Effect of Operating Temperature, Pressure and Potassium Loading on the Performance of Silica-Supported Cobalt Catalyst in CO2 Hydrogenation to Hydrocarbon Fuel

Abstract
:1. Introduction
2. Results and Discussion
2.1. Surface Area and Porosity
2.2. X-ray Diffraction
2.3. Temperature-Programmed Reduction (TPR)
2.4. CO2 Temperature-Programmed Desorption (CO2-TPD)
2.5. X-ray Photoelectron Spectroscopy (XPS)
2.6. Catalyst Testing
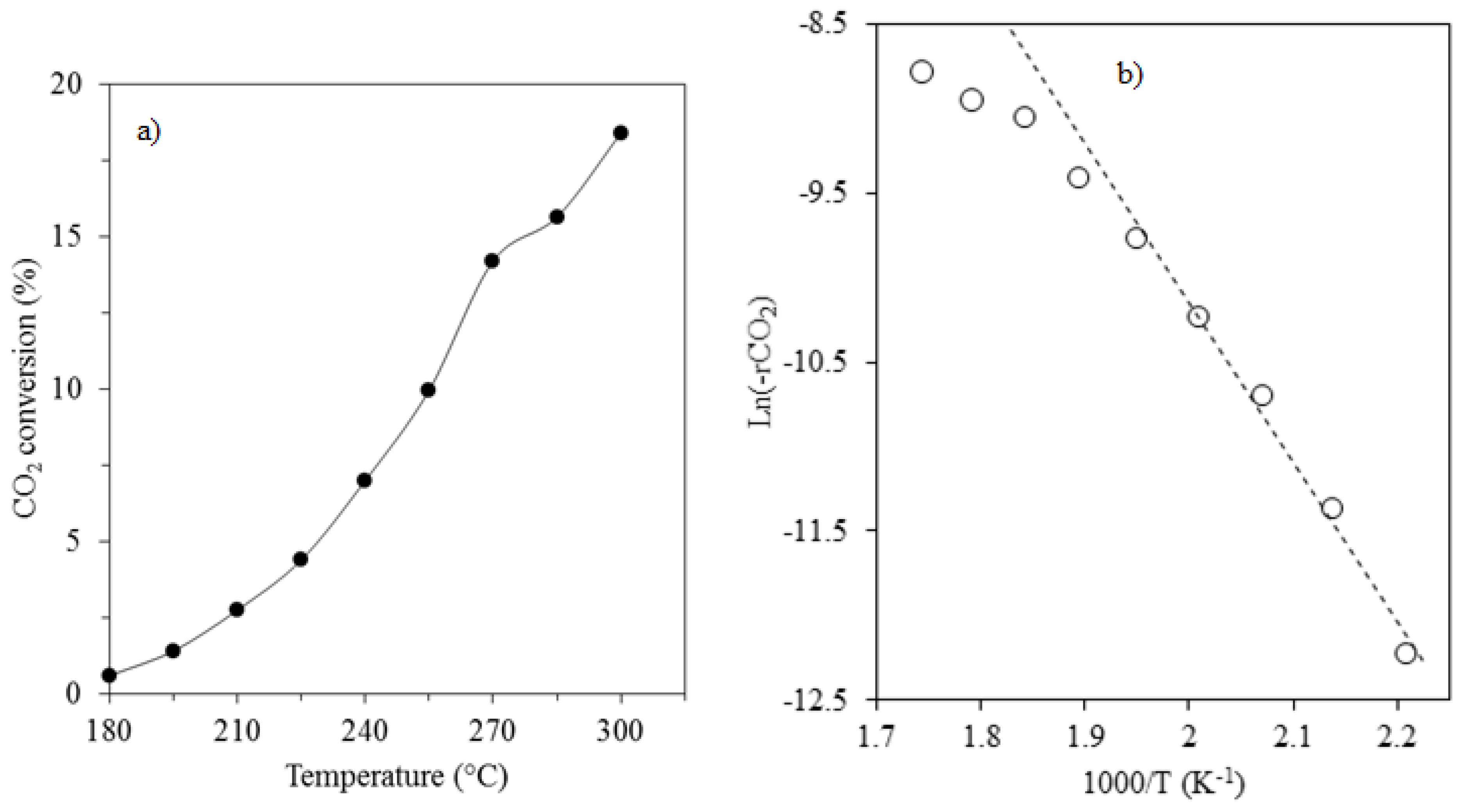
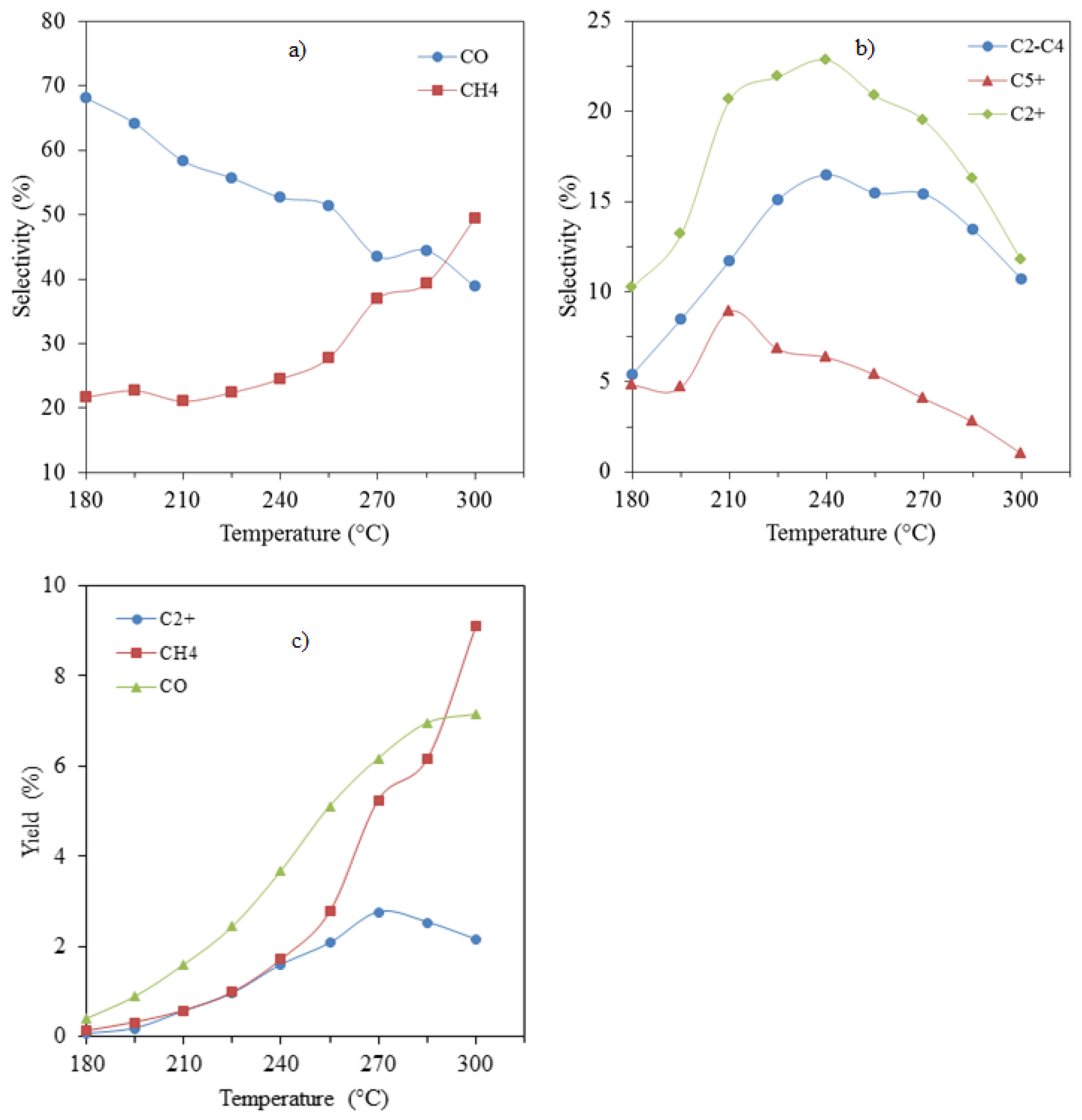
2.6.1. Effect of Temperature
2.6.2. Effect of Pressure
2.6.3. Effect of Potassium Addition
3. Materials and Methods
3.1. Catalyst Synthesis
3.2. Catalyst Characterization
3.3. Catalyst Testing
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Espinal, R.; Taboada, E.; Molins, E.; Chimentao, R.J.; Medina, F.; Llorca, J. Cobalt hydrotalcites as catalysts for bioethanol steam reforming. The promoting effect of potassium on catalyst activity and long-term stability. Appl. Catal. B Environ. 2012, 127, 59–67. [Google Scholar] [CrossRef]
- Xie, X.; Yin, H.; Dou, B.; Huo, J. Characterization of a potassium-promoted cobalt-molybdenum/alumina water-gas shift catalyst. Appl. Catal. 1991, 77, 187–198. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B Environ. 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Trépanier, M.; Tavasoli, A.; Dalai, A.K.; Abatzoglou, N. Co, Ru and K loadings effects on the activity and selectivity of carbon nanotubes supported cobalt catalyst in Fischer–Tropsch synthesis. Appl. Catal. A Gen. 2009, 353, 193–202. [Google Scholar] [CrossRef]
- Tavasoli, A.H.M.A.D.; Khodadadi, A.; Mortazavi, Y.; Sadaghiani, K.; Ahangari, M.G. Lowering methane and raising distillates yields in Fischer–Tropsch synthesis by using promoted and unpromoted cobalt catalysts in a dual bed reactor. Fuel Process. Technol. 2006, 87, 641–647. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer–Tropsch synthesis: Support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- Calafat, A.; Vivas, F.; Brito, J.L. Effects of phase composition and of potassium promotion on cobalt molybdate catalysts for the synthesis of alcohols from CO2 and H2. Appl. Catal. A Gen. 1998, 172, 217–224. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, H.; Gao, P.; Li, X.; Zhong, L.; Wang, H.; Liu, H.; Wei, W.; Sun, Y. Direct conversion of CO2 to long-chain hydrocarbon fuels over K–promoted CoCu/TiO2 catalysts. Catal. Today 2018, 311, 65–73. [Google Scholar] [CrossRef]
- Petala, A.; Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: I. Effect of alkali additives on catalytic activity and selectivity. Appl. Catal. B Environ. 2018, 224, 919–927. [Google Scholar] [CrossRef]
- Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: II. Effect of alkali additives on the reaction pathway. Appl. Catal. B Environ. 2018, 236, 162–170. [Google Scholar] [CrossRef]
- Numpilai, T.; Witoon, T.; Chanlek, N.; Limphirat, W.; Bonura, G.; Chareonpanich, M.; Limtrakul, J. Structure–activity relationships of Fe-Co/K-Al2O3 catalysts calcined at different temperatures for CO2 hydrogenation to light olefins. Appl. Catal. A Gen. 2017, 547, 219–229. [Google Scholar] [CrossRef]
- Russell, W.W.; Miller, G.H. Catalytic hydrogenation of carbon dioxide to higher hydrocarbons. J. Am. Chem. Soc. 1950, 72, 2446–2454. [Google Scholar] [CrossRef]
- Owen, R.E.; O’Byrne, J.P.; Mattia, D.; Plucinski, P.; Pascu, S.I.; Jones, M.D. Cobalt catalysts for the conversion of CO2 to light hydrocarbons at atmospheric pressure. Chem. Commun. 2013, 49, 11683–11685. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Bensaddik, A.; Hilaire, L.; Chaumette, P.; Kiennemann, A. Study on a cobalt silica catalyst during reduction and Fischer-Tropsch reaction: In situ EXAFS compared to XPS and XRD. Catal. Today 1998, 39, 329–341. [Google Scholar] [CrossRef]
- Huffman, G.P.; Shah, N.; Zhao, J.M.; Huggins, F.E.; Hoost, T.E.; Halvorsen, S.; Goodwin, J.G. In-situ XAFS investigation of K-promoted Co catalysts. J. Catal. 1995, 151, 17–25. [Google Scholar] [CrossRef]
- De la Osa, A.; De Lucas, A.; Valverde, J.L.; Romero, A.; Monteagudo, I.; Coca, P.; Sánchez, P. Influence of alkali promoters on synthetic diesel production over Co catalyst. Catal. Today 2011, 167, 96–106. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Hu, X.; Dong, D.; Shao, X.; Zhang, L.; Lu, G. Steam reforming of acetic acid over cobalt catalysts: Effects of Zr, Mg and K addition. Int. J. Hydrog. Energy 2017, 42, 4793–4803. [Google Scholar] [CrossRef]
