Cofactor F420-Dependent Enzymes: An Under-Explored Resource for Asymmetric Redox Biocatalysis

 , , ,
, , ,
Abstract
1. Introduction
2. Families of F420-Dependent Enzymes Relevant to Biocatalysis
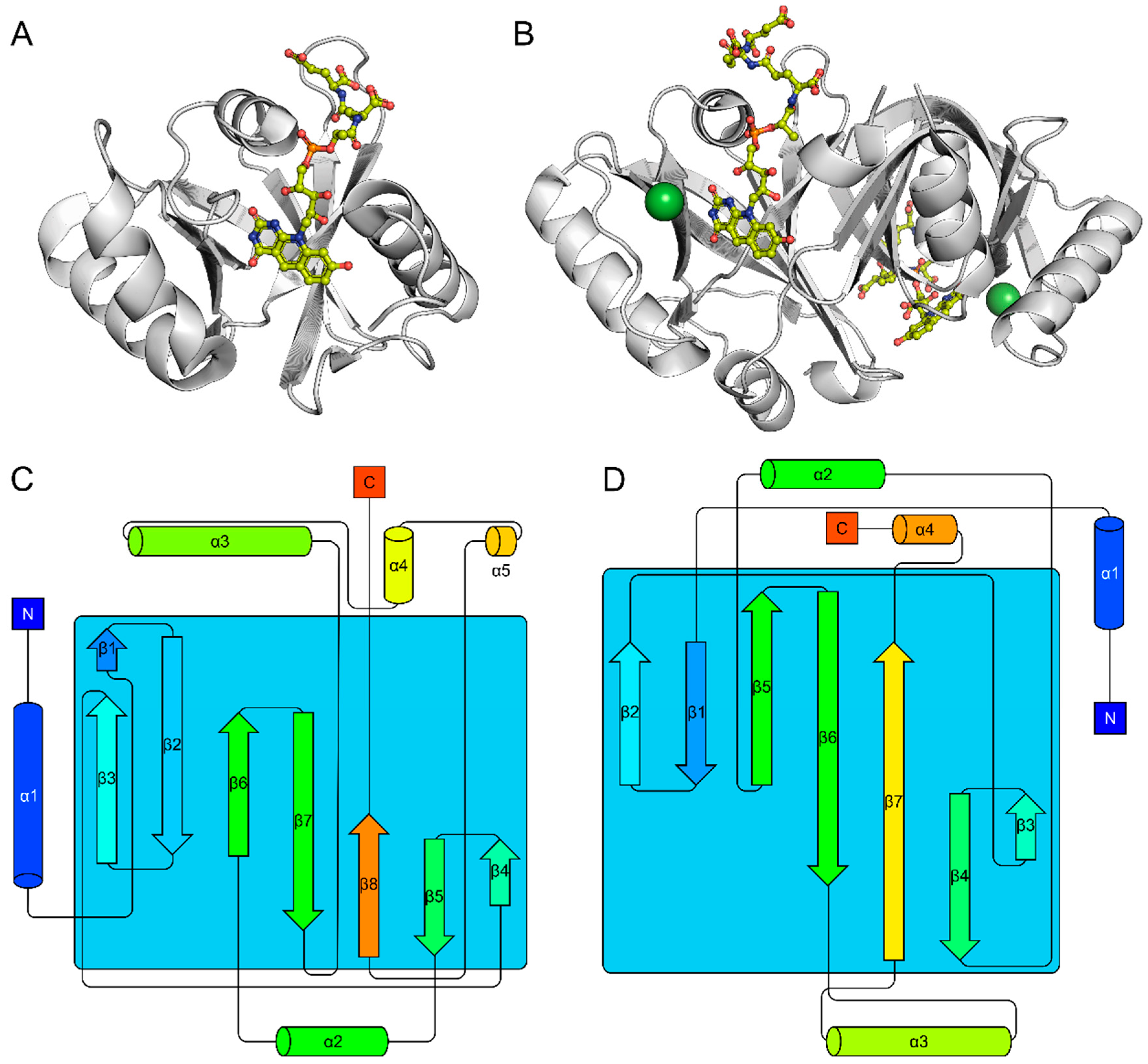
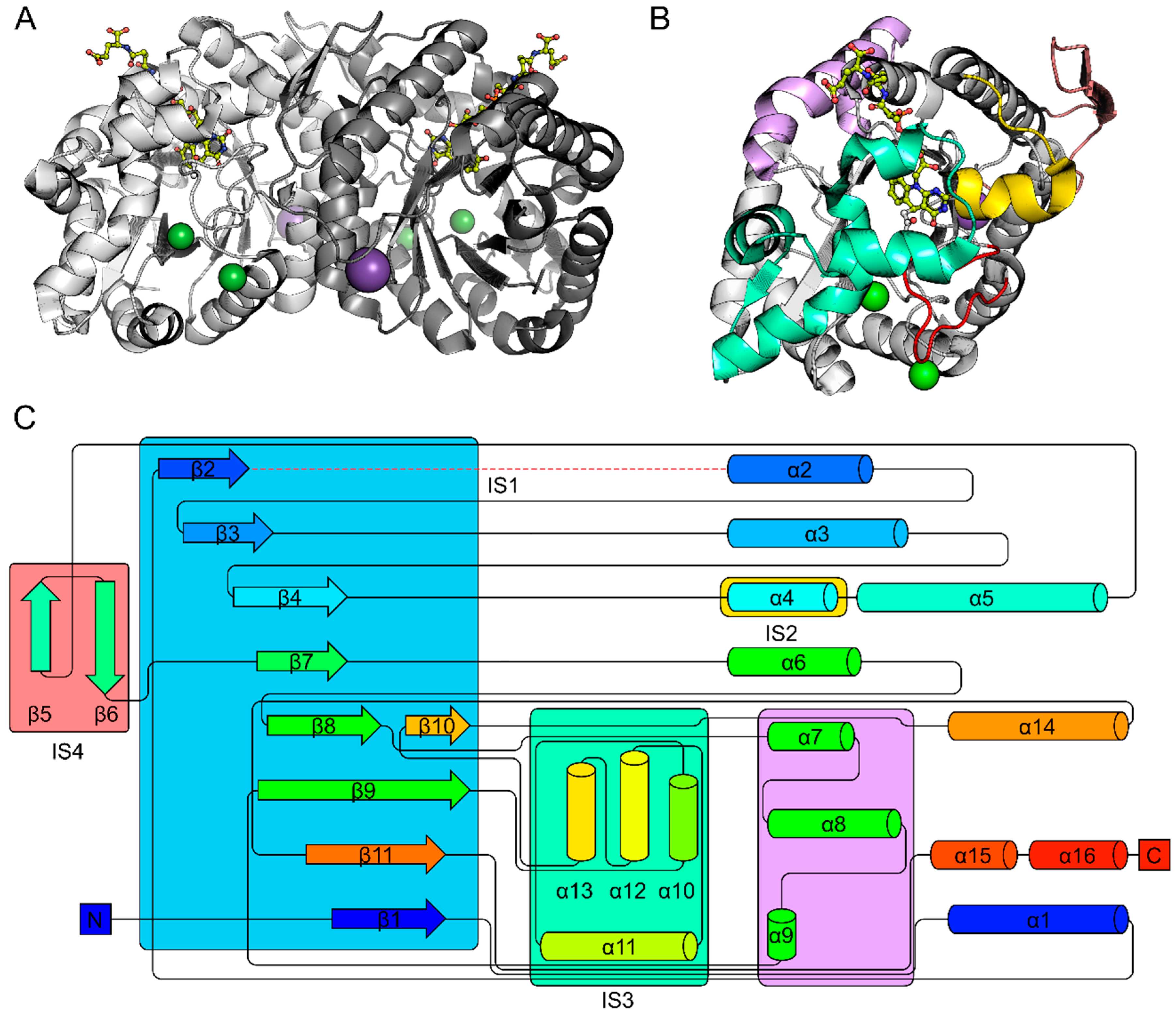
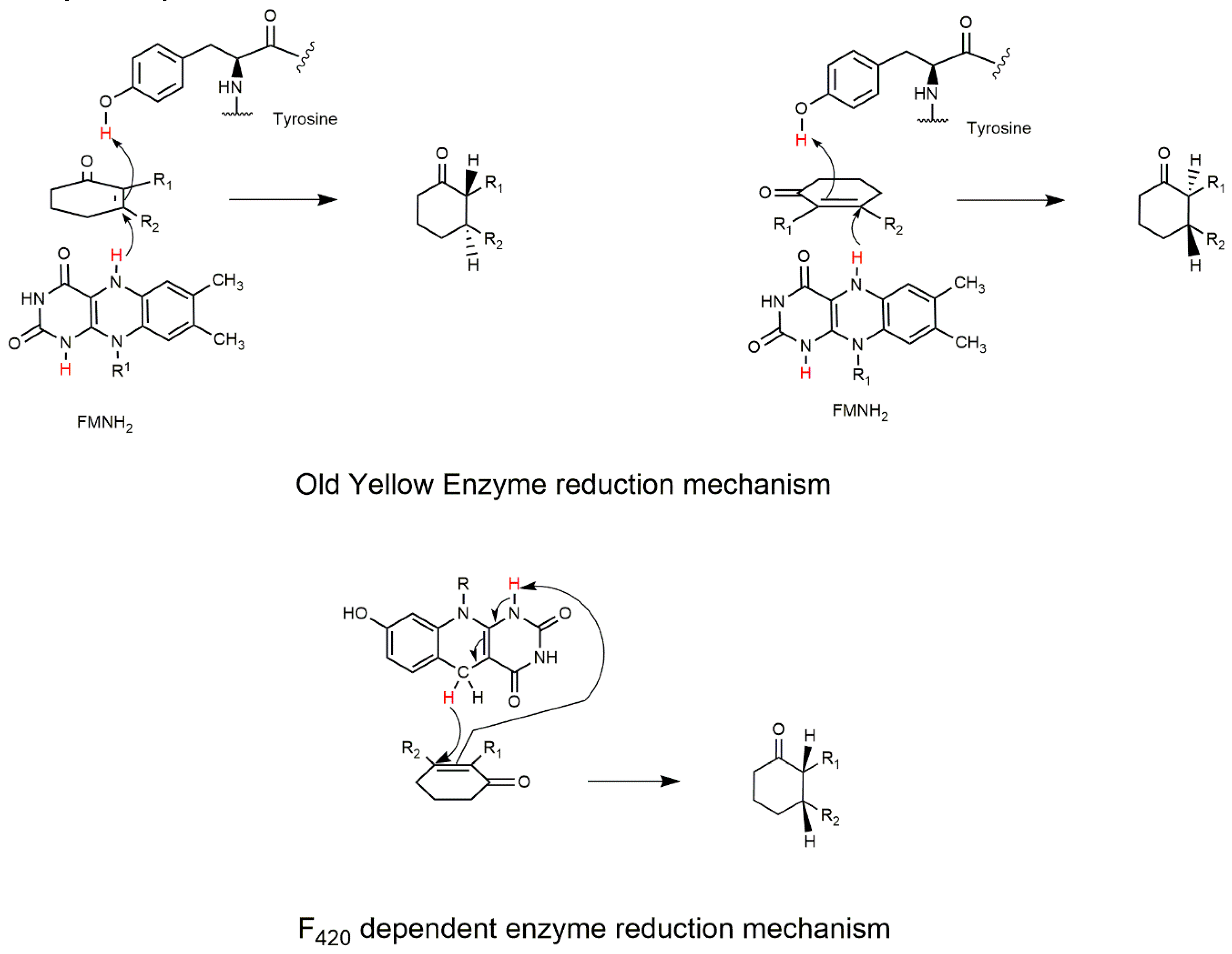
2.1. The FDOR Superfamily
2.2. The LLHT Family:
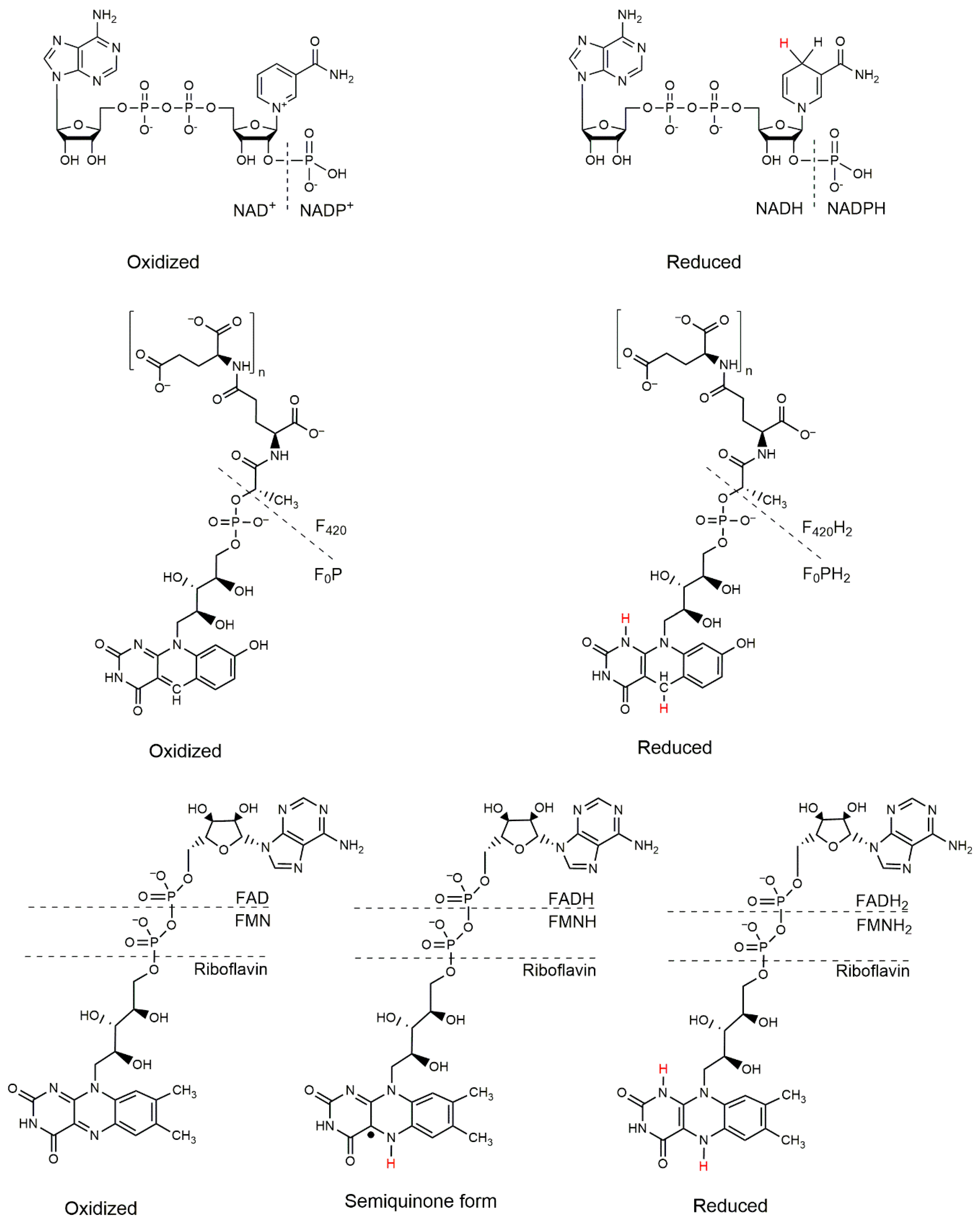
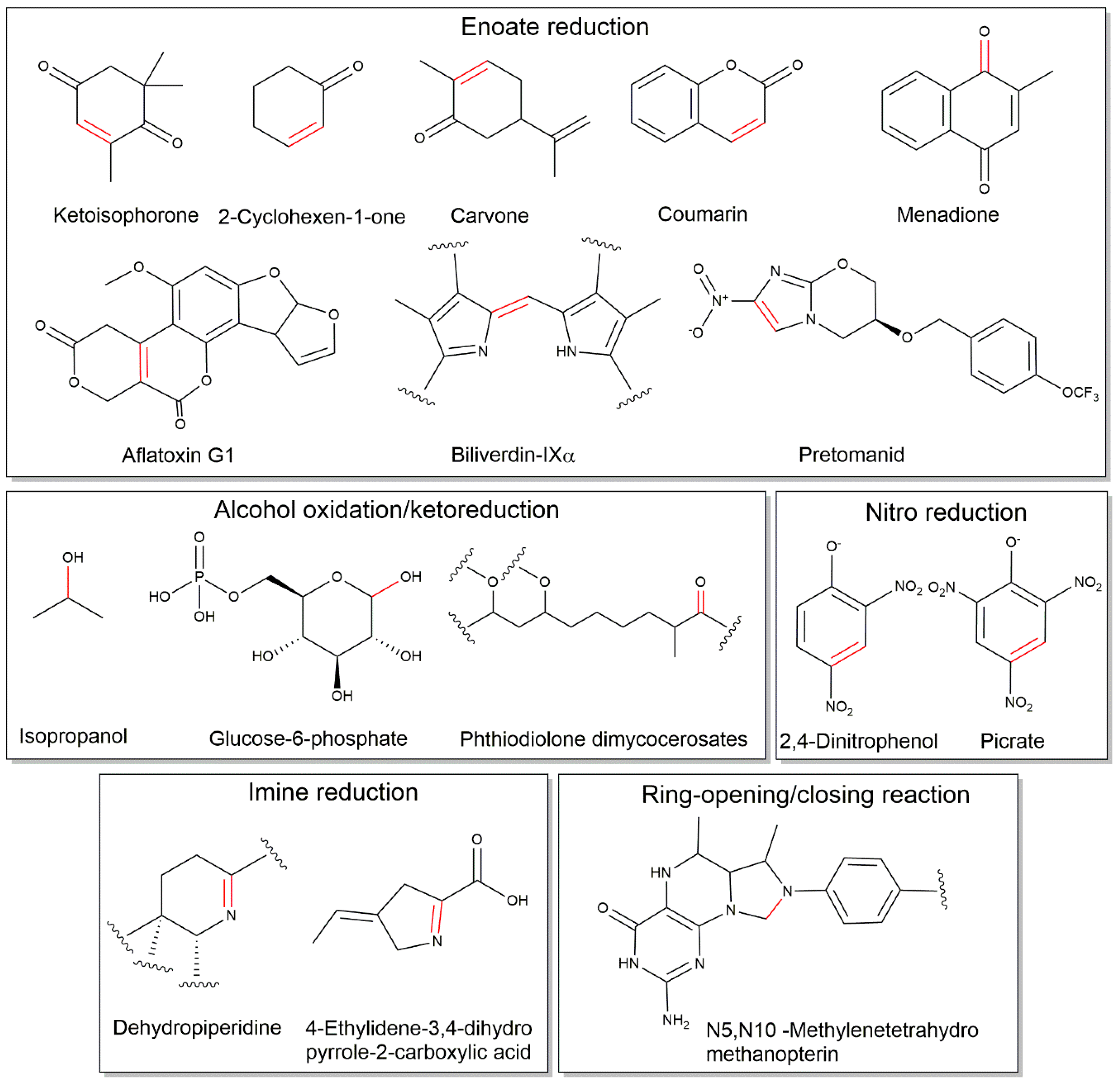
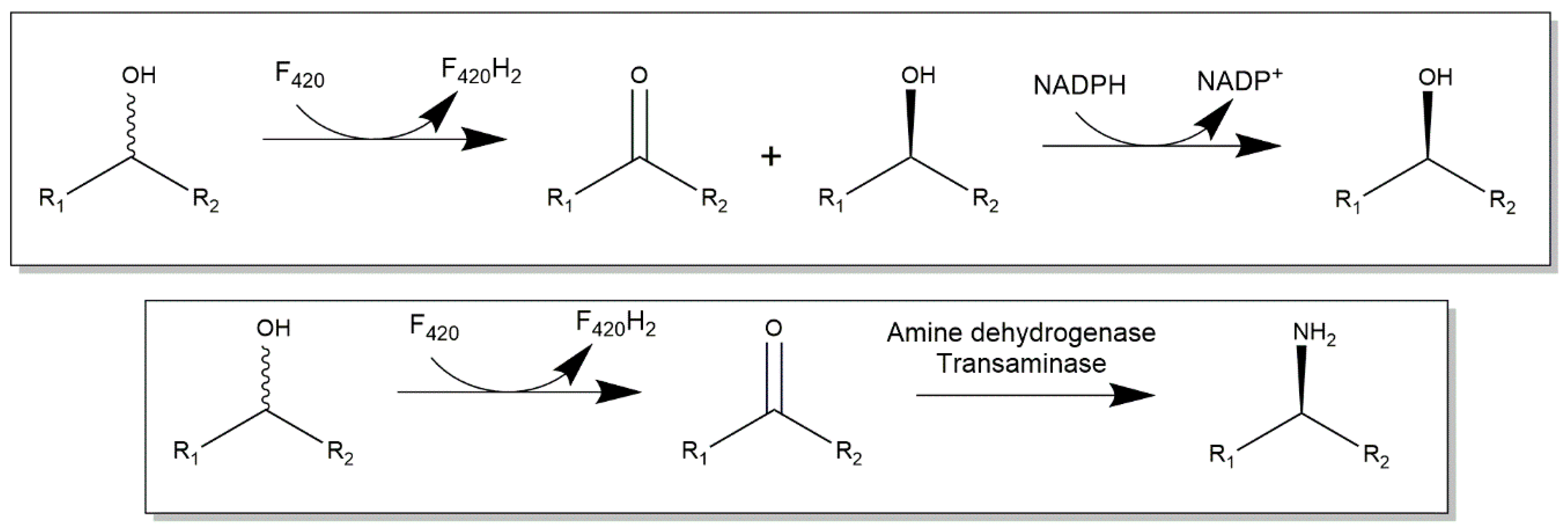
2.3. Cofactor F420-Dependent Reactions with Relevance to Biocatalysis
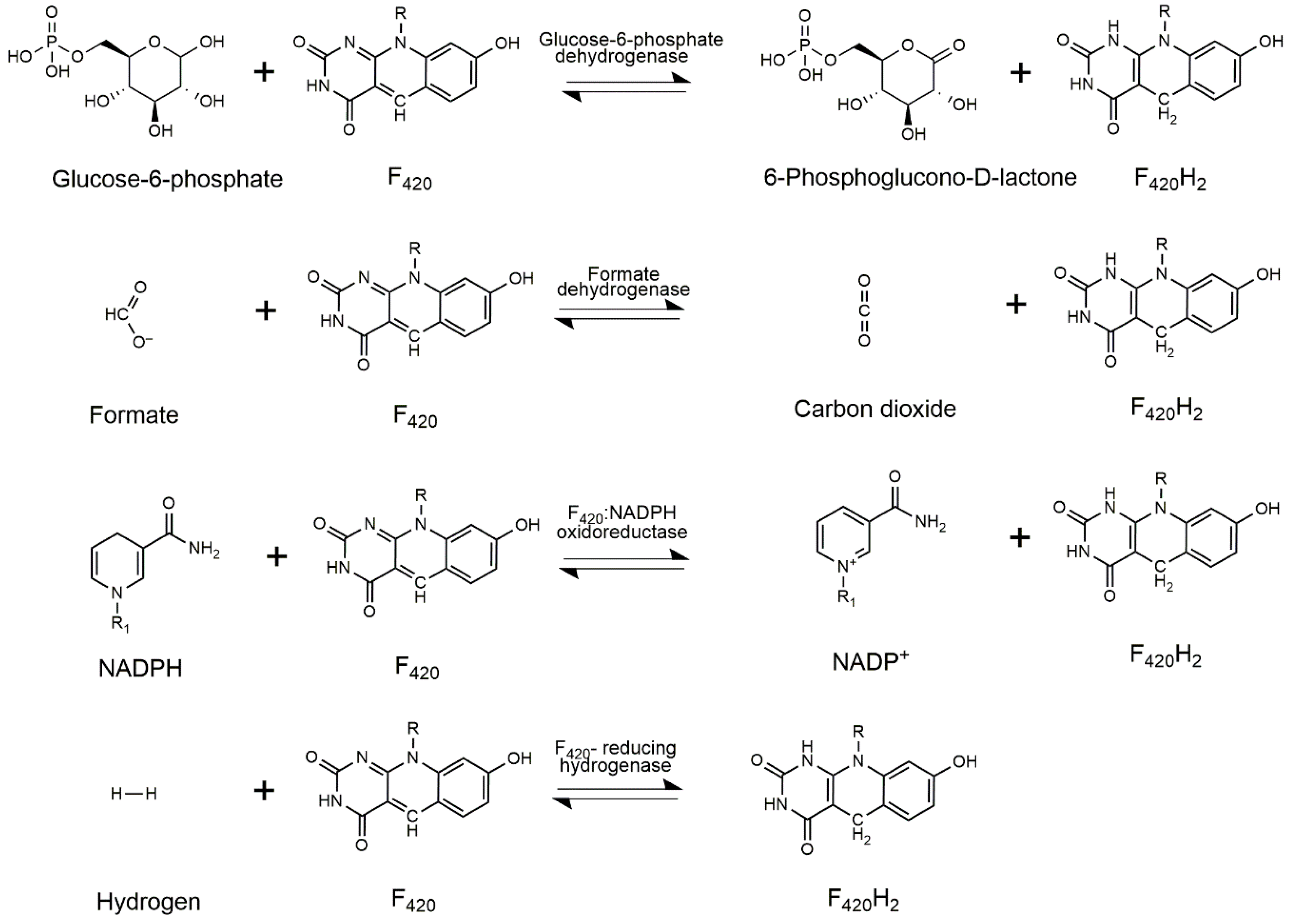
3. Cofactor Recycling for Cofactor F420
4. Cofactor Production
5. Prospects
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
Appendix A. Thermodynamics of F420 Biosynthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Reaction a | ΔrGt (kJ) b |
|---|---|---|
| CofC/FbiD | PEP + GTP → EPPG + PPi d | +71.27(±67) |
| CofG/FbiC | 5ARPD + Tyr + SAMe → 5ARPD4HB + ImAcet + Met + 5AD | −1192.39(±0) c |
| CofH/FbiC | 5ARPD4HB + SAMe → FO + NH4+ + Met + 5AD | +71.90(±36) c |
| CofD/FbiA | FO + EPPG → dF420-0 + GMP | −31.3(±128) |
| CofX/FbiB | dF420-0 + FMNH2 → F420-0 + FMN | −74.59(±87) |
| CofE/FbiB | F420-0 + GTP + Glu → F420-1 + GDP + Pi | −7.50(±24) |
| CofE/FbiB | F420-1 + GTP + Glu → F420-2 + GDP + Pi | −39.44(±35) |
| CofE/FbiB | F420-2 + GTP + Glu → F420-3 + GDP + Pi | −21.99(±38) |
| Overall | PEP + 5ARPD + Tyr + (2) SAMe + FMNH2 + (3) Glu + (4) GTP | −1224.05(±82) |
| → F420-3 + (2) Met + (2) 5AD + ImAcet + NH4+ + FMN + (3) GDP + (3) Pi + GMP + PPi |
References
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Oxidoreductase-catalyzed synthesis of chiral amines. ACS Catal. 2018, 8, 10985–11015. [Google Scholar] [CrossRef]
