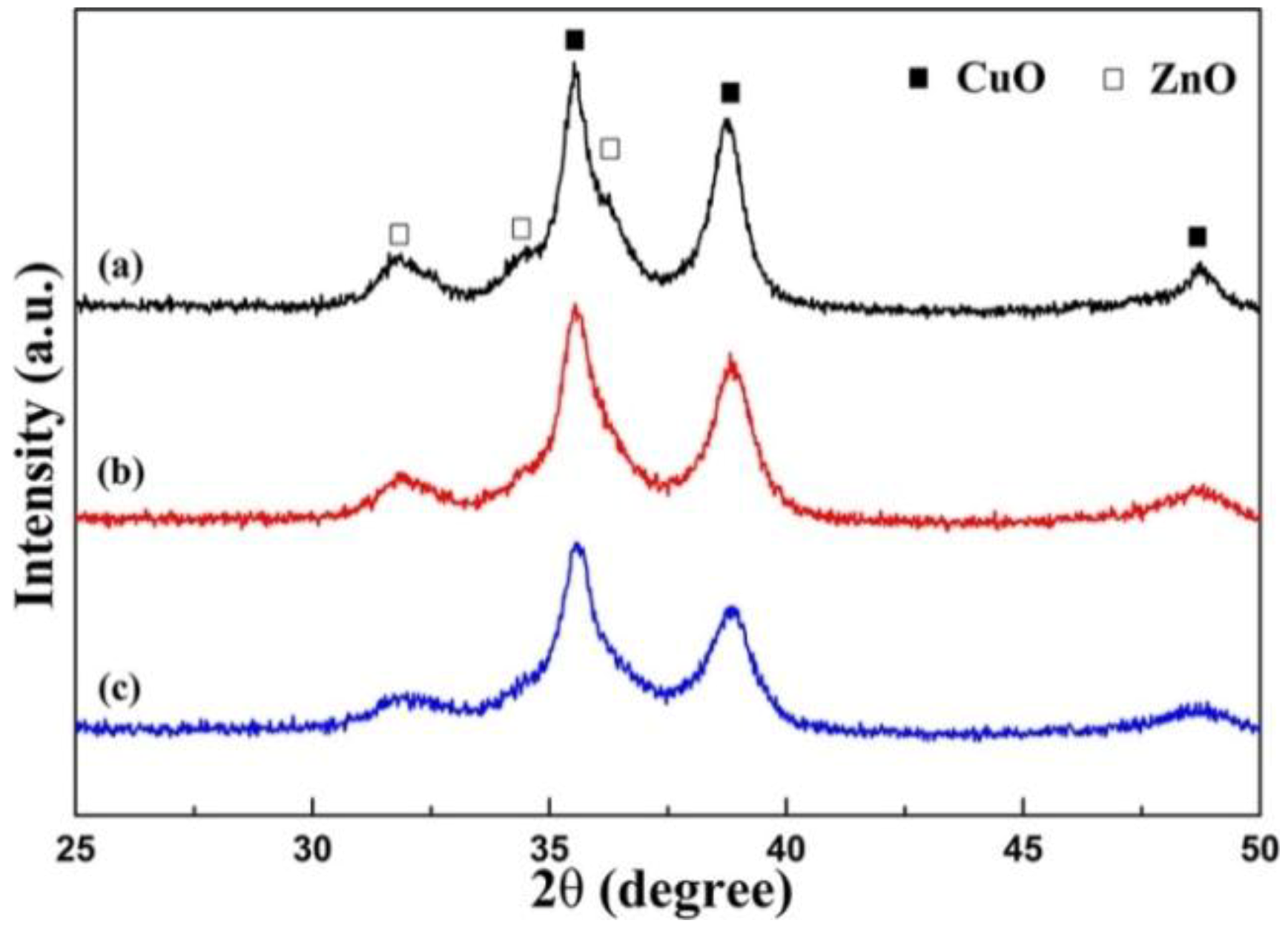
To overcome such problem, very recently a new mechanical-force-driven, solid-state ion-exchange procedure has been refined [129]. It was found that the mutual substitution between Cu2+ and Zn2+ was improved with increasing mechanical ball-milling speed from 200 to 400 rpm, leading to significant enhancement of the catalytic activity. The XRD characterization shown in Figure 18 provided evidence of the presence of peaks related either to the CuO phase or the ZnO phase. Such peaks become broader and at the same time decrease in intensity upon the increase of the milling speed, indicating a decrease in crystallinity and crystal size. The absence of the peaks related to the presence of the Al2O3 phase was explained by the alumina change into an amorphous state after ball-milling [126,129].
The results of TEM measurements performed on the CZA400 catalyst reported in Figure 19 indicated a close cross-distribution of CuO and ZnO due to the occurrence of the solid-state ion exchange, in which ZnO NPs act as spacers, thereby avoiding the CuO agglomeration (as shown in Figure 19b).
Such alternation of CuO and ZnO NPs represents the result of the complete ion exchange between Cu2+ and Zn2+ in the precursors. For the CZA400 catalyst, impressive 59.5% CO2 conversion and 73.4% CH3OH selectivity were obtained at 240 °C and 4 MPa, resulting in a noticeable yield to CH3OH of 43.7%. Therefore, this mechanical-force-driven, solid-state ion-exchange method proved to be a valuable alternative to synthesizing CZA catalysts with improved practical application potential for chemical hydrogenation of CO2 to CH3OH.
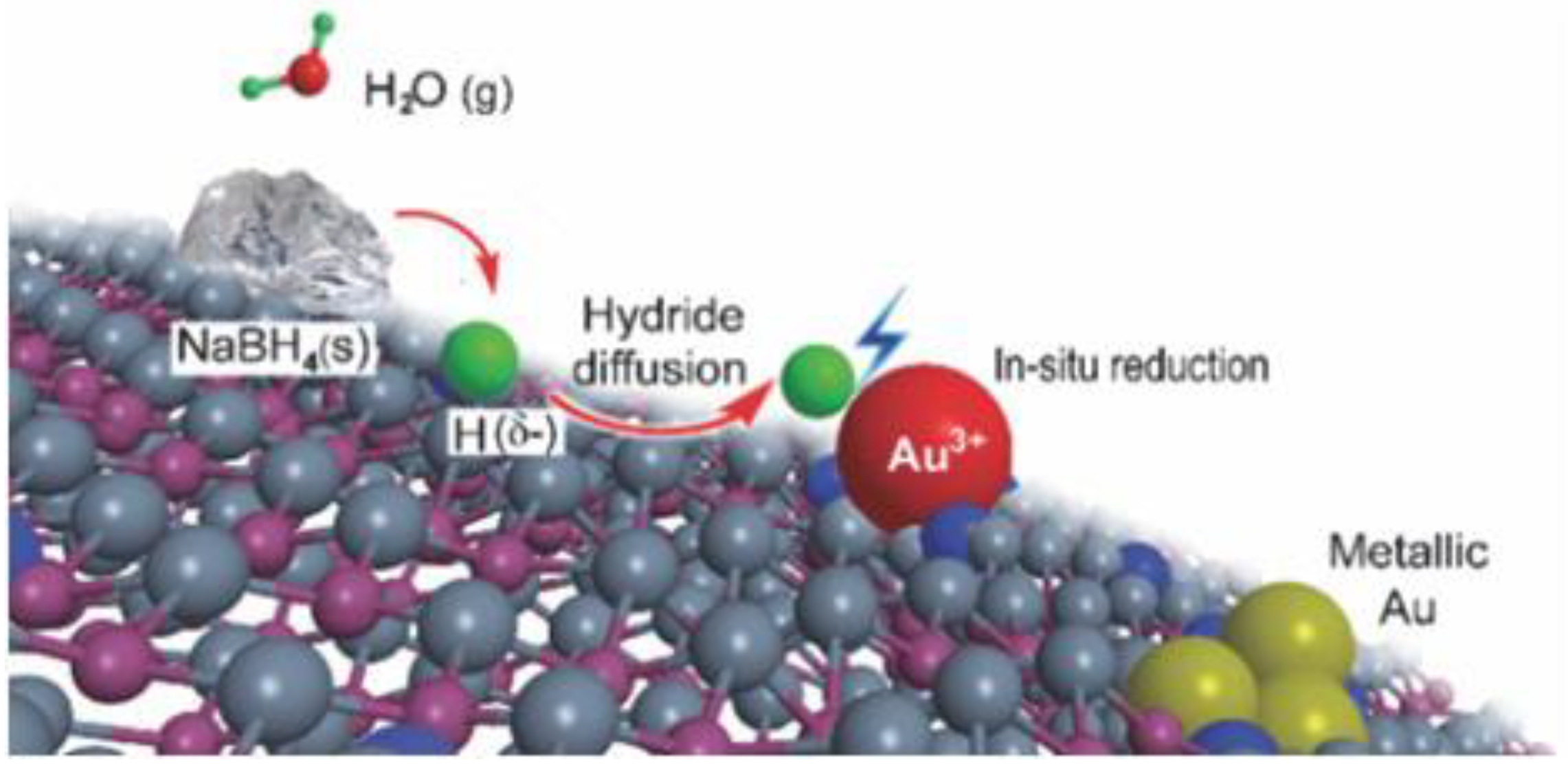
In another very recent study, Au clusters were directly deposited onto a functionalized support by newly developed, milling-mediated solid reduction procedure [130]. The proposed method involved the use of HAuCl4 as Au precursor, which was pre-adsorbed on the Schiff base modified silica surface by electrostatic interaction in water. After grinding the dried carrier with solid NaBH4, according to Scheme 10, the Au precursor can be reduced in situ by atomic H coming from the NaBH4 reducing agent in the solid state. Depending on the gold loading, highly dispersed clusters and isolated Au atoms can be obtained by solid grinding.
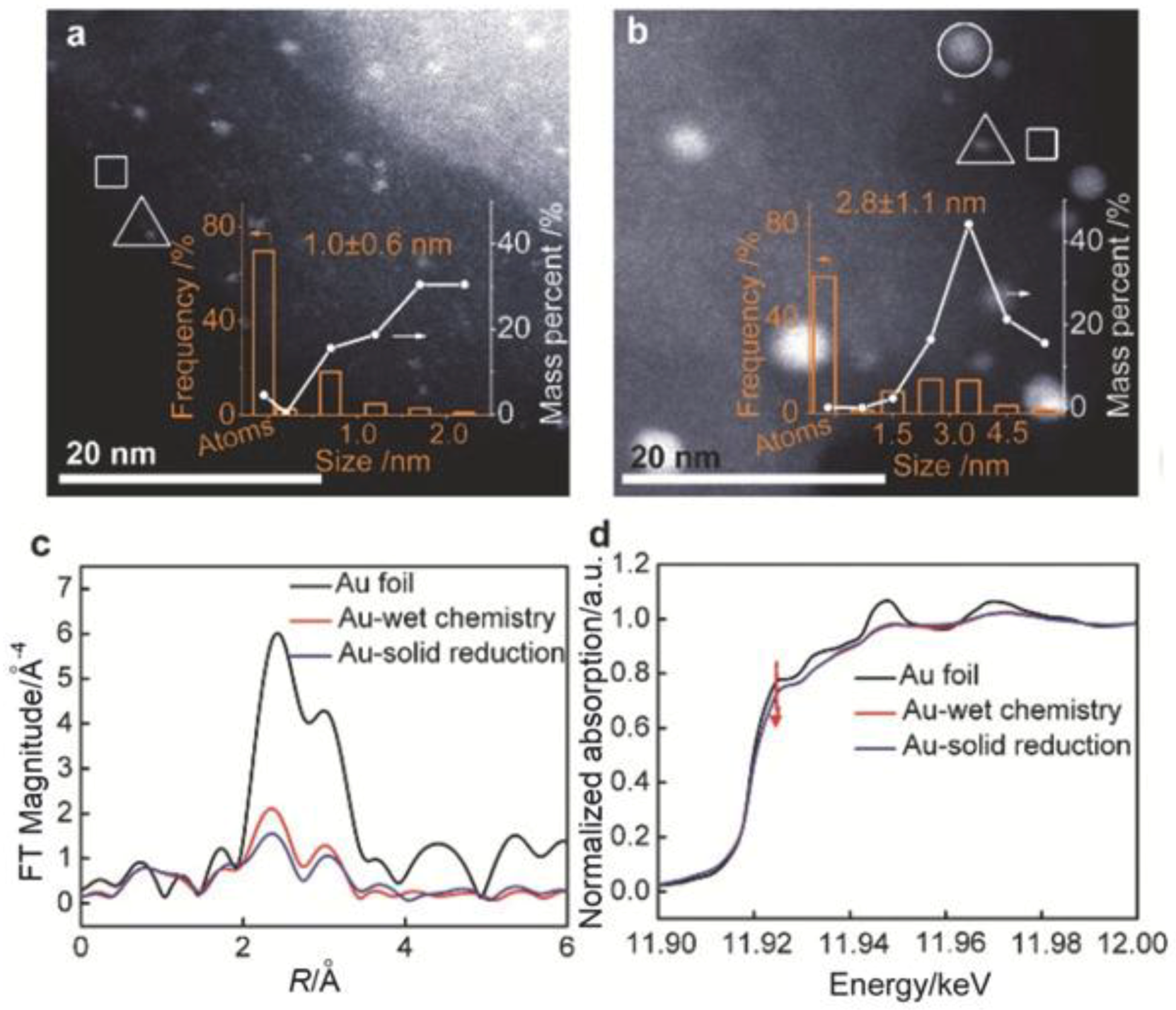
Following such activation, it was found that the Au species were dispersed as 1.0 nm nanoclusters (Figure 20a), as determined by the aberration-corrected, HAADF-STEM (high-angle annular dark-field scanning transmission electron microscopy) analysis. At the same time, a lot of isolated Au atoms were detected, even if as a minor fraction of the total metal content. It was proposed that such Au subnanoclusters were effectively stabilized by the Schiff base groups present on the silica surface by providing strong electronic interaction. Differently, a variety of Au shapes and sizes were observed on the support (Figure 20b) when an Au-containing solution was reduced by NaBH4. The obtained statistical particle size distributions were in agreement with the L3-edge extended X-ray absorption fine structure (EXAFS, Figure 20c), revealing an increase in intensity of the peak related to the first shell, as well as in the Au coordination number when the conventional wet method was employed. In addition, a decrease in the white line intensity compared with the Au foil in the X-ray absorption near-edge structure (XANES) spectra obtained for the Au NPs (Figure 20d) is an indication that the functionalization of the silica surface with nitrogen allowed one to optimize the Au d-electron density [131].
These features put in evidence that the opportunity to use the solvent-free solid reduction synthesis allows the prevention of Au growth; indeed, crystal nucleation was hampered, giving rise to either subnanoclusters or single-atoms, depending on the Au content. The catalysts were tested in the hydrogenation of CO2 to formate, and a marked dependence on the Au size, due to the different electronic structure of the metal species, was observed. The authors claimed that the milling-mediated solid reduction procedure could be considered as a general methodology to achieve basic knowledge ion the catalysis by Au, providing new insights for supported catalyst design for reactions involving the CO2 transformation.
5. Final Remarks
The main purpose of this review was to point out the possibilities offered by microwave, ultrasound, and mechanochemical protocols in materials engineering at a nanoscale level for catalytic applications.
Summarising, concerning the synthesis of CO2 reduction catalysts, assistance of microwave, ultrasound, and mechanochemistry allows one to obtain highly dispersed active sites and controlled particle dimension, and in most cases reinforces the active site/support interaction, giving rise to synergistic effects that help CO2 reduction and improve catalyst stability. The procedures reported are usually simple, and require relatively short reaction times (minutes) without implying many steps. Moreover, stronger and/or more abundant basic sites occurring at the support surface favour CO2 adsorption, a key step for its further reduction. Complex systems (hierarchical structures, composites, bi-functional catalysts, etc.) may be obtained by relatively simple methods.
Finally, a further crucial aspect concerns the effect of microwave irradiation during reaction, whilst carbon deposition usually leads to catalyst deactivation, under microwave the same carbon deposits may act as hot spots per se favouring the reaction.
Funding
This research was funded by the Università degli Studi di Torino (Progetto Ricerca Locale 2016), and Politecnico di Torino (Progetto Ricerca di base 2017).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, C.; Miller, B.G.; Scaroni, A.W. Adsorption separation of carbon dioxide from flue gas of natural gas-fired boiler by a novel nanoporous “molecular basket” adsorbent. Fuel Process. Technol. 2005, 86, 1457–1472. [Google Scholar] [CrossRef]
- Choi, S.; Watanabe, T.; Bae, T.H.; Sholl, D.S.; Jones, C.W. Modification of the Mg/DOBDC MOF with amines to enhance CO2 adsorption from ultradilute gases. J. Phys. Chem. Lett. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.Y.; Brunelli, N.A.; Yucelen, G.I.; Venkatasubramanian, A.; Zang, J.; Leisen, J.; Hesketh, P.J.; Jones, C.W.; Nair, S. Direct synthesis of single-walled aminoaluminosilicate nanotubes with enhanced molecular adsorption selectivity. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Aaron, D.; Tsouris, C. Separation of CO2 from flue gas: A review. Sep. Sci. Technol. 2005, 40, 321–348. [Google Scholar] [CrossRef]
- Hinkov, I.; Darkrim Lamari, F.; Langlois, P.; Dicko, M.; Chilev, C.; Pentchev, I. Carbon Dioxide Capture by Adsorption (Review). J. Chem. Technol. Metall. 2016, 51, 609–626. [Google Scholar]
- An, J.; Rosi, N.L. Tuning MOF CO2 Adsorption Properties via Cation Exchange. J. Am. Chem. Soc. 2010. [Google Scholar] [CrossRef] [PubMed]
- Webley, P.A. Adsorption technology for CO2 separation and capture: A perspective. Adsorption 2014. [Google Scholar] [CrossRef]
- Yu, C.H.; Huang, C.H.; Tan, C.S. A review of CO2 capture by absorption and adsorption. Aerosol Air Qual. Res. 2012. [Google Scholar] [CrossRef]
- Gibson, J.A.A.; Mangano, E.; Shiko, E.; Greenaway, A.G.; Gromov, A.V.; Lozinska, M.M.; Friedrich, D.; Campbell, E.E.B.; Wright, P.A.; Brandani, S. Adsorption Materials and Processes for Carbon Capture from Gas-Fired Power Plants: AMPGas. Ind. Eng. Chem. Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Milani, D.; Khalilpour, R.; Zahedi, G.; Abbas, A. A model-based analysis of CO2 utilization in methanol synthesis plant. J. CO2 Util. 2015, 10, 12–22. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vesselli, E.; Monachino, E.; Rizzi, M.; Furlan, S.; Duan, X.; Dri, C.; Peronio, A.; Africh, C.; Lacovig, P.; Baldereschi, A.; et al. Steering the chemistry of carbon oxides on a NiCu catalyst. ACS Catal. 2013. [Google Scholar] [CrossRef]
- Li, K.; An, X.; Park, K.H.; Khraisheh, M.; Tang, J. A critical review of CO2 photoconversion: Catalysts and reactors. Catal. Today 2014. [Google Scholar] [CrossRef]
- Low, J.; Cheng, B.; Yu, J. Surface modification and enhanced photocatalytic CO2 reduction performance of TiO2: A review. Appl. Surf. Sci. 2017, 392, 658–686. [Google Scholar] [CrossRef]
- White, J.L.; Baruch, M.F.; Pander, J.E.; Hu, Y.; Fortmeyer, I.C.; Park, J.E.; Zhang, T.; Liao, K.; Gu, J.; Yan, Y.; et al. Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes. Chem. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nahar, S.; Zain, M.; Kadhum, A.; Hasan, H.; Hasan, M. Advances in Photocatalytic CO2 Reduction with Water: A Review. Materials 2017, 10, 629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kawamura, S.; Tamba, M.; Kojima, T.; Yoshiba, M.; Izumi, Y. Is water more reactive than H2 in photocatalytic CO2 conversion into fuels using semiconductor catalysts under high reaction pressures? J. Catal. 2017. [Google Scholar] [CrossRef]
- Kim, D.; Xie, C.; Becknell, N.; Yu, Y.; Karamad, M.; Chan, K.; Crumlin, E.J.; Nørskov, J.K.; Yang, P. Electrochemical Activation of CO2 through Atomic Ordering Transformations of AuCu Nanoparticles. J. Am. Chem. Soc. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lum, Y.; Yue, B.; Lobaccaro, P.; Bell, A.T.; Ager, J.W. Optimizing C-C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction. J. Phys. Chem. C 2017. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Malik, K.; Singh, S.; Basu, S.; Verma, A. Electrochemical reduction of CO2 for synthesis of green fuel. Wiley Interdiscip. Rev. Energy Environ. 2017. [Google Scholar] [CrossRef]
- Li, C.W.; Kanan, M.W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Shen, Q.; Liu, J.; Yang, N.; Zhao, G. A CO2 adsorption-enhanced semiconductor/metal-complex hybrid photoelectrocatalytic interface for efficient formate production. Energy Environ. Sci. 2016. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Heterogeneous Catalytic Reactions with CO2: Status and Perspectives. Stud. Surf. Sci. Catal. 2004, 153, 1–8. [Google Scholar] [CrossRef]
- Windle, C.D.; Perutz, R.N. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. [Google Scholar] [CrossRef]
- Gray, H.B. Powering the planet with solar fuel. Nat. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Windle, C.; Reisner, E. Heterogenised Molecular Catalysts for the Reduction of CO2 to Fuels. Chim. Int. J. Chem. 2015, 69, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.C.J.; Vannice, M.A. CO2 Reforming of CH4. Catal. Rev. 1999, 41, 1–42. [Google Scholar] [CrossRef]
- Ahmadi, M.; Mistry, H.; Roldan Cuenya, B. Tailoring the Catalytic Properties of Metal Nanoparticles via Support Interactions. J. Phys. Chem. Lett. 2016, 7, 3519–3533. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-K.; Zhao, G.-X.; Huang, X.; Wang, X. Progress in the catalyst exploration for heterogeneous CO2 reduction and utilization: A critical review. J. Mater. Chem. A 2017, 5, 21625–21649. [Google Scholar] [CrossRef]
- Sahoo, T.R.; Armandi, M.; Arletti, R.; Piumetti, M.; Bensaid, S.; Manzoli, M.; Panda, S.R.; Bonelli, B. Pure and Fe-doped CeO2 nanoparticles obtained by microwave assisted combustion synthesis: Physico-chemical properties ruling their catalytic activity towards CO oxidation and soot combustion. Appl. Catal. B Environ. 2017, 211. [Google Scholar] [CrossRef]