- Kono, E.; Tamura, S.; Yamamuro, K.; Ogo, S.; Sekine, Y. Pd/K/Co-oxide catalyst for water gas shift. Appl. Catal. A Gen. 2015, 489, 247–254. [Google Scholar] [CrossRef]
- Petitto, S.C.; Langell, M.A. Surface composition and structure of Co3O4 (110) and the effect of impurity segregation. J. Vac. Sci. Technol. A 2004, 22, 1690–1696. [Google Scholar] [CrossRef]
- Melaet, G.; Lindeman, A.E.; Somorjai, G.A. Cobalt particle size effects in the Fischer–Tropsch synthesis and in the hydrogenation of CO2 studied with nanoparticle model catalysts on silica. Top. Catal. 2014, 57, 500–507. [Google Scholar] [CrossRef]
- Iablokov, V.; Beaumont, S.K.; Alayoglu, S.; Pushkarev, V.V.; Specht, C.; Gao, J.; Alivisatos, A.P.; Kruse, N.; Somorjai, G.A. Size-controlled model Co nanoparticle catalysts for CO2 hydrogenation: Synthesis, characterization, and catalytic reactions. Nano Lett. 2012, 12, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on group VIII metals: IV. Specific activities and selectivities of silica-supported Co, Fe, and Ru. J. Catal. 1984, 87, 352–362. [Google Scholar] [CrossRef]
- Mutschler, R.; Moioli, E.; Luo, W.; Gallandat, N.; Züttel, A. CO2 hydrogenation reaction over pristine Fe, Co, Ni, Cu and Al2O3 supported Ru: Comparison and determination of the activation energies. J. Catal. 2018, 366, 139–149. [Google Scholar] [CrossRef]
- Guerrero-Ruiz, A.; Rodriguez-Ramos, I. Hydrogenation of CO2 on carbon-supported nickel and cobalt. React. Kinet. Catal. Lett. 1985, 29, 93–99. [Google Scholar] [CrossRef]
- Fernández-Morales, I.; Guerrero-Ruiz, A.; López-Garzón, F.J.; Rodríguez-Ramos, I.; Moreno-Castilla, C. Hydrogenolysis of n-butane and hydrogenation of carbon monoxide on Ni and Co catalysts supported on saran carbons. Appl. Catal. 1985, 14, 159–172. [Google Scholar] [CrossRef]
