Synthesis and Catalytic Application of Knölker-Type Iron Complexes with a Novel Asymmetric Cyclopentadienone Ligand Design


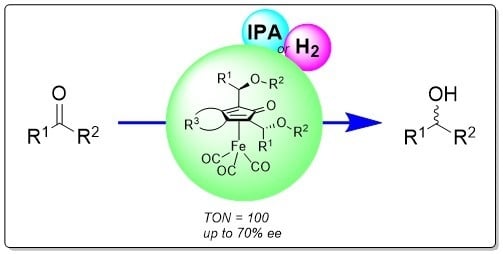
Abstract
:
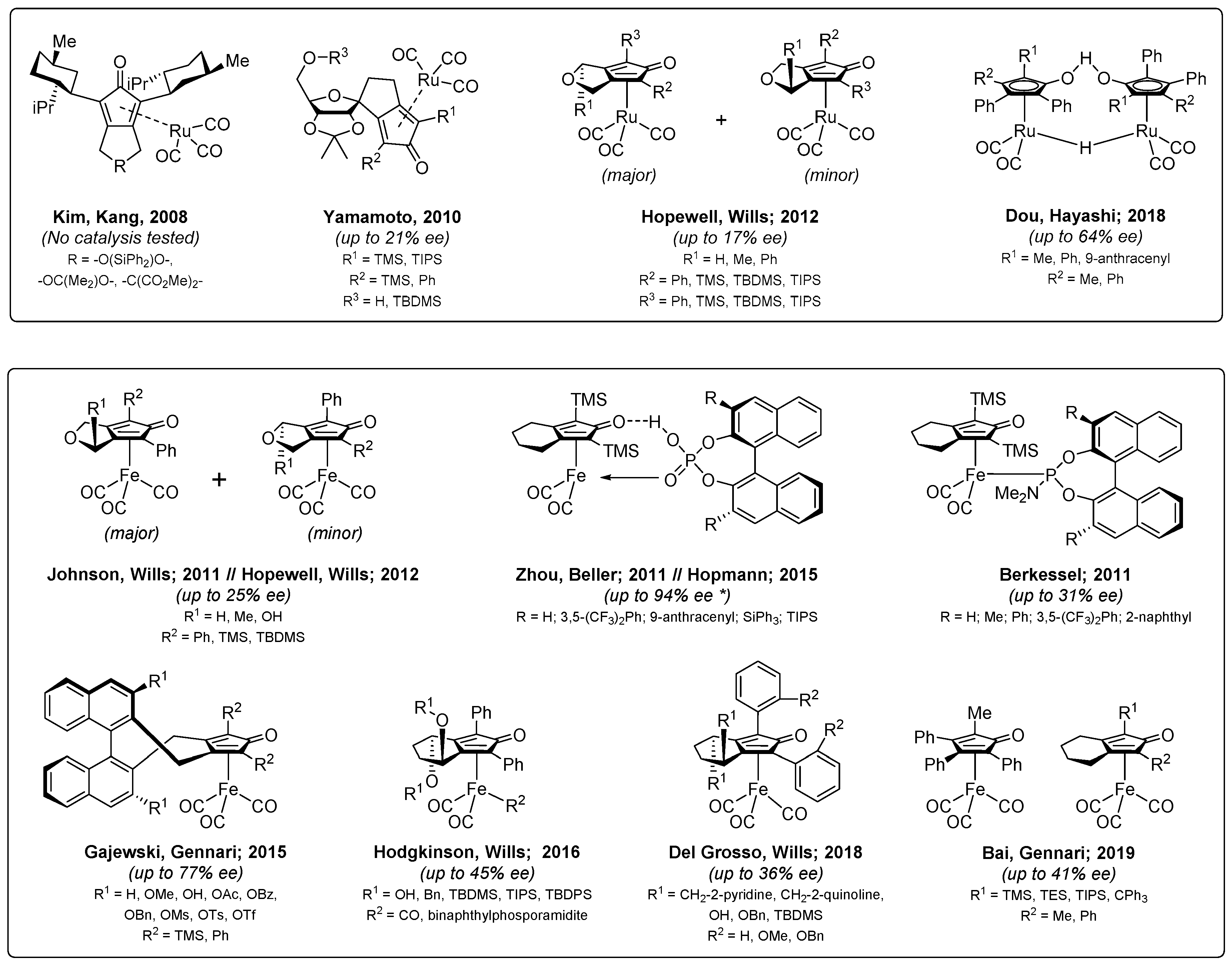
1. Introduction
2. Results and Discussion
2.1. Design, Synthesis and Characterization of the Pre-Ligands and Catalysts
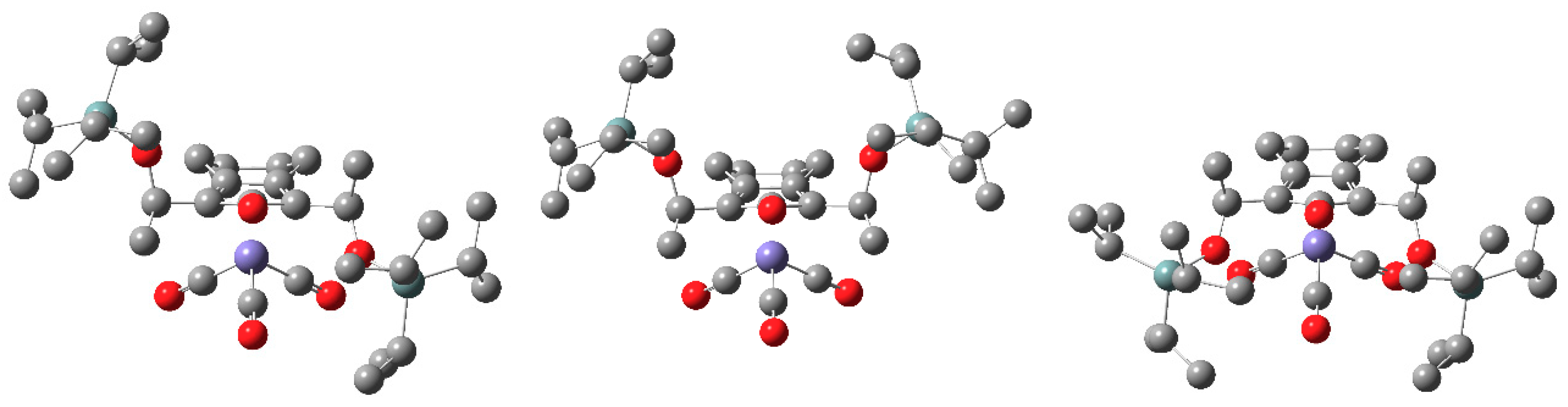
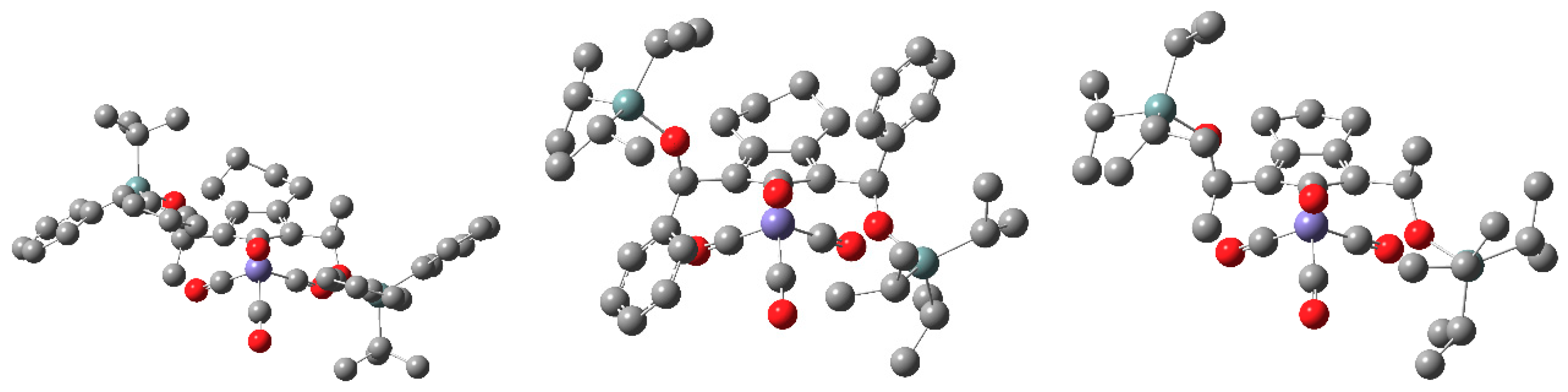
2.2. Computational Structure Assessment of the Novel Catalyst Design
2.3. Performance of the Catalysts in Asymmetric Hydrogenation
3. Materials and Methods
3.1. General Statements
3.2. General Procedure for the Synthesis of Alkynic Silyl Ethers 12a–c
3.3. General Procedure for the Synthesis of Pre-Ligands 13a–e
3.4. General Procedure for the Synthesis of Iron Complexes 14a–e
3.5. General Procedure for Transfer–Hydrogenation
3.6. General Procedure for Pressure–Hydrogenation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Knowles, W.S. Asymmetric Hydrogenations (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 1998–2007. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Knowles, W.S. Application of Organometallic Catalysis to the Commercial Production of L-DOPA. J. Chem. Ed. 1986, 63, 222–225. [Google Scholar] [CrossRef]
- Enthaler, S.; Junge, K.; Beller, M. Sustainable Metal Catalysis with Iron: From Rust to a Rising Star? Angew. Chem. Int. Ed. 2008, 47, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Kirchner, K.A. Well-Defined Bifunctional Iron Catalysts for the Hydrogenation of Ketones: Iron, the New Ruthenium. Angew. Chem. Int. Ed. 2011, 50, 5798–5800. [Google Scholar] [CrossRef] [PubMed]
- Darwish, M.; Wills, M. Asymmetric catalysis using iron complexes—‘Ruthenium Lite’? Catal. Schi. Technol. 2012, 2, 243–255. [Google Scholar] [CrossRef]
- Li, Y.; Yu, S.; Wu, X.; Xiao, J.; Shen, W.; Dong, Z.; Gao, J. Iron Catalyzed Asymmetric Hydrogenation of Ketones. J. Am. Chem. Soc. 2014, 136, 4031–4039. [Google Scholar] [CrossRef]
- Sues, P.E.; Demmans, K.Z.; Morris, R.H. Rational development of iron catalysts for asymmetric transfer hydrogenation. Dalton Trans. 2014, 43, 7650–7667. [Google Scholar] [CrossRef] [Green Version]
- Obligacion, J.V.; Chirik, P.J. Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2018, 2, 15–34. [Google Scholar] [CrossRef]
- Zell, T.; Milstein, D. Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal-Ligand Cooperation by Aromatization/Dearomatization. Acc. Chem. Res. 2015, 48, 1979–1994. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef] [PubMed]
- Reppe, W.; Vetter, H. Carbonylierung VI. Synthesen mit Metallcarbonylwasserstoffen. Liebigs Ann. Chem. 1953, 582, 133–161. [Google Scholar] [CrossRef]