- Devaiah, D.; Reddy, L.H.; Park, S.-E.; Reddy, B.M. Ceria-zirconia mixed oxides: Synthetic methods and applications. Catal. Rev. 2018. [Google Scholar] [CrossRef]
- Ganesh, I.; Johnson, R.; Rao, G.V.N.; Mahajan, Y.R.; Madavendra, S.S.; Reddy, B.M. Microwave-assisted combustion synthesis of nanocrystalline MgAl2O4 spinel powder. Ceram. Int. 2005, 31, 67–74. [Google Scholar] [CrossRef]
- Ai, L.; Zeng, Y.; Jiang, J. Hierarchical porous BiOI architectures: Facile microwave nonaqueous synthesis, characterization and application in the removal of Congo red from aqueous solution. Chem. Eng. J. 2014. [Google Scholar] [CrossRef]
- Cai, W.; De La Piscina, P.R.; Toyir, J.; Homs, N. CO2 hydrogenation to methanol over CuZnGa catalysts prepared using microwave-assisted methods. Catal. Today 2015. [Google Scholar] [CrossRef]
- Huang, C.; Mao, D.; Guo, X.; Yu, J. Microwave-Assisted Hydrothermal Synthesis of CuO-ZnO-ZrO2 as Catalyst for Direct Synthesis of Methanol by Carbon Dioxide Hydrogenation. Energy Technol. 2017, 5, 2100–2107. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Yu, Z. CO2 Methanation: The Effect of Catalysts and Reaction Conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- He, S.; Li, C.; Chen, H.; Su, D.; Zhang, B.; Cao, X.; Wang, B.; Wei, M.; Evans, D.G.; Duan, X. A Surface Defect-Promoted Ni Nanocatalyst with Simultaneously Enhanced Activity and Stability. Chem. Mater. 2013, 25, 1040–1046. [Google Scholar] [CrossRef]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Kesavan, J.K.; Luisetto, I.; Tuti, S.; Meneghini, C.; Iucci, G.; Battocchio, C.; Mobilio, S.; Casciardi, S.; Sisto, R. Nickel supported on YSZ: The effect of Ni particle size on the catalytic activity for CO2 methanation. J. CO2 Util. 2018, 23, 200–211. [Google Scholar] [CrossRef]
- Du, G.; Lim, S.; Yang, Y.; Wang, C.; Pfefferle, L.; Haller, G.L. Methanation of carbon dioxide on Ni-incorporated MCM-41 catalysts: The influence of catalyst pretreatment and study of steady-state reaction. J. Catal. 2007, 249, 370–379. [Google Scholar] [CrossRef]
- Hu, L.; Urakawa, A. Continuous CO2 capture and reduction in one process: CO2 methanation over unpromoted and promoted Ni/ZrO2. J. CO2 Util. 2018, 25, 323–329. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Mukti, R.R.; Taufiq-Yap, Y.H.; Sazegar, M.R. Highly active Ni-promoted mesostructured silica nanoparticles for CO2 methanation. Appl. Catal. B Environ. 2014, 147, 359–368. [Google Scholar] [CrossRef]
- Song, F.; Zhong, Q.; Yu, Y.; Shi, M.; Wu, Y.; Hu, J.; Song, Y. Obtaining well-dispersed Ni/Al2O3 catalyst for CO2 methanation with a microwave-assisted method. Int. J. Hydrogen Energy 2017, 42, 4174–4183. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Westermann, A.; Ribeiro, M.F.; Lopes, J.M.; Henriques, C. CO2 Hydrogenation Over Ni-Based Zeolites: Effect of Catalysts Preparation and Pre-reduction Conditions on Methanation Performance. Top. Catal. 2016, 59, 314–325. [Google Scholar] [CrossRef]
- Dong, X.; Jin, B.; Sun, Y.; Shi, K.; Yu, L. Re-promoted Ni-Mn bifunctional catalysts prepared by microwave heating for partial methanation coupling with water gas shift under low H2/CO conditions. Appl. Catal. A Gen. 2018, 552, 105–116. [Google Scholar] [CrossRef]
- Fidalgo, B.; Arenillas, A.; Menéndez, J.A. Mixtures of carbon and Ni/Al2O3 as catalysts for the microwave-assisted CO2 reforming of CH4. Fuel Process. Technol. 2011, 92, 1531–1536. [Google Scholar] [CrossRef]
- Barros, B.S.; Melo, D.M.A.; Libs, S.; Kiennemann, A. CO2 reforming of methane over La2NiO4/α-Al2O3 prepared by microwave assisted self-combustion method. Appl. Catal. A Gen. 2010. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, S.; Su, X.; Fan, S.; Ma, Q.; Zhao, T. Selective formation of light olefins from CO2 hydrogenation over Fe–Zn–K catalysts. Biochem. Pharmacol. 2015, 12, 95–100. [Google Scholar] [CrossRef]
- Hou, W.; Hung, W.H.; Pavaskar, P.; Goeppert, A.; Aykol, M.; Cronin, S.B. Photocatalytic conversion of CO2 to hydrocarbon fuels via plasmon-enhanced absorption and metallic interband transitions. ACS Catal. 2011. [Google Scholar] [CrossRef]
- Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q. A facile strategy to fabricate plasmonic Cu modified TiO2 nano-flower films for photocatalytic reduction of CO2 to methanol. Mater. Res. Bull. 2015. [Google Scholar] [CrossRef]
- Shown, I.; Hsu, H.-C.; Chang, Y.-C.; Lin, C.-H.; Roy, P.K.; Ganguly, A.; Wang, C.-H.; Chang, J.-K.; Wu, C.-I.; Chen, L.-C.; et al. Highly Efficient Visible Light Photocatalytic Reduction of CO2 to Hydrocarbon Fuels by Cu-Nanoparticle Decorated Graphene Oxide. Nano Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Li, K.; Tang, J. Cu2O/reduced graphene oxide composites for the photocatalytic conversion of CO2. ChemSusChem 2014. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y. Photocatalytic reduction of CO2 to CO over copper decorated g-C3N4nanosheets with enhanced yield and selectivity. Appl. Surf. Sci. 2018. [Google Scholar] [CrossRef]
- Yu, J.; Jin, J.; Cheng, B.; Jaroniec, M. A noble metal-free reduced graphene oxide-CdS nanorod composite for the enhanced visible-light photocatalytic reduction of CO2 to solar fuel. J. Mater. Chem. A 2014. [Google Scholar] [CrossRef]
- Fenton, T.; Gillingham, S.; Jin, T.; Li, G. Microwave-assisted deposition of a highly active cobalt catalyst on mesoporous silica for photochemical CO2 reduction. Dalton Trans. 2017, 46, 10721–10726. [Google Scholar] [CrossRef] [PubMed]
- Hart, E.J.; Henglein, A. Free radical and free atom reactions in the sonolysis of aqueous iodide and formate solutions. J. Phys. Chem. 1985, 89, 4342–4347. [Google Scholar] [CrossRef]
- Luo, S.; Wu, J.; Toyir, J.; Saito, M.; Takeuchi, M.; Watanabe, T. Optimization of preparation conditions and improvement of stability of Cu/ZnO-based multicomponent catalysts for methanol synthesis from CO2 and H2. Stud. Surf. Sci. Catal. 1998, 114, 549–552. [Google Scholar] [CrossRef]
- Vafaeian, Y.; Haghighi, M.; Aghamohammadi, S. Ultrasound assisted dispersion of different amount of Ni over ZSM-5 used as nanostructured catalyst for hydrogen production via CO2 reforming of methane. Energy Convers. Manag. 2013, 76, 1093–1103. [Google Scholar] [CrossRef]
- Gaudino, E.C.; Rinaldi, L.; Rotolo, L.; Carnaroglio, D.; Pirola, C.; Cravotto, G.; Radoiu, M.; Eynde, J.J. Vanden Heterogeneous phase microwave-assisted reactions under CO2 or CO pressure. Molecules 2016, 21, 253. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, M.A.; Lee, S.J.; Jung, J.; Jang, H.M.; Upare, P.P.; Hwang, Y.K.; Chang, J.-S.; Park, J.K. Preparation and characterization of carbon-encapsulated iron nanoparticles and their catalytic activity in the hydrogenation of levulinic acid. J. Mater. Sci. 2015, 50, 334–343. [Google Scholar] [CrossRef]
- Pirola, C.; Bianchi, C.L.; Di Michele, A.; Diodati, P.; Boffito, D.; Ragaini, V. Ultrasound and microwave assisted synthesis of high loading Fe-supported Fischer-Tropsch catalysts. Ultrason. Sonochem. 2010, 17, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Chinchen, G.C.; Denny, P.J.; Jennings, J.R.; Spencer, M.S.; Waugh, K.C. Synthesis of Methanol: Part 1. Catalysts and Kinetics. Appl. Catal. 1988, 36, 1–65. [Google Scholar] [CrossRef]
- Sun, K.; Lu, W.; Qiu, F.; Liu, S.; Xu, X. Direct synthesis of DME over bifunctional catalyst: Surface properties and catalytic performance. Appl. Catal. A Gen. 2003, 252, 243–249. [Google Scholar] [CrossRef]
- Chen, H.Y.; Chen, L.; Lin, J.; Tan, K.L.; Li, J. Copper Sites in Copper-Exchanged ZSM-5 for CO Activation and Methanol Synthesis: XPS and FTIR Studies. Inorg. Chem. 1997, 36, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Liu, Y.; Wang, F.; Zhou, S.; Chen, A.; Yang, Y.; Fang, W. Promoting effect of a Cu–Zn binary precursor on a ternary Cu–Zn–Al catalyst for methanol synthesis from synthesis gas. RSC Adv. 2014, 4. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Kozłowska, A.; Olszewski, P.; Stoch, J.; Skrzypek, J.; Lachowska, M. Catalytic activity of the M/(3ZnO·ZrO2) system (M = Cu, Ag, Au) in the hydrogenation of CO2 to methanol. Appl. Catal. A Gen. 2004, 278, 11–23. [Google Scholar] [CrossRef]
- Hansen, P.L.; Wagner, J.B.; Helveg, S.; Rostrup-Nielsen, J.R.; Clausen, B.S.; Topsøe, H. Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science 2002, 295, 2053–2055. [Google Scholar] [CrossRef] [PubMed]
- Ovesen, C.V.; Clausen, B.S.; Schiøtz, J.; Stoltze, P.; Topsøe, H.; Nørskov, J.K. Kinetic Implications of Dynamical Changes in Catalyst Morphology during Methanol Synthesis over Cu/ZnO Catalysts. J. Catal. 1997, 168, 133–142. [Google Scholar] [CrossRef]
- Choi, Y.; Futagami, K.; Fujitani, T.; Nakamura, J. The role of ZnO in Cu/ZnO methanol synthesis catalysts—Morphology effect or active site model? Appl. Catal. A Gen. 2001, 208, 163–167. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Kozłowska, A.; Olszewski, P.; Lachowska, M.; Skrzypek, J.; Stoch, J. Effect of Mg and Mn oxide additions on structural and adsorptive properties of Cu/ZnO/ZrO2 catalysts for the methanol synthesis from CO2. Appl. Catal. A Gen. 2003, 249, 129–138. [Google Scholar] [CrossRef]
- Omata, K.; Watanabe, Y.; Umegaki, T.; Ishiguro, G.; Yamada, M. Low-pressure DME synthesis with Cu-based hybrid catalysts using temperature-gradient reactor. Fuel 2002, 81, 1605–1609. [Google Scholar] [CrossRef]
- Melián-Cabrera, I.; Granados, M.L.; Fierro, J.L.G. Pd-Modified Cu–Zn Catalysts for Methanol Synthesis from CO2/H2 Mixtures: Catalytic Structures and Performance. J. Catal. 2002, 210, 285–294. [Google Scholar] [CrossRef]
- Pokrovski, K.A.; Rhodes, M.D.; Bell, A.T. Effects of cerium incorporation into zirconia on the activity of Cu/ZrO2 for methanol synthesis via CO hydrogenation. J. Catal. 2005, 235, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Znak, L.; Stołecki, K.; Zieliński, J. The effect of cerium, lanthanum and zirconium on nickel/alumina catalysts for the hydrogenation of carbon oxides. Catal. Today 2005, 101, 65–71. [Google Scholar] [CrossRef]
- Köppel, R.A.; Stöcker, C.; Baiker, A. Copper- and Silver-Zirconia Aerogels: Preparation, Structural Properties and Catalytic Behavior in Methanol Synthesis from Carbon Dioxide. J. Catal. 1998, 179, 515–527. [Google Scholar] [CrossRef]
- Sun, Y.; Sermon, P.A. Evidence of a metal-support interaction in sol-gel derived Cu-ZrO2 catalysts for CO hydrogenation. Catal. Lett. 1994, 29, 361–369. [Google Scholar] [CrossRef]
- Jingfa, D.; Qi, S.; Yulong, Z.; Songying, C.; Dong, W. A novel process for preparation of a Cu/ZnO/Al2O3 ultrafine catalyst for methanol synthesis from CO2 + H2: Comparison of various preparation methods. Appl. Catal. A Gen. 1996, 139, 75–85. [Google Scholar] [CrossRef]
- Coteron, A.; Hayhurst, A.N. Methanol synthesis by amorphous copper-based catalysts prepared by spark-erosion. Appl. Catal. A Gen. 1993, 101, 151–165. [Google Scholar] [CrossRef]
- Arena, F.; Barbera, K.; Italiano, G.; Bonura, G.; Spadaro, L.; Frusteri, F. Synthesis, characterization and activity pattern of Cu–ZnO/ZrO2 catalysts in the hydrogenation of carbon dioxide to methanol. J. Catal. 2007, 249, 185–194. [Google Scholar] [CrossRef]
- Chen, H.-J.; Fan, C.-W.; Yu, C.-S. Analysis, synthesis, and design of a one-step dimethyl ether production via a thermodynamic approach. Appl. Energy 2013, 101, 449–456. [Google Scholar] [CrossRef]
