The Promotional Effects of ZrO2 and Au on the CuZnO Catalyst Regarding the Durability and Activity of the Partial Oxidation of Methanol


Department of Biomedical Engineering & Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(9), 345; https://doi.org/10.3390/catal8090345
Submission received: 2 July 2018
/
Revised: 21 August 2018
/
Accepted: 21 August 2018
/
Published: 24 August 2018
Abstract
:The promoter ZrO2 was applied to prevent Cu crystallites from sintering over CZ (ca. Cu 30 wt.% and Zn 70 wt.%) under partial oxidation of the methanol (POM) reaction. Gold was selected to promote the performance of CZrZ (ca. Cu 31 wt.%, Zr 16 wt.%, and Zn 53 wt.%) catalyst to overcome a high ignition temperature of 175 °C and CO selectivity (SCO) (>10% at T. > 200 °C). Experimentally, the deactivation rate constant of A5CZrZ (ca. Au 5 wt.%, Cu 31 wt.%, Zr 17 wt.%, and Zn 47 wt.%) and CZrZ was 1.7 times better than A5CZ (ca. Au 5 wt.%, Cu 31 wt.%, and Zn 64 wt.%) and CZ. The methanol conversion of CZrZ and A5CZrZ catalysts was kept higher than 70% for 12 h in an accelerated aging process. Meanwhile, the Au prompted more methoxy species oxidizing to formate on Cu+-rich A5CZrZ surface at lower temperature, and also improved CO transfer from formate reacting with moveable oxygen to form CO2. The SCO can lower to ca. 6% at 200 °C after adding 3–5% of gold promoter. These features all prove that the CZ catalyst with ZrO2 and Au promoters could enhance catalytic activity, lower the SCO and ignition temperature, and maintain good durability in the POM reaction.

1. Introduction
Hydrogen as a clean energy source has been an important focus of research for more than a decade. Partial oxidation of methanol (POM), an exothermic reaction, is one of the reactions employed in producing hydrogen at low temperatures [1,2,3]. The low reaction temperature of POM can simplify the reactor design. In addition, the exothermic property and higher reaction rate at lower temperatures can shorten the start-up time and reach working temperatures more quickly. In addition, no heat supply is required if the reaction reaches steady-state [2,4,5]. Thus, the POM reaction is considered to be more energy-efficient than the steam reforming of methanol (SRM) reaction; thus, it can produce hydrogen affordably on an industrial scale. The POM reaction is shown as Equation (1):
CH3OH + 1/2O2 → 2H2 + CO2, ΔH0 = −192 kJ·mol−1
Copper-zinc oxide (CuZnO) catalysts have been widely used for POM due to high methanol conversion, high H2 selectivity, and low cost. However, significantly higher CO selectivity (SCO), higher ignition temperature (Ti) (~185 °C) [3,6], and poor durability typically restrict the application of CuZnO-based catalysts [7,8]. Generally, the sintering of copper is the main factor for the deactivation of the catalyst. Thus, more-suitable promoters have been tested to improve catalyst activity and durability of CuZnO-based catalyst in POM reaction. Li and Lin [9] used Al2O3, MgO, and ZrO2 to promote the CuZnO-based catalysts in POM and found that Cu/Zn/ZrO2 performed with the slowest deactivation rate, as well as with better methanol conversion in POM. Sanches [10] indicated that the preparation methods and promoter influenced the structural and textural features of the catalyst. Compared with CuZnO catalysts prepared by coprecipitation (CP) and homogeneous precipitation (HP) methods, the CuZnO-based catalyst with Zr promoter prepared using the CP method had the highest Cu surface and lattice constant, and better methanol conversion in SRM. CuZnO-based catalysts contain ZrO2, which apparently has improved the Brunauer–Emmett–Teller (BET) surface area up to 2–3 times [10,11,12,13] and enhanced dispersion and reducibility in the methanol steam reforming (MSR) reaction [14]. In addition, the Cu-based catalysts with ZrO2 extended the Cu2O phase after MSR reaction. The degree of Cu2O aggregation is assumed to be smaller than Cu metal, which is expected to have better durability [15]. Amorphous ZrO2 particles, which work to stabilize the catalyst structurally and hamper the aggregation of Cu and ZnO particles, delay the deactivation of CuZnO [13,16,17].
In 1987, Au nanoparticles (<5 nm) were found to be very effective catalysts [18]. Afterwards, several applications, such as CO oxidation, utilized nano-gold particles as the catalysts [19,20,21,22,23,24,25,26,27]. As reported, Au particles could lower reaction temperature [22,24,25]. Konova et al. found that the reaction temperature in the CO oxidation over Au/TiO2 lowers to −60 °C, and conversion was higher than 95% at 0 °C [26]. The conversion of CO (above 98%) over Au/ZnO was observed at 50 °C [27]. In a previous study, methanol decomposed quickly on gold-promoted catalysts [23]. The interaction between the Au particles and the support might significantly affect catalytic performance [28]. Based on these features, gold was selected in this study to promote the CZrZ catalyst for lowering ignition temperature, improving catalytic activity, and decreasing CO selectivity in POM reaction. Moreover, the catalytic reaction mechanism was evaluated by in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), and X-ray diffraction (XRD) spectroscopy were applied to characterize the electronic states of copper and crystallized sizes in the POM reaction.
2. Results and Discussion
2.1. Durability of Catalysts
Generally, thermal sintering has been regarded as the main factor leading to CZ catalyst deactivation in methanol-reforming reactions [29]. To improve the poor durability of CZ catalyst, the textural promoter ZrO2 was used to prevent the sintering of Cu crystallites under POM reaction. Figure 1 shows that methanol conversion (CMeOH) and H2 selectivity (SH2) of CZrZ performed better than that of CZ during the POM reaction. The time-on-stream of POM reaction at 250 °C for 12 h in an accelerated aging condition over CZ, and CZrZ in Figure 2, also show that CZrZ catalyst could maintain higher methanol conversion compared to CZ catalysts. However, a higher ignition temperature (Ti at 175 °C) and higher SCO (ca. 13% at 200 °C) should be overcome. In this case, gold was used as a promoter to improve the performance of catalysts. The CMeOH and SH2 were over 80% and 90%, respectively, at 125 °C on A5CZ and A5CZrZ catalysts. Compared with CZ, the Ti of A1CZrZ, A3CZrZ, A5CZrZ, and A5CZ was reduced from 180 °C to 170 °C, 145 °C, 120 °C, and 125 °C, respectively. In addition, the SCO could be lowered to ca. 6% at 200 °C after adding 3–5% of gold promoter. However, compared with CZ only, the addition of gold as a promoter could not improve its durability. The A5CZ catalyst decayed very quickly. In contrast, CZrZ with the addition of gold not only kept excellent durability but also promoted higher methanol conversion, lower CO selectivity, and lower ignition temperature. The methanol conversion of CZrZ and A5CZrZ catalysts stayed higher than 70% after 12 h accelerated aging, whilst the CZ and A5CZ were down to approximately 60% of methanol conversion. In order to quantitate the deactivation rate, the decay curve based on the hyperbolic form with second order (Equation (S1)) [30,31] was applied and is shown in Figure S1 and Table 1. The rate constant k is a measurement of how rapidly a catalyst is deactivating. The order of deactivation rate constant of catalyst was A5CZ = CZ > CZrZ = A5CZrZ. The deactivation rate constant of A5CZrZ and CZrZ catalyst was 1.7 times lower than CZ catalyst. Co-existence of Au and ZrO does not induce negative effect in the POM reaction. However, optimized conditions cannot be reached with the existence of only Au or ZrO.
Although the above behaviors were observed, the interactions between Au particles and the CZrZ might significantly affect catalytic performance and should be explored. For these purposes, the characteristics of AxCZrZ (x = 1, 3, 5) catalysts are discussed as follows.