- Yao, Y.; Hildebrandt, D.; Glasser, D.; Liu, X. Fischer−Tropsch synthesis using H2/CO/CO2 syngas mixtures over a cobalt catalyst. Ind. Eng. Chem. Res. 2010, 49, 11061–11066. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Shafer, W.D.; Sparks, D.E.; Davis, B.H. Fischer–Tropsch synthesis: Effect of CO2 containing syngas over Pt promoted Co/γ-Al2O3 and K-promoted Fe catalysts. Catal. Commun. 2011, 12, 936–939. [Google Scholar] [CrossRef]
- Habazaki, H.; Yamasaki, M.; Zhang, B.P.; Kawashima, A.; Kohno, S.; Takai, T.; Hashimoto, K. Co-methanation of carbon monoxide and carbon dioxide on supported nickel and cobalt catalysts prepared from amorphous alloys. Appl. Catal. A Gen. 1998, 172, 131–140. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Davis, B.H.; Willauer, H.D. Influence of Gas Feed Composition and Pressure on the Catalytic Conversion of CO2 to Hydrocarbons Using a Traditional Cobalt-Based Fischer-Tropsch Catalyst. Energy Fuels 2009, 23, 4190–4195. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Shafer, W.D.; Sparks, D.; Davis, B.H. Fischer–Tropsch synthesis: Deuterium kinetic isotope study for hydrogenation of carbon oxides over cobalt and iron catalysts. Catal. Lett. 2011, 141, 1420–1428. [Google Scholar] [CrossRef]
- Fröhlich, G.; Kestel, U.; Łojewska, J.; Łojewski, T.; Meyer, G.; Voß, M.; Borgmann, D.; Dziembaj, R.; Wedler, G. Activation and deactivation of cobalt catalysts in the hydrogenation of carbon dioxide. Appl. Catal. A Gen. 1996, 134, 1–19. [Google Scholar] [CrossRef]
- Lahtinen, J.; Anraku, T.; Somorjai, G.A. C, CO and CO2 hydrogenation on cobalt foil model catalysts: Evidence for the need of CoO reduction. Catal. Lett. 1994, 25, 241–255. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; Mattia, D.; Jones, M.D.; Plucinski, P.K. Formation of hydrocarbons via CO2 hydrogenation–A thermodynamic study. J. CO2 Util. 2014, 6, 34–39. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Melaet, G.; Ralston, W.T.; Li, C.S.; Alayoglu, S.; An, K.; Musselwhite, N.; Kalkan, B.; Somorjai, G.A. Evidence of highly active cobalt oxide catalyst for the Fischer–Tropsch synthesis and CO2 hydrogenation. J. Am. Chem. Soc. 2014, 136, 2260–2263. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.; Claeys, M.; Schulz, H.; Schaub, G.; Nam, S.-S.; Jun, K.-W.; Choi, M.-J.; Kishan, G.; Lee, K.-W. Comparative study of Fischer–Tropsch synthesis with H2/CO and H2/CO2 syngas using Fe- and Co-based catalysts. Appl. Catal. A Gen. 1999, 186, 201–213. [Google Scholar] [CrossRef]