- Chen, H.-B.; Liao, D.-W.; Yu, L.-J.; Lin, Y.-J.; Yi, J.; Zhang, H.-B.; Tsai, K.-R. Influence of trivalent metal ions on the surface structure of a copper-based catalyst for methanol synthesis. Appl. Surf. Sci. 1999, 147, 85–93. [Google Scholar] [CrossRef]
- Yang, R.; Yu, X.; Zhang, Y.; Li, W.; Tsubaki, N. A new method of low-temperature methanol synthesis on Cu/ZnO/Al2O3 catalysts from CO/CO2/H2. Fuel 2008, 87, 443–450. [Google Scholar] [CrossRef]
- Tokay, K.C.; Dogu, T.; Dogu, G. Dimethyl ether synthesis over alumina based catalysts. Chem. Eng. J. 2012, 184, 278–285. [Google Scholar] [CrossRef]
- Sai Prasad, P.S.; Bae, J.W.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Single-step synthesis of DME from syngas on Cu–ZnO–Al2O3/zeolite bifunctional catalysts: The superiority of ferrierite over the other zeolites. Fuel Process. Technol. 2008, 89, 1281–1286. [Google Scholar] [CrossRef]
- Khoshbin, R.; Haghighi, M. Direct syngas to DME as a clean fuel: The beneficial use of ultrasound for the preparation of CuO–ZnO–Al2O3/HZSM-5 nanocatalyst. Chem. Eng. Res. Des. 2013, 91, 1111–1122. [Google Scholar] [CrossRef]
- Khoshbin, R.; Haghighi, M. Direct conversion of syngas to dimethyl ether as a green fuel over ultrasound-assisted synthesized CuO–ZnO–Al2O3/HZSM-5 nanocatalyst: Effect of active phase ratio on physicochemical and catalytic properties at different process conditions. Catal. Sci. Technol. 2014, 4, 1779–1792. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A.; Qavam Saeedi, H. Direct synthesis of dimethyl ether as a green fuel from syngas over nanostructured CuO–ZnO–Al2O3/HZSM-5 catalyst: Influence of irradiation time on nanocatalyst properties and catalytic performance. J. Power Sources 2014, 272, 929–939. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S. Ultrasound assisted co-precipitation of nanostructured CuO–ZnO–Al2O3 over HZSM-5: Effect of precursor and irradiation power on nanocatalyst properties and catalytic performance for direct syngas to DME. Ultrason. Sonochem. 2014, 21, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Pinjari, D.V.; Pandit, A.B.; Mhaske, S.T. Synthesis of zirconium dioxide by ultrasound assisted precipitation: Effect of calcination temperature. Ultrason. Sonochem. 2011, 18, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Zha, F.; Tian, H.; Yan, J.; Chang, Y. Multi-walled carbon nanotubes as catalyst promoter for dimethyl ether synthesis from CO2 hydrogenation. Appl. Surf. Sci. 2013, 285, 945–951. [Google Scholar] [CrossRef]
- Stark, W.J.; Stoessel, P.R.; Wohlleben, W.; Hafner, A. Industrial applications of nanoparticles. Chem. Soc. Rev. 2015, 44, 5793–5805. [Google Scholar] [CrossRef] [PubMed]
- Behrens, S. Preparation of functional magnetic nanocomposites and hybrid materials: Recent progress and future directions. Nanoscale 2011, 3, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Singamaneni, S.; Bliznyuk, V.N.; Binek, C.; Tsymbal, E.Y. Magnetic nanoparticles: Recent advances in synthesis, self-assembly and applications. J. Mater. Chem. 2011, 21, 16819. [Google Scholar] [CrossRef]
- Gual, A.; Godard, C.; Castillón, S.; Curulla-Ferré, D.; Claver, C. Colloidal Ru, Co and Fe-nanoparticles. Synthesis and application as nanocatalysts in the Fischer-Tropsch process. Catal. Today 2012, 183, 154–171. [Google Scholar] [CrossRef]
- Burda, C.; Chen, X.; Narayanan, R.; El-Sayed, M.A. Chemistry and Properties of Nanocrystals of Different Shapes. Chem. Rev. 2005. [Google Scholar] [CrossRef] [PubMed]
- Vargas, E.; Romero-Sáez, M.; Denardin, J.C.; Gracia, F. The ultrasound-assisted synthesis of effective monodisperse nickel nanoparticles: Magnetic characterization and its catalytic activity in CO2 methanation. New J. Chem. 2016, 40, 7307–7310. [Google Scholar] [CrossRef]
- Yamasaki, M.; Habazaki, H.; Asami, K.; Izumiya, K.; Hashimoto, K. Effect of tetragonal ZrO2 on the catalytic activity of Ni/ZrO2 catalyst prepared from amorphous Ni–Zr alloys. Catal. Commun. 2006, 7, 24–28. [Google Scholar] [CrossRef]
- Yamasaki, M.; Komori, M.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Asami, K.; Hashimoto, K. CO2 methanation catalysts prepared from amorphous Ni–Zr–Sm and Ni–Zr–misch metal alloy precursors. Mater. Sci. Eng. A 1999, 267, 220–226. [Google Scholar] [CrossRef]
- Men, Y.; Kolb, G.; Zapf, R.; Löwe, H. Selective methanation of carbon oxides in a microchannel reactor—Primary screening and impact of gas additives. Catal. Today 2007, 125, 81–87. [Google Scholar] [CrossRef]
- Sominski, E.; Gedanken, A.; Perkas, N.; Buchkremer, H.P.; Menzler, N.H.; Zhang, L.Z.; Yu, J.C. The sonochemical preparation of a mesoporous NiO/yttria stabilized zirconia composite. Microporous Mesoporous Mater. 2003, 60, 91–97. [Google Scholar] [CrossRef]
- Perkas, N.; Amirian, G.; Zhong, Z.; Teo, J.; Gofer, Y.; Gedanken, A. Methanation of Carbon Dioxide on Ni Catalysts on Mesoporous ZrO2 Doped with Rare Earth Oxides. Catal. Lett. 2009, 130, 455–462. [Google Scholar] [CrossRef]
- Daroughegi, R.; Meshkani, F.; Rezaei, M. Enhanced activity of CO2 methanation over mesoporous nanocrystalline Ni–Al2O3 catalysts prepared by ultrasound-assisted co-precipitation method. Int. J. Hydrogen Energy 2017, 42, 15115–15125. [Google Scholar] [CrossRef]
- Kruatim, J.; Jantasee, S.; Jongsomjit, B. Improvement of cobalt dispersion on Co/SBA-15 and Co/SBA-16 catalysts by ultrasound and vacuum treatments during post-impregnation step. Eng. J. 2017, 21, 17–28. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, G.; Wang, L.; Xu, Y.; Sun, Y. Selective methanation of carbon monoxide over Ru-based catalysts in H2-rich gases. J. Ind. Eng. Chem. 2012, 18, 1590–1597. [Google Scholar] [CrossRef]
- Xu, C.; De, S.; Balu, A.M.; Ojeda, M.; Luque, R. Mechanochemical synthesis of advanced nanomaterials for catalytic applications. Chem. Commun. 2015, 51, 6698–6713. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.A.P.; Frizzo, C.P.; Moreira, D.N.; Buriol, L.; Machado, P. Solvent-Free Heterocyclic Synthesis. Chem. Rev. 2009, 109, 4140–4182. [Google Scholar] [CrossRef] [PubMed]
- Mitchenko, S.A. Mechanochemistry in heterogeneous catalysis. Theor. Exp. Chem. 2007, 43, 211–228. [Google Scholar] [CrossRef]
- Mori, S.; Xu, W.-C.; Ishidzuki, T.; Ogasawara, N.; Imai, J.; Kobayashi, K. Mechanochemical activation of catalysts for CO2 methanation. Appl. Catal. A Gen. 1996, 137, 255–268. [Google Scholar] [CrossRef]
- Morozova, O.S.; Streletskii, A.N.; Berestetskaya, I.V.; Borunova, A.B. Co and Co2 hydrogenation under mechanochemical treatment. Catal. Today 1997, 38, 107–113. [Google Scholar] [CrossRef]
- Trovarelli, A.; Matteazzi, P.; Dolcetti, G.; Lutman, A.; Miani, F. Nanophase iron carbides as catalysts for carbon dioxide hydrogenation. Appl. Catal. A Gen. 1993, 95, L9–L13. [Google Scholar] [CrossRef]
- Guczi, L.; Takács, L.; Stefler, G.; Koppány, Z.; Borkó, L. Re–Co/Al2O3 bimetallic catalysts prepared by mechanical treatment: CO hydrogenation and CH4 conversion. Catal. Today 2002, 77, 237–243. [Google Scholar] [CrossRef]
- Delogu, F.; Mulas, G.; Garroni, S. Hydrogenation of carbon monoxide under mechanical activation conditions. Appl. Catal. A Gen. 2009, 366, 201–205. [Google Scholar] [CrossRef]
- Meunier, F.C. Mixing Copper Nanoparticles and ZnO Nanocrystals: A Route towards Understanding the Hydrogenation of CO2 to Methanol? Angew. Chem. Int. Ed. 2011, 50, 4053–4054. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Li, F.; Zhan, H.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Wang, H.; Sun, Y. Influence of Zr on the performance of Cu/Zn/Al/Zr catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. J. Catal. 2013, 298, 51–60. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C. Morphology-Dependent Interactions of ZnO with Cu Nanoparticles at the Materials’ Interface in Selective Hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-M.; Lu, G.Q.; Yan, Z.-F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003. [Google Scholar] [CrossRef]
- Raudaskoski, R.; Turpeinen, E.; Lenkkeri, R.; Pongrácz, E.; Keiski, R.L. Catalytic activation of CO2: Use of secondary CO2 for the production of synthesis gas and for methanol synthesis over copper-based zirconia-containing catalysts. Catal. Today 2009, 144, 318–323. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, J. The effect of ZnO in methanol synthesis catalysts on Cu dispersion and the specific activity. Catal. Lett. 1998, 56, 119–124. [Google Scholar] [CrossRef]
- Behrens, M.; Zander, S.; Kurr, P.; Jacobsen, N.; Senker, J.; Koch, G.; Ressler, T.; Fischer, R.W.; Schlögl, R. Performance Improvement of Nanocatalysts by Promoter-Induced Defects in the Support Material: Methanol Synthesis over Cu/ZnO:Al. J. Am. Chem. Soc. 2013, 135, 6061–6068. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Italiano, G.; Barbera, K.; Bonura, G.; Spadaro, L.; Frusteri, F. Basic evidences for methanol-synthesis catalyst design. Catal. Today 2009, 143, 80–85. [Google Scholar] [CrossRef]
- Behrens, M. Meso- and nano-structuring of industrial Cu/ZnO/(Al2O3) catalysts. J. Catal. 2009, 267, 24–29. [Google Scholar] [CrossRef]
- Baltes, C.; Vukojević, S.; Schüth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Behrens, M.; Schlögl, R. How to Prepare a Good Cu/ZnO Catalyst or the Role of Solid State Chemistry for the Synthesis of Nanostructured Catalysts. Z. Anorg. Allg. Chem. 2013, 639, 2683–2695. [Google Scholar] [CrossRef]
- Li, Z.; Yan, S.; Fan, H. Enhancement of stability and activity of Cu/ZnO/Al2O3 catalysts by microwave irradiation for liquid phase methanol synthesis. Fuel 2013, 106, 178–186. [Google Scholar] [CrossRef]
- Wu, W.; Xie, K.; Sun, D.; Li, X.; Fang, F. CuO/ZnO/Al2O3 Catalyst Prepared by Mechanical-Force-Driven Solid-State Ion Exchange and Its Excellent Catalytic Activity under Internal Cooling Condition. Ind. Eng. Chem. Res. 2017, 56, 8216–8223. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Ren, Y.; Yang, X.; Wu, Z.; Liu, X.; Li, L.; Miao, S.; Su, Y.; Li, Y.; et al. Synthesis of Subnanometer-Sized Gold Clusters by a Simple Milling-Mediated Solid Reduction Method. Chin. J. Chem. 2018, 36, 329–332. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, P. Tailoring the local structure and electronic property of AuPd nanoparticles by selecting capping molecules. Appl. Phys. Lett. 2010, 96, 043105. [Google Scholar] [CrossRef]
Scheme 1.
Some products of CO2 hydrogenation. Reproduced from Ref. [1] with permission of The Royal Society of Chemistry.
Scheme 1.
Some products of CO2 hydrogenation. Reproduced from Ref. [1] with permission of The Royal Society of Chemistry.
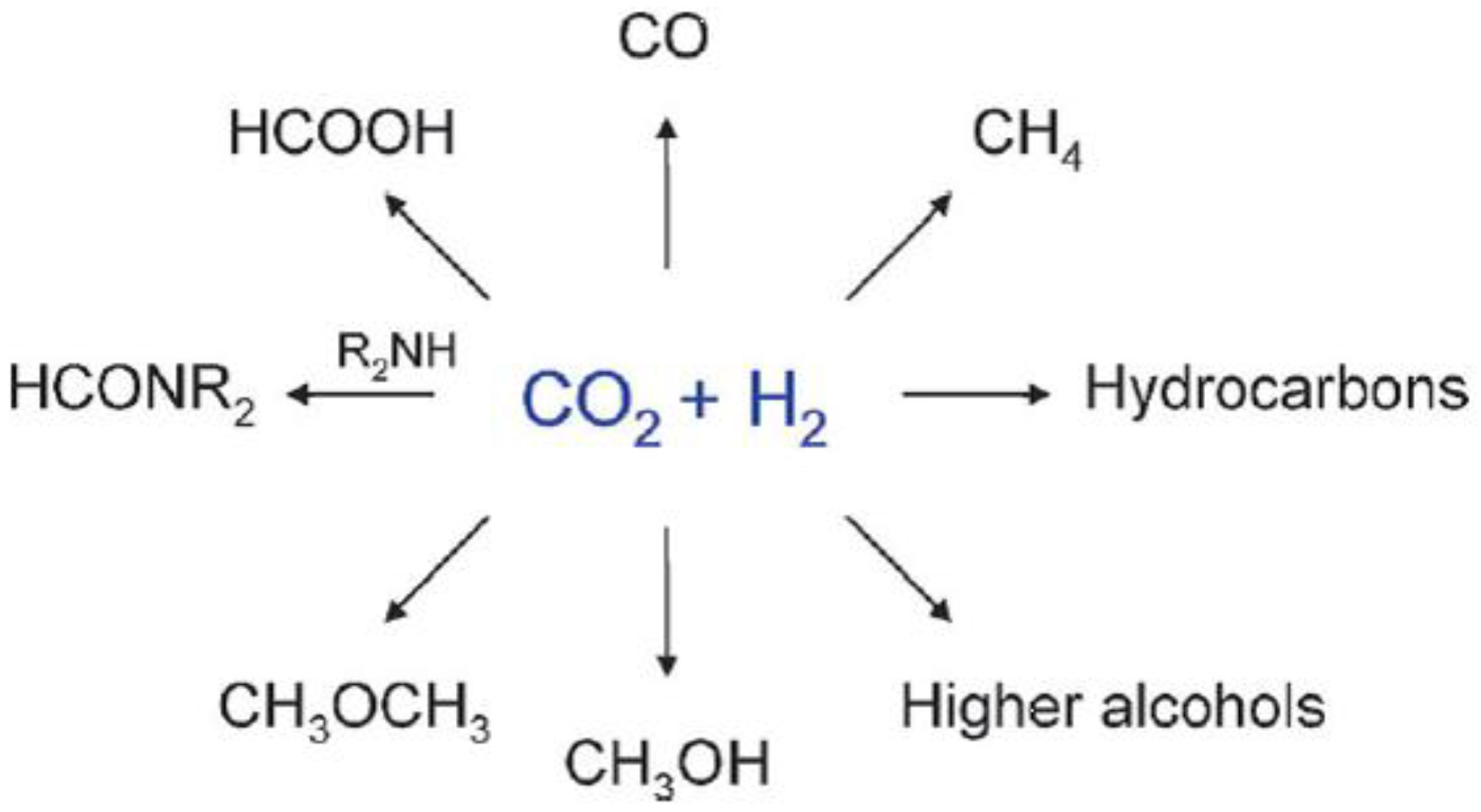
Figure 1.
SEM (a,b) and TEM (c,d) images of BiOI flower-like spheres (dimensions in the 500 nm−1 µm range) made up by nanosheets as building blocks. Reprinted from Chem. Eng. J., 235 Ai, L.; Zeng, Y.; Jiang, J., Hierarchical Porous BiOI Architectures: facile Microwave Nonaqueous Synthesis, Characterization, and Application in the Removal of Congo Red from Aqueous Solution, 331–339. Copyright (2014) with permission from Elsevier.
Figure 1.
SEM (a,b) and TEM (c,d) images of BiOI flower-like spheres (dimensions in the 500 nm−1 µm range) made up by nanosheets as building blocks. Reprinted from Chem. Eng. J., 235 Ai, L.; Zeng, Y.; Jiang, J., Hierarchical Porous BiOI Architectures: facile Microwave Nonaqueous Synthesis, Characterization, and Application in the Removal of Congo Red from Aqueous Solution, 331–339. Copyright (2014) with permission from Elsevier.

