Photooxidation of Cyclohexane by Visible and Near-UV Light Catalyzed by Tetraethylammonium Tetrachloroferrate

 , ,
, ,
Abstract
:
1. Introduction
2. Results
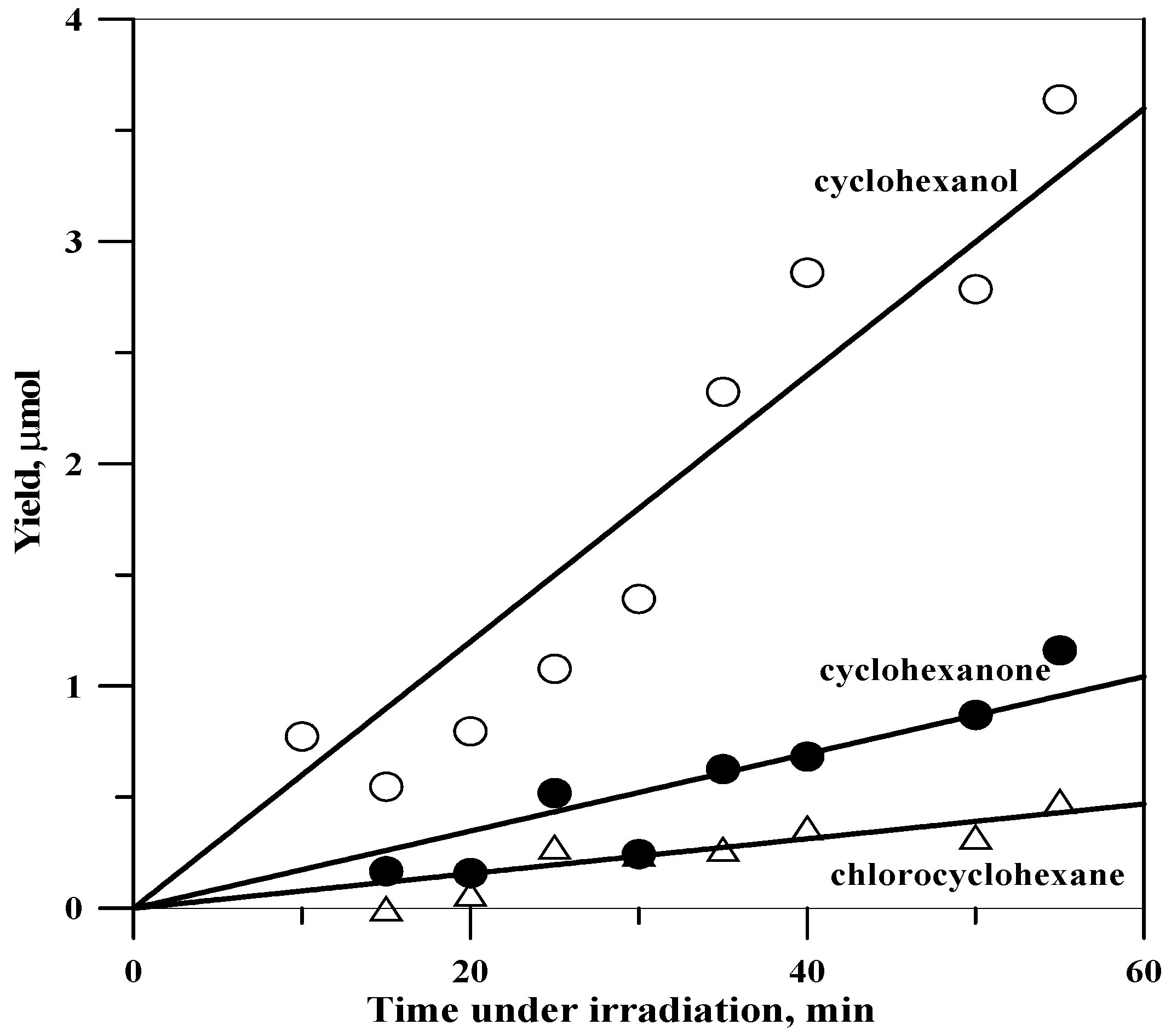
2.1. Development of Products with Time
2.2. Polar Accelerants