- Quintard, A.; Rodriquez, J. Iron Cyclopentadienone Complexes: Discovery, Properties, and Catalytic Reactivity. Angew. Chem. Int. Ed. 2014, 53, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Knölker, H.-J.; Heber, J.; Mahler, C.H. Transition Metal-Diene Complexes in Organic Synthesis, Part 14. Regioselective Iron-Mediated [2 + 2 + 1] Cycloadditions of Alkynes and Carbon Monoxide: Synthesis of Substituted Cyclopentadienones. Synlett 1992, 12, 1002–1004. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Heber, J. Transition Metal-Diene Complexes in Organic Synthesis, Part 18. Iron-Mediated [2 + 2 + 1] Cycloadditions of Diynes and Carbon Monoxide: Selective Demetalation Reactions. Synlett 1993, 12, 924–926. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Baum, E.; Klauss, R. Transition Metal-Diene Complexes in Organic Synthesis, Part 25. Cycloadditions of Annulated 2,5-Bis(trimethylsilyl)cyclopentadienones. Tetrahedron Lett. 1995, 36, 7647–7650. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Baum, E.; Goesmann, R.; Klauss, R. A Novel Method for the Demetalation of Tricarbonyliron–Diene Complexes by a Photolytically Induced Ligand Exchange Reaction with Acetonitrile. Angew. Chem. Int. Ed. 1999, 38, 702–705. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Goesmann, R.; Klauss, R. Demetalation of Tricarbonyl(cyclopentadienone)iron Complexes Initiated by a Ligand Exchange Reaction with NaOH—X-Ray Analysis of a Complex with Nearly Square-Planar Coordinated Sodium. Angew. Chem. Int. Ed. 1999, 38, 2064–2066. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Braier, A.; Bröcher, D.J.; Cämmerer, S.; Fröhner, W.; Gonser, P.; Hermann, H.; Herzberg, D.; Reddy, K.R.; Rohde, G. Recent applications of tricarbonyliron-diene complexes to organic synthesis. Pure Appl. Chem. 2001, 73, 1075–1086. [Google Scholar] [CrossRef]
- Casey, C.P.; Guan, H. An Efficient and Chemoselective Iron Catalyst for the Hydrogenation of Ketones. J. Am. Chem. Soc. 2007, 129, 5816–5817. [Google Scholar] [CrossRef]
- Casey, C.P.; Guan, H. Cyclopentadienone Iron Alcohol Complexes: Synthesis, Reactivity, and Implications for the Mechanism of Iron-Catalyzed Hydrogenation of Aldehydes. J. Am. Chem. Soc. 2009, 131, 2499–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von der Höh, A.; Berkessel, A. Insights into the Mechanism of Dihydrogen-Heterolysis at Cyclopentadienone Iron Complexes and Subsequent C=X Hydrogenation. ChemCatChem 2011, 3, 861–867. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, Y.; Yun, P.; Zhang, M.; Li, T. The mechanism for the hydrogenation of ketones catalyzed by Knölker’s iron-catalyst. Org. Biomol. Chem. 2013, 11, 5264–5277. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Reichau, S.; von der Höh, A.; Leconte, N.; Neudörfl, J.-M. Light-Induced Enantioselective Hydrogenation Using Chiral Derivatives of Casey’s Iron–Cyclopentadienone Catalyst. Organometallics 2011, 30, 3880–3887. [Google Scholar] [CrossRef]