- Visconti, C.G.; Lietti, L.; Tronconi, E.; Forzatti, P.; Zennaro, R.; Finocchio, E. Fischer–Tropsch synthesis on a Co/Al2O3 catalyst with CO2 containing syngas. Appl. Catal. A Gen. 2009, 355, 61–68. [Google Scholar] [CrossRef]
- Janlamool, J.; Praserthdam, P.; Jongsomjit, B. Ti-Si composite oxide-supported cobalt catalysts for CO2 hydrogenation. J. Nat. Gas Chem. 2011, 20, 558–564. [Google Scholar] [CrossRef]
- Das, T.; Deo, G. Synthesis, characterization and in situ DRIFTS during the CO2 hydrogenation reaction over supported cobalt catalysts. J. Mol. Catal. A Chem. 2011, 350, 75–82. [Google Scholar] [CrossRef]
- Srisawad, N.; Chaitree, W.; Mekasuwandumrong, O.; Shotipruk, A.; Jongsomjit, B.; Panpranot, J. CO2 hydrogenation over Co/Al2O3 catalysts prepared via a solid-state reaction of fine gibbsite and cobalt precursors. React. Kinet. Mech. Catal. 2012, 107, 179–188. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Diameter (nm) |
|---|---|---|---|
| SiO2 | 186.6 | 1.5 | 31.2 |
| 15%Co/SiO2 | 133.1 | 1.0 | 30.2 |
| 15%Co-1%K/SiO2 | 137.8 | 1.2 | 33.4 |
| 15%Co-3%K/SiO2 | 123.4 | 1.1 | 35.5 |
| 15%Co-5%K/SiO2 | 70.0 | 0.8 | 45.1 |
| Catalyst | Freshly Calcined a | Reduced and Passivated a | |
|---|---|---|---|
| Co3O4 | CoO | Co | |
| 15%Co/SiO2 | 18.16 | 9.36 | 9.44 |
| 15%Co-1%K/SiO2 | 23.52 | 7.30 | 7.34 |
| 15%Co-3%K/SiO2 | 24.35 | 6.70 | 6.72 |
| 15%Co-5%K/SiO2 | 19.13 | 2.46 | 8.85 |
| Catalyst | H2/CO2 | P (atm.) | T (°C) | Ea (kJ/mol) | References |
|---|---|---|---|---|---|
| 15%Co/3%K/SiO2 | 3/1 | 1 | 180–240 | 78 | This work |
| Pristine Co | 4/1 | 1 | 207–237 | 77 | [24] |
| 100% Co | 4/1 | 1 | 190–230 | 79 | [25] |
| 4.5%Co/S1 * | 4/1 | 1 | 210–260 | 79 | [25] |
| 4.6%Co/S3 * | 4/1 | 1 | 200–240 | 76 | [25] |
| 15%Co/SiO2 | 4/1 | 1 | 183–203 | 93 | [23] |
| 15%Co/SiO2 | 4/1 | 11 | 180–222 | 171 | [23] |
| 3%Co/SiO2 | 4/1 | 1 | 227–277 | 79 | [23] |