- Toogood, H.S.; Scrutton, N.S. New developments in ‘ene’-reductase catalysed biological hydrogenations. Curr. Opin. Chem. Biol. 2014, 19, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, S.C.; Brzezniak, A.; France, S.P.; Ramsden, J.I.; Mangas-Sanchez, J.; Montgomery, S.L.; Heath, R.S.; Turner, N.J. Imine reductases, reductive aminases, and amine oxidases for the synthesis of chiral amines: Discovery, characterization, and synthetic applications. In Enzymes in Synthetic Biology; Scrutton, N., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2018; Volume 608, pp. 131–149. [Google Scholar]
- Bai, D.Y.; He, J.Y.; Ouyang, B.; Huang, J.; Wang, P. Biocatalytic asymmetric synthesis of chiral aryl alcohols. Prog. Chem. 2017, 29, 491–501. [Google Scholar]
- Taylor, M.; Scott, C.; Grogan, G. F-420-dependent enzymes-potential for applications in biotechnology. Trends Biotechnol. 2013, 31, 63–64. [Google Scholar] [CrossRef]
- Mathew, S.; Trajkovic, M.; Kumar, H.; Nguyen, Q.-T.; Fraaije, M.W. Enantio- and regioselective ene-reductions using F420H2-dependent enzymes. Chem. Commun. 2018, 54, 11208–11211. [Google Scholar] [CrossRef]
- Greening, C.; Ahmed, F.H.; Mohamed, A.E.; Lee, B.M.; Pandey, G.; Warden, A.C.; Scott, C.; Oakeshott, J.G.; Taylor, M.C.; Jackson, C.J. Physiology, biochemistry, and applications of F420- and Fo-dependent redox reactions. Microbiol. Mol. Biol. Rev. 2016, 80, 451–493. [Google Scholar] [CrossRef]
- Tzing, S.F.; Bryant, M.P.; Wolfe, R.S. Factor 420-dependent pyridine nucleotide-linked formate metabolism of Methanobacterium ruminantium. J. Bacteriol. 1975, 121, 192–196. [Google Scholar]
- Eirich, L.D.; Vogels, G.D.; Wolfe, R.S. Distribution of coenzyme F420 and properties of its hydrolytic fragments. J. Bacteriol. 1979, 140, 20–27. [Google Scholar]
- Wang, P.; Bashiri, G.; Gao, X.; Sawaya, M.R.; Tang, Y. Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J. Am. Chem. Soc. 2013, 135, 7138–7141. [Google Scholar] [CrossRef]