2.2. Physiochemical Properties of the CZ Catalysts with ZrO2 and Gold Promoter
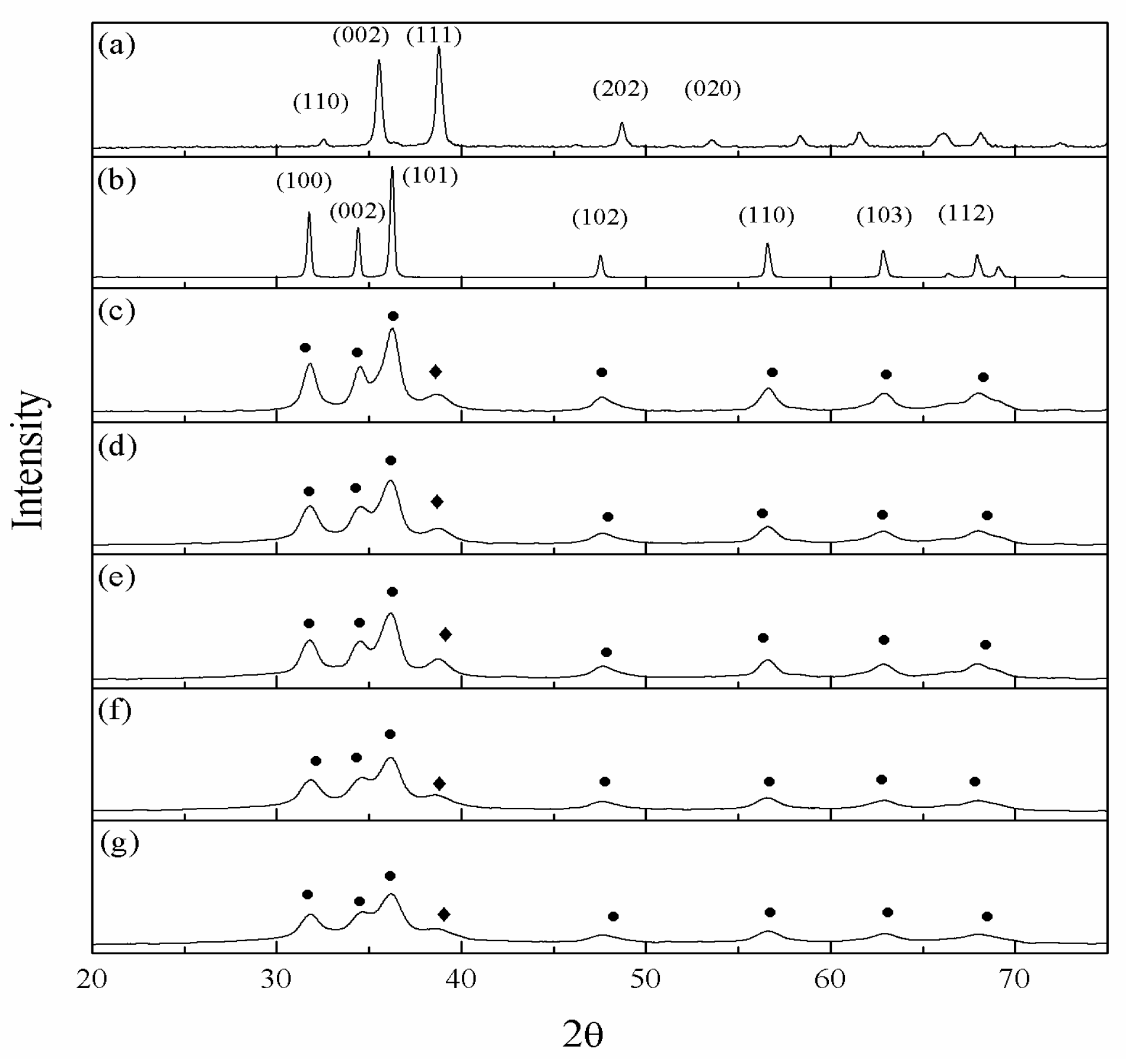
The physiochemical properties of catalysts are listed in Table 2. The metallic composition is similar to the stoichiometric calculation. The XRD patterns of fresh and reduced catalysts are shown in Figure 3c–g and Figure S2, respectively. ZrO2 diffraction peaks were not observed in all catalysts, which indicated that the ZrO2 might be the amorphous or poorer crystallization forms of ZrO2. Figure S3 shows that the tetragonal structure (t-ZrO2) and tetragonal with monoclinic structure (m-ZrO2) were formed when the calcination temperature was above 550 °C and 750 °C, respectively. The Raman spectrum of CZrZ in Figure S4b shows that no band corresponding to t-ZrO2 and m-ZrO2 was detected, which also indicates that the ZrO2 was amorphous [32]. Moreover, the diffraction peak of Cu2ZnZr (110) (JCPDS no. 65-6823) at 2θ of 42.14° was not observed. Thus, the ZrO2 was amorphous, and no mixed oxides (Cu, Zn, and Zr) were formed over CZrZ after 400 °C calcination. The variation of surface area might vary with ZrO2 or/and gold promoters. Generally, the amorphous phase of zirconium exhibits a high surface area [12]. The surface area of CZ with Zr or Zr/Au increased from 34.59 to 77.52 m2/g, and from 64.06 to 77.03 m2/g, respectively. It seems that gold had minor effect on surface area. Table 2 also shows that the Cu dispersion was improved with incremental Au loading, e.g., the A5CZrZ was 7% higher than the CZ and CZrZ catalysts. The sizes of CuO, Cu, and ZnO were estimated by CuO (111) XRD peak at 38.7°, Cu (200) XRD peak at 50.3° and ZnO (101) XRD peak at 36.2°, respectively, through the Debye-Scherer formula. The results show that CuO and Cu sizes decreased with the addition of ZrO2 (lowering from 5.65 nm to 4.49 nm and 6.25 nm to 5.4 nm, respectively) and also led to the formation of the smaller ZnO crystallite (from 9.03 nm to 6.42 nm). The weaker and broader copper diffraction peak of CuO (111) was observed from the CZrZ catalyst with the addition of gold. The CuO size decreased from 4.49 nm to 3.48 nm, and the size of Cu of reduced catalysts was down from 5.4 nm to 3.97 nm. The composition of catalysts surfaces calculated by XPS and Energy Dispersive Spectrometer (EDS) (Table 2 and Table S1) indicated a small amount of ZrO2 on the surface. In addition, the Au proportion was higher than the stoichiometric calculation, which indicated that most of the gold was on the catalyst surface. The EDS spectra are listed in Figure S5.
Transmission electron microscope (TEM) images of AxCZrZ catalysts shown in Figure 4a–c also show that gold particles were well-dispersed on catalysts with sizes around 2–5 nm. The mean sizes of Au of A1CZrZ, A3CZrZ, A5CZrZ are 2.66 nm, 2.67 nm, and 3.09 nm, respectively. Generally, 3–10 nm-sized catalysts can perform higher activity due to more interface and higher surface area [33]. In addition, the solid black points are Au nanoparticles. The presence of Au (111) with 0.23 nm lattice fringes reportedly could exhibit high activity for oxidation reactions [34,35], and 0.25 nm corresponds to the wurtzite ZnO planes (101) of lattice fringes (shown in the high resolution TEM (HR-TEM) images in Figure 5a–c). Figure 5d shows the selected area electron diffraction (SAED) patterns of A5CZrZ. Au (111), CuO (111), ZnO (100), ZnO (101), and ZnO (102) were detected, but no tetragonal ZrO2 (101) was observed, which indicated the existence of amorphous phase ZrO2. This result corresponds to XRD profiles in Figure 3.
In summary, the size, Cu dispersion, and surface area of the fresh CZ could be improved by the addition of Zr or Au/Zr promoters. Copper with small particle size and high dispersion could improve methanol conversion [36,37]. However, it is very important to evaluate whether those properties can be maintained after POM reaction. The XRD profiles of catalysts after reduction pretreatment and after the POM reaction were utilized to estimate the variation of Cu size. The results, shown in Table 1, were calculated based on a Cu (200) peak at 50.3° through Debye-Scherer formula. As a matter of fact, the CZ and A5CZ catalysts show particle dimension increasing to more than 200%, while on CZrZ and A5CZrZ samples, the corresponding value is about 120%. After POM reaction, the XRD results indicate that the catalyst with zirconium would maintain high durability due to amorphous ZrO functioning as textural promoter, which limited the growth of copper, stabilized the structure, and hampered the aggregation of Cu particles [38]. Coke formation during the reaction might also be a factor contributing to the loss of catalytic activity. Low carbon balance was due to the possibility that carbon was deposited on the catalyst [30]. The carbon balance of CZ, CZrZ, and A5CZrZ in an accelerated aging condition for 12 h (Figure S6) was close to 100%, so the coke was not the main factor on the deactivation of catalysts.
2.3. Reducibility of Catalyst
The reducibility of catalyst can be changed by adding different promoters, which might induce various catalytic performances. Temperature-programmed reduction (TPR) was used to evaluate whether the reducibility of CZ could be improved by adding ZrO2 and Au promoters. For all the catalysts, the H2/Cu ratio is ca. 1.0 (1.02–1.00), which indicates full reduction of CuO to Cu0 [39]. Figure 6a is the TPR profile of CuO, which shows a board reduction profile at 200–250 °C, with a main reduction peak α at around 233 °C. Sloczynski et al. [40] and Velu et al. [41] indicated α peak might be formed from the reduction of Cu+ or crystallized copper oxide to metallic Cu. The reduction profile of CZ, shown in Figure 6b, shifts to a lower temperature with α peak at 205 °C and a front shoulder β peak at 188 °C. Fierro et al. [42] hypothesized that the β peak resulted when the well-dispersed copper came into contact with ZnO particle, which lowered the reduction temperature. The improved reducibility of CuO was contributed to ZnO, which enhanced the spillover of hydrogen atoms [43,44]. The addition of zirconium had significant effect on the peaks of α (at 190 °C) and β (at 174 °C). Wang et al. [45] indicated that smaller copper particles can exist on catalysts with Zr prompter, and Tada et al. [46] illustrated that highly dispersed Cu in close contact with amorphous ZrO2 improved catalytic activity. Figure 6d–f shows the reduction profiles of catalysts containing gold that shifted to lower temperature. As the gold content increases from 1 to 5%, the β peak shifts from 172 °C to 158 °C and the concomitant α peak shifts from 186 °C to 170 °C. This obviously shows that the more gold content it had, the greater shifting effect could be observed. Some research has indicated that gold particles could adsorb and then dissociate hydrogen [47]; hence, increasing gold content might enhance H2 adsorption and dissociation at lower temperature. Dissociated hydrogen spilled over to neighboring copper oxide and led to a lower reduction temperature. Moreover, the fraction ratios of β peak to α peak (Table 3) on AxCZrZ are lower than CZrZ and CZ, which may be due to a smaller amount of ZnO on the surface. In summary, the TPR data show that the catalysts with Zr/Au promoter really present a better redox ability, which might provide a lower ignition temperature and better methanol conversion.
2.4. Oxygen Mobility of Catalyst
From the physiochemical properties and TPR data, we know that the particle size, Cu dispersion, surface area, and reducibility of CZ have been improved by ZrO2 and Au promoters. However, it is interesting to see what properties induce lower SCO. For the SCO, the CZrZ catalysts showed the highest CO selectivity compared with other catalysts. The AxCZrZ catalysts were able to lower the SCO to less than 10% at 200 °C, the effect of which was proportional to gold content. Gold prompter, as has been reported, can improve oxygen mobility [23]. We assume that oxygen mobility might be the reason for the SCO decrement.