Figure 2.
Linear relation between the CH3OH % yield and the surface of Cu particles (SCu). Reprinted from Huang, C.; Mao, D.; Guo, X.; Yu, J., Microwave-Assisted Hydrothermal Synthesis of CuO-ZnO-ZrO2 as Catalyst for Direct Synthesis of Methanol by Carbon Dioxide Hydrogenation. Energy Technol. Copyright (2017) with permission from Wiley.
Figure 2.
Linear relation between the CH3OH % yield and the surface of Cu particles (SCu). Reprinted from Huang, C.; Mao, D.; Guo, X.; Yu, J., Microwave-Assisted Hydrothermal Synthesis of CuO-ZnO-ZrO2 as Catalyst for Direct Synthesis of Methanol by Carbon Dioxide Hydrogenation. Energy Technol. Copyright (2017) with permission from Wiley.

Figure 3.
TEM, HRTEM, and SAED patterns of Ni/Al2O3 prepared under MW radiation (a–c) and without MW irradiation (d–f). Reprinted from Int. J. Hydrogen Energy, 42, Song, F.; Zhong, Q.; Yu, Y.; Shi, M.; Wu, Y.; Hu, J.; Song, Y., Obtaining Well-Dispersed Ni/Al2O3 Catalyst for CO2 Methanation with a Microwave-Assisted Method, 4174–4183. Copyright (2017) with permission from Elsevier.
Figure 3.
TEM, HRTEM, and SAED patterns of Ni/Al2O3 prepared under MW radiation (a–c) and without MW irradiation (d–f). Reprinted from Int. J. Hydrogen Energy, 42, Song, F.; Zhong, Q.; Yu, Y.; Shi, M.; Wu, Y.; Hu, J.; Song, Y., Obtaining Well-Dispersed Ni/Al2O3 Catalyst for CO2 Methanation with a Microwave-Assisted Method, 4174–4183. Copyright (2017) with permission from Elsevier.
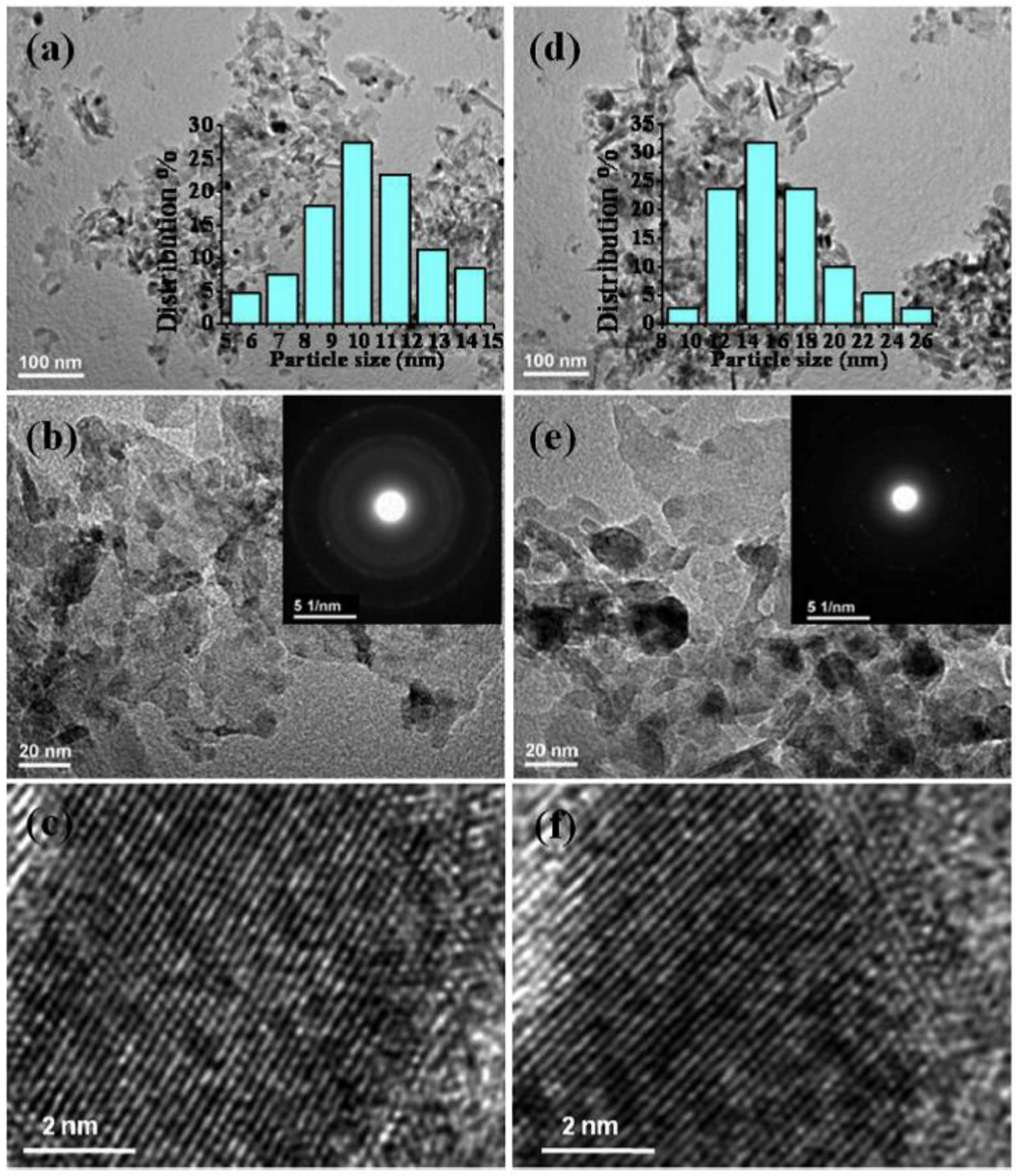
Scheme 2.
Scheme of the processes occurring at the surface of Re-promoted Ni-Mn bimetallic particles on Al2O3 support. Reprinted from Appl. Catal. A Gen., 552, Dong, X.; Jin, B.; Sun, Y.; Shi, K.; Yu, L., Re-Promoted Ni-Mn Bifunctional Catalysts Prepared by Microwave Heating for Partial Methanation Coupling with Water Gas Shift under Low H2/CO Conditions. Copyright (2018) with permission from Elsevier.
Scheme 2.
Scheme of the processes occurring at the surface of Re-promoted Ni-Mn bimetallic particles on Al2O3 support. Reprinted from Appl. Catal. A Gen., 552, Dong, X.; Jin, B.; Sun, Y.; Shi, K.; Yu, L., Re-Promoted Ni-Mn Bifunctional Catalysts Prepared by Microwave Heating for Partial Methanation Coupling with Water Gas Shift under Low H2/CO Conditions. Copyright (2018) with permission from Elsevier.