| Entry | Catalyst | Prep. Method | H2:CO2 | T [K] | P [bar] | SV | Conv. [%] | %Selectivity | References | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | CO | C2+ | C5+ | |||||||||
| 1 | 100Co/5Cu | Coprecip. | 3:1 | 473 | 0.2 L/gCat/h a | [12] | ||||||
| 2 | 100Co/5Cu/2K2CO3 | Coprecip. | 3:1 | 498 | 0.16 | 49 | 1.12 b | |||||
| 3 | 100Co/5Cu/2K2CO3 | Coprecip. | 3:1 | 498 | 0.16 | 56 | 2.32 | |||||
| 4 | 100Co/5Cu/2K2CO3 | Coprecip. | 3:1 | 473 | 0.16 | 40 | 1.91 | |||||
| 5 | 100Co/5Cu/2K2CO3 | Coprecip. | 3:1 | 498 | 0.16 | 22 | 2.81 | |||||
| 6 | 100Co/5Cu/2K2CO3 | Coprecip. | 3:1 | 473 | 0.16 | 10 | 0.19 | |||||
| 7 | 100Co/5Cu/2K2CO3 | Coprecip. | 3:1 | 473 | 0.16 | 44 | 1.59 | |||||
| 8 | 100Co/5Cu/5CeO2/2K2CO3 | Coprecip. | 3:1 | 498 | 0.16 | 54 | 1.43 | |||||
| 9 | 100Co/5Cu/1CeO2/2K2CO3 | Coprecip. | 2:1 | 498 | 0.15 | 34 | 2.11 | |||||
| 10 | 100Co/5Cu/1CeO2 | Coprecip. | 2:1 | 498 | 0.15 | 40 | 0.10 | |||||
| 11 | 100Co/5Cu/1CeO2/3K3PO4 | Coprecip. | 3:1 | 498 | 0.16 | 40 | 1.42 | |||||
| 12 | 100Co/5Cu/1CeO2/4.5K2CO3/100MgO | Coprecip. | 2:1 | 513 | 0.3 | 21 | 0.29 | |||||
| 13 | 100Co/5Cu/1CeO2/4.5K2CO3/100MgO | Coprecip. | 2:1 | 498 | 0.15 | 8 | 0.51 | |||||
| 14 | 100Co/5Cu/1CeO2/6K2CO3/100H.S.C. | Coprecip. | 2:1 | 518 | 0.15 | 19 | 2.71 | |||||
| 15 | 100Co/5Cu/100CeO2/7K2CO3 | Coprecip. | 2:1 | 523 | 0.15 | 23 | 1.61 | |||||
| 16 | 100Co/5Cu/1CeO2/4.5K2CO3/100F.C. | Coprecip. | 2:1 | 513 | 0.075 | 23 | 0.24 | |||||
| 17 | 100Co/5Cu/1CeO2/3.8K2CO3/50H.S.C. | Coprecip. | 2:1 | 498 | 0.12 | 22 | 1.90 | |||||
| 18 | 3%Co/SiO2 | Impregnation | 4:1, 95%N2 | 500 | 1.4 | 4340/h | 9.6 | 71 | 25 | 4.6 | [23] | |
| 19 | 500 | 8480 | 6.5 | 54 | 35 | 11 | ||||||
| 20 | 525 | 8480 | 12.3 | 59 | 33 | 8.2 | ||||||
| 21 | 525 | 16,400 | 9.4 | 42 | 49 | 8.9 | ||||||
| 22 | 550 | 16,400 | 13.7 | 42 | 52 | 5.9 | ||||||
| 23 | 550 | 24,600 | 12 | 32 | 52 | 17 | ||||||
| 24 | 15%Co/SiO2 | Impregnation | 4:1, no N2 | 476 | 1 | 2050–3850 | 10.5 | 86.9 | 12.6 | 0.7 | 0 | |
| 25 | 478 | 11 | 450–9620 | 11.2 | 89 | 10.7 | 0.34 | 0 | ||||
| 26 | 100%Co | Reduction | 4:1 | 493 | 1 | 500–3000 h−1 | 1.9 | 98 | 2 | [25] | ||