- Hopewell, J.P.; Martins, J.E.D.; Johnson, T.C.; Godfrey, J.; Wills, M. Developing asymmetric iron and ruthenium-based cyclone complexes; complex factors influence the asymmetric induction in the transfer hydrogenation of ketones. Org. Biomol. Chem. 2012, 10, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Natte, K.; Li, W.; Zhou, S.; Neumann, H.; Wu, X.-F. Iron-catalyzed reduction of aromatic aldehydes with paraformaldehyde and H2O as the hydrogen source. Tetrahedron Lett. 2015, 56, 1118–1121. [Google Scholar] [CrossRef]
- Coleman, M.G.; Brown, A.N.; Bolton, B.A.; Guan, H. Iron-Catalyzed Oppenauer-Type Oxidation of Alcohols. Adv. Synth. Catal. 2010, 352, 967–970. [Google Scholar] [CrossRef]
- Pagnoux-Ozherelyeva, A.; Pannetier, N.; Mbaye, M.D.; Gaillard, S.; Renaud, J.-L. Knölker’s Iron Complex: An Efficient in Situ Generated Catalyst for Reductive Amination of Alkyl Aldehydes and Amines. Angew. Chem. Int. Ed. 2012, 124, 5060–5064. [Google Scholar] [CrossRef]
- Tlili, A.; Schrank, J.; Neumann, H.; Beller, M. Discrete Iron Complexes for the Selective Catalytic Reduction of Aromatic, Aliphatic, and α, β-Unsaturated Aldehydes under Water-Gas Shift Conditions. Chem. Eur. J. 2012, 18, 15935–15939. [Google Scholar] [CrossRef]
- Rosas-Hernández, A.; Junge, H.; Beller, M.; Roemelt, M.; Francke, R. Cyclopentadienone iron complexes as efficient and selective catalysts for the electroreduction of CO2 to CO. Catal. Sci. Technol. 2017, 7, 459–465. [Google Scholar] [CrossRef]
- Elangovan, S.; Sortais, J.-B.; Beller, M.; Darcel, C. Iron-Catalyzed α-Alkylation of Ketones with Alcohols. Angew. Chem. Int. Ed. 2015, 54, 14483–14486. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Y.; Wang, H.; Chen, N.-Y.; Lennox, A.J.J.; Friedrich, A.; Xia, L.-M.; Lochbrunner, S.; Junge, H.; Beller, M.; Zhou, S.; et al. Efficient Photocatalytic Water Reduction Using in Situ Generated Knölker’s Iron Complexes. ChemCatChem 2016, 8, 2340–2344. [Google Scholar] [CrossRef]
- Quintard, A.; Constantieux, T.; Rodriquez, J. An Iron/Amine-catalyzed Cascade Process for the Enantioselective Functionalization of Allylic Alcohols. Angew. Chem. 2013, 125, 13121–13125. [Google Scholar] [CrossRef]
- Roudier, M.; Constantieux, T.; Quintard, A.; Rodriquez, J. Enantioselective Cascade Formal Reductive Insertion of Allylic Alcohols into the C(O)-C Bond of 1,3-Diketones: Ready Access to Synthetically Valuable 3-Alkylpentanol Units. Org. Lett. 2014, 16, 2802–2805. [Google Scholar] [CrossRef] [PubMed]
- Roudier, M.; Constantieux, T.; Quintard, A.; Rodriquez, J. Triple Iron/Copper/Iminium Activation for the Efficient Redox Neutral Catalytic Enantioselective Functionalization of Allylic Alcohols. ACS Catal. 2016, 6, 5236–5244. [Google Scholar] [CrossRef]
- Quintard, A.; Rodriquez, J. A Step into an eco-Compatible Future: Iron- and Cobalt-catalyzed Borrowing Hydrogen Transformation. ChemSusChem 2016, 9, 28–30. [Google Scholar] [CrossRef]