- Li, W.; Chou, S.C.; Khullar, A.; Gerratana, B. Cloning and characterization of the biosynthetic gene cluster for tomaymycin, an SJG-136 monomeric analog. Appl. Environ. Microbiol. 2009, 75, 2958–2963. [Google Scholar] [CrossRef]
- Ahmed, F.H.; Carr, P.D.; Lee, B.M.; Afriat-Jurnou, L.; Mohamed, A.E.; Hong, N.-S.; Flanagan, J.; Taylor, M.C.; Greening, C.; Jackson, C.J. Sequence–structure–function classification of a catalytically diverse oxidoreductase superfamily in mycobacteria. J. Mol. Biol. 2015, 427, 3554–3571. [Google Scholar] [CrossRef] [PubMed]
- Selengut, J.D.; Haft, D.H. Unexpected abundance of coenzyme F420-dependent enzymes in Mycobacterium tuberculosis and other actinobacteria. J. Bacteriol. 2010, 192, 5788–5798. [Google Scholar] [CrossRef] [PubMed]
- Lapalikar, G.V.; Taylor, M.C.; Warden, A.C.; Onagi, H.; Hennessy, J.E.; Mulder, R.J.; Scott, C.; Brown, S.E.; Russell, R.J.; Easton, C.J.; et al. Cofactor promiscuity among F420-dependent reductases enables them to catalyse both oxidation and reduction of the same substrate. Catal. Sci. Technol. 2012, 2, 1560–1567. [Google Scholar] [CrossRef]
- Harold, L.K.; Antoney, J.; Ahmed, F.H.; Hards, K.; Carr, P.D.; Rapson, T.; Greening, C.; Jackson, C.J.; Cook, G.M. FAD-sequestering proteins protect Mycobacteria against hypoxic and oxidative stress. J. Biol. Chem. 2019, 294, 2903–2912. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.H.; Mohamed, A.E.; Carr, P.D.; Lee, B.M.; Condic-Jurkic, K.; O’Mara, M.L.; Jackson, C.J. Rv2074 is a novel F420H2-dependent biliverdin reductase in Mycobacterium tuberculosis. Protein Sci. 2016, 25, 1692–1709. [Google Scholar] [CrossRef] [PubMed]