We used X-ray photoelectron spectroscopy (XPS), temperature program desorption of oxygen (O2-TPD), and X-ray absorption near-edge structure (XANES) to confirm oxygen mobility. Figure 7 shows the XPS spectra of O1s for fresh CZ, CZrZ, and A5CZrZ catalysts fitted to two peaks (OI and OII). The higher energy peak (OII) (with binding energy within 531–532 eV) is commonly present as non-lattice oxygen [48,49]. The order of area ratio of OII/OI is A5CZrZ (4.85) > CZ (2.22) > CZrZ (1.42). The amorphous structure of Zr in CZ catalyst may be the main factor for the decrement of OII. Conversely, CZrZ with Au prompter could overcome this issue, and the A5CZrZ obviously increased the proportion of OII in O1s peak. Furthermore, the O2-TPD was utilized to test oxygen chemisorption ability. Figure 8 shows the O2-TPD profiles of CZ, CZrZ, and A5CZrZ catalysts. The CZ catalyst had desorption peaks at around 225–300 °C and 400–450 °C. The CZrZ showed a minor peak at 250–300 °C and large peak at 350–450 °C. The A5CZrZ catalyst started to desorb oxygen at 125 °C and apparently had an oxygen consumption peak at 200–450 °C. Clearly, the oxygen mobility is related to the ratio of OII/OI in XPS. When a higher OII/OI is present, a lower oxygen desorption temperature is found. Non-lattice oxygen might tend to form superoxide (O2−), which would enhance CO oxidation reaction [50,51]. In addition, the order of oxygen adsorption is A5CZrZ > CZrZ > CZ. However, there are more adsorbed oxygen molecules on the CZrZ than on the CZ, and a higher desorption temperature might limit the mobility of oxygen and result in high Sco on CZrZ.
Furthermore, to better realize the properties of gold for inducing a change in reducibility and in oxygen mobility, the electron states of gold and copper species on fresh A5CZrZ and after POM reaction were measured by XPS and XAS. The XPS Au4f profiles of AxCZrZ catalysts in fresh and after POM reaction are shown in Figures S7 and S8, respectively. All catalysts exhibit peaks assigned as Au0 and Au3+, and the ratio of Au0/Au3+ is listed in Table 2. The Au0/Au3+ ratio increases as the Au loading amount increases (0.65–1.37). The metallic gold worked as the active site when Au deposited on irreducible oxide, such as Silica or ZnO (in this study), which indicated a weaker metal-support interaction between Au and support [52]. Thus, a higher portion of Au0 ions might induce higher catalytic activity [53,54,55]. Moreover, higher electrical capacity and lower Fermi level of gold can induce more electrons to go through the copper and transferring to Au [56], which may result in a smaller amount of Cu0 on A5CZrZ. The XPS results (Figure S8, Table 2) also show that the Au0/Au3+ ratio of A5CZrZ after POM reaction (1.43) is higher than fresh (1.37). For copper species, XANES in Figure 9a,d shows that the fresh CZ and A5CZrZ catalysts consisted of 100% Cu2+, a state that is non-active toward reforming [1]. All catalysts were pre-reduced to Cu0 before POM reaction. Due to higher oxygen mobility, gold deposited on CZrZ catalyst can adsorb/desorb more oxygen atoms and spill them over to copper and keep more partially reduced copper [23], which results in the more active species Cu+ on the catalytic surface after POM reaction. Figure 9b,e indicates that the A5CZrZ had a higher percentage of Cu2O (47.3%) compared with CZ (39.4%) and less Cu0 (A5CZrZ 52.73%, CZ 60.6%). Oguchi et al. [57] indicate that Cu2O was the major active site for methanol reforming as a result of higher oxidation and reduction ability. However, after an extended duration test (over four months) (Figure 9c,f), a large volume of oxygen diffused into the lattice of copper species, and the content of Cu2+ increased to 53.6%, higher than the sum of Cu+ and Cu0 on CZ. While less Cu2+ (37.8%) formed, Cu0 and Cu+ were the main species on A5CZrZ. This indicates that the Au promoter enhanced the mobility of oxygen and reduced the accumulation of oxygen on the copper-active site. As the result, XPS, O2-TPD, and XANES all indicated that the Au promoter could enhance the oxygen mobility.
2.5. In-Situ DRIFTS
To better understand how the Au promoter affects the intermediates during the POM reaction, in-situ DRIFTS was used to examine the POM mechanism. Figure 10a–f presents the FT-IR spectra of A5CZrZ catalyst under the reaction temperatures of 100 °C, 125 °C, 175 °C, 200 °C, 250 °C, and 300 °C. At 100 °C, the C–H stretching peaks at 2930 and 2819 cm−1. Also C–O stretching peaks at 1446, 1076 cm−1 of methoxy group [58] was found, and 1153 cm−1 was assigned as the on-top methoxy C–O stretching on Zr4+ [59]. The electron affinity of Au enhanced the adsorption of CH3OH, and methoxy species formed on the surface of the catalyst. Hydrogen from methanol reacted with oxygen, thus forming the hydroxy group, which showed a broad band at 3100–3500 cm−1. The peaks at 1592 and 1367 cm−1 were assigned as mono-dentate formate (OCO asymmetric and symmetric stretching), which was formed from the methoxy group interacting with the oxygen atom adsorbed onto the catalyst surface [58]. After temperatures higher than ignition temperature (>120 °C), the DRIFTS spectra were obviously different in the region of C–H stretching (3000–2700 cm−1). The bi-dentate formate, which peaks at 2964, 2865, and 2739 cm−1 [58,60,61], formed at 125 °C. The formate is an intermediate in the formation of H2, CO and CO2. The bidentate formate was reported to decompose to COO* (*: metal site), H*, or CO* and OH*, and produced CO2 and CO [61]. At 175 °C, the bi-dentate formate decomposed very quickly, and minor peaks were observed. Furthermore, the methoxy and mono-dentate formate peaks decreased and almost disappeared at temperatures higher than 200 °C. At 300 °C, only carbonate species (CO3*), CO2(g), and H2O(g), were observed.
Figure 11 (I) a–c shows the DRIFTS spectra from 3400–2600 cm−1 of CZ, CZrZ, and A5CZrZ catalysts during POM reaction at 125 °C. Methoxy group was observed on the surface of CZ and CZrZ, but only the formate and –OH group could be detected on A5CZrZ. Literature [58,62] indicates that methanol dehydrogenated to CH3O, then afterwards dehydrogenated to CH2O (formaldehyde) in the POM reaction mechanism. The CH2O preferred to react with oxygen to form CH2OO and dehydrogenate to formate (HCOO). However, the rapid oxidation of formaldehyde led to difficulty in detecting the formaldehyde peaks in the DRIFTS spectra [61]. Kulkarni et al. [63] indicated that CH3OH favored dehydrogenation to methoxy on the Cu0 surface, and the Cu+-rich surface tended toward oxidization of the methoxy species to formate. The A5CZrZ with higher Cu+/Cu0 ratio and better oxygen mobility could speed up the formation of formate at lower temperatures. The formate transferred to H2, H2O, CO2, and CO very quickly, while the adsorbed formate peaks almost disappeared on FT-IR spectra of A5CZrZ at temperatures higher than 200 °C. Obviously, the dehydrogenation and oxidation of methoxy-formaldehyde-formate sequentially reacted rapidly on A5CZrZ surface with the presence of active oxygen.
To further confirm the effect of Au promoter, the in-situ DRIFTS spectra of CO chemisorption are shown in Figure 11 (II). CO gas displayed two peaks located at 2173 and 2115 cm−1, respectively. When CO gas adsorbed onto metal surface (COads), rotational freedom was lost, and COads shows only one peak. Meanwhile, only A5CZrZ adsorbed CO at 25 °C, and COads peaks were observed at around 2110 cm−1. The CO preferentially bonded onto Au at room temperature [64,65]. Lee et al. [66] indicated that moveable oxygen tended to catalyze COads to CO2. Integrating the XAS and in-situ DRIFTS results, the suggested mechanism of POM reaction on A5CZrZ catalyst is depicted in Scheme 1. The dehydrogenation of CH3OH formed a methoxy group, which absorbed onto Cu0 surface. The moveable oxygen enhanced the formation of methoxy-formaldehyde-formate sequential reaction on Cu+-rich A5CZrZ surface. Monodentate formed at a temperature lower than Ti (120 °C). After ignition, bidentate formate was observed, which may have decomposed quickly. The H2O and H2 were generated from H* and OH*, and the CO* reacted with moveable oxygen to form CO2. These features all prove that the A5CZrZ with good oxygen mobility could start the reaction at low temperature and lower CO selection.
3. Materials and Methods
3.1. Preparation of Catalyst
The catalysts of CZ (ca. Cu 30 wt.% and Zn 70 wt.%) and CZrZ (ca. Cu 30 wt.%, Zr 15 wt.% and Zn 55 wt.%) were prepared using the coprecipitation (CP) technique. The precursors of Cu(NO3)2·3H2O, ZrO(NO3)2·xH2O, and Zn(NO3)2·6H2O were dissolved in pre-heated deionized water with vigorous stirring at 70 °C. The solution pH was kept to 7 by 1N Na2CO3 for one hour, followed by filtration of the CZ and CZrZ precipitates. The precipitates were washed with 2 L of deionized water and dried at 105 °C overnight. Afterwards, the catalysts were calcined in 100 mL/minute air flow at 400 °C for 4 h.