Figure 4.
MW heating curves of different catalyst precursors: (1) Ni-Mn/Al2O3, (2) Ni-Mn-Re/Al2O3, (3) Re-Al2O3, and (4) Ni-Mn/Re-Al2O3. Reprinted from Appl. Catal. A Gen., 552, Dong, X.; Jin, B.; Sun, Y.; Shi, K.; Yu, L., Re-Promoted Ni-Mn Bifunctional Catalysts Prepared by Microwave Heating for Partial Methanation Coupling with Water Gas Shift under Low H2/CO Conditions. Copyright (2018) with permission from Elsevier.
Figure 4.
MW heating curves of different catalyst precursors: (1) Ni-Mn/Al2O3, (2) Ni-Mn-Re/Al2O3, (3) Re-Al2O3, and (4) Ni-Mn/Re-Al2O3. Reprinted from Appl. Catal. A Gen., 552, Dong, X.; Jin, B.; Sun, Y.; Shi, K.; Yu, L., Re-Promoted Ni-Mn Bifunctional Catalysts Prepared by Microwave Heating for Partial Methanation Coupling with Water Gas Shift under Low H2/CO Conditions. Copyright (2018) with permission from Elsevier.

Figure 5.
TEM pictures of the catalysts prepared by MW-assisted CS (a) and impregnation (b) after reaction. Reprinted from Appl. Catal. A Gen., 378, Barros, B. S.; Melo, D. M. A.; Libs, S.; Kiennemann, A., CO2 Reforming of Methane Over La2NiO4/α-Al2O3 Prepared by Microwave-Assisted Self-Combustion Method, 69–75. Copyright (2010) with permission from Elsevier.
Figure 5.
TEM pictures of the catalysts prepared by MW-assisted CS (a) and impregnation (b) after reaction. Reprinted from Appl. Catal. A Gen., 378, Barros, B. S.; Melo, D. M. A.; Libs, S.; Kiennemann, A., CO2 Reforming of Methane Over La2NiO4/α-Al2O3 Prepared by Microwave-Assisted Self-Combustion Method, 69–75. Copyright (2010) with permission from Elsevier.