- Quintard, A.; Rodriquez, J. Catalytic enantioselective OFF↔ON activation processes initiated by hydrogen transfer: Concepts and challenges. Chem. Commun. 2016, 52, 10456–10473. [Google Scholar] [CrossRef]
- Rodriquez, J.; Quintard, A. Discovery of Eco-compatible Synthetic Paths by a Multi-catalysis Approach. Chimia 2018, 72, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Piarulli, U.; Vailati Fachini, S.; Pignataro, L. Enantioselective Reductions Promoted by (Cyclopentadienone)iron Complexes. Chimia 2017, 71, 580–585. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Akagi, M.; Shimanuki, K.; Kuwahara, S.; Watanabe, M.; Harada, N. A general method for the synthesis of enantiopure aliphatic chain alcohols with established absolute configurations. Part 1. Application of the MαNP acid method to acetylene alcohols. Tetrahedron Asym. 2010, 25, 1456–1465. [Google Scholar] [CrossRef]
- Johnson, T.C.; Clarkson, G.J.; Wills, M. (Cyclopentadienone)iron Shvo Complexes: Synthesis and Application to Hydrogen Transfer Reactions. Organometallics 2011, 30, 1850–1868. [Google Scholar] [CrossRef]
- Hodgkinson, R.; Del Grosso, A.; Clarkson, G.; Wills, M. Iron cyclopentadienone complexes derived from C2-symmetric bis-propargylic alcohols; preparation and application to catalysis. Dalton Trans. 2016, 45, 3992–4005. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, A.; Chamberlain, A.E.; Clarkson, G.J.; Wills, M. Synthesis and applications to catalysis of novel cyclopentadienone iron tricarbonyl complexes. Dalton Trans. 2018, 47, 1451–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, P.; Renom-Carrasco, M.; Vailati Facchini, S.; Pignataro, L.; Lefort, L.; de Vries, J.G.; Ferraccioli, R.; Forni, A.; Piarulli, U.; Gennari, C. Chiral (Cyclopentadienone)iron Complexes for the Catalytic Asymmetric Hydrogenation of Ketones. Eur. J. Org. Chem. 2015, 9, 1887–1893. [Google Scholar] [CrossRef]
- Gajewski, P.; Renom-Carrasco, M.; Vailati Facchini, S.; Pignataro, L.; Lefort, L.; de Vries, J.G.; Ferraccioli, R.; Piarulli, U.; Gennari, C. Synthesis of (R)-BINOL-Derived (Cyclopentadienone)iron Complexes and Their Application in the Catalytic Asymmetric Hydrogenation of Ketones. Eur. J. Org. Chem. 2015, 25, 5526–5536. [Google Scholar] [CrossRef]
- Cettolin, M.; Bai, X.; Lübken, D.; Gatti, M.; Vailati Facchini, S.; Piarulli, U.; Pignataro, L.; Gennari, C. Improving C = N Bond Reductions with (Cyclopentadienone)iron Complexes: Scope and Limitations. Eur. J. Org. Chem. 2019, 4, 647–654. [Google Scholar] [CrossRef]
- Dou, X.; Hayashi, T. Synthesis of Planar Chiral Shvo Catalysts for Asymmetric Transfer Hydrogenation. Adv. Synth. Catal. 2016, 358, 1054–1058. [Google Scholar] [CrossRef]