| 27 | 4.5%Co/S1 | Impregnation | 493 | 1.8 | 40 | 60 | ||||||
| 28 | 4.6%Co/S3 | Impregnation | 493 | 6.3 | 66 | 34 | ||||||
| 29 | 100 Co/60 MnO/147 SiO2/0.15Pt | Precip. and Impregnation | 2:1 | 463 | 10 | 30 mL/min/g of Co | 18 | 95 | [37] | |||
| 30 | 15%Co/Al2O3 | Impregnation | 2.45:1 | 493 | 20 | 4800 cm3(STP)/h/gcat | 33 | >90 | [38] | |||
| 31 | 20%Co/SSP | Impregnation | 20:2 | 493 | 1 | 18 L/gcat/h | 27 | 89.5 | 10.5 | [39] | ||
| 32 | 20%Co/MCM-41 | 28 | 91.4 | 8.6 | ||||||||
| 33 | 20%Co/TiSSP | 16 | 92.1 | 7.9 | ||||||||
| 34 | Co/TiMCM-41 | 34 | 94.9 | 5.1 | ||||||||
| 35 | 0.5% Pt–25% Co/γ-Al2O3 | Impregnation | 3:1 | 493 | 19.9 | 5.0 L/g cat/h | 93.3 | 6.66 | 5.16 | [28] | ||
| 36 | 5%Co/Al2O3 c | Impregnation | 6:1 | 533 | 1 | 13.5 mL/min/(63 to 70 mg of cat) | 0.21 | 35.7 | [40] | |||
| 37 | 10%Co/Al2O3 c | 0.91 | 74.2 | |||||||||
| 38 | 15%Co/Al2O3 c | 2.45 | 87.8 | |||||||||
| 39 | 20%Co/Al2O3 c | 2.1 | 85.7 | |||||||||
| 40 | Co/Al2O3 | Solid state reaction of gibbsite and CoNT | 10:1 | 543 | 1 | 150 mL/min/gcat | 76 | 82.2 | 17.8 | [41] | ||
| 41 | Co/Al2O3 | Solid state reaction of gibbsite and CoAc | 48.7 | 76.7 | 23.3 | |||||||
| 42 | Co/Al2O3 | Solid state reaction of gibbsite and CoAA | 20.3 | 76.4 | 23.6 | |||||||
| 43 | Co/Al2O3 | Solid state reaction of gibbsite and CoCL | 6.1 | 100 | 0 | |||||||
| 44 | Co/Al2O3 | Impregnation using CoNT | 32.2 | 86.5 | 13.5 | |||||||
| 45 | 20%Co/SiO2 | 3:1 | 643 | Atmospheric | 67.4 | 95.3 | 4.2 | 0.6 | 0 | [13] | ||
| 46 | 20%Co/1%Pd/SiO2 | 50.7 | 93.4 | 6.3 | 0.3 | 0 | ||||||
| 47 | 10%Co/1%Pd/1%K/SiO2 | 36.4 | 89.3 | 8.0 | 2.8 | 0 | ||||||
| 48 | 20%Co/1%Pd/1%K/SiO2 | 63.4 | 80.3 | 13.9 | 5.9 | 0 | ||||||
| 49 | 10%Co/1%Pd/1%K/SiO2 | 39.1 | 82.9 | 9.5 | 7.6 | 0.09 | ||||||
| 50 | 20%Co/1%Pd/0.5%K/SiO2 | 62.8 | 76.0 | 15.3 | 8.8 | 0 | ||||||
| 51 | 20%Co/1%Pd/1.5%K/SiO2 | 59.1 | 64.7 | 16.2 | 19.1 | 1.26 | ||||||
| 52 | 20%Co/1%Pd/3%K/SiO2 | 43.2 | 53.1 | 24.3 | 22.6 | 2.73 | ||||||
| 53 | 20%Co/1%K/SiO2 | 36.1 | 45.3 | 16.9 | 37.8 | 7.87 | ||||||
| 54 | 20%Co/1%Pt/1%K/SiO2 | 36.5 | 41.5 | 20.8 | 37.7 | 9.58 | ||||||
| 55 | 20%Co/1%Ru/1%K/SiO2 | 45.1 | 52.6 | 12.6 | 34.8 | 5.68 | ||||||
| 56 | 20%Co/1%Pd/1%Li/SiO2 | 39.5 | 56.1 | 19.2 | 24.6 | 1.94 | ||||||
| 57 | 20%Co/1%Pd/1%Na/SiO2 | 41.9 | 48.4 | 20.3 | 31.3 | 7.33 | ||||||
| 58 | 20%Co/1%Li/SiO2 | 39.3 | 58.4 | 21.4 | 20.2 | 0.47 | ||||||
| 59 | 20%Co/1%Na/SiO2 | 51.2 | 42.1 | 21.7 | 36.3 | 5.01 | ||||||
| 60 | 20%Co/1%K/SiO2 | 47.6 | 50.1 | 17.0 | 32.9 | 3.65 | ||||||
| 61 | 20%Co/1%Mo/SiO2 | 64.8 | 88.7 | 6.5 | 4.8 | 0 | ||||||
| 62 | 20%Co/1%Cr/SiO2 | 60.9 | 75.9 | 22.8 | 1.2 | 0 | ||||||
| 63 | 20%Co/1%Mn/SiO2 | 62 | 91.1 | 6.9 | 2.0 | 0 | ||||||
| 64 | 20%Co/1%Na/1%Mn/SiO2 | 42.7 | 58.2 | 19.7 | 22.2 | 0.80 | ||||||
| 65 | 20%Co/1%Na/1%Mo/SiO2 | 43.9 | 38.3 | 15.7 | 45.9 | 8.76 | ||||||
| 66 | CoCu/TiO2 | Deposition-precipitation | 73:24 | 523 | 50 | 3000 mL/g/h | 23.1 | 87.0 | 1.3 | 10.2 | 4.76 | [8] |
| 67 | 1.5 K–CoCu/TiO2 | 21.2 | 59.3 | 4.7 | 36.5 | 13.21 | ||||||
| 68 | 2.0 K–CoCu/TiO2 | 13.8 | 37.1 | 19.7 | 44.6 | 17.39 | ||||||
| 69 | 2.5 K–CoCu/TiO2 | 13 | 22.4 | 35.1 | 43.3 | 23.08 | ||||||
| 70 | 3.0 K–CoCu/TiO2 | 12.8 | 21.9 | 35.9 | 41.5 | 19.53 | ||||||
| 71 | 3.5 K–CoCu/TiO2 | 11.9 | 18.9 | 45.9 | 35.1 | 16.81 | ||||||
| 72 | 15%Co-1%K/SiO2 | Impregnation | 3:1 | 543 | 1 bar | 0.92 NL/gcat/h | 16 | 37.6 | 31.9 | 30.5 | 7.8 | This study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iloy, R.A.; Jalama, K. Effect of Operating Temperature, Pressure and Potassium Loading on the Performance of Silica-Supported Cobalt Catalyst in CO2 Hydrogenation to Hydrocarbon Fuel. Catalysts 2019, 9, 807. https://doi.org/10.3390/catal9100807
Iloy RA, Jalama K. Effect of Operating Temperature, Pressure and Potassium Loading on the Performance of Silica-Supported Cobalt Catalyst in CO2 Hydrogenation to Hydrocarbon Fuel. Catalysts. 2019; 9(10):807. https://doi.org/10.3390/catal9100807
Chicago/Turabian StyleIloy, Rama Achtar, and Kalala Jalama. 2019. "Effect of Operating Temperature, Pressure and Potassium Loading on the Performance of Silica-Supported Cobalt Catalyst in CO2 Hydrogenation to Hydrocarbon Fuel" Catalysts 9, no. 10: 807. https://doi.org/10.3390/catal9100807