The catalysts of AxCZrZ (x = 1, 3, 5 wt.% of Au, Cu 30 wt.%, Zr 15 wt.%, and Zn 55−x wt.%) and A5CZ (ca. Au 5 wt.%, Cu 30 wt.%, and Zn 65 wt.%) were prepared using the deposition precipitation (DP) technique. The dried CZrZ and CZ catalysts were suspended in 500 mL deionized water with vigorous stirring at 70 °C. The 0.01 M HAuCl4·3H2O solution was added to the CZrZ and CZ solution, and kept at pH 7–7.5 for one hour by using 10% HCl solution. After filtrating, the AxCZrZ and A5CZ precipitates were washed with 2 L deionized water, then dried at 105 °C overnight. Afterwards, the catalysts were calcined in 100 mL/minute air flow at 400 °C for 4 h.
3.2. Characterization of Catalysts
The ICP-MS (Perkin Elmer-SCIEX ELAN 5000, Waltham, MA, USA) was manipulated to evaluate the metallic compositions of the catalysts.
XRD analysis was measured by Rigaku RINT1100 diffractometers (Tokyo, Japan) with Copper-Kα1 (λ = 1.54056 Å). The radiation scanning 2θ angle rates ranged from 25° to 70° and 3°/minute. The fresh catalyst reduced by 10% H2/N2 at 250 °C for 30 min before reduction state XRD measurement.
The surface area of catalysts was obtained by Micromeritics ASAP-2020 (Norcross, GA, USA). The catalysts were pre-treated under vacuum at 300 °C. The N2 adsorption/desorption isotherms were measured at −196 °C. Surface area calculation was performed according to the Brunauer–Emmett–Teller (BET) method [67].
Raman spectra were collected by Thermo Scientific DXR Microscopy Raman (Madison, WI, USA). The excitation source was 532 nm Laser. The spectra were collected 30 times at laser power 10 mW with 5 s exposure.
HR-TEM images were taken on a JEOL-2100 (Tokyo, Japan) with a LaB6 electron gun source and operated at 200 kV. The sample preparation procedures included putting 1 mg of catalyst into 2 mL ethanol, which was ultra-sonicated for 120 min to produce a well-suspended sample solution. Then, 1 mL suspended solution was deposited onto carbon-coated nickel grids. The grids with catalyst were placed into a vacuum oven at 60 °C overnight to completely evaporate the ethanol.
The energy-dispersive X-ray spectroscopy (EDS) was measured by Thermo Phenom ProX scanning electron microscope (SEM) equipped with EDS (EDAX, Mahwah, NJ, USA). The electron gun was a CeB6 and was operated at 15 kV. The energy resolution of EDS by silicon drift detector (SDD) at Mn Kα was less than 137 eV. The samples were grinded and mounted with conductive carbon tape for measurement.
In the temperature programmed reduction (TPR) experiment, a 4 mm inner diameter U-shaped tube reactor was filled with 55 mg of catalyst. The catalyst was reduced by 10% H2/N2 at a flow rate of 30 mL/min with a heating rate at 7 °C/min from ambient temperature to 300 °C, and the consumption of hydrogen was recorded by thermal couple detector (TCD). The TPR profile was deconvoluted to multiple peaks by using Origin software.
Copper dispersions were analyzed by nitrous oxide chemisorption [68]. After the TPR process, the reactor was cooled to room temperature. N2O gas in 30 mL/min of flow rate was introduced into the reactor for 30 min and then purged with N2 for 15 min. The second TPR (S-TPR) test was processed and detected by TCD. The Cu dispersion was calculated by multiplying two times of the area of S-TPR and dividing by the TPR area.
Temperature program desorption of oxygen (O2-TPD) was utilized to identify the oxygen adsorption-desorption ability of the catalysts. The 55 mg catalysts were pre-reduced at 250 °C by 10% H2/90% N2 mixed gas in a U-shape tube reactor for one hour and then were cooled down to room temperature in flowing He (99.9995%). After that, the catalyst was treated in O2 (99.999%) at 200 °C for one hour and then was cooled down to room temperature under flowing O2. The O2 gas was removed by flowing He for one hour. In order to ensure the saturation of O2 chemisorption on catalysts, the O2 treatment procedure was applied one more time [66]. Afterwards, the reactor temperature was raised from room temperature to 450 °C at a heating rate of 7 °C/min. Oxygen desorption during the heating range was determined by TCD.
X-ray photoelectron spectroscopy (XPS) spectra were recorded on a photoelectron instrument via Kratos Axis Ultra DLD (Kratos Analytical Ltd, Manchester, UK) under ultra-high vacuum. The binding energies were referred to C1s line of adventitious carbon 285 eV.
XAS (X-ray absorption spectroscopy) spectra, studied for the electronic states of copper catalysts, were recorded on the Wiggler beam-line at Synchrotron Radiation Research Center (SRRC), Taiwan. Absorption of Cu K-edge (8.979 KeV) measured by transmission mode was used for XANES (X-ray absorption near-edge structure) analysis, and the data was calculated by using WinXAS software (V.3.0).
The in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) infrared spectra were collected by Thermo Scientific Nicolet 6700 spectrometer with a MCT-A detector (Madison, WI, USA). The 100 mg catalyst was packed into the in-situ high-temperature reaction chamber, which was installed in the HARRICK diffuse reflectance accessory. The total flow gas through the in-situ cell was set at 30 mL/minute and 100 mL/minute for CO adsorption and POM reaction, respectively. The spectra scan number was 64 with a resolution of 4 cm−1.
3.3. Catalytic Activity and Durability
The freshly calcined catalysts were ground into fine powders and sieved to 60–80 mesh for catalytic activity testing, then 100 mg of catalyst was packed into a 4-mm inner diameter quartz fixed-bed reactor and reduced by 10% H2/N2 at 250 °C for 30 min before measurement. The conditions of POM experiment consisted of O2/CH3OH ratio was 0.5 with 60 K h−1 Gas Hourly Space Velocity (GHSV) and 9.48 h−1 Weight Hourly Space Velocity (WHSV), respectively. Meanwhile, durability was tested at 250 °C for 12 h under an accelerated aging condition (60 Kh−1 of GHSV and 47.4 h−1 of WHSV). Catalytic activity analyzed by on-line GC with two TCD detectors equipped with Porapak Q (H2, CO2, and CO) and Molecular Sieve 5A (CO2, H2O, and CH3OH) columns. The calculation equations for CH3OH conversion (CMeOH), H2 selectivity (SH2), and CO selectivity (SCO) were as follows:
CMeOH = (nMeOH,in − nMeOH,out)/nMeOH,in × 100%
SH2 = nH2/(nH2 + nH2O) × 100%
SCO = nCO/(nCO2 + nCO) × 100%
4. Conclusions
The poor durability of CZ catalyst was improved by the addition of ZrO2, which prevented catalyst sintering during POM reaction. Meanwhile, the Au promoter was utilized to lower the Ti and SCO of CZrZ in POM. The deactivation rate constant of A5CZrZ was similar to CZrZ and 1.7 times better than A5CZ and CZ, which indicated that ZrO2 was the main factor in maintaining durability. The A5CZrZ catalyst with better reducibility, oxygen mobility, and higher Cu+/Cu0 ratio could lower the Ti to 120 °C and showed more than 90% of CMeOH and SH2 at 125 °C. It was observed with in-situ DRIFTS of A5CZrZ that the methoxy transferred to formate at around 125 °C. The methoxy-formaldehyde-formate sequence processed rapidly with more Cu+ and the presence of active oxygen. The Au promoter not only enhanced the affinity to adsorb CO but also increased moveable oxygen to react with CO on the catalyst surface. The SCO could be lowered to 6% at 200 °C. Based on these features, the AxCZrZ catalyst produced a good catalytic performance, reduced Sco and ignition temperature, and still maintained good durability in POM reaction.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/8/9/345/s1, Figure S1: Deactivation rate in POM reaction: (■) CZ, (●) CZrZ, (★) Au5CZrZ, and (◊) Au5CZ at 250 °C for 12 h in an accelerated aging condition (60 Kh−1 of GHSV and 47.4 h−1 of WHSV). Figure S2: XRD profiles of different catalysts (reduction state): (a) CZ, (b) CZrZ, (c) A1CZrZ, (d) A3CZrZ, and (e) A5CZrZ. (◊) Cu, (●) ZnO. Figure S3: XRD profiles of CZrZ catalysts after calcination for 4 h at (a) 400 °C, (b) 550 °C, and (c) 750 °C. (◆) CuO, (●) ZnO, (o) ZrO2-tetragonal, (□) ZrO2- monoclinic. Figure S4: Raman spectra of (a) CZ, and (b) CZrZ. Table S1: Composition of catalyst surfaces measured by EDS (%). Figure S5: The EDS element mapping profile: (a) CZ, (b) CZrZ, (c) A1CZrZ, (d) A3CZrZ, and (e) A5CZrZ. Figure S6: Mole fractions of carbon components at 250 °C for 12 h in an accelerated aging condition: (■) CO, (●) CO2, (▲) MeOHout, (∇) Carbon balance. (a) CZ, (b) CZrZ, and (c) A5CZrZ. Figure S7: XPS of Au4f (BE in 82–97 ev) (Fresh): (a) A1CZrZ, (b) A3CZrZ, and (c) A5CZrZ (blue lines are assigned to BE in Au0; green lines are assigned to BE in Au3+) and Figure S8: XPS of Au4f (BE in 82–97 ev): (a) A1CZrZ, (b) A3CZrZ, and (c) A5CZrZ, after POM at 250 °C for 24 h. (Blue lines are assigned to BE in Au0; green lines are assigned to BE in Au3+).