Figure 6.
SEM picture of pure TiO2 film (a) and Back Scattered Electrons images of TiO2 NFFs containing increasing amounts of Cu from (b–d). Reprinted from Mater. Res. Bull., 68, Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q., A Facile Strategy to Fabricate Plasmonic, Cu-Modified TiO2 Nano-flower Films for Photocatalytic Reduction of CO2 to Methanol, 203–209. Copyright (2015) with permission from Elsevier.
Figure 6.
SEM picture of pure TiO2 film (a) and Back Scattered Electrons images of TiO2 NFFs containing increasing amounts of Cu from (b–d). Reprinted from Mater. Res. Bull., 68, Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q., A Facile Strategy to Fabricate Plasmonic, Cu-Modified TiO2 Nano-flower Films for Photocatalytic Reduction of CO2 to Methanol, 203–209. Copyright (2015) with permission from Elsevier.

Figure 7.
CH3OH production rate by photocatalytic reduction of CO2 as obtained under UV light with both pure TiO2 film and a Cu/NFF obtained by starting from 0.5 mM Cu2+ solution (0.5 Cu/TiO2 film). Reprinted from Mater. Res. Bull., 68, Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q., A Facile Strategy to Fabricate Plasmonic, Cu-Modified TiO2 Nano-Flower Films for Photocatalytic Reduction of CO2 to Methanol, 203–209. Copyright (2015) with permission from Elsevier.
Figure 7.
CH3OH production rate by photocatalytic reduction of CO2 as obtained under UV light with both pure TiO2 film and a Cu/NFF obtained by starting from 0.5 mM Cu2+ solution (0.5 Cu/TiO2 film). Reprinted from Mater. Res. Bull., 68, Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q., A Facile Strategy to Fabricate Plasmonic, Cu-Modified TiO2 Nano-Flower Films for Photocatalytic Reduction of CO2 to Methanol, 203–209. Copyright (2015) with permission from Elsevier.

Scheme 3.
The synergistic mechanism proposed for the photocatalytic reduction of CO2 to CH3OH with Cu/TiO2 NFFs. Reprinted from Mater. Res. Bull., 68, Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q., A Facile Strategy to Fabricate Plasmonic, Cu-Modified TiO2 Nano-Flower Films for Photocatalytic Reduction of CO2 to Methanol, 203–209. Copyright (2015) with permission from Elsevier.
Scheme 3.
The synergistic mechanism proposed for the photocatalytic reduction of CO2 to CH3OH with Cu/TiO2 NFFs. Reprinted from Mater. Res. Bull., 68, Liu, E.; Qi, L.; Bian, J.; Chen, Y.; Hu, X.; Fan, J.; Liu, H.; Zhu, C.; Wang, Q., A Facile Strategy to Fabricate Plasmonic, Cu-Modified TiO2 Nano-Flower Films for Photocatalytic Reduction of CO2 to Methanol, 203–209. Copyright (2015) with permission from Elsevier.

Figure 8.
Scheme of the MW assisted synthesis process adopted to decorate GO with Cu NPs (a). Sections (b−d): TEM images of Cu/GO hybrids with 5, 10, and 15 wt % Cu, respectively. Inset: HRTEM image of single Cu-NP of the respective Cu/GO hybrid. Sections (e–g): size distribution of Cu-NPs in Cu/GO hybrids with 5, 10, and 15 wt % Cu, respectively. Representative TEM image of pure GO (h). Reprinted with permission from Shown, I.; Hsu, H.-C.; Chang, Y.-C.; Lin, C.-H.; Roy, P.K.; Ganguly, A.; Wang, C.-H.; Chang, J.-K.; Wu, C.-I.; Chen, L.-C.; Chen, K.-H., Highly Efficient Visible Light Photocatalytic Reduction of CO2 to Hydrocarbon Fuels by Cu-Nanoparticle Decorated Graphene Oxide. Nano Lett. 14, 6097–6103. Copyright (2014) American Chemical Society.
Figure 8.
Scheme of the MW assisted synthesis process adopted to decorate GO with Cu NPs (a). Sections (b−d): TEM images of Cu/GO hybrids with 5, 10, and 15 wt % Cu, respectively. Inset: HRTEM image of single Cu-NP of the respective Cu/GO hybrid. Sections (e–g): size distribution of Cu-NPs in Cu/GO hybrids with 5, 10, and 15 wt % Cu, respectively. Representative TEM image of pure GO (h). Reprinted with permission from Shown, I.; Hsu, H.-C.; Chang, Y.-C.; Lin, C.-H.; Roy, P.K.; Ganguly, A.; Wang, C.-H.; Chang, J.-K.; Wu, C.-I.; Chen, L.-C.; Chen, K.-H., Highly Efficient Visible Light Photocatalytic Reduction of CO2 to Hydrocarbon Fuels by Cu-Nanoparticle Decorated Graphene Oxide. Nano Lett. 14, 6097–6103. Copyright (2014) American Chemical Society.

Scheme 4.
Section (a): work functions of pure GO and of Cu/GO hybrids with 5, 10, and 15 wt % Cu (Cu/GO-1, Cu/GO-2, and Cu/GO-3, respectively). Section (b): band-edge positions of pure GO and Cu/GO hybrids in comparison with CO2/CH3OH and CO2/CH3CHO formation potentials. Section (c): proposed photocatalytic reaction mechanism under solar irradiation. Reprinted with permission from Shown, I.; Hsu, H.-C.; Chang, Y.-C.; Lin, C.-H.; Roy, P.K.; Ganguly, A.; Wang, C.-H.; Chang, J.-K.; Wu, C.-I.; Chen, L.-C.; Chen, K.-H. Highly Efficient Visible Light Photocatalytic Reduction of CO2 to Hydrocarbon Fuels by Cu-Nanoparticle Decorated Graphene Oxide. Nano Lett. 14, 6097–6103. Copyright (2014) American Chemical Society.
Scheme 4.
Section (a): work functions of pure GO and of Cu/GO hybrids with 5, 10, and 15 wt % Cu (Cu/GO-1, Cu/GO-2, and Cu/GO-3, respectively). Section (b): band-edge positions of pure GO and Cu/GO hybrids in comparison with CO2/CH3OH and CO2/CH3CHO formation potentials. Section (c): proposed photocatalytic reaction mechanism under solar irradiation. Reprinted with permission from Shown, I.; Hsu, H.-C.; Chang, Y.-C.; Lin, C.-H.; Roy, P.K.; Ganguly, A.; Wang, C.-H.; Chang, J.-K.; Wu, C.-I.; Chen, L.-C.; Chen, K.-H. Highly Efficient Visible Light Photocatalytic Reduction of CO2 to Hydrocarbon Fuels by Cu-Nanoparticle Decorated Graphene Oxide. Nano Lett. 14, 6097–6103. Copyright (2014) American Chemical Society.

Scheme 5.
Schematic picture of the charge transfer process in Cu2O/RGO composites during CO2 photoreduction to CO in water. Reprinted from Ref. [56]. Copyright 2014 with permission of Wiley.
Scheme 5.
Schematic picture of the charge transfer process in Cu2O/RGO composites during CO2 photoreduction to CO in water. Reprinted from Ref. [56]. Copyright 2014 with permission of Wiley.
Scheme 6.
Schematic picture of the synthesis of Cu decorated g-C3N4 nanosheets. Reprinted from Appl. Surf. Sci., 427, Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y., Photocatalytic Reduction of CO2 to CO Over Copper-Decorated g-C3N4 Nanosheets with Enhanced Yield and Selectivity, 1165–1173. Copyright (2014) with permission from Elsevier.
Scheme 6.
Schematic picture of the synthesis of Cu decorated g-C3N4 nanosheets. Reprinted from Appl. Surf. Sci., 427, Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y., Photocatalytic Reduction of CO2 to CO Over Copper-Decorated g-C3N4 Nanosheets with Enhanced Yield and Selectivity, 1165–1173. Copyright (2014) with permission from Elsevier.

Figure 9.
TEM images of g-C3N4 at low (a) and high (b) magnification and TEM image of Cu decorated g-C3N4 nanosheets (c). Reprinted from Appl. Surf. Sci., 427, Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y., Photocatalytic Reduction of CO2 to CO Over Copper-Decorated g-C3N4 Nanosheets with Enhanced Yield and Selectivity, 1165–1173. Copyright (2014) with permission from Elsevier.
Figure 9.
TEM images of g-C3N4 at low (a) and high (b) magnification and TEM image of Cu decorated g-C3N4 nanosheets (c). Reprinted from Appl. Surf. Sci., 427, Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y., Photocatalytic Reduction of CO2 to CO Over Copper-Decorated g-C3N4 Nanosheets with Enhanced Yield and Selectivity, 1165–1173. Copyright (2014) with permission from Elsevier.