- Mashalidis, E.H.; Mukherjee, T.; Śledź, P.; Matak-Vinković, D.; Boshoff, H.I.; Abell, C.; Barry, C.E. Rv2607 from Mycobacterium tuberculosis is a pyridoxine 5′-phosphate oxidase with unusual substrate specificity. PLoS ONE 2011, 6, e27643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, M.C.; Jackson, C.J.; Tattersall, D.B.; French, N.; Peat, T.S.; Newman, J.; Briggs, L.J.; Lapalikar, G.V.; Campbell, P.M.; Scott, C.; et al. Identification and characterization of two families of F420H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol. Microbiol. 2010, 78, 561–575. [Google Scholar] [CrossRef]
- Cellitti, S.E.; Shaffer, J.; Jones, D.H.; Mukherjee, T.; Gurumurthy, M.; Bursulaya, B.; Boshoff, H.I.; Choi, I.; Nayyar, A.; Lee, Y.S.; et al. Structure of DDN, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 2012, 20, 101–112. [Google Scholar] [CrossRef]
- De Souza, G.A.; Leversen, N.A.; Målen, H.; Wiker, H.G. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteom. 2011, 75, 502–510. [Google Scholar] [CrossRef]
- He, Z.; De Buck, J. Cell wall proteome analysis of Mycobacterium smegmatis strain mc2 155. BMC Microbiol. 2010, 10, 121. [Google Scholar] [CrossRef]
- Sinha, S.; Kosalai, K.; Arora, S.; Namane, A.; Sharma, P.; Gaikwad, A.N.; Brodin, P.; Cole, S.T. Immunogenic membrane-associated proteins of Mycobacterium tuberculosis revealed by proteomics. Microbiology 2005, 151, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Aufhammer, S.W.; Warkentin, E.; Ermler, U.; Hagemeier, C.H.; Thauer, R.K.; Shima, S. Crystal structure of methylenetetrahydromethanopterin reductase (MER) in complex with coenzyme F420: Architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Sci. 2005, 14, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Aufhammer, S.W.; Warkentin, E.; Berk, H.; Shima, S.; Thauer, R.K.; Ermler, U. Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure 2004, 12, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Bashiri, G.; Squire, C.J.; Moreland, N.J.; Baker, E.N. Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding. J. Biol. Chem. 2008, 283, 17531–17541. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.T.; Trinco, G.; Binda, C.; Mattevi, A.; Fraaije, M.W. Discovery and characterization of an F420-dependent glucose-6-phosphate dehydrogenase (Rh-FGD1) from rhodococcus jostii rha1. Appl. Microbiol. Biotechnol. 2017, 101, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Mascotti, M.L.; Kumar, H.; Nguyen, Q.-T.; Ayub, M.J.; Fraaije, M.W. Reconstructing the evolutionary history of F420-dependent dehydrogenases. Sci. Rep. 2018, 8, 17571. [Google Scholar] [CrossRef] [PubMed]
- Ceh, K.; Demmer, U.; Warkentin, E.; Moll, J.; Thauer, R.K.; Shima, S.; Ermler, U. Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry 2009, 48, 10098–10105. [Google Scholar] [CrossRef]
- Shima, S.; Warkentin, E.; Grabarse, W.; Sordel, M.; Wicke, M.; Thauer, R.K.; Ermler, U. Structure of coenzyme F420 dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. J. Mol. Biol. 2000, 300, 935–950. [Google Scholar] [CrossRef]
- Vaupel, M.; Thauer, R.K. Coenzyme F420 dependent N-5, N-10-methylenetetrahydromethanopterin reductase (MER) from Methanobacterium thermautotrophicum strain marburg: Cloning, sequencing, transcriptional analysis and functional expression in Escherichia coli of the mer gene. Eur. J. Biochem. 1995, 231, 773–778. [Google Scholar]
- Purwantini, E.; Mukhopadhyay, B. Rv0132c of Mycobacterium tuberculosis encodes a coenzyme F420-dependent hydroxymycolic acid dehydrogenase. PLoS ONE 2013, 8, e81985. [Google Scholar] [CrossRef]
- Fida, T.T.; Palamuru, S.; Pandey, G.; Spain, J.C. Aerobic biodegradation of 2,4-dinitroanisole by Nocardioides sp. Strain js1661. Appl. Environ. Microbiol. 2014, 80, 7725–7731. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.; Rieger, P.G.; Knackmuss, H.J. Function of coenzyme f420 in aerobic catabolism of 2,4,6-trinitrophenol and 2,4-dinitrophenol by Nocardioides simplex FJ2-1A. J. Bacteriol. 1999, 181, 2669–2674. [Google Scholar] [PubMed]