2.3. Salt Addition
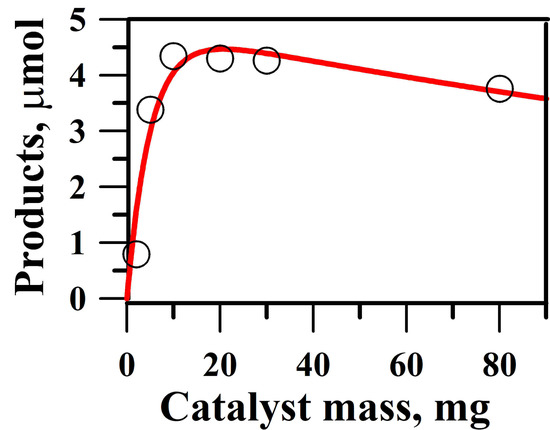
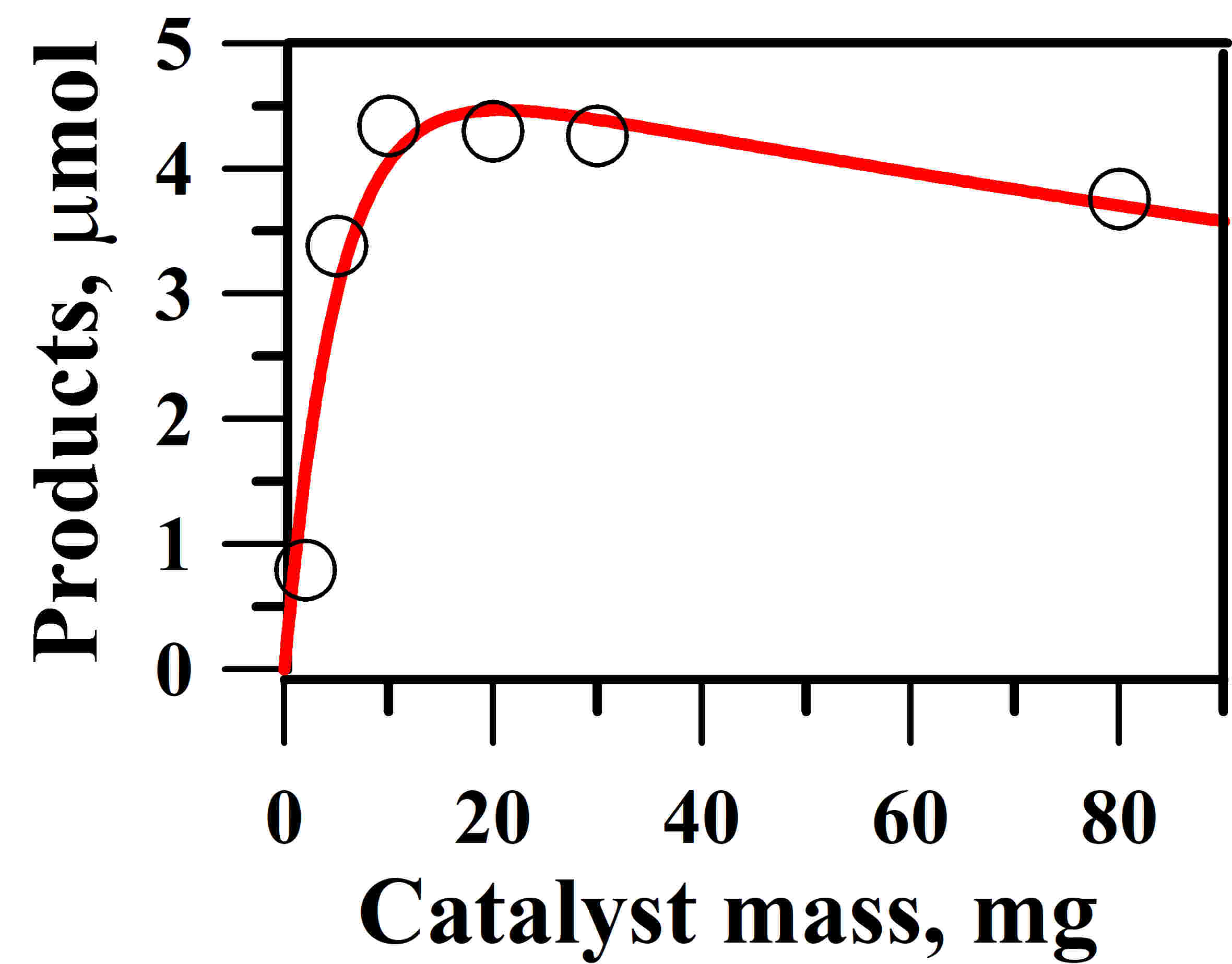
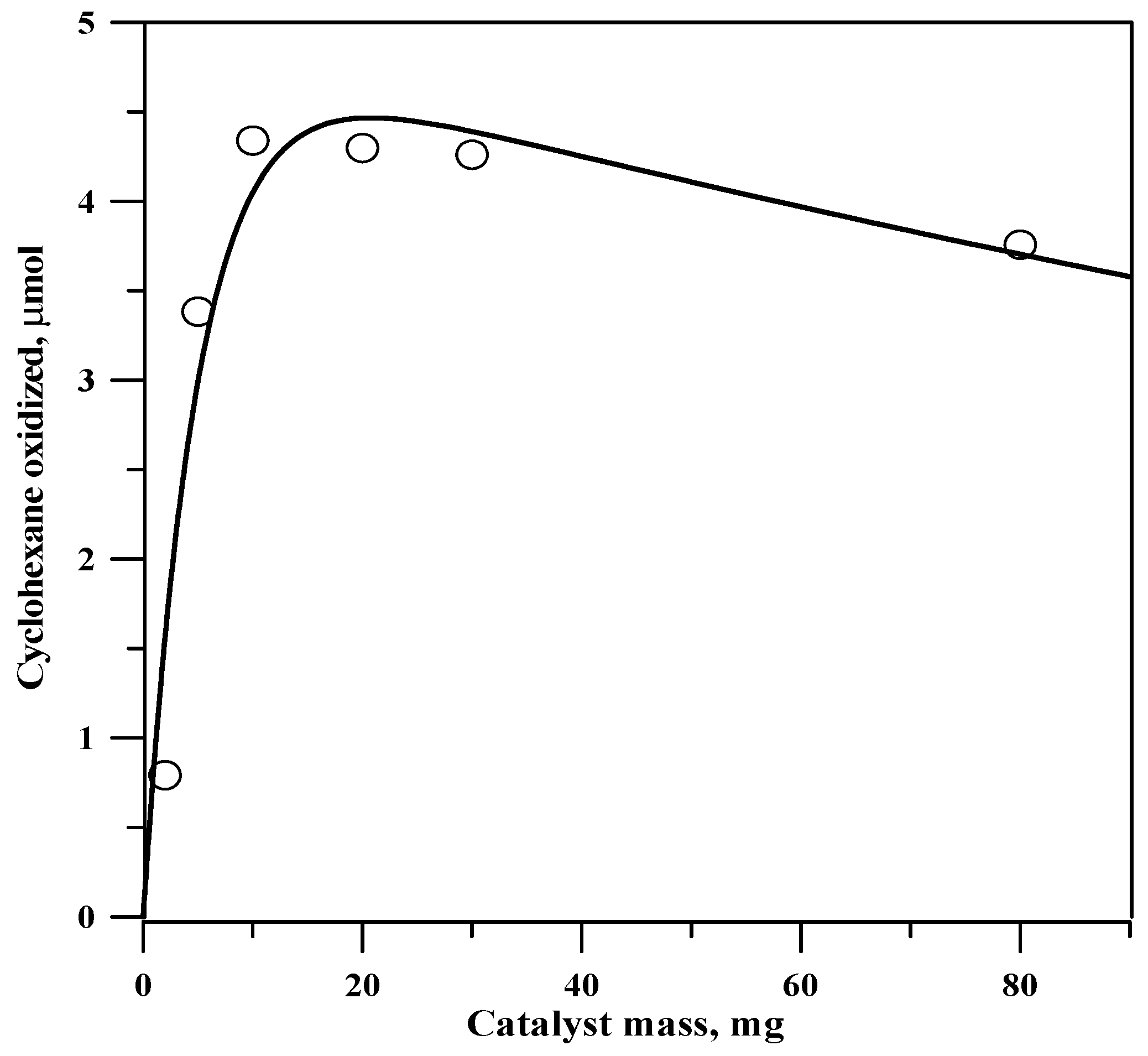
2.4. Optimum Amount of Catalyst
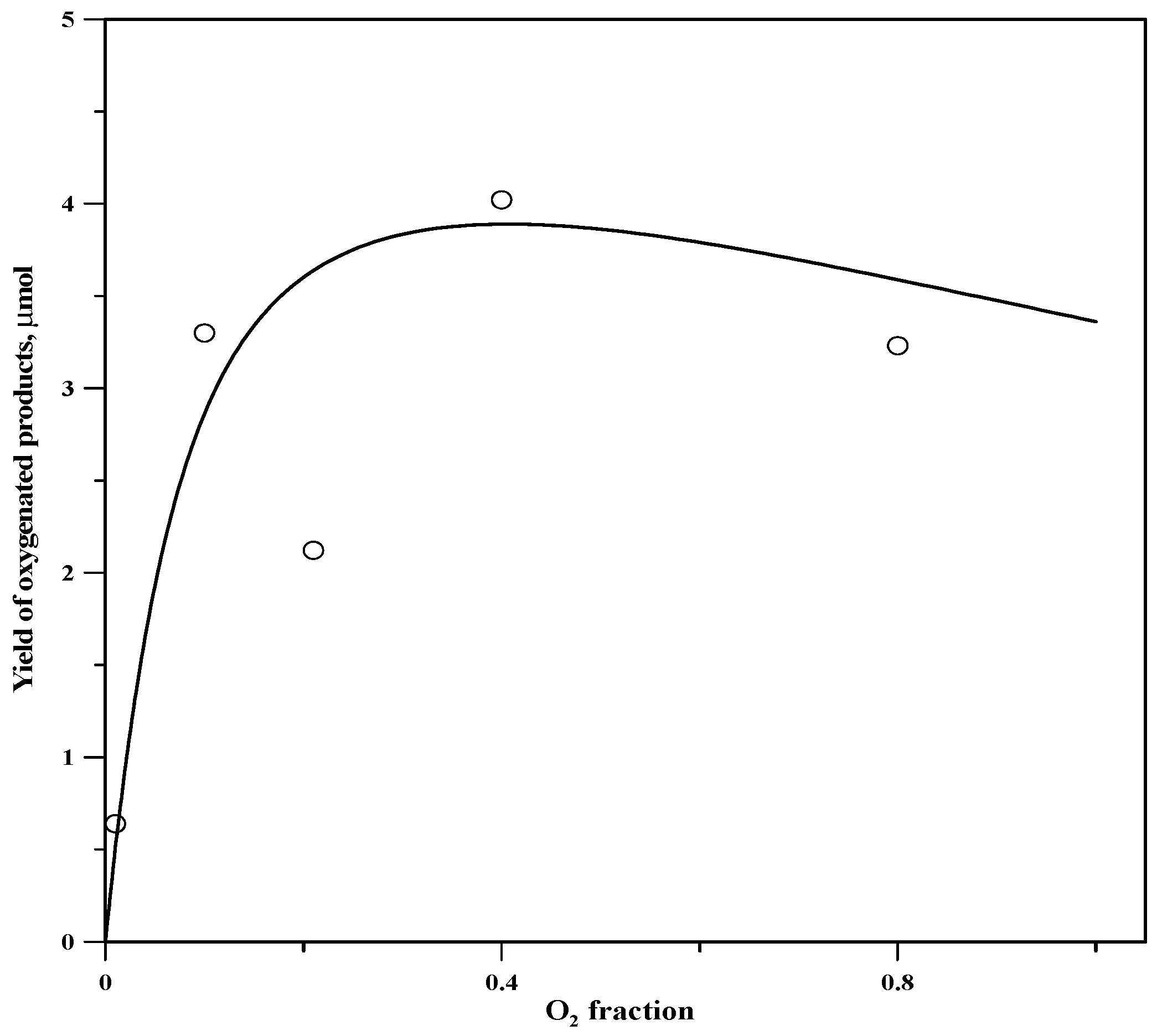
2.5. Variation of Yield with Fraction of Oxygen above the Reaction Mixture
2.6. Yield and Cutoff Wavelength
2.7. Photonic Efficiency
2.8. Catalyst Reuse
2.9. Longer Irradiation Times
2.10. Comparison of Et4NFeCl4 with Dissolved FeCl3 and FeCl3 on Silica Gel
2.11. Homogeneous Catalysis with Et4N[FeCl4] and Other Compounds
2.12. Sunlight
2.13. Comparison with Other Studies
2.14. Direct Comparison with Titanium Dioxide
2.15. Selectivity
2.16. Mechanistic Considerations
- In neat C6H12 catalysis by Et4N[FeCl4] usually yielded an A/K ratio of about 4.