- Bai, X.; Cettolin, M.; Mazzoccanti, G.; Pierini, M.; Piarulli, U.; Colombo, V.; Dal Corso, A.; Pignataro, L.; Gennari, C. Chiral (cyclopentadienone)iron complexes with a stereogenic plane as pre-catalysts for the asymmetric hydrogenation of polar double bonds. Tetrahedron 2019, 75, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Fleischer, S.; Junge, K.; Beller, M. Cooperative Transition-Metal and Chiral Brønsted Acid Catalysis: Enantioselective Hydrogenation of Imines to Form Amines. Angew. Chem. 2011, 50, 5120–5124. [Google Scholar] [CrossRef]
- Fleischer, S.; Werkmeister, S.; Zhou, S.; Junge, K.; Beller, M. Consecutive Intermolecular Reductive Hydroamination: Cooperative Transition-Metal and Chiral Brønsted Acid Catalysis. Chem. Eur. J. 2012, 18, 9005–9010. [Google Scholar] [CrossRef]
- Zhou, S.; Fleischer, S.; Jiao, H.; Junge, K.; Beller, M. Cooperative Catalysis with Iron and a Chiral Brønsted Acid for Asymmetric Reductive Amination of Ketones. Adv. Synth. Catal. 2014, 356, 3451–3455. [Google Scholar] [CrossRef]
- Hopmann, K. Iron/Brønsted Acid Catalyzed Asymmetric Hydrogenation: Mechanism and Selectivity-Determining Interactions. Chem. Eur. J. 2015, 21, 10020–10030. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, S.; Zhou, S.; Werkmeister, S.; Junge, K.; Beller, M. Cooperative Iron-Brønsted Acid Catalysis: Enantioselective Hydrogenation of Quinoxalines and 2H-1,4-Benzoxazines. Chem. Eur. J. 2013, 19, 4997–5003. [Google Scholar] [CrossRef] [PubMed]
- El-Sepelgy, O.; Brzozowska, A.; Rueping, M. Asymmetric Chemoenzymatic Reductive Acylation of Ketones by a Combined Iron-Catalyzed Hydrogenation-Racemization and Enzymatic Resolution Cascade. ChemSusChem 2017, 10, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, K.P.J.; Gudmundsson, A.; Lewis, K.; Bäckvall, J.-E. Chemoenzymatic Dynamic Kinetic Resolution of Secondary Alcohols Using and Air- and Moisture-Stable Iron Racemization Catalyst. Chem. Eur. J. 2017, 23, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Mérel, D.S.; Gaillard, S.; Ward, T.R.; Renaud, J.-L. Achiral Cyclopentadienone Iron Tricarbonyl Complexes Embedded in Streptadivin: An Access to Artificial Iron Hydrogenases and Application in Asymmetric Hydrogenation. Catal. Lett. 2016, 146, 564–569. [Google Scholar] [CrossRef]
- Kim, M.; Lee, J.W.; Lee, J.E.; Kang, J. Synthesis of Enantiopure Ruthenium Tricarbonyl Complexes of a Bicyclic Cyclopentadienone Derivative. Eur. J. Inorg. Chem. 2008, 16, 2510–2513. [Google Scholar] [CrossRef]
- Funk, T.W.; Mahoney, A.R.; Sponenburg, R.A.; Zimmerman, K.P.; Kim, D.K.; Harrison, E.E. Synthesis and Catalytic Activity of (3,4-Diphenylcyclopentadienone)Iron Tricarbonyl Compounds in Transfer Hydrogenations and Dehydrogentations. Organometallics 2018, 37, 1133–1140. [Google Scholar] [CrossRef]
- Cesari, C.; Sambri, L.; Zacchini, S.; Zanotti, V.; Mazzoni, R. Microwave-Assisted Synthesis of Functionalized Shvo-Type Complexes. Organometallics 2014, 33, 2814–2819. [Google Scholar] [CrossRef]