- Lapalikar, G.V.; Taylor, M.C.; Warden, A.C.; Scott, C.; Russell, R.J.; Oakeshott, J.G. F420H2-dependent degradation of aflatoxin and other furanocoumarins is widespread throughout the Actinomycetales. PLoS ONE 2012, 7, e30114. [Google Scholar] [CrossRef] [PubMed]
- Greening, C.; Jirapanjawat, T.; Afroze, S.; Ney, B.; Scott, C.; Pandey, G.; Lee, B.M.; Russell, R.J.; Jackson, C.J.; Oakeshott, J.G.; et al. Mycobacterial F420H2-dependent reductases promiscuously reduce diverse compounds through a common mechanism. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Gurumurthy, M.; Rao, M.; Mukherjee, T.; Rao, S.P.S.; Boshoff, H.I.; Dick, T.; Barry, C.E.; Manjunatha, U.H. A novel F420-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol. Microbiol. 2013, 87, 744–755. [Google Scholar] [CrossRef]
- Mohamed, A.E.; Ahmed, F.H.; Arulmozhiraja, S.; Lin, C.Y.; Taylor, M.C.; Krausz, E.R.; Jackson, C.J.; Coote, M.L. Protonation state of F420H2 in the prodrug-activating deazaflavin dependent nitroreductase (DDN) from Mycobacterium tuberculosis. Mol. BioSys. 2016, 12, 1110–1113. [Google Scholar] [CrossRef]
- Drenth, J.; Trajkovic, M.; Fraaije, M.W. Chemoenzymatic synthesis of an unnatural deazaflavin cofactor that can fuel F420-dependent enzymes. ACS Catal. 2019, 9, 6435–6443. [Google Scholar] [CrossRef]
- Purwantini, E.; Daniels, L. Molecular analysis of the gene encoding F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J. Bacteriol. 1998, 180, 2212–2219. [Google Scholar]
- Bleicher, K.; Winter, J. Purification and properties of F420-and NADP+-dependent alcohol dehydrogenases of Methanogenium liminatans and Methanobacterium palustre, specific for secondary alcohols. Europ. J. Biochem. 1991, 200, 43–51. [Google Scholar] [CrossRef]