Scheme 7.
Scheme of the photocatalytic reduction of CO2 to CO and simultaneous H2O oxidation to O2 in the presence of Cu-decorated g-C3N4 nanosheets. Reprinted from Appl. Surf. Sci., 427, Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y., Photocatalytic Reduction of CO2 to CO Over Copper-Decorated g-C3N4 Nanosheets with Enhanced Yield and Selectivity, 1165–1173. Copyright (2014) with permission from Elsevier.
Scheme 7.
Scheme of the photocatalytic reduction of CO2 to CO and simultaneous H2O oxidation to O2 in the presence of Cu-decorated g-C3N4 nanosheets. Reprinted from Appl. Surf. Sci., 427, Shi, G.; Yang, L.; Liu, Z.; Chen, X.; Zhou, J.; Yu, Y., Photocatalytic Reduction of CO2 to CO Over Copper-Decorated g-C3N4 Nanosheets with Enhanced Yield and Selectivity, 1165–1173. Copyright (2014) with permission from Elsevier.

Figure 10.
TEM images of pure GO (a) and pure CdS nanorods (b). TEM (c) and HRTEM (d) images of RGO-CdS nanorods composites. Reproduced from Ref. [58]. Copyright 2014 with permission from The Royal Society of Chemistry.
Figure 10.
TEM images of pure GO (a) and pure CdS nanorods (b). TEM (c) and HRTEM (d) images of RGO-CdS nanorods composites. Reproduced from Ref. [58]. Copyright 2014 with permission from The Royal Society of Chemistry.

Scheme 8.
Scheme of the mechanism proposed for the photocatalytic reduction of CO2 to CH4 by RGO-CdS nanorods composites. Reprinted from Ref. [58]. Copyright 2014 with permission from The Royal Society of Chemistry.
Scheme 8.
Scheme of the mechanism proposed for the photocatalytic reduction of CO2 to CH4 by RGO-CdS nanorods composites. Reprinted from Ref. [58]. Copyright 2014 with permission from The Royal Society of Chemistry.

Figure 11.
CO production by CO2 photochemical reduction after 4 h reaction with 2.0 mM p-terphenyl with Co(III) cyclam deposited by MW-assisted synthesis and conventional reflux method. Reprinted from Ref. [59]. Copyright 2017 with permission from The Royal Society of Chemistry.
Figure 11.
CO production by CO2 photochemical reduction after 4 h reaction with 2.0 mM p-terphenyl with Co(III) cyclam deposited by MW-assisted synthesis and conventional reflux method. Reprinted from Ref. [59]. Copyright 2017 with permission from The Royal Society of Chemistry.
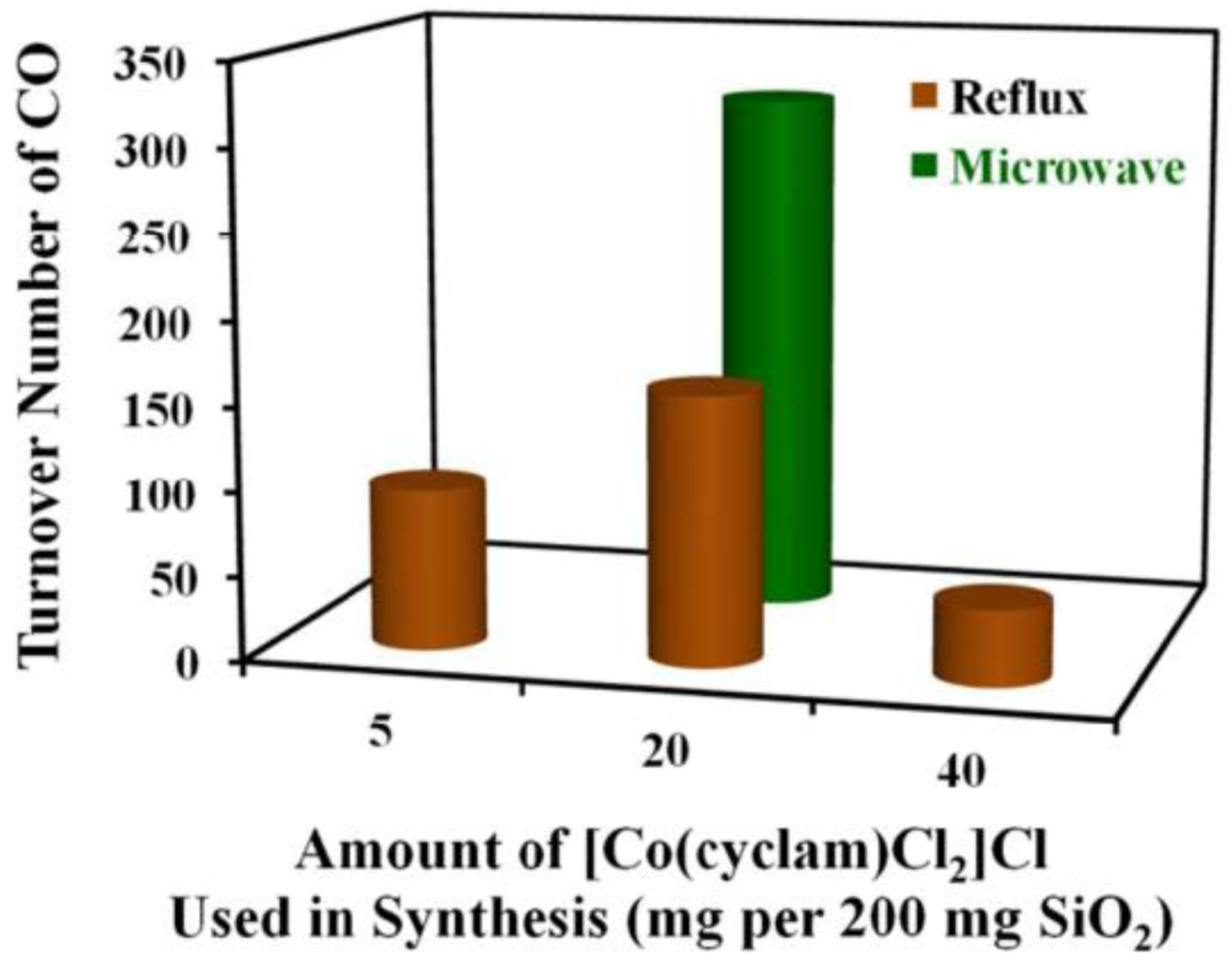
Figure 12.
Schematic flow chart for preparation steps of CZA/HZSM-5 catalysts synthesized via combined co-precipitation-ultrasound method with different active phase to support ratios. Reprinted from Ref. [90]. Copyright 2014 with permission of The Royal Society of Chemistry.
Figure 12.
Schematic flow chart for preparation steps of CZA/HZSM-5 catalysts synthesized via combined co-precipitation-ultrasound method with different active phase to support ratios. Reprinted from Ref. [90]. Copyright 2014 with permission of The Royal Society of Chemistry.

Figure 13.
Experimental setup for dispersion of CZA over HZSM-5 using US energy. Reprinted from Ref. [90]. Copyright 2014 with permission of The Royal Society of Chemistry.
Figure 13.
Experimental setup for dispersion of CZA over HZSM-5 using US energy. Reprinted from Ref. [90]. Copyright 2014 with permission of The Royal Society of Chemistry.

Figure 14.
Surface particle size distribution histogram of synthesized CZA/HZSM-5 nanocatalyst using acetate precursor and ultrasound irradiation power of 150 W (CZAZ-AU150). Reprinted from Ultrason. Sonochem., 21, Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S., Ultrasound-Assisted Co-Precipitation of Nanostructured CuO-ZnO-Al2O3 Over HZSM-5: Effect of Precursor and Irradiation Power on Nanocatalyst Properties and Catalytic Performance for Direct Syngas to DME, 663–673. Copyright (2014) with permission from Elsevier.
Figure 14.
Surface particle size distribution histogram of synthesized CZA/HZSM-5 nanocatalyst using acetate precursor and ultrasound irradiation power of 150 W (CZAZ-AU150). Reprinted from Ultrason. Sonochem., 21, Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S., Ultrasound-Assisted Co-Precipitation of Nanostructured CuO-ZnO-Al2O3 Over HZSM-5: Effect of Precursor and Irradiation Power on Nanocatalyst Properties and Catalytic Performance for Direct Syngas to DME, 663–673. Copyright (2014) with permission from Elsevier.

Figure 15.
Reaction pathway for synthesis of ZnO in CZA nanocatalyst by ultrasound-assisted co-precipitation of zinc acetate. Reprinted from Ultrason. Sonochem., 21, Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S., Ultrasound assisted co-precipitation of nanostructured CuO-ZnO-Al2O3 over HZSM-5: Effect of Precursor and Irradiation Power on Nanocatalyst Properties and Catalytic Performance for Direct Syngas to Dimethyl Ether, 663–673. Copyright (2014) with permission from Elsevier.
Figure 15.
Reaction pathway for synthesis of ZnO in CZA nanocatalyst by ultrasound-assisted co-precipitation of zinc acetate. Reprinted from Ultrason. Sonochem., 21, Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S., Ultrasound assisted co-precipitation of nanostructured CuO-ZnO-Al2O3 over HZSM-5: Effect of Precursor and Irradiation Power on Nanocatalyst Properties and Catalytic Performance for Direct Syngas to Dimethyl Ether, 663–673. Copyright (2014) with permission from Elsevier.

Figure 16.
TEM images of CZA supported on multi-walled carbon nanotubes pretreated with a H2SO4/HNO3 mixture. Reprinted from Appl. Surf. Sci., 285, Zha, F.; Tian, H.; Yan, J.; Chang, Y., Multi-Walled Carbon Nanotubes as Catalyst Promoter for Dimethyl Ether Synthesis from CO2 Hydrogenation, 945–951. Copyright (2013) with permission from Elsevier.
Figure 16.
TEM images of CZA supported on multi-walled carbon nanotubes pretreated with a H2SO4/HNO3 mixture. Reprinted from Appl. Surf. Sci., 285, Zha, F.; Tian, H.; Yan, J.; Chang, Y., Multi-Walled Carbon Nanotubes as Catalyst Promoter for Dimethyl Ether Synthesis from CO2 Hydrogenation, 945–951. Copyright (2013) with permission from Elsevier.

Figure 17.
TEM images of nickel nanoparticles developed using hydrazine (A), high magnification image (inset), and sodium borohydride (B). Hydrodynamic diameter (C), colloidal systems images (inset), and XRD patterns (D) of samples. Reproduced from [100] with permission of The Royal Society of Chemistry.
Figure 17.
TEM images of nickel nanoparticles developed using hydrazine (A), high magnification image (inset), and sodium borohydride (B). Hydrodynamic diameter (C), colloidal systems images (inset), and XRD patterns (D) of samples. Reproduced from [100] with permission of The Royal Society of Chemistry.

Figure 18.
XRD patterns of CZAx catalysts (x = 200 (a), 300 (b), and 400 (c)). Reprinted from Wu, W.; Xie, K.; Sun, D.; Li, X.; Fang, F. CuO/ZnO/Al2O3 Catalyst Prepared by Mechanical-Force-Driven Solid-State Ion Exchange and Its Excellent Catalytic Activity under Internal Cooling Condition. Ind. Eng. Chem. Res. 56, 8216–8223. Copyright (2017) American Chemical Society.
Figure 18.
XRD patterns of CZAx catalysts (x = 200 (a), 300 (b), and 400 (c)). Reprinted from Wu, W.; Xie, K.; Sun, D.; Li, X.; Fang, F. CuO/ZnO/Al2O3 Catalyst Prepared by Mechanical-Force-Driven Solid-State Ion Exchange and Its Excellent Catalytic Activity under Internal Cooling Condition. Ind. Eng. Chem. Res. 56, 8216–8223. Copyright (2017) American Chemical Society.

Scheme 9.
Pictorial representation of different types of MC synthesized (nano)materials. From supported metal NPs, composite nanomaterials to metal oxide NPs and metal organic frameworks (MOFs) including covalent organic frameworks (COF). Examples of relevant materials include zinc-based ZIF-8 structures (BIT-11), Cu(INA)2, and Cu-containing HKUST-1, as well as supported noble metals on aluminosilicates, graphene, etc. Reprinted from Ref. [109]. Copyright 2015 with permission of the Royal Society of Chemistry.
Scheme 9.
Pictorial representation of different types of MC synthesized (nano)materials. From supported metal NPs, composite nanomaterials to metal oxide NPs and metal organic frameworks (MOFs) including covalent organic frameworks (COF). Examples of relevant materials include zinc-based ZIF-8 structures (BIT-11), Cu(INA)2, and Cu-containing HKUST-1, as well as supported noble metals on aluminosilicates, graphene, etc. Reprinted from Ref. [109]. Copyright 2015 with permission of the Royal Society of Chemistry.
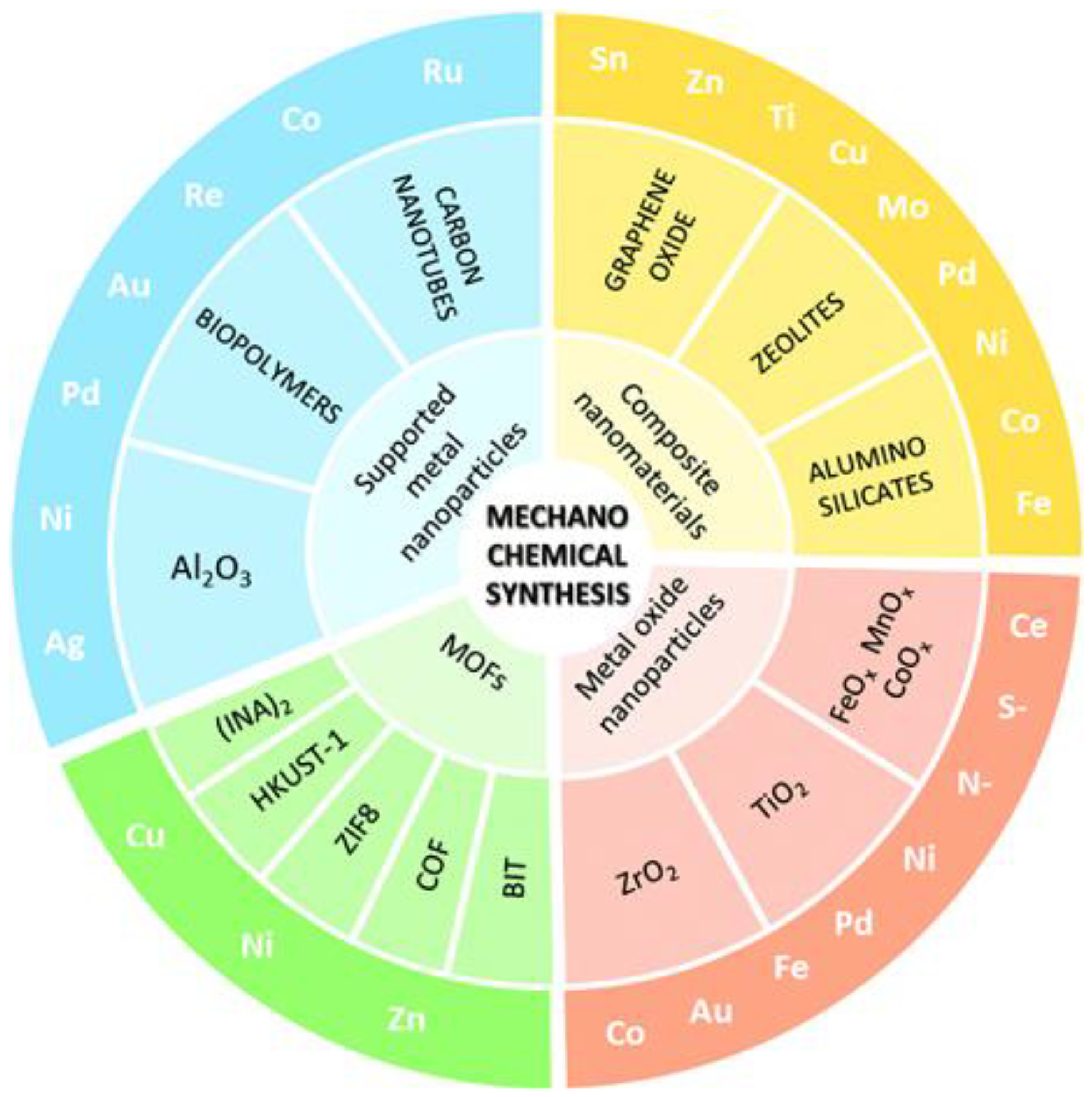
Figure 19.
TEM (a) and HRTEM (b) images of CuO/ZnO/Al2O3 catalyst prepared at 400 rpm. Reprinted from Wu, W.; Xie, K.; Sun, D.; Li, X.; Fang, F. CuO/ZnO/Al2O3 Catalyst Prepared by Mechanical-Force-Driven Solid-State Ion Exchange and Its Excellent Catalytic Activity under Internal Cooling Condition. Ind. Eng. Chem. Res. 56, 8216–8223. Copyright (2017) American Chemical Society.
Figure 19.
TEM (a) and HRTEM (b) images of CuO/ZnO/Al2O3 catalyst prepared at 400 rpm. Reprinted from Wu, W.; Xie, K.; Sun, D.; Li, X.; Fang, F. CuO/ZnO/Al2O3 Catalyst Prepared by Mechanical-Force-Driven Solid-State Ion Exchange and Its Excellent Catalytic Activity under Internal Cooling Condition. Ind. Eng. Chem. Res. 56, 8216–8223. Copyright (2017) American Chemical Society.

Scheme 10.
Illustration of the in situ construction of ultrafine Au clusters on a nitrogenous carrier using the solid reduction method. Reprinted from Ref. [130]. Copyright 2018 with permission of Wiley.
Scheme 10.
Illustration of the in situ construction of ultrafine Au clusters on a nitrogenous carrier using the solid reduction method. Reprinted from Ref. [130]. Copyright 2018 with permission of Wiley.
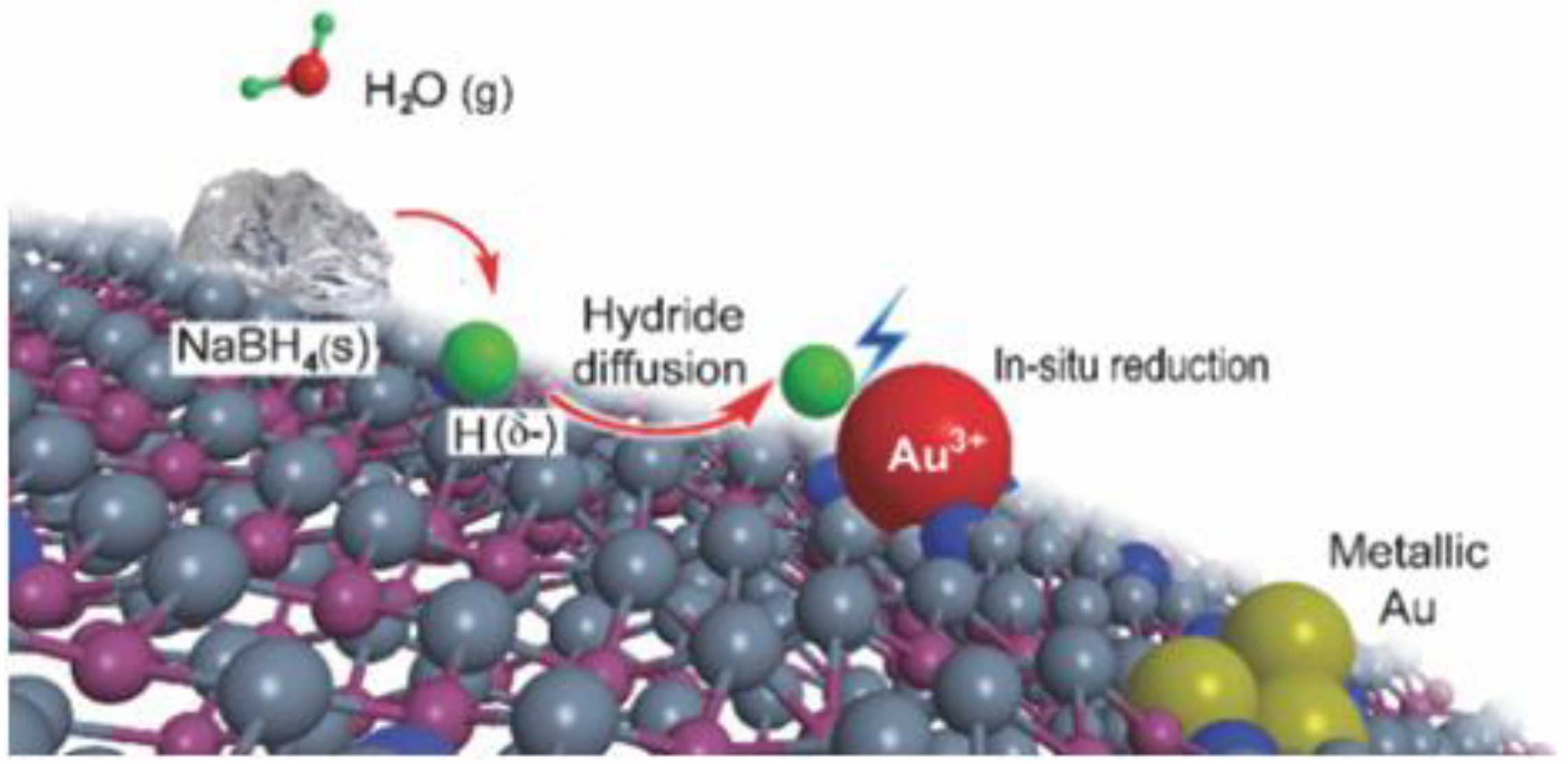
Figure 20.
Aberration-corrected HAADF-STEM images of Au/SiO2-Schiff prepared by (a) solid reduction method and (b) wet chemistry method, respectively. The NPs, subnanoclusters, and isolated Au atoms were indicated by white circles, triangles, and squares, respectively. (c) Fourier transform of k3-weighted EXAFS spectra and (d) normalized Au L3-edge XANES spectra of Au/SiO2-Schiff catalysts prepared by wet chemistry and solid reduction method, respectively. Reprinted from Ref. [130]. Copyright 2018 with permission of Wiley.
Figure 20.
Aberration-corrected HAADF-STEM images of Au/SiO2-Schiff prepared by (a) solid reduction method and (b) wet chemistry method, respectively. The NPs, subnanoclusters, and isolated Au atoms were indicated by white circles, triangles, and squares, respectively. (c) Fourier transform of k3-weighted EXAFS spectra and (d) normalized Au L3-edge XANES spectra of Au/SiO2-Schiff catalysts prepared by wet chemistry and solid reduction method, respectively. Reprinted from Ref. [130]. Copyright 2018 with permission of Wiley.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Manzoli, M.; Bonelli, B. Microwave, Ultrasound, and Mechanochemistry: Unconventional Tools that Are Used to Obtain “Smart” Catalysts for CO2 Hydrogenation. Catalysts 2018, 8, 262. https://doi.org/10.3390/catal8070262
AMA Style
Manzoli M, Bonelli B. Microwave, Ultrasound, and Mechanochemistry: Unconventional Tools that Are Used to Obtain “Smart” Catalysts for CO2 Hydrogenation. Catalysts. 2018; 8(7):262. https://doi.org/10.3390/catal8070262
Chicago/Turabian StyleManzoli, Maela, and Barbara Bonelli. 2018. "Microwave, Ultrasound, and Mechanochemistry: Unconventional Tools that Are Used to Obtain “Smart” Catalysts for CO2 Hydrogenation" Catalysts 8, no. 7: 262. https://doi.org/10.3390/catal8070262
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.