- Moulin, S.; Dentel, H.; Pagnoux-Ozherelyeva, A.; Gaillard, S.; Poater, A.; Cavallo, L.; Lohier, J.-F.; Renaud, J.-L. Bifunctional (Cyclopentadienone)Iron-Tricarbonyl Complexes: Synthesis, Computational Studies and Application in Reductive Amination. Chem. Eur. J. 2013, 19, 17881–17890. [Google Scholar] [CrossRef]
- Boss, C.; Keese, R. Synthesis of Cycloalkadiynes of Various Ring Size. Tetrahedron 1997, 53, 3111–3122. [Google Scholar] [CrossRef]
- Weaving, R.; Roulland, E.; Monneret, C.; Florent, J.-C. A rapid acces to chiral alkylidene cyclopentenone prostaglandins involving ring-closing methathesis reaction. Tetrahedron Lett. 2003, 44, 2579–2581. [Google Scholar] [CrossRef]
- Melikyan, G.G.; Voorhees, E.; Wild, C.; Spencer, R.; Molnar, J. Carbon tether rigidity as a stereochemical tool directing intramolecular cyclizations. Tetrahedron Lett. 2010, 51, 2287–2290. [Google Scholar] [CrossRef]
- Matsuya, Y.; Ihara, D.; Fukuchi, M.; Honma, D.; Itoh, K.; Tabuchi, A.; Nemoto, H.; Tsuda, M. Synthesis and biological evaluation of pyrethroid insecticide-derivatives as a chemical inducer for Bdnf mRNA expression in neurons. Bioorg. Med. Chem. 2012, 20, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Šebesta, R.; Pizzuti, M.G.; Minnaard, A.J.; Feringa, B.L. Copper-Catalyzed Enantioselective Conjugate Addition of Organometallic Reagents to Acyclic Dienones. Adv. Synth. Catal. 2007, 349, 1931–1937. [Google Scholar] [CrossRef]
- Pan, L.; Yang, K.; Li, G.; Ge, H. Palladium-catalyzed site-selective arylation of aliphatic ketones enabled by a transient ligand. Chem. Commun. 2018, 54, 2759–2762. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.J.; Macmillan, D.; Hogarth, G. Microwave-assisted synthesis of cyclopentadienone iron tricarbonyl complexes: Molecular structures of [{η4-C4R2C(O)C4H8}Fe(CO)3] (R = Ph, 2,4-F2C6H3, 4-MeOC6H4) and attempts to prepare Fe(II) hydroxycyclopentadienyl-hydride complexes. Transit. Met. Chem. 2018, 43, 421–430. [Google Scholar] [CrossRef]
- Corey, E.J.; Venkateswarlu, A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar] [CrossRef]
- Johnson, C.S.; Mottley, C. Theory of the temperature dependence of the NMR spectra of tunneling methyl groups. J. Phys. C Solid State Phys. 1976, 9, 2789–2795. [Google Scholar] [CrossRef]
- Sobieski, J.W. Assessing Steric Bulk of Protecting Groups Via A Computational Determination of Exact Cone Angle (θ°) and Exact Solid Cone Angle (Θ°). Ph.D. Thesis, Kent State University Honors College, Kent, OH, USA, May 2018. [Google Scholar]
- Xu, L.; Li, S.; Jiang, L.; Zhang, G.; Zhang, W. Electronic and steric effects of substituents in 1,3-diphenylprop-2-yn-1-one during ist reaction with Ru3(CO)12. RSC Adv. 2018, 8, 4354–4361. [Google Scholar] [CrossRef]