- Knaus, T.; Cariati, L.; Masman, M.F.; Mutti, F.G. In vitro biocatalytic pathway design: Orthogonal network for the quantitative and stereospecific amination of alcohols. Org. Biomol. Chem. 2017, 15, 8313–8325. [Google Scholar]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef]
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G.D. Biocatalysis: A pharma perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef]
- Musa, M.M.; Hollmann, F.; Mutti, F.G. Synthesis of enantiomerically pure alcohols and amines via biocatalytic deracemisation methods. Cat. Sci. Technol. 2019, 9, 10–1039. [Google Scholar] [CrossRef]
- Ichikawa, H.; Bashiri, G.; Kelly, W.L. Biosynthesis of the thiopeptins and identification of an F420H2-dependent dehydropiperidine reductase. J. Am. Chem. Soc. 2018, 140, 10749–10756. [Google Scholar] [CrossRef]
- Miller, A.F.; Park, J.T.; Ferguson, K.L.; Pitsawong, W.; Bommarius, A.S. Informing efforts to develop nitroreductase for amine production. Molecules 2018, 23, 22. [Google Scholar] [CrossRef]
- Heiss, G.; Hofmann, K.W.; Trachtmann, N.; Walters, D.M.; Rouvière, P.; Knackmuss, H.J. Npd gene functions of Rhodococcus (opacus) erythropolis Hl PM-1 in the initial steps of 2,4,6-trinitrophenol degradation. Microbiology 2002, 148, 799–806. [Google Scholar] [CrossRef][Green Version]
- Xu, J.; Green, A.P.; Turner, N.J. Chemo-enzymatic synthesis of pyrazines and pyrroles. Angew. Chem.-Int. Ed. 2018, 57, 16760–16763. [Google Scholar] [CrossRef]
- Busacca, C.A.; Fandrick, D.R.; Song, J.J.; Senanayake, C.H. The growing impact of catalysis in the pharmaceutical industry. Adv. Synth. Catal. 2011, 353, 1825–1864. [Google Scholar] [CrossRef]
- Heiss, G.; Trachtmann, N.; Abe, Y.; Takeo, M.; Knackmuss, H.J. Homologous npdgi genes in 2,4-dinitrophenol- and 4-nitrophenol-degrading Rhodococcus spp. Appl. Environ. Microbiol. 2003, 69, 2748–2754. [Google Scholar] [CrossRef]