- In mixed acetone/cyclohexane solutions catalyzed homogeneously by Et4N[FeCl4] the A/K ratio was about 10.
- In mixed dichloromethane/cyclohexane solutions catalyzed homogeneously by Et4N[FeCl4] the A/K ratio was approximately 1.
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maldotti, A.; Molinari, A.; Amadelli, R. Photocatalysis with organized systems for the oxofunctionalization of hydrocarbons by O2. Chem. Rev. 2002, 102, 3811–3836. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, G.; Po-Lock, Y.; Kutal, C. Photocatalytic oxidation of cyclohexane over TiO2 nanoparticles by molecular oxygen under mild conditions. J. Chem. Technol. Biotechnol. 2003, 78, 1246–1251. [Google Scholar] [CrossRef]
- Maldotti, A.; Varani, G.; Molinari, A. Photo-assisted chlorination of cycloalkanes with iron chloride heterogenized with Amberlite. Photochem. Photobiol. Sci. 2006, 5, 993–995. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, P.E.; Gruber, M.; Vogler, A. The photolysis of iron(III) chloride in chloroform. Inorg. Chim. Acta 2003, 346, 137–142. [Google Scholar] [CrossRef]
- Shulpin, G.B.; Kats, M.M. Ferric chloride catalyzed photooxidation of alkanes by air in organic solvents. React. Kinet. Catal. Lett. 1990, 41, 239–243. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V.; Kozlov, Y.N. Photochemical aerobic oxidation of alkanes promoted by iron complexes. New J. Chem. 1996, 20, 1243–1256. [Google Scholar]
- The H−Cl Bond Energy Is 432 kJ/mol, While that of H−OH Is 497 kJ/mol. “Bond-Dissociation Energy”. Available online: https://en.wikipedia.org/wiki/Bond-dissociation_energy (accessed on 18 September 2018).
- Hoggard, P.E.; Maldotti, A. Catalysis of the photodecomposition of carbon tetrachloride in ethanol by an Amberlite anion exchange resin. J. Catal. 2010, 275, 243–249. [Google Scholar] [CrossRef]
- Solis Montiel, E.; Solano, J.A. Spectrophotometric analysis for chlorine by the extraction of triiodide formed in chloroform solution of tetrabutylammonium perchlorate. Ingenieria y Ciencia Quimica 1986, 10, 45–48. [Google Scholar]
- Chan, A.M.; Harvey, B.M.; Hoggard, P.E. Photodecomposition of dichloromethane catalyzed by tetrachloroferrate(III) supported on a Dowex anion exchange resin. Photochem. Photobiol. Sci. 2013, 12, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, P.E.; Cohen, L.R.; Peña, L.A.; Harvey, B.M.; Chan, A.M. The dependence of photocatalytic reaction yields on catalyst mass in solid-liquid suspensions. Curr. Catal. 2013, 2, 2–6. [Google Scholar] [CrossRef]
- Loddo, V.; Addamo, M.; Augugliaro, V.; Palmisano, L.; Schiavello, M.; Garrone, E. Optical properties and quantum yield determination in photocatalytic suspensions. AIChE J. 2006, 52, 2565–2574. [Google Scholar] [CrossRef]
- Barnard, K.R.; Bright, V.R.; Enright, R.J.; Fahy, K.M.; Liu, A.C.; Hoggard, P.E. Photooxidation of toluene by visible and near-UV light catalyzed by tetraethylammonium tetrachloroferrate. Catalysts 2018, 8, 79. [Google Scholar] [CrossRef]