- Stevens, P.J.; Devlin, F.J.; Chablowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Huzinaga, S.; Anzelm, J.; Klobukowski, M.; Radzio-Andzelm, E.; Sakai, Y.; Tatewaki, H. Gaussian Basis Sets for Molecular Calculations; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Hach, V. Meerwein-Ponndorf-Verley Reduction of Mono- and Bicyclic Ketones. Rate of Reaction. J. Org. Chem. 1973, 38, 293–299. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Fridrich, B.; Dibó, G.; Székely, E.; Mika, L.T. Direct asymmetric reduction of levulinic acid to gamma-valerolactone: Synthesis of a chiral platform molecule. Green Chem. 2015, 17, 5189–5195. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | R3 | Isolated Yield (%) | Enantiomeric Configuration | Specific Rotation |
|---|---|---|---|---|---|---|
| 11a | Me | - | - | n/a | R | n/d |
| 11b | Ph | - | - | n/a | R | n/d |
| 11c | Me | - | - | n/a | rac | 0 |
| 12a | Me | TIPS | - | 92.7 | R | +96.1 |
| 12b | Me | TBDPS | - | 91.3 | R | +288.3 |
| 12c | Ph | TIPS | - | 85.5 | R | −14.8 |
| 12d | Me | TIPS | - | 76.3 | rac | 0 |
| 13a | Me | TIPS | (CH2)4 | 89.1 | R,R | +122.6 |
| 13b | Me | TBDPS | (CH2)4 | 38.7 | R,R | +364.1 |
| 13c | Ph | TIPS | (CH2)4 | 60.8 | R,R | −37.0 |
| 13d | Me | TIPS | (CH2)3 | 70.3 | R,R | +131.9 |
| 13e | Me | TIPS | (CH2)4 | 66.8 | rac | 0 |
| 14a | Me | TIPS | (CH2)4 | 51.4 | R,R | +16.5 |
| 14b | Me | TBDPS | (CH2)4 | 33.8 | R,R | +62.5 |
| 14c | Ph | TIPS | (CH2)4 | 18.2 | R,R | −107.5 |
| 14d | Me | TIPS | (CH2)3 | 51.9 | R,R | +8.4 |
| 14e 1 | Me | TIPS | (CH2)4 | 48.2 | rac | 0 |
| Complex | 1H-NMR | |||
|---|---|---|---|---|
| Methine | Methyl | |||
| δ (ppm) | Δδ (Hz) | δ (ppm) | Δδ (Hz) | |
| 14a | 4.99 | 66.3 | 1.47 | 6.4 |
| 14b | 4.82 | 85.7 | 1.32 | 6.4 |
| 14c | 5.94 | 52.7 | - | - |
| 14d | 4.89 | 28.1 | 1.47 | 18.8 |
| Complex | 13C-NMR | |||||||
|---|---|---|---|---|---|---|---|---|
| 3,4-Cp | 2,5-Cp | Methine | Methyl | |||||
| δ (ppm) | Δδ (Hz) | δ (ppm) | Δδ (Hz) | δ (ppm) | Δδ (Hz) | δ (ppm) | Δδ (Hz) | |
| 14a | 100.5 | 113.9 | 87.2 | 61.2 | 64.1 | 194.6 | 25.2 | 261.2 |
| 14b | 100.6 | 74.5 | 86.0 | 110.4 | 64.3 | 98.0 | 25.1 | 237.1 |
| 14c | 99.8 | 156.4 | 87.7 | 248.0 | 69.2 | 223.6 | - | - |
| 14d | 105.5 | 60.6 | 88.4 | 28.0 | 64.5 | 52.6 | 27.6 | 5.2 |
| Entry | LFe(CO)3 (14) | LFe(CO)2[vac] (15) | (LH)FeH(CO)2 (16) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| θ (°) | φ (°) | |θ − φ| | θ (°) | φ (°) | |θ − φ| | θ (°) | φ (°) | |θ − φ| | |
| a | −133.2 | −76.6 | 56.6 | −135.6 | −80.7 | 54.9 | −168.7 | −80.8 | 87.9 |
| b | −142.5 | −78.1 | 64.3 | −127.6 | −84.6 | 43.0 | −169.2 | −86.6 | 82.6 |
| c’ | −159.2 | −90.8 | 68.4 | −155.0 | −92.8 | 62.3 | −169.0 | −96.5 | 72.5 |
| d | −132.5 | −77.5 | 54.9 | −129.8 | −80.0 | 49.8 | −170.1 | −82.4 | 87.7 |
| f | 75.4 | −75.4 | 0.0 | n/a | n/a | ||||
| g | −135.2 | 134.9 | 0.3 | ||||||

| Entry | Solvent | equiv. iPrOH | Temperature (°C) | Conversion (%) | Ee (%) | Enantiomeric Configuration |
|---|---|---|---|---|---|---|
| 1 1 | none | 2.5 | 80 | 24 | 36 | S |