- Purwantini, E.; Daniels, L.; Mukhopadhyay, B. F420H2 is required for phthiocerol dimycocerosate synthesis in Mycobacteria. J. Bacteriol. 2016, 198, 2020–2028. [Google Scholar] [CrossRef]
- Wichmann, R.; Vasic-Racki, D. Cofactor regeneration at the lab scale. In Technology Transfer in Biotechnology; Springer: Berlin, Germany, 2005; pp. 225–260. [Google Scholar]
- Tishkov, V.I.; Popov, V.O. Catalytic mechanism and application of formate dehydrogenase. Biochemistry 2004, 69, 1252. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Kuge, Y.; Inoue, K.; Yoshikawa, N.; Mochida, K.; Uwajima, T. NADPH regeneration by glucose dehydrogenase from Gluconobacter scleroides for L-leucovorin synthesis. Biosci. Biotechnol. Biochem. 1992, 56, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.S.; Talpur, F.N.; Sopaci, B.; Kohring, G.-W.; Celik, A. Selective oxidation and reduction reactions with cofactor regeneration mediated by galactitol-, lactate-, and formate dehydrogenases immobilized on magnetic nanoparticles. J. Biotechnol. 2011, 152, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-H.; Whitesides, G.M. Enzyme-catalyzed organic synthesis: NAD(P)H cofactor regeneration by using glucose-6-phosphate and the glucose-5-phosphate dehydrogenase from Leuconostoc mesenteroides. J. Am. Chem. Soc. 1981, 103, 4890–4899. [Google Scholar] [CrossRef]
- Lee, W.-H.; Park, J.-B.; Park, K.; Kim, M.-D.; Seo, J.-H. Enhanced production of ɛ-caprolactone by overexpression of NADPH-regenerating glucose 6-phosphate dehydrogenase in recombinant Escherichia coli harboring cyclohexanone monooxygenase gene. Appl. Microbiol. Biotechnol. 2007, 76, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Berrios-Rivera, S.J.; Bennett, G.N.; San, K.Y. Metabolic engineering of Escherichia coli: Increase of NADH availability by overexpressing an NAD+-dependent formate dehydrogenase. Metab. Eng. 2002, 4, 217–229. [Google Scholar] [CrossRef]
- Bