Methane and Ethane Steam Reforming over MgAl2O4-Supported Rh and Ir Catalysts: Catalytic Implications for Natural Gas Reforming Application

 ,
,
Abstract
:1. Introduction
- They must be active over a wide range of operating temperatures, which are largely dictated by efficiency of the solar collector and local solar irradiance.
- To facilitate small reactor footprints, they must operate at high throughput rates.
- They must work under continuous shutdown cycles (i.e., sun/shade).
- To reduce energy requirements for steam vaporization, they must operate with reduced steam concentrations (e.g., low S/C molar ratios).
- They must tolerate temperature and gas composition changes caused by abrupt variations in local conditions.
2. Results and Discussion
2.1. Catalyst Characterization
- STEM characterization shows that high dispersion that can be achieved with Rh and Ir catalysts supported on MgAl2O4 compared to Ni.
- TPR characterization showed that, associated with a greater dispersion, reducibility is enhanced, particularly for Rh where TPR profiles indicate a narrower particle size distribution and full reduction at low temperatures (broad single peak centered at 112 °C).
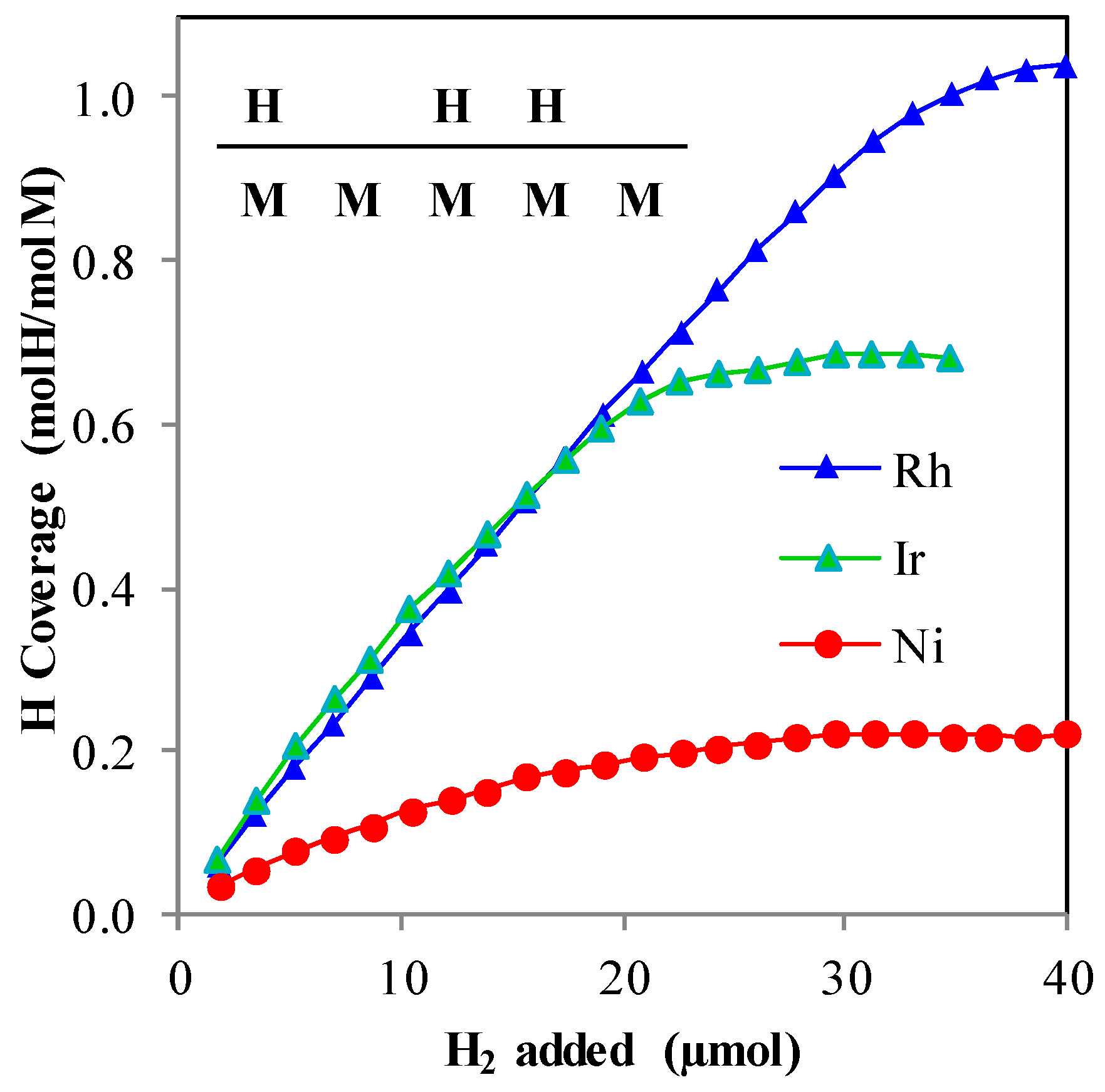
- H2 adsorption demonstrated that surface stoichiometry for H2 on metal surfaces can vary greatly under reforming conditions.