- Ohkubo, K.; Hirose, K.; Fukuzumi, S. Photooxygenation of alkanes by dioxygen with p-benzoquinone derivatives with high quantum yields. Photochem. Photobiol. Sci. 2016, 15, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, K.; Hirose, K.; Fukuzumi, S. Solvent-free photooxidation of alkanes by dioxygen with 2,3-dichloro-5,6-dicyano-p-benzoquinone via. photoinduced electron transfer. Chem. Asian J. 2016, 11, 2255–2259. [Google Scholar] [CrossRef] [PubMed]
- Peña, L.A.; Segura, R.E.; Chan, A.M.; Hoggard, P.E. Photodegradation of dichloromethane catalyzed by iron(III) chloride on silica gel. Curr. Catal. 2014, 3, 35–38. [Google Scholar] [CrossRef]
- Boarini, P.; Carassiti, V.; Maldotti, A.; Amadelli, R. Photocatalytic oxygenation of cyclohexane on titanium dioxide suspensions: Effect of the solvent and of oxygen. Langmuir 1998, 14, 2080–2085. [Google Scholar] [CrossRef]
- Brusa, M.A.; Di Iorio, Y.; Churio, M.S.; Grela, M.A. Photocatalytic air oxidation of cyclohexane in CH2Cl2-C6H12 mixtures over TiO2 particles. An attempt to rationalize the positive effect of dichloromethane on the yields of valuable oxygenates. J. Mol. Catal. A Chem. 2007, 268, 29–35. [Google Scholar] [CrossRef]
- Almquist, C.B.; Biswas, P. The photo-oxidation of cyclohexane on titanium dioxide: An investigation of competitive adsorption and its effects on product formation and selectivity. Appl. Catal. A 2001, 214, 259–271. [Google Scholar] [CrossRef]
- Molinari, A.; Amadelli, R.; Andreotti, L.; Maldotti, A. Heterogeneous photocatalysis for synthetic purposes: Oxygenation of cyclohexane with H3PW12O40 and (nBu4N)4W10O32 supported on silica. Dalton Trans. 1999, 1203–1204. [Google Scholar] [CrossRef]
- Molinari, A.; Maldotti, A.; Amadelli, R.; Sgobino, A.; Carassiti, V. Integrated photocatalysts for hydrocarbon oxidation: Polyoxotungstates/iron porphyrins systems in the reductive activation of molecular oxygen. Inorg. Chim. Acta 1998, 272, 197–203. [Google Scholar] [CrossRef]
- Simic, M.; Hayon, E. Spectroscopic investigation of cyclohexanol and cyclohexyl radicals and their corresponding peroxy radicals. J. Phys. Chem. 1971, 75, 1677–1680. [Google Scholar]
- Howard, J.A.; Ingold, K.U. Absolute rate constants for hydrocarbon autoxidation. XV. The induced decomposition of some t-hydroperoxides. Can. J. Chem. 1969, 47, 3797–3801. [Google Scholar] [CrossRef]
- Hermans, I.; Jacobs, P.A.; Peeters, J. Understanding the autoxidation of hydrocarbons at the molecular level and consequences for catalysis. J. Mol. Catal. A Chem. 2006, 251, 221–228. [Google Scholar] [CrossRef]
- Hermans, I.; Nguyen, T.L.; Jacobs, P.A.; Peeters, J. Autoxidation of cyclohexane: Conventional views challenged by theory and experiment. ChemPhysChem 2005, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Hermans, I.; Peeters, J.; Jacobs, P.A. Autoxidation of ethylbenzene: The mechanism elucidated. J. Org. Chem. 2007, 72, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Maldotti, A.; Molinari, A.; Varani, G.; Lenarda, M.; Storaro, L.; Bigi, F.; Mazzacani, A.; Sartori, G. Immobilization of (n-Bu4N)4W10O32 on mesoporous MCM-41 and amorphous silicas for photocatalytic oxidation of cycloalkanes with molecular oxygen. J. Catal. 2002, 209, 210–216. [Google Scholar] [CrossRef]
- Maldotti, A.; Amadelli, R.; Carassiti, V.; Molinari, A. Catalytic oxygenation of cyclohexane by photoexcited (nBu4N)4W10O32: The role of radicals. Inorg. Chim. Acta 1997, 256, 309–312. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acetic Acid Content | Oxidized Products, µmol |
|---|---|
| 0 | 1.8 |
| 0.2% | 15.7 |
| 0.5% | 28.8 |
| 2.0% | 12.3 |
| Bu4NBr | Chx−OH | Chx=O | Chx−Cl | Total |
|---|---|---|---|---|
| 0 | 6.1 | 1.7 | 1.4 | 9.2 |
| 10 mg | 9.9 | 4.3 | 6.8 | 21.0 |
| 20 mg | 7.5 | 4.0 | 9.1 | 20.5 |
| O2 Fraction | Chx−OH | Chx=O | Chx−Cl |
|---|---|---|---|
| 0.01 | 0.6 | 0.1 | 0.2 |
| 0.10 | 2.7 | 0.6 | 1.5 |
| 0.21 | 1.9 | 0.2 | 2.7 |
| 0.40 | 3.3 | 0.7 | 1.8 |
| 0.80 | 2.5 | 0.8 | 4.3 |
| Cutoff Filter | Chx−OH, μmol | Chx=O, μmol | Chx−Cl, μmol | Total Chx Reacted |
|---|---|---|---|---|
| None a | 7.4 | 1.0 | 10.8 | 19.1 |
| 320 nm | 4.6 | 1.5 | 3.3 | 9.4 |
| 360 nm | 5.5 | 1.4 | 2.0 | 8.9 |
| 395 nm | 2.0 | 0.4 | 0.7 | 3.1 |
| Catalyst Mass | Chx−OH, μmol | Chx=O, μmol | Chx−Cl, μmol |
|---|---|---|---|
| 60 mg | 5.0 | 3.9 | 6.3 |
| 50 mg | 3.8 | 1.0 | 1.9 |
| 32 mg | 2.8 | 0.7 | 1.6 |
| 11 mg | 3.9 | 1.0 | 1.9 |
| Experiment | Chx−OH | Chx=O | Chx−Cl | % Reaction |
|---|---|---|---|---|
| 1 mL Chx, 60 mg Et4EtFeCl4, 500 W lamp, water filter, 1 h | 121 | 28 | 50 | 2.1% |
| 3 mL Chx, 50 mg Et4NFeCl4, 500 W lamp, λ > 395 nm, 2 h | 82 | 20 | 3 | 0.4% |
| 3 mL Chx, 50 mg Et4NFeCl4, 500 W lamp, λ > 360 nm, 2 h | 559 | 173 | 17 | 2.6% |
| 250 mL Chx, 500 mg Et4NFeCl4, 200 W immersion lamp, 3 h | 727 | 326 | 88 | 0.05% |
| Catalyst | mg | Chx−OH | Chx=O | Chx−Cl | Oxy Products |
|---|---|---|---|---|---|
| Et4N[FeCl4] | 20 | 6.1 | 1.7 | 1.3 | 7.8 |
| FeCl3 5% on SiO2 | 20 | 1.0 | 0.3 | 1.0 | 1.3 |
| FeCl3 (dissolved) | 5 | 2.7 | 2.9 | 13.7 | 5.6 |
| Catalyst | Chx Environment | Catalyst Conc. | Light Source | Irrad Time, min | Chx−OH+Chx=O μmol | Reference |
|---|---|---|---|---|---|---|
| FeCl3 | 0.5 M in CH3CN | 5 × 10−4 M | 150 W b | 120 | 6 | [5] |
| FeCl3 | 0.5 M in acetone | 5 × 10−4 M | 150 W b | 300 | 225 | [5] |
| Bu4N[FeCl4] | 40% CH2Cl2 10% CH3CN | 3 × 10−4 M | 400 W Hg | 240 | 67 | [3] |
| Et4N[FeCl4] | 50% acetone a | 0.06 M | 100 W Hg λ > 360 nm | 20 | 48 | c |
| Catalyst | Catalyst Amount | Sample Volume | Light Source | λ | Irrad Time | Chx−OH+Chx=O μmol | Reference |
|---|---|---|---|---|---|---|---|
| PXQ a | 2 × 10−3 M | 3 mL | 500 W Xe | >390 nm | 26 h | 225 d | [14] |
| Et4N[FeCl4] b | 50 mg | 3 mL | 500 W Hg | >395 nm | 2 h | 102 | c |
| Experiment | Chx−OH | Chx=O | Chx−Cl | % Oxidation |
|---|---|---|---|---|
| 6 mL, Erlenmeyer, 150 mg Et4NFeCl4, no balloon, 3 h | 103 | 53 | 7 | 0.3% |
| 6 mL, Erlenmeyer, 200 mg FeCl3/SiO2, no balloon, 3 h | 18 | 0 | 76 | 0.2% |
| 1.5 mL, 1-cm rect. cuvette, 300 mg Et4NFeCl4, 60% O2, 6 h | 27 | 7 | 14 | 0.3% |
| 1 mL, 1-cm triangular cuvette, 80 mg Et4NFeCl4, 60% O2, 6 h | 27 | 11 | 6 | 0.5% |
| 3 mL, 4-cm rectangular cuvette, 200 mg Et4NFeCl4, 60% O2, 4 h | 60 | 10 | 17 | 0.3% |
| Product | Yield, μmol |
|---|---|
| Chx−OH | 993 |
| Chx=O | 902 |
| Chx−Cl | 22 |
| Catalyst | Ref | Sample Volume mL | Catalyst Mass mg | Gas Above rxn | Radiation Source | Irrad Time, min | Yield Chx OH μmol | Yield Chx=O μmol | % Chx−Cl | % Chx Reacted |
|---|---|---|---|---|---|---|---|---|---|---|
| TiO2 | [17] | 2.5 a | 10 | air | 150 W Hg λ > 360 nm | 270 | <1 | 18 | n/a | 0.08% |
| TiO2 | [19] | 20 | 20 | air | 450 W Xe no filter | 150 | 45 | 105 | n/a | 0.08% |
| TiO2 | [18] | 1.0 | 2 | air | not stated λ = 303 nm | 45 | 0.7 | 2 | n/a | 0.03% |
| W10O324− b | [20] | 3.0 | 45 | O2 | 150 W Hg λ > 280 nm | 90 | 11 | 11 | n/a | 0.1% |
| FeCl4− on Amberlite | [3] | 2.5 | 625 | air | 400 W Hg λ > 300 nm | 240 | <0.1 | <0.1 | 9 μmol 100% | 0.04% |
| Et4NFeCl4 | c | 1.0 | 50 | 60%O2 | 500 W Hg glass filter e | 120 | 121 | 28 | 25% | 2.1% |
| Et4NFeCl4 | d | 3.0 | 50 | air | 500 W Hg λ > 360 nm | 120 | 559 | 173 | 2% | 2.7% |
| Et4NFeCl4 | d | 3.0 | 50 | air | 500 W Hg λ > 395 nm | 120 | 82 | 20 | 3% | 0.4% |
| Catalyst | λcutoff, nm | Chx=O, µmol | Chx−OH, µmol |
|---|---|---|---|
| TiO2 | 360 | 5.1 | 1.8 |
| Et4N[FeCl4] | 360 | 2.4 | 0.7 |
| TiO2 | 375 | 2.8 | 1.5 |
| Et4N[FeCl4] | 375 | 1.6 | 0.3 |
| TiO2 | 385 | 0.0 | 0.0 |
| Et4N[FeCl4] | 385 | 1.9 | 0.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahy, K.M.; Liu, A.C.; Barnard, K.R.; Bright, V.R.; Enright, R.J.; Hoggard, P.E. Photooxidation of Cyclohexane by Visible and Near-UV Light Catalyzed by Tetraethylammonium Tetrachloroferrate. Catalysts 2018, 8, 403. https://doi.org/10.3390/catal8090403
Fahy KM, Liu AC, Barnard KR, Bright VR, Enright RJ, Hoggard PE. Photooxidation of Cyclohexane by Visible and Near-UV Light Catalyzed by Tetraethylammonium Tetrachloroferrate. Catalysts. 2018; 8(9):403. https://doi.org/10.3390/catal8090403
Chicago/Turabian StyleFahy, Kira M., Adam C. Liu, Kelsie R. Barnard, Valerie R. Bright, Robert J. Enright, and Patrick E. Hoggard. 2018. "Photooxidation of Cyclohexane by Visible and Near-UV Light Catalyzed by Tetraethylammonium Tetrachloroferrate" Catalysts 8, no. 9: 403. https://doi.org/10.3390/catal8090403
APA StyleFahy, K. M., Liu, A. C., Barnard, K. R., Bright, V. R., Enright, R. J., & Hoggard, P. E. (2018). Photooxidation of Cyclohexane by Visible and Near-UV Light Catalyzed by Tetraethylammonium Tetrachloroferrate. Catalysts, 8(9), 403. https://doi.org/10.3390/catal8090403