Author Contributions
Y.-J.H. coordinated the whole study and revised this manuscript. H.-Y.H. and H.-I.C. prepared and processed the experiment. H.-Y.H. analyzed XRD, IR, XPS, and XANES data, and H.-I.C. measured the physicochemical properties of the catalysts and calculated the deactivation rates constant. H.-Y.H. wrote this manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors are grateful for the financial support of this work from the Ministry of Science and Technology of Taiwan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alejo, L.; Lago, R.; Peña, M.A.; Fierro, J.L.G. Partial oxidation of methanol to produce hydrogen over CnZn-based catalysts. Appl. Catal. A: Gen. 1997, 162, 281–297. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Osaki, T. Selective production of hydrogen by partial oxidation of methanol over catalysts derived from CuZnAl-layered double hydroxides. Catal. Lett. 1999, 62, 159–167. [Google Scholar] [CrossRef]
- Agrell, J.; Hasselbo, K.; Jansson, K.; Järås, S.G.; Boutonnet, M. Production of hydrogen by partial oxidation of methanol over Cu/ZnO catalysts prepared by microemulsion technique. Appl. Catal. A: Gen. 2001, 211, 239–250. [Google Scholar] [CrossRef]
- Mo, L.; Zheng, X.; Yeh, C.-T. Selective production of hydrogen from partial oxidation of methanol over silver catalysts at low temperatures. Chem. Commun. 2004, 1426–1427. [Google Scholar] [CrossRef] [PubMed]
- Wan, A.; Yeh, C.-T. Ignition of methanol partial oxidation over supported platinum catalyst. Catal. Today 2007, 129, 293–296. [Google Scholar] [CrossRef]
- Huang, T.-J.; Wang, S.-W. Hydrogen production via partial oxidation of methanol over copper-zinc catalysts. Appl. Catal. 1986, 24, 287–297. [Google Scholar] [CrossRef]
- Shishido, T.; Yamamoto, Y.; Morioka, H.; Takaki, K.; Takehira, K. Active Cu/ZnO and Cu/ZnO/Al2O3 catalysts prepared by homogeneous precipitation method in steam reforming of methanol. Appl. Catal. A: Gen. 2004, 263, 249–253. [Google Scholar] [CrossRef]
- Shishido, T.; Yamamoto, Y.; Morioka, H.; Takehira, K. Production of hydrogen from methanol over Cu/ZnO and Cu/ZnO/Al2O3 catalysts prepared by homogeneous precipitation: Steam reforming and oxidative steam reforming. J. Mol. Catal. A: Chem. 2007, 268, 185–194. [Google Scholar] [CrossRef]
- Li, C.-L.; Lin, Y.-C. Catalytic partial oxidation of methanol over copper–zinc based catalysts: A comparative study of alumina, zirconia, and magnesia as promoters. Catal. Lett. 2010, 140, 69–76. [Google Scholar] [CrossRef]
- Sanches, S.G.; Flores, J.H.; de Avillez, R.R.; Pais da Silva, M.I. Influence of preparation methods and Zr and Y promoters on Cu/ZnO catalysts used for methanol steam reforming. Int. J. Hydrog. Energy 2012, 37, 6572–6579. [Google Scholar] [CrossRef]
- Zhang, X.R.; Shi, P.; Zhao, J.; Zhao, M.; Liu, C. Production of hydrogen for fuel cells by steam reforming of methanol on Cu/ZrO2/Al2O3 catalysts. Fuel Process. Technol. 2003, 83, 183–192. [Google Scholar] [CrossRef]
- Matter, P.H.; Braden, D.J.; Ozkan, U.S. Steam reforming of methanol to H2 over nonreduced Zr-containing CuO/ZnO catalysts. J. Catal. 2004, 223, 340–351. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ishibe, H. High temperature steam reforming of methanol over Cu/ZnO/ZrO2 catalysts. Appl. Catal. B: Environ. 2009, 91, 524–532. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, L.; Ni, C.; Sun, T.; Zhao, S.; Wang, S.; Wang, A.; Hu, Y. CeO2–ZrO2-promoted CuO/ZnoO catalyst for methanol steam reforming. Int. J. Hydrog. Energy 2013, 38, 4397–4406. [Google Scholar] [CrossRef]
- Oguchi, H.; Nishiguchi, T.; Matsumoto, T.; Kanai, H.; Utani, K.; Matsumura, Y.; Imamura, S. Steam reforming of methanol over Cu/CeO2/ZrO2 catalysts. Appl. Catal. A: Gen. 2005, 281, 69–73. [Google Scholar] [CrossRef]
- Park, J.E.; Yim, S.-D.; Kim, C.S.; Park, E.D. Steam reforming of methanol over Cu/ZnO/ZrO2/Al2O3 catalyst. Int. J. Hydrog. Energy 2014, 39, 11517–11527. [Google Scholar] [CrossRef]
- Chang, C.-C.; Chang, C.-T.; Chiang, S.-J.; Liaw, B.-J.; Chen, Y.-Z. Oxidative steam reforming of methanol over CuO/ZnO/CeO2/ZrO2/Al2O3 catalysts. Int. J. Hydrog. Energy 2010, 35, 7675–7683. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Grisel, R.J.H.; Nieuwenhuys, B.E. A comparative study of the oxidation of CO and CH4 over Au/Mox/Al2O3 catalysts. Catal. Today 2001, 64, 69–81. [Google Scholar] [CrossRef]
- Haruta, M. Catalysis of gold nanoparticles deposited on metal oxides. Cattech 2002, 6, 102–115. [Google Scholar] [CrossRef]
- Wolf, A.; Schüth, F. A systematic study of the synthesis conditions for the preparation of highly active gold catalysts. Appl. Catal. A: Gen. 2002, 226, 1–13. [Google Scholar] [CrossRef]
- Hvolbæk, B.; Janssens, T.V.W.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Nørskov, J.K. Catalytic activity of Au nanoparticles. Nano Today 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Ng, K.L.; Huang, H.-Y. The effect of gold on the copper-zinc oxides catalyst during the partial oxidation of methanol reaction. Int. J. Hydrog. Energy 2011, 36, 15203–15211. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Kim, D.H.; Kung, M.C.; Kozlova, A.; Yuan, S.D.; Kung, H.H. Synergism between Pt/Al2O3 and Au/TiO2 in the low temperature oxidation of propene. Catal. Lett. 2004, 98, 11–15. [Google Scholar] [CrossRef]
- Konova, P.; Naydenov, A.; Venkov, C.; Mehandjiev, D.; Andreeva, D.; Tabakova, T. Activity and deactivation of Au/TiO2 catalyst in CO oxidation. J. Mol. Catal. A: Chem. 2004, 213, 235–240. [Google Scholar] [CrossRef]
- Naknam, P.; Luengnaruemitchai, A.; Wongkasemjit, S. Au/ZnO and Au/ZnO−Fe2O3 prepared by deposition−precipitation and their activity in the preferential oxidation of CO. Energy Fuels 2009, 23, 5084–5091. [Google Scholar] [CrossRef]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; Imperial College Press: London, UK, 2006. [Google Scholar]
- Lee, H.H. Deactivation of catalysts by R. Hughes, academic press (London), June 1984; 265p. AIChE J. 1985, 31, 523. [Google Scholar] [CrossRef]
- Schrum, E.D.; Reitz, T.L.; Kung, H.H. Deactivation of CuO/ZnO and CuO/ZrO2 catalysts for oxidative methanol reforming. Stud. Surf. Sci. Catal. 2001, 139, 229–235. [Google Scholar]
- Fogler, H.S. Elements of Chemical Reaction Engineering; Prentice-Hall International, Inc.: Upper Saddle River, NJ, USA, 2006. [Google Scholar]
- Xie, S.; Iglesia, E.; Bell, A.T. Water-assisted tetragonal-to-monoclinic phase transformation of ZrO2 at low temperatures. Chem. Mater. 2000, 12, 2442–2447. [Google Scholar] [CrossRef]
- Petkov, V.; Ren, Y.; Shan, S.Y.; Luo, J.; Zhong, C.J. A distinct atomic structure-catalytic activity relationship in 3–10 nm supported Au particles. Nanoscale 2014, 6, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Huo, Z.; Tsung, C.-K.; Huang, W.; Zhang, X.; Yang, P. Sub-two nanometer single crystal Au nanowires. Nano Lett. 2008, 8, 2041–2044. [Google Scholar] [CrossRef] [PubMed]
- Min, B.K.; Alemozafar, A.R.; Biener, M.M.; Biener, J.; Friend, C.M. Reaction of Au (111) with sulfur and oxygen: Scanning tunneling microscopic study. Top. Catal. 2005, 36, 77–90. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Wang, J.-H.; Shen, C.-C.; Yeh, C.-T. Promotion of a copper–zinc catalyst with rare earth for the steam reforming of methanol at low temperatures. J. Catal. 2011, 279, 241–245. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ishibe, H. Effect of zirconium oxide added to Cu/ZnO catalyst for steam reforming of methanol to hydrogen. J. Mol. Catal. A: Chem. 2011, 345, 44–53. [Google Scholar] [CrossRef]