2.2. Steam Reforming Activity Comparison Using Natural Gas Simulant
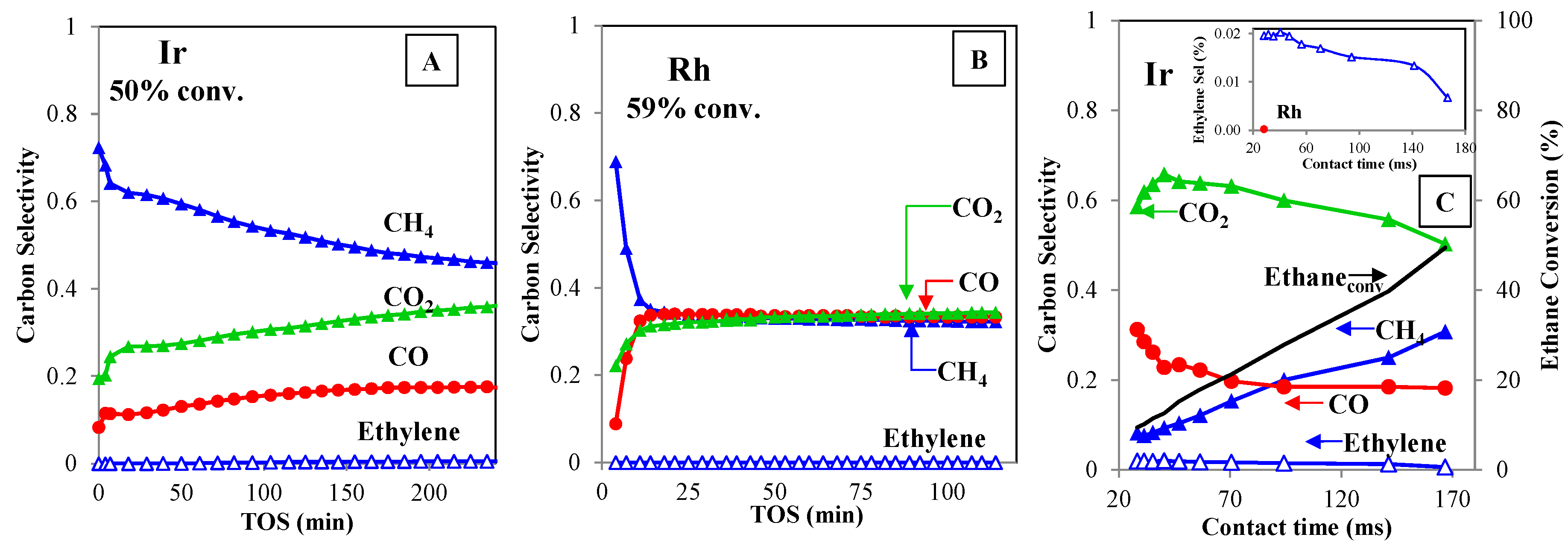
2.3. Methane and Ethane Steam Reforming Activity Comparison
2.4. Ethane Steam Reforming over Rh vs. Ir: Mechanistic Insights
- (1)
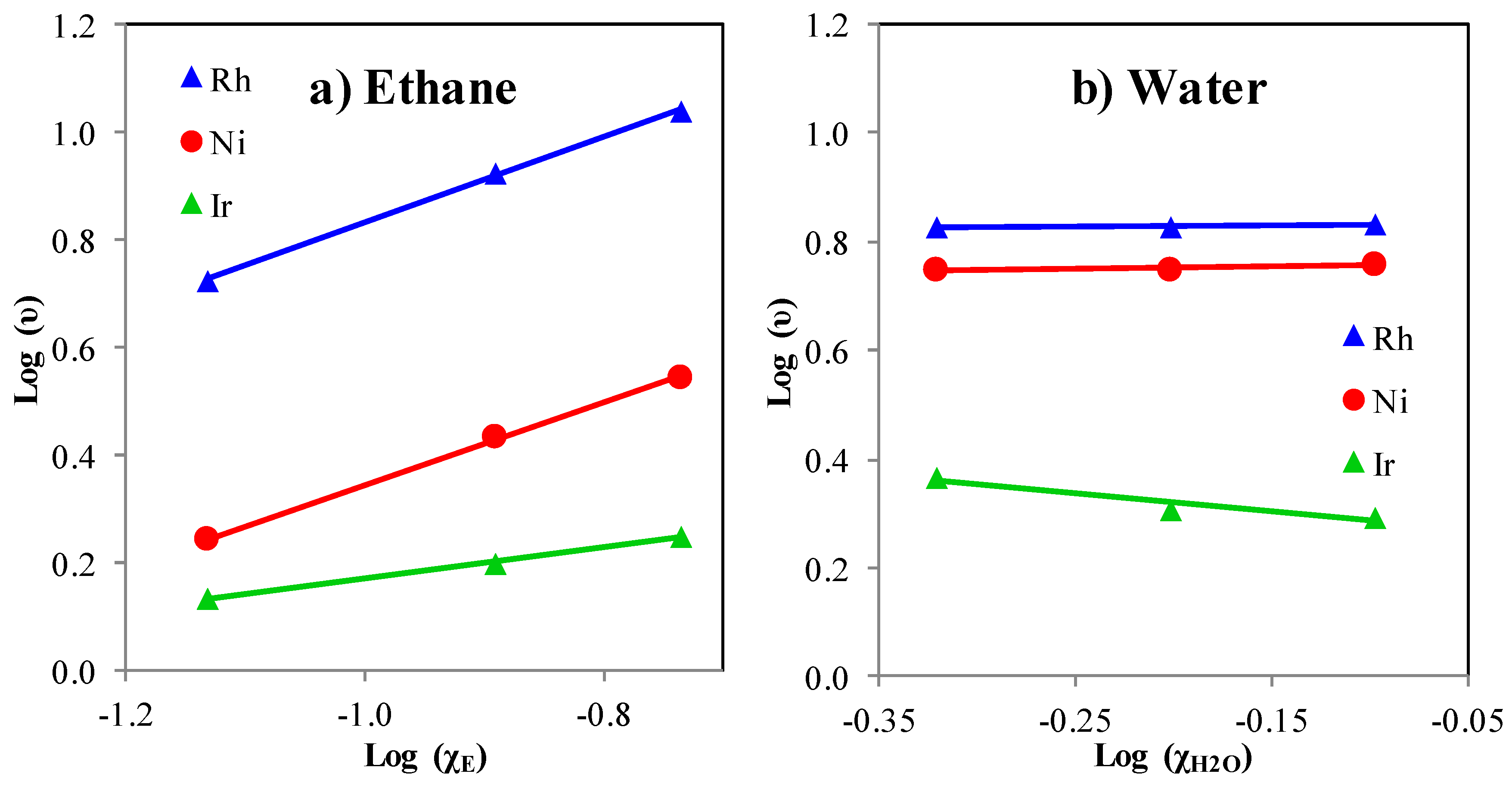
- Ethane steam reforming rate does not increase with higher S/C ratios. A wide range of S/C ratios were used to eliminate oxidation (reforming steps) as being responsible for the lower reaction rate on Ir (S/C up to 10). This is illustrated in Figure 6. This result is consistent with kinetic literature studies reporting that the reforming reaction is independent of the partial water pressure (zero order with respect to water) [36].
- (2)
- Low activity over Ir catalyst is not caused by coke formation. In spite of higher carbon content on Ir spent catalyst (Table 2), coke (or at least hard coke) is not formed. A series of control experiments showed that high activity is consistently low for ethane reforming after repeated methane/ethane cycles (see Table S1).
- (3)
- Lower ethane partial pressure increases catalytic conversion. This is consistent with hydrocarbon hydrogenolysis literature reporting that hydrocarbons equilibrate with metal surfaces by sequential dehydrogenation steps. As a consequence, a lower ethane partial pressure (from 14 to 8 mol %) causes a lower coverage that increases the number of active sites (ethane conversion increases from 10 to 47%).