| 2 1 | none | 10 | 80 | 15 | 41 | S |
| 3 1 | none | 17.5 | 80 | 9 | 42 | S |
| 4 1 | none | 25 | 80 | 8 | 42 | S |
| 5 2 | toluene | 10 | 80 | 7 | 39 | S |
| 6 2 | tBuOH | 10 | 80 | 3 | 42 | S |
| 7 | none | 10 | 100 | 25 | 40 | S |
| 8 | none | 10 | 60 | 11 | 45 | S |
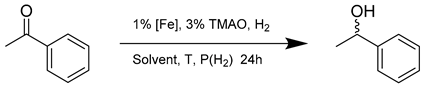
| Entry | Catalyst | Solvent | Temperature (°C) | H2 Pressure (bar) | Conversion (%) | Ee (%) | Enantiomeric Configuration |
|---|---|---|---|---|---|---|---|
| 1 | 14a | iPrOH | 80 | 50 | 99 | 33 | S |
| 2 | 14a | MeOH | 80 | 50 | 5 | 30 | S |
| 3 | 14a | EtOH | 80 | 50 | 99 | 33 | S |
| 4 | 14a | toluene | 80 | 50 | 99 | 33 | S |
| 5 | 14a | 2-MeTHF | 80 | 50 | 65 | 30 | S |
| 6 | 14b | iPrOH | 80 | 50 | 99 | 21 | S |
| 7 | 14c | iPrOH | 80 | 50 | 99 | 41 | R |
| 8 | 14d | iPrOH | 80 | 50 | 99 | 21 | S |
| 9 | 14e | iPrOH | 80 | 50 | 99 | 0 | - |
| 10 | 14a | iPrOH | 60 | 50 | 74 | 40 | S |
| 11 | 14b | iPrOH | 60 | 50 | 29 | 36 | S |
| 12 | 14c | iPrOH | 60 | 50 | 14 | 62 | R |
| 13 | 14d | iPrOH | 60 | 50 | 97 | 34 | S |
| 14 1 | 14a | iPrOH | 60 | 1 | 45 | 44 | S |
| 15 1 | 14a | iPrOH | 22 | 1 | 4 | 60 | S |
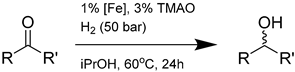
| Entry | Substrate | Catalyst | Conversion (%) | Ee (%) | Enantiomeric Configuration |
|---|---|---|---|---|---|
| 1 |  | 14a | 99 | 29 | S |
| 2 |  | 14a | 66 | 26 | S |
| 3 |  | 14a | 97 | 29 | S |
| 4 | 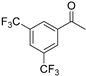 | 14a | 97 | 38 | S |
| 14c | 93 | 56 | R | ||
| 14d | 72 | 34 | S | ||
| 5 | 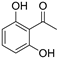 | 14a | 99 | 47 | S |
| 6 |  | 14a | 69 | 26 | S |
| 14c | 32 | 63 | R | ||
| 14d | 95 | 23 | S | ||
| 7 | 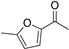 | 14a | 100 | 14 | S |
| 14c | 41 | 28 | R | ||
| 8 |  | 14a | 100 | 3 | S |
| 14c | 2 | 22 | R | ||
| 9 |  | 14a | 98 | 47 | S |
| 14c | 53 | 70 | R | ||
| 14d | 68 | 38 | S | ||
| 10 1 | 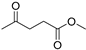 | 14a | 50 | 1 | S |
| 14c | 32 | 4 | R |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Slagmaat, C.A.M.R.; Chou, K.C.; Morick, L.; Hadavi, D.; Blom, B.; De Wildeman, S.M.A. Synthesis and Catalytic Application of Knölker-Type Iron Complexes with a Novel Asymmetric Cyclopentadienone Ligand Design. Catalysts 2019, 9, 790. https://doi.org/10.3390/catal9100790
van Slagmaat CAMR, Chou KC, Morick L, Hadavi D, Blom B, De Wildeman SMA. Synthesis and Catalytic Application of Knölker-Type Iron Complexes with a Novel Asymmetric Cyclopentadienone Ligand Design. Catalysts. 2019; 9(10):790. https://doi.org/10.3390/catal9100790
Chicago/Turabian Stylevan Slagmaat, Christian A. M. R., Khi Chhay Chou, Lukas Morick, Darya Hadavi, Burgert Blom, and Stefaan M. A. De Wildeman. 2019. "Synthesis and Catalytic Application of Knölker-Type Iron Complexes with a Novel Asymmetric Cyclopentadienone Ligand Design" Catalysts 9, no. 10: 790. https://doi.org/10.3390/catal9100790