- Koeppel, R.A.; Baiker, A.; Wokaun, A. Copper/zirconia catalysts for the synthesis of methanol from carbon dioxide: Influence of preparation variables on structural and catalytic properties of catalysts. Appl. Catal. A: Gen. 1992, 84, 77–102. [Google Scholar] [CrossRef]
- Wang, Y.H.; Gao, W.G.; Wang, H.; Zheng, Y.E.; Na, W.; Li, K.Z. Structure–activity relationships of Cu–ZrO2 catalysts for CO2 hydrogenation to methanol: Interaction effects and reaction mechanism. RSC Adv. 2017, 7, 8709–8717. [Google Scholar] [CrossRef]
- Sloczynski, J.; Grabowski, R.; Kozlowska, A.; Olszewski, P.K.; Stoch, J. Reduction kinetics of CuO in CuO/ZnO/ZrO2 systems. Phys. Chem. Chem. Phys. 2003, 5, 4631–4640. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Okazaki, M.; Kapoor, M.P.; Osaki, T.; Ohashi, F. Oxidative steam reforming of methanol over CuZnAl(Zr)-oxide catalysts for the selective production of hydrogen for fuel cells: Catalyst characterization and performance evaluation. J. Catal. 2000, 194, 373–384. [Google Scholar] [CrossRef]
- Fierro, G.; Lo Jacono, M.; Inversi, M.; Porta, P.; Cioci, F.; Lavecchia, R. Study of the reducibility of copper in CuO-ZnO catalysts by temperature-programmed reduction. Appl. Catal. A: Gen. 1996, 137, 327–348. [Google Scholar] [CrossRef]
- Burch, R.; Golunski, S.E.; Spencer, M.S. The role of hydrogen in methanol synthesis over copper catalysts. Catal. Lett. 1990, 5, 55–60. [Google Scholar] [CrossRef]
- Burch, R.; Golunski, S.E.; Spencer, M.S. The role of copper and zinc oxide in methanol synthesis catalysts. J. Chem. Soc. Faraday Trans. 1990, 86, 2683–2691. [Google Scholar] [CrossRef]
- Wang, L.-C.; Liu, Q.; Chen, M.; Liu, Y.-M.; Cao, Y.; Fan, K.-N. Structural evolution and catalytic properties of nanostructured Cu/ZrO2 catalysts prepared by oxalate gel-coprecipitation technique. J. Phys. Chem. C 2007, 111, 16549–16557. [Google Scholar] [CrossRef]
- Tada, S.; Katagiri, A.; Kiyota, K.; Honma, T.; Kamei, H.; Nariyuki, A.; Uchida, S.; Satokawa, S. Cu species incorporated into amorphous ZrO2 with high activity and selectivity in CO2-to-methanol hydrogenation. J. Phys. Chem. C 2018, 122, 5430–5442. [Google Scholar] [CrossRef]
- Kartusch, C.; van Bokhoven, J.A. Hydrogenation over gold catalysts: The interaction of gold with hydrogen. Gold Bull. 2009, 42, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Law, J.B.K.; Thong, J.T.L. Improving the NH3 gas sensitivity of ZnO nanowire sensors by reducing the carrier concentration. Nanotechnology 2008, 19, 205502. [Google Scholar] [CrossRef] [PubMed]
- Sinhamahapatra, A.; Jeon, J.-P.; Kang, J.; Han, B.; Yu, J.-S. Oxygen-deficient zirconia (ZrO(2−x)): A new material for solar light absorption. Sci. Rep. 2016, 6, 27218. [Google Scholar] [CrossRef] [PubMed]
- Bozo, C.; Guilhaume, N.; Herrmann, J.-M. Role of the ceria–zirconia support in the reactivity of platinum and palladium catalysts for methane total oxidation under lean conditions. J. Catal. 2001, 203, 393–406. [Google Scholar] [CrossRef]
- Pu, Z.-Y.; Liu, X.-S.; Jia, A.-P.; Xie, Y.-L.; Lu, J.-Q.; Luo, M.-F. Enhanced activity for CO oxidation over Pr- and Cu-doped CeO2 catalysts: Effect of oxygen vacancies. J. Phys. Chem. C 2008, 112, 15045–15051. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, S.; Zhu, H.; Dai, S.; Overbury, S.H. Oxygen-assisted reduction of Au species on Au/SiO2 catalyst in room temperature CO oxidation. Chem. Commun. 2008, 0, 3308–3310. [Google Scholar] [CrossRef] [PubMed]
- Cutrufello, M.G.; Rombi, E.; Cannas, C.; Casu, M.; Virga, A.; Fiorilli, S.; Onida, B.; Ferino, I. Synthesis, characterization and catalytic activity of Au supported on functionalized SBA-15 for low temperature CO oxidation. J. Mater. Sci. 2009, 44, 6644. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, S.; Zhu, H.; Dai, S.; Overbury, S.H. DRIFTS-QMS study of room temperature CO oxidation on Au/SiO2 catalyst: Nature and role of different Au species. J. Phys. Chem. C 2009, 113, 3726–3734. [Google Scholar] [CrossRef]
- Simakov, A.; Tuzovskaya, I.; Pestryakov, A.; Bogdanchikova, N.; Gurin, V.; Avalos, M.; Farías, M.H. On the nature of active gold species in zeolites in CO oxidation. Appl. Catal. A: Gen. 2007, 331, 121–128. [Google Scholar] [CrossRef]
- Liao, Y.-C.; Huang, H.-Y.; Huang, Y.-J. Photo-triggered catalytic reforming of methanol over gold-promoted, copper-zinc catalyst at low ignition temperature. Appl. Catal. B: Environ. 2018, 220, 264–271. [Google Scholar] [CrossRef]
- Oguchi, H.; Kanai, H.; Utani, K.; Matsumura, Y.; Imamura, S. Cu2O as active species in the steam reforming of methanol by CuO/ZrO2 catalysts. Appl. Catal. A: Gen. 2005, 293, 64–70. [Google Scholar] [CrossRef]
- Millar, G.J.; Rochester, C.H.; Waugh, K.C. Evidence for the adsorption of molecules at special sites located at copper/zinc oxide interfaces. Part 2. –A fourier-transform infrared spectroscopy study of methanol adsorption on reduced and oxidised Cu/ZnO/SiO2 catalysts. J. Chem. Soc. Faraday Trans. 1992, 88, 2257–2261. [Google Scholar] [CrossRef]
- Finocchio, E.; Daturi, M.; Binet, C.; Lavalley, J.C.; Blanchard, G. Thermal evolution of the adsorbed methoxy species on CexZr1−xO2 solid solution samples: A FT-IR study. Catal. Today 1999, 52, 53–63. [Google Scholar] [CrossRef]
- Lin, S.D.; Cheng, H.; Hsiao, T.C. In situ DRIFTS study on the methanol oxidation by lattice oxygen over Cu/ZnO catalyst. J. Mol. Catal. A: Chem. 2011, 342–343, 35–40. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Shen, C.-C.; Huang, Y.-J. Enhancement of the partial oxidation of methanol reaction over CuZn catalyst by Mn promoter. Ind. Eng. Chem. Res. 2014, 53, 12622–12630. [Google Scholar] [CrossRef]
- Zuo, Z.-J.; Gao, X.-Y.; Han, P.-D.; Liu, S.-Z.; Huang, W. Density functional theory (DFT) and kinetic monte carlo (KMC) study of the reaction mechanism of hydrogen production from methanol on ZnCu (111). J. Phys. Chem. C 2016, 120, 27500–27508. [Google Scholar] [CrossRef]
- Kulkarni, G.U.; Rao, C.N.R. EXAFS and XPS investigations of Cu/ZnO catalysts and their interaction with CO and methanol. Top. Catal. 2003, 22, 183–189. [Google Scholar] [CrossRef]
- Liu, Z.-P.; Gong, X.-Q.; Kohanoff, J.; Sanchez, C.; Hu, P. Catalytic role of metal oxides in gold-based catalysts: A first principles study of CO oxidation on TiO2 supported Au. Phys. Rev. Lett. 2003, 91, 266102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kim, H.Y.; Henkelman, G. CO oxidation at the Au–Cu interface of bimetallic nanoclusters supported on CeO2 (111). J. Phys. Chem. Lett. 2013, 4, 2943–2947. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Huang, Y.-J. Low CO generation on tunable oxygen vacancies of non-precious metallic Cu/ZnO catalysts for partial oxidation of methanol reaction. Appl. Catal. B: Environ. 2014, 150–151, 506–514. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Gervasini, A.; Bennici, S. Dispersion and surface states of copper catalysts by temperature-programmed-reduction of oxidized surfaces (s-TPR). Appl. Catal. A: Gen. 2005, 281, 199–205. [Google Scholar] [CrossRef]
Figure 1.
Temperature profiles of catalyst performance: (a) conversion of methanol (CMeOH), (b) selectivity of hydrogen (SH2), (c) selectivity of carbon monoxide (SCO) in POM reaction. (■) CZ, (●) CZrZ, (▲) A1CZrZ, (▼) A3CZrZ, (★) A5CZrZ, and (◊) A5CZ at 60 Kh−1 Gas Hourly Space Velocity (GHSV) and 9.48 h−1 Weight Hourly Space Velocity (WHSV) with O2/MeOH = 0.5.
Figure 1.
Temperature profiles of catalyst performance: (a) conversion of methanol (CMeOH), (b) selectivity of hydrogen (SH2), (c) selectivity of carbon monoxide (SCO) in POM reaction. (■) CZ, (●) CZrZ, (▲) A1CZrZ, (▼) A3CZrZ, (★) A5CZrZ, and (◊) A5CZ at 60 Kh−1 Gas Hourly Space Velocity (GHSV) and 9.48 h−1 Weight Hourly Space Velocity (WHSV) with O2/MeOH = 0.5.

Figure 2.
Conversion of methanol as a function of time-on-stream of POM reaction over (■) CZ, (●) CZrZ, (★) A5CZrZ, and (◊) A5CZ at 250 °C for 12 h in an accelerated aging condition (60 Kh−1 of GHSV and 47.4 h−1 of WHSV).
Figure 2.
Conversion of methanol as a function of time-on-stream of POM reaction over (■) CZ, (●) CZrZ, (★) A5CZrZ, and (◊) A5CZ at 250 °C for 12 h in an accelerated aging condition (60 Kh−1 of GHSV and 47.4 h−1 of WHSV).

Figure 3.
XRD profiles of fresh catalysts: (a) CuO, (b) ZnO, (c) CZ, (d) CZrZ, (e) A1CZrZ, (f) A3CZrZ, and (g) A5CZrZ. (◆) CuO (●) ZnO.
Figure 3.
XRD profiles of fresh catalysts: (a) CuO, (b) ZnO, (c) CZ, (d) CZrZ, (e) A1CZrZ, (f) A3CZrZ, and (g) A5CZrZ. (◆) CuO (●) ZnO.

Figure 4.
TEM images and mean size of (a) A1CZrZ, (b) A3CZrZ, and (c) A5CZrZ.

Figure 5.
HR-TEM images of (a) A1CZrZ, (b) A3CZrZ, (c) A5CZrZ, and (d) SAED of A5CZrZ.
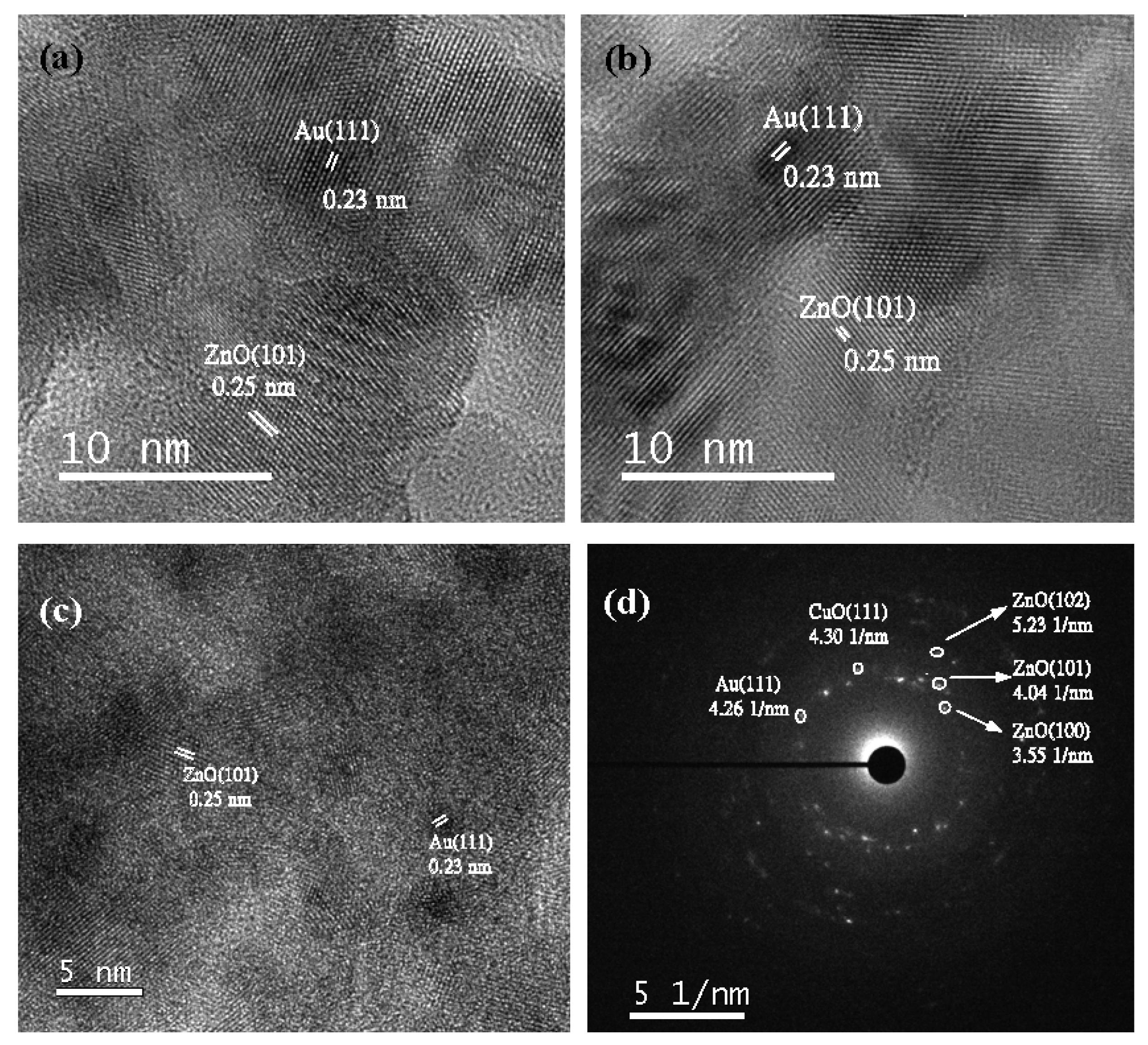
Figure 6.
Hydrogen temperature programmed reduction profiles: (a) CuO, (b) CZ, (c) CZrZ, (d) A1CZrZ, (e) A3CZrZ, and (f) A5CZrZ.
Figure 6.
Hydrogen temperature programmed reduction profiles: (a) CuO, (b) CZ, (c) CZrZ, (d) A1CZrZ, (e) A3CZrZ, and (f) A5CZrZ.

Figure 7.
XPS spectra of O1s: (a) CZ, (b) CZrZ, and (c) A5CZrZ.

Figure 8.
The O2-TPD of reduced catalysts: (a) CZ, (b) CZrZ, and (c) A5CZrZ.
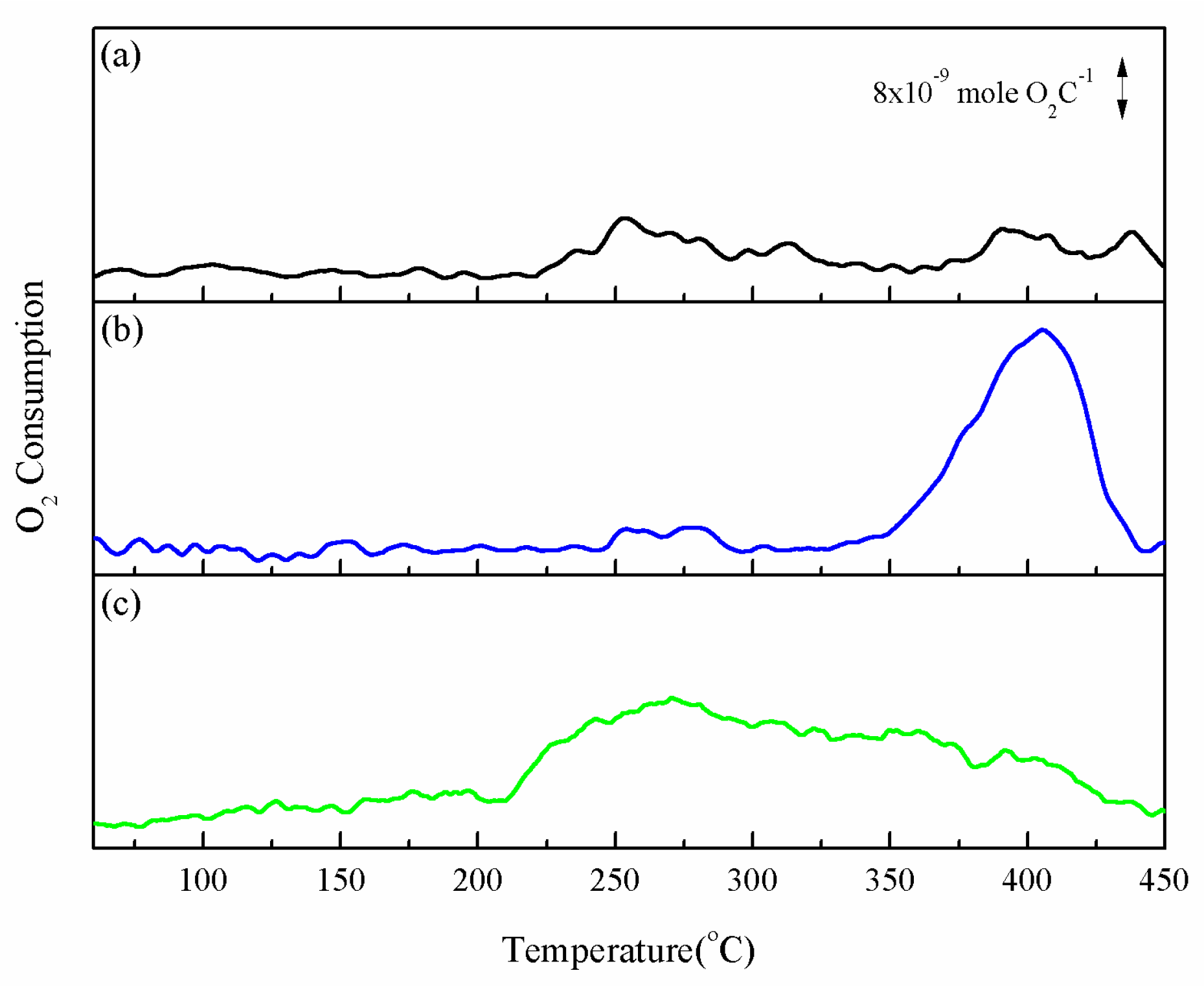
Figure 9.
The compound fit XANES spectra of Cu species on (a) fresh CZ, (b) CZ during POM at 250 °C for 24 h, (c) CZ after POM over 4 months, (d) fresh A5CZrZ, (e) A5CZrZ during POM at 250 °C for 24 h, and (f) A5CZrZ after POM over 4 months.
Figure 9.
The compound fit XANES spectra of Cu species on (a) fresh CZ, (b) CZ during POM at 250 °C for 24 h, (c) CZ after POM over 4 months, (d) fresh A5CZrZ, (e) A5CZrZ during POM at 250 °C for 24 h, and (f) A5CZrZ after POM over 4 months.
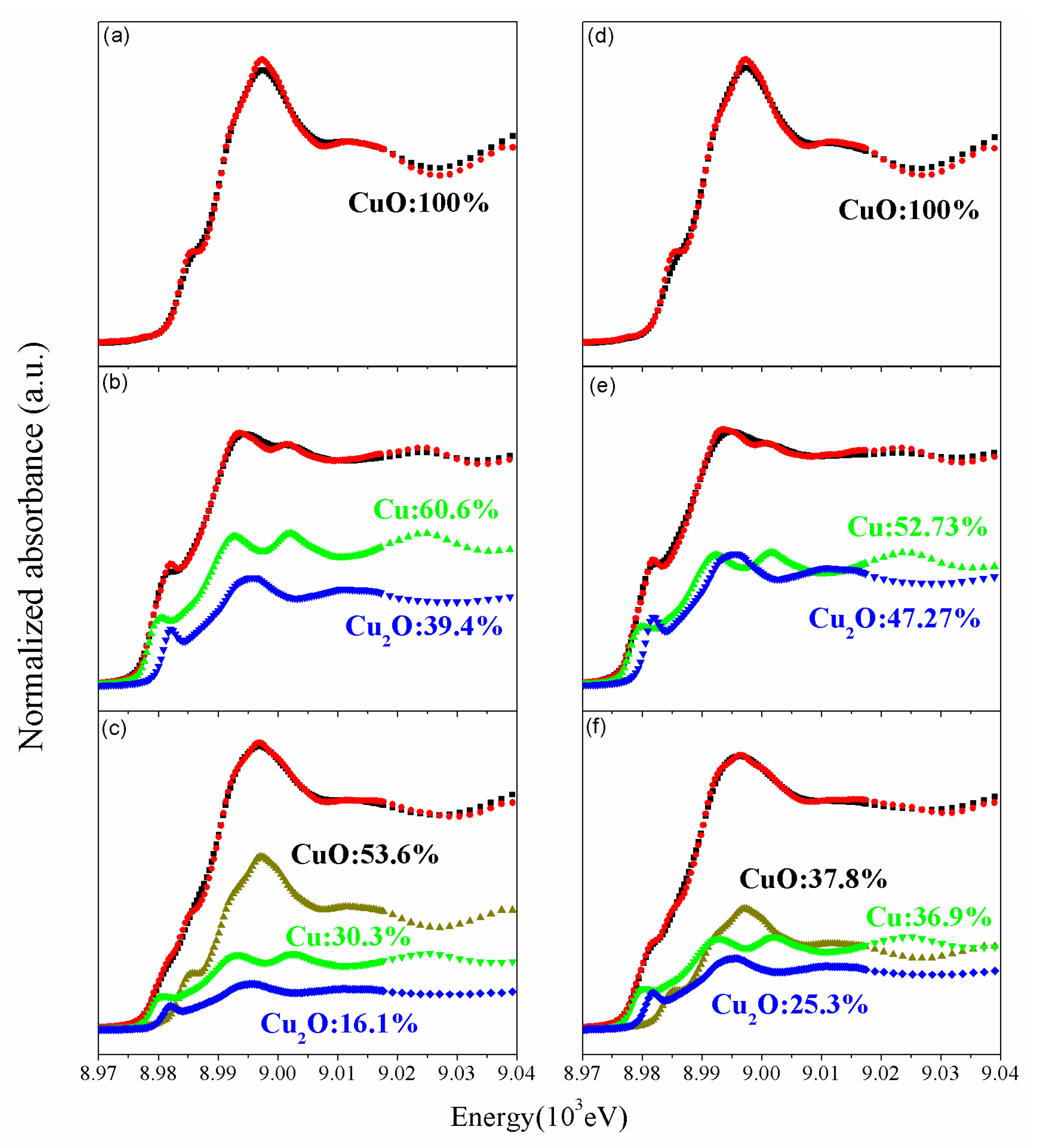
Figure 10.
DRIFT spectra of A5CZrZ during POM reaction at (a) 100 °C, (b) 125 °C, (c) 175 °C, (d) 200 °C, (e) 250 °C, and (f) 300 °C.
Figure 10.
DRIFT spectra of A5CZrZ during POM reaction at (a) 100 °C, (b) 125 °C, (c) 175 °C, (d) 200 °C, (e) 250 °C, and (f) 300 °C.

Figure 11.
In-situ DRIFT spectra of (I) POM reaction at 125 °C (II) CO chemisorption at 25 °C over (a) CZ, (b) CZrZ, and (c) A5CZrZ.
Figure 11.
In-situ DRIFT spectra of (I) POM reaction at 125 °C (II) CO chemisorption at 25 °C over (a) CZ, (b) CZrZ, and (c) A5CZrZ.

Scheme 1.
Suggested mechanism of POM reaction on A5CZrZ catalyst. * indicted as metal on catalysts.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The ignition temperature, Cu size, and deactivation rate constant.
| Sample | Ti | Reduction State d(Cu) a nm | After POM Reaction d(Cu) a nm | d(Cu) Increase % | Deactivation Rate Constant b ± CI c (hr−1) |
|---|---|---|---|---|---|
| CZ | 180 | 6.25 | 19.50 | 212 | 0.358 ± 0.0238 |
| CZrZ | 175 | 5.40 | 12.24 | 127 | 0.202 ± 0.0248 |
| A5CZrZ | 120 | 3.97 | 9.00 | 127 | 0.191 ± 0.0253 |
| A5CZ | 125 | 3.30 | 11.78 | 257 | 0.351 ± 0.0256 |
a Normal diameter estimated from XRD data using the Debye-Scherrer equation. b Deactivation rate constant calculated by Equation S1. c 95% Confidence interval (CI).
Table 2.
Physicochemical properties of the catalysts measured by various methods.
| Sample Name | Metallic Composition a | Cu Dispersion (%) b | BET | dCuO | dCu | dZnO | Composition of Catalyst Surfaces d (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (wt.%) | Surface Area | (nm) c | (nm) c | (nm) c | |||||||||||
| Au | Cu | Zr | Zn | (m²/g) | Cu (111) | Cu (200) | Zn (101) | Au4f | Cu2p | Zr3d | Zn2p | Au0/Au3+ | |||
| Fresh | After POM | ||||||||||||||
| CZ | – | 29.8 | – | 70.2 | 20.51 | 34.59 | 5.65 | 6.25 | 9.03 | – | 26.24 | – | 73.76 | – | – |
| CZrZ | – | 31.2 | 15.9 | 52.9 | 21.02 | 77.52 | 4.49 | 5.40 | 6.42 | – | 27.85 | 3.38 | 68.77 | – | – |
| A1CZrZ | 1.0 | 31.4 | 15.4 | 52.2 | 26.93 | 64.06 | 4.81 | 4.33 | 6.96 | 10.07 | 27.45 | 4.39 | 58.09 | 0.65 | 0.83 |
| A3CZrZ | 3.1 | 31.6 | 15.6 | 49.7 | 28.32 | 73.21 | 4.09 | 4.28 | 5.98 | 10.18 | 28.29 | 4.89 | 56.64 | 0.85 | 1.11 |
| A5CZrZ | 4.5 | 31.7 | 16.7 | 47.1 | 27.92 | 77.03 | 3.48 | 3.97 | 5.69 | 11.59 | 26.35 | 3.72 | 58.34 | 1.37 | 1.43 |
a Calculated Metallic Composition by inductively coupled plasma mass spectrometry (ICP-MS). b Calculated Cu dispersion of catalysts by N2O chemisorptions. c Normal diameter estimated from XRD data using the Debye-Scherrer equation. d Calculated from XPS.
Table 3.
Fractions of TPR area.
| Catalyst | CZ | CZrZ | A1CZrZ | A3CZrZ | A5CZrZ | |
|---|---|---|---|---|---|---|
| Fractions of TPR Area (%) | β | 56 | 61 | 50 | 51 | 48 |
| α | 44 | 39 | 50 | 49 | 53 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, H.-Y.; Chen, H.-I.; Huang, Y.-J. The Promotional Effects of ZrO2 and Au on the CuZnO Catalyst Regarding the Durability and Activity of the Partial Oxidation of Methanol. Catalysts 2018, 8, 345. https://doi.org/10.3390/catal8090345
AMA Style
Huang H-Y, Chen H-I, Huang Y-J. The Promotional Effects of ZrO2 and Au on the CuZnO Catalyst Regarding the Durability and Activity of the Partial Oxidation of Methanol. Catalysts. 2018; 8(9):345. https://doi.org/10.3390/catal8090345
Chicago/Turabian StyleHuang, Hsiao-Yu, Hao-I Chen, and Yuh-Jeen Huang. 2018. "The Promotional Effects of ZrO2 and Au on the CuZnO Catalyst Regarding the Durability and Activity of the Partial Oxidation of Methanol" Catalysts 8, no. 9: 345. https://doi.org/10.3390/catal8090345
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.