- (4)
- Ethane reforming over Ir supported on Al2O3 shows similar trends. Ir supported by Al2O3 is active for methane reforming; however, ethane steam reforming is severely hampered. Ir supported on alumina has a bigger particle size (4 nm) [20].
2.5. Ethane Reforming Kinetic Measurements
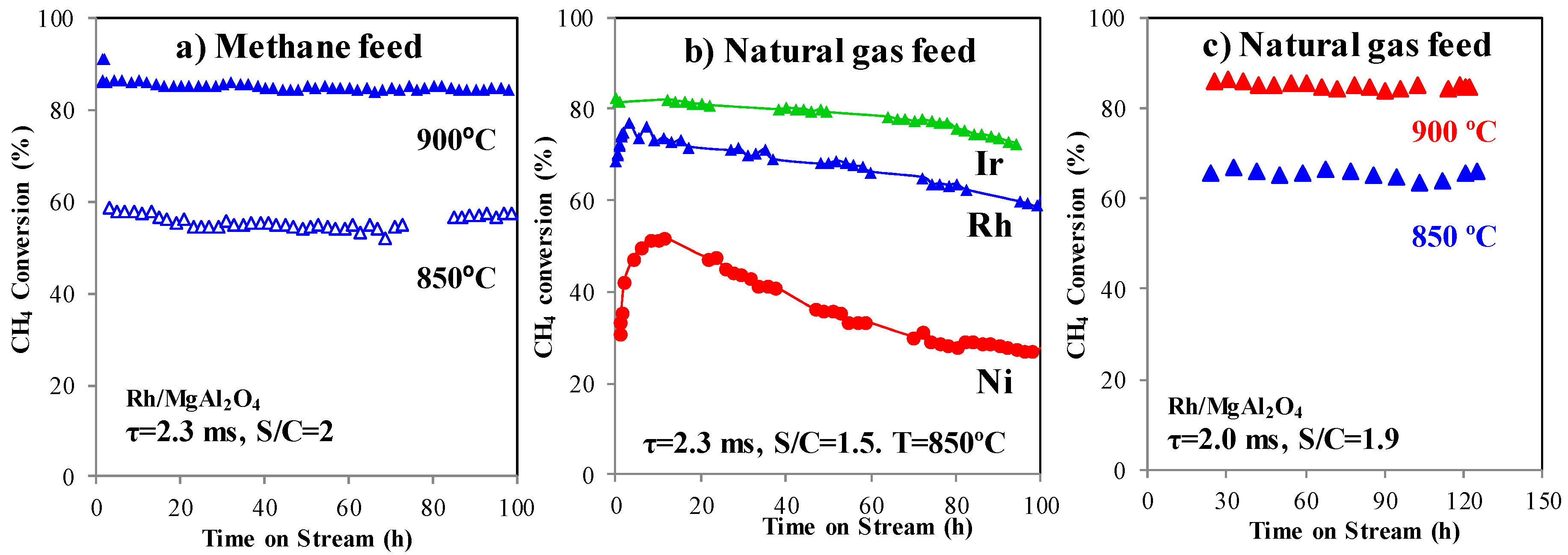
2.6. Catalyst Stability
3. Experimental Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Activity Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- DOE. Hydrogen Production: Natural Gas Reforming, Washington, DC, USA. Available online: https://www.energy.gov/eere/fuelcells/hydrogen-production-natural-gas-reforming (accessed on 23 April 2019).
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, M.R. Rahimpour, Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. Process Intensif. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Wood, D.A.; Nwaoha, C.; Towler, B.F. Gas-to-liquids (GTL): A review of an industry offering several routes for monetizing natural gas. J. Nat. Gas Sci. Eng. 2012, 9, 196–208. [Google Scholar] [CrossRef]
- Glasser, D.; Hildebrandt, D.; Liu, X.; Lu, X.; Masuku, C.M. Recent advances in understanding the Fischer–Tropsch synthesis (FTS) reaction. Curr. Opin. Chem. Eng. 2012, 1, 296–302. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Jakobsen, J.; Shim, S.; Kleis, J.; Andersson, M.; Rossmeisl, J.; Abildpedersen, F.; Bligaard, T.; Helveg, S.; Hinnemann, B.; et al. First principles calculations and experimental insight into methane steam reforming over transition metal catalysts. J. Catal. 2008, 259, 147–160. [Google Scholar] [CrossRef]
- Zheng, R.; Diver, R.; Caldwell, D.; Fritz, B.; Cameron, R.; Humble, P.; TeGrotenhuis, W.; Dagle, R.; Wegeng, R. Integrated Solar Thermochemical Reaction System for Steam Methane Reforming. Energy Procedia 2015, 69, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Simakov, D.S.A.; Wright, M.M.; Ahmed, S.; Mokheimer, E.M.A.; Román-Leshkov, Y. Solar thermal catalytic reforming of natural gas: A review on chemistry, catalysis and system design. Catal. Sci. Technol. 2015, 5, 1991–2016. [Google Scholar] [CrossRef]
- Steinfel, A. Solar thermochemical production of hydrogen––A review. Sol. Energy 2005, 78, 603–615. [Google Scholar] [CrossRef]
- Wang, Y.; Chin, Y.H.; Rozmiarek, R.T.; Johnson, B.R.; Gao, Y.; Watson, J.; Tonkovich, A.Y.L.; Wiel, D.P.V. Highly active and stable Rh/MgOAl2O3 catalysts for methane steam reforming. Catal. Today 2004, 98, 575–581. [Google Scholar] [CrossRef]
- Palo, D.R.; Stenkamp, V.S.; Dagle, R.A.; Jovanovic, G.N. Industrial Applications of Microchannel Process Technology in the United States. In Micro Process Engineering; Kockmann, N., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 387–414. [Google Scholar]
- Kehres, J.; Andreasen, J.W.; Fløystad, J.B.; Liu, H.; Molenbroek, A.; Jakobsen, J.G.; Chorkendorff, I.; Nielsen, J.H.; Høydalsvik, K.; Breiby, D.W.; et al. Reduction of a Ni/Spinel Catalyst for Methane Reforming. J. Phys. Chem. C 2015, 119, 1424–1432. [Google Scholar] [CrossRef]
- Salhi, N.; Boulahouache, A.; Petit, C.; Kiennemann, A.; Rabia, C. Steam reforming of methane to syngas over NiAl2O4 spinel catalysts. Int. J. Hydrog. Energy 2011, 36, 11433–11439. [Google Scholar] [CrossRef]
- Boukha, Z.; Jiménez-González, C.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Synthesis, characterisation and performance evaluation of spinel-derived Ni/Al2O3 catalysts for various methane reforming reactions. Appl. Catal. B Environ. 2014, 158–159, 190–201. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst design for dry reforming of methane: Analysis review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Ligthart, D.A.J.M.; van Santen, R.A.; Hensen, E.J.M. Influence of particle size on the activity and stability in steam methane reforming of supported Rh nanoparticles. J. Catal. 2011, 280, 206–220. [Google Scholar] [CrossRef]
- Duarte, R.B.; Krumeich, F.; van Bokhoven, J.A. Structure, Activity, and Stability of Atomically Dispersed Rh in Methane Steam Reforming. ACS Catal. 2014, 4, 1279–1286. [Google Scholar] [CrossRef]
- Mei, D.; Lebarbier, V.M.; Rousseau, R.; Glezakou, V.-A.; Albrecht, K.O.; Kovarik, L.; Flake, M.; Dagle, R.A. Comparative Investigation of Benzene Steam Reforming over Spinel Supported Rh and Ir Catalysts. ACS Catal. 2013, 3, 1133–1143. [Google Scholar] [CrossRef]
- Mei, D.; Glezakou, V.-A.; Lebarbier, V.; Kovarik, L.; Wan, H.; Albrecht, K.O.; Gerber, M.; Rousseau, R.; Dagle, R.A. Highly active and stable MgAl2O4-supported Rh and Ir catalysts for methane steam reforming: A combined experimental and theoretical study. J. Catal. 2014, 316, 11–23. [Google Scholar] [CrossRef]
- Dagle, V.L.; Dagle, R.; Kovarik, L.; Genc, A.; Wang, Y.-G.; Bowden, M.; Wan, H.; Flake, M.; Glezakou, V.-A.; King, D.L.; et al. Steam reforming of hydrocarbons from biomass-derived syngas over MgAl2O4-supported transition metals and bimetallic IrNi catalysts. Appl. Catal. B Environ. 2016, 184, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Speight, J.G. Composition and Properties, Natural Gas—A Basic Handbook; Gulf Publishing Company: Houston, TX, USA, 2007. [Google Scholar]
- U.S.E.I. Administration. Ethane Production Growth Led to Record U.S. Natural Gas Plant Liquids Production in 2017, 2018; US Energy Initiatives Corporation: Santa Clarita, CA, USA, 2018. [Google Scholar]
- Adesina, A.A.; Trimm, D.L.; Cant, N.W. Kinetic study of iso-octane steam reforming over a nickel-based catalyst. Chem. Eng. J. 2004, 99, 131–136. [Google Scholar]
- Sperle, T.; Chen, D.; Lødeng, R.; Holmen, A. Pre-reforming of natural gas on a Ni catalyst. Appl. Catal. A Gen. 2005, 282, 195–204. [Google Scholar] [CrossRef]
- Takeguchi, T.; Kani, Y.; Yano, T.; Kikuchi, R.; Eguchi, K.; Tsujimoto, K.; Uchida, Y.; Ueno, A.; Omoshiki, K.; Aizawa, M. Study on steam reforming of CH4 and C2 hydrocarbons and carbon deposition on Ni-YSZ cermets. J. Power Sources 2002, 112, 588–595. [Google Scholar] [CrossRef]
- Jeong, H.; Kang, M. Hydrogen production from butane steam reforming over Ni/Ag loaded MgAl2O4 catalyst. Appl. Catal. B Environ. 2010, 95, 446–455. [Google Scholar] [CrossRef]
- Baek, B.; Aboiralor, A.; Wang, S.; Kharidehal, P.; Grabow, L.C.; Massa, J.D. Strategy to improve catalytic trend predictions for methane oxidation and reforming. AIChE J. 2017, 63, 66–77. [Google Scholar] [CrossRef]
- Angeli, S.D.; Monteleone, G.; Giaconia, A.; Lemonidou, A.A. State-of-the-art catalysts for CH4 steam reforming at low temperature. Int. J. Hydrog. Energy 2014, 39, 1979–1997. [Google Scholar] [CrossRef]
- Halabi, M.H.; de Croon, M.H.J.M.; van der Schaaf, J.; Cobden, P.D.; Schouten, J.C. Low temperature catalytic methane steam reforming over ceria–zirconia supported rhodium. Appl. Catal. A Gen. 2010, 389, 68–79. [Google Scholar] [CrossRef]
- Morlanés, N. Reaction mechanism of naphtha steam reforming on nickel-based catalysts, and FTIR spectroscopy with CO adsorption to elucidate real active sites. Int. J. Hydrog. Energy 2013, 38, 3588–3596. [Google Scholar] [CrossRef]
- Wei, J. Structural requirements and reaction pathways in methane activation and chemical conversion catalyzed by rhodium. J. Catal. 2004, 225, 116–127. [Google Scholar] [CrossRef]
- Kneale, B.; Ross, J.R.H. The steam reforming of ethane over nickel/alumina catalysts. Faraday Discuss. Chem. Soc. 1981, 72, 157–171. [Google Scholar] [CrossRef]
- Graf, P.O.; Mojet, B.L.; van Ommen, J.G.; Lefferts, L. Comparative study of steam reforming of methane, ethane and ethylene on Pt, Rh and Pd supported on yttrium-stabilized zirconia. Appl. Catal. A Gen. 2007, 332, 310–317. [Google Scholar] [CrossRef]
- Schädel, B.T.; Duisberg, M.; Deutschmann, O. Steam reforming of methane, ethane, propane, butane, and natural gas over a rhodium-based catalyst. Catal. Today 2009, 142, 42–51. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Mechanism and Site Requirements for Activation and Chemical Conversion of Methane on Supported Pt Clusters and Turnover Rate Comparisons among Noble Metals. J. Phys. Chem. B 2004, 108, 4094–4103. [Google Scholar] [CrossRef]
- Mei, D.; Dagle, V.L.; Xing, R.; Albrecht, K.O.; Dagle, R.A. Steam Reforming of Ethylene Glycol over MgAl2O4 Supported Rh, Ni, and Co Catalysts. ACS Catal. 2016, 6, 315–325. [Google Scholar] [CrossRef]
- Xing, R.; Dagle, V.L.; Flake, M.; Kovarik, L.; Albrecht, K.O.; Deshmane, C.; Dagle, R.A. Steam reforming of fast pyrolysis-derived aqueous phase oxygenates over Co, Ni, and Rh metals supported on MgAl2O4. Catal. Today 2016, 269, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Ma, Z.; Miao, C.; Yue, Y.; Hua, W.; Gao, Z. Catalytic decomposition of N2O over Rh/Zn–Al2O3 catalysts. RSC Adv. 2017, 7, 4243–4252. [Google Scholar] [CrossRef]
- Mizuno, T.; Matsumura, Y.; Nakajima, T.; Mishima, S. Effect of support on catalytic properties of Rh catalysts for steam reforming of 2-propanol. Int. J. Hydrog. Energy 2003, 28, 1393–1399. [Google Scholar] [CrossRef]
- Flaherty, D.W.; Hibbitts, D.D.; Gürbüz, E.I.; Iglesia, E. Theoretical and kinetic assessment of the mechanism of ethane hydrogenolysis on metal surfaces saturated with chemisorbed hydrogen. J. Catal. 2014, 311, 350–356. [Google Scholar] [CrossRef]
- Hernández-Cristóbal, O.; Díaz, G.; Gómez-Cortés, A. Effect of the Reduction Temperature on the Activity and Selectivity of Titania-Supported Iridium Nanoparticles for Methylcyclopentane Reaction. Ind. Eng. Chem. Res. 2014, 53, 10097–10104. [Google Scholar] [CrossRef]
- Kip, B.J.; van Grondelle, J.; Martens, J.H.A.; Prins, R. Preparation and characterization of very highly dispersed iridium on Al2O3 and SiO2. Appl. Catal. 1986, 26, 353–373. [Google Scholar] [CrossRef] [Green Version]
- Nuernberg, G.D.B.; Fajardo, H.V.; Foletto, E.L.; Hickel-Probst, S.M.; Carreño, N.L.V.; Probst, L.F.D.; Barrault, J. Methane conversion to hydrogen and nanotubes on Pt/Ni catalysts supported over spinel MgAl2O4. Catal. Today 2011, 176, 465–469. [Google Scholar] [CrossRef]
- Horváth, A.; Guczi, L.; Kocsonya, A.; Sáfrán, G.; la Parola, V.; Liotta, L.F.; Pantaleo, G.; Venezia, A.M. Sol-derived AuNi/MgAl2O4 catalysts: Formation, structure and activity in dry reforming of methane. Appl. Catal. A Gen. 2013, 468, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Gillan, C.; Fowles, M.; French, S.; Jackson, S.D. Ethane Steam Reforming over a Platinum/Alumina Catalyst: Effect of Sulfur Poisoning. Ind. Eng. Chem. Res. 2013, 52, 13350–13356. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kulkarni, A.R.; Aljama, H.; Montoya, J.H.; Yoo, J.S.; Tsai, C.; Abild-Pedersen, F.; Studt, F.; Nørskov, J.K. Understanding trends in C–H bond activation in heterogeneous catalysis. Nat. Mater. 2016, 16, 225. [Google Scholar] [CrossRef] [PubMed]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Burch, R.; Loader, P.K.; Urbano, F.J. Some aspects of hydrocarbon activation on platinum group metal combustion catalysts. Catal. Today 1996, 27, 243–248. [Google Scholar] [CrossRef]
- Sinfelt, J.H.; Yates, D.J.C. Catalytic hydrogenolysis of ethane over the noble metals of Group VIII. J. Catal. 1967, 8, 82–90. [Google Scholar] [CrossRef]
- Bond, G.C. Kinetics of alkane reactions on metal catalysts: Activation energies and the compensation effect. Catal. Today 1999, 49, 41–48. [Google Scholar] [CrossRef]
- Kuz’min, I.V.; Zeigarnik, A.V. Microkinetic Modeling of Ethane Hydrogenolysis on Metals. Kinet. Catal. 2004, 45, 561–569. [Google Scholar] [CrossRef]
- Vincent, R.S.; Lindstedt, R.P.; Malik, N.A.; Reid, I.A.B.; Messenger, B.E. The chemistry of ethane dehydrogenation over a supported platinum catalyst. J. Catal. 2008, 260, 37–64. [Google Scholar] [CrossRef]
- Goddard, S.A.; Amiridis, M.D.; Rekoske, J.E.; Cardona-Martinez, N.; Dumesic, J.A. Kinetic simulation of heterogeneous catalytic processes: Ethane hydrogenolysis over supported group VIII metals. J. Catal. 1989, 117, 155–169. [Google Scholar] [CrossRef]
- Mark, M.F.; Maier, W.F. CO2-Reforming of Methane on Supported Rh and Ir Catalysts. J. Catal. 1996, 164, 122–130. [Google Scholar] [CrossRef]
- Sinfelt, J.H. Kinetics of ethane hydrogenolysis. J. Catal. 1972, 27, 468–471. [Google Scholar] [CrossRef]
- Sinfelt, J.H. Specificity in Catalytic Hydrogenolysis by Metals. In Advances in Catalysis; Eley, D.D., Pines, H., Weisz, P.B., Eds.; Academic Press: Cambridge, MA, USA, 1973; pp. 91–119. [Google Scholar]
- Guczi, L.; Gudkov, B.S.; Tétényi, P. The mechanism of catalytic hydrogenolysis of ethane over nickel. J. Catal. 1972, 24, 187–196. [Google Scholar] [CrossRef]
- Kahle, L.C.S.; Roussière, T.; Maier, L.; Delgado, K.H.; Wasserschaff, G.; Schunk, S.A.; Deutschmann, O. Methane Dry Reforming at High Temperature and Elevated Pressure: Impact of Gas-Phase Reactions. Ind. Eng. Chem. Res. 2013, 52, 11920–11930. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Metal Loading (wt.%) | Metal Particle Size TEM (nm) | Catalyst Surface Area BET a (m2/g) |
|---|---|---|---|
| 5Ir/MgAl2O4 [20] | 5.0 | 1.0 | 133 |
| 5Rh/MgAl2O4 [20] | 5.0 | 2.0 | 117 |
| 15Ni/MgAl2O4 [21] | 15.0 | 6.6 | 86 |
| Condition | 5%Rh/MgAl2O4 | 5%Ir/MgAl2O4 |
|---|---|---|
| Metal on catalyst surface (µmol) a | 23.5 | 25.2 |
| Conversion after 1 h | 60.0 | 8.3 |
| Conversion after 2 h | 58.9 | 8.1 |
| Deactivation rate (%) | 1.8 | 2.4 |
| ppm ethylene formed | 50 | 2000 |
| Selectivity towards methane (mol C %) | 32 | 9.6 |
| Carbon on spent catalyst (wt.%) | bdl b | 2.8 c |
| Catalyst | nEthane | nwater | Ea(kJ/mol) a |
|---|---|---|---|
| Rh/MgAl2O4 | 0.79 | 0 | 26.8 |
| Ni/MgAl2O4 | 0.76 | 0 | 79.1 |
| Ir/MgAl2O4 | 0.29 | −0.3 | 95.7 |
| Catalyst | Metal Loading (wt.%) | Metal Dispersion (%) | Metal Particle Size (nm) | H Stoichiometry H/Msurf (mol/mol) | nEthane | nwater | Ea (kJ/mol) |
|---|---|---|---|---|---|---|---|
| 5Rh/MgAl2O4 | 5 | 50 | 2.0 | 1:1 | 0.79 | 0 | 26.8 |
| 5Ir/MgAl2O4 | 5 | 100 | 1.0 | 0.7:1 | 0.29 | −0.3 | 95.7 |
| 15Ni/MgAl2O4 | 15 | 15.4 | 6.5 | 0.2:1 | 0.76 | 0 | 79.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saavedra Lopez, J.; Lebarbier Dagle, V.; Deshmane, C.A.; Kovarik, L.; Wegeng, R.S.; Dagle, R.A. Methane and Ethane Steam Reforming over MgAl2O4-Supported Rh and Ir Catalysts: Catalytic Implications for Natural Gas Reforming Application. Catalysts 2019, 9, 801. https://doi.org/10.3390/catal9100801
Saavedra Lopez J, Lebarbier Dagle V, Deshmane CA, Kovarik L, Wegeng RS, Dagle RA. Methane and Ethane Steam Reforming over MgAl2O4-Supported Rh and Ir Catalysts: Catalytic Implications for Natural Gas Reforming Application. Catalysts. 2019; 9(10):801. https://doi.org/10.3390/catal9100801
Chicago/Turabian StyleSaavedra Lopez, Johnny, Vanessa Lebarbier Dagle, Chinmay A. Deshmane, Libor Kovarik, Robert S. Wegeng, and Robert A. Dagle. 2019. "Methane and Ethane Steam Reforming over MgAl2O4-Supported Rh and Ir Catalysts: Catalytic Implications for Natural Gas Reforming Application" Catalysts 9, no. 10: 801. https://doi.org/10.3390/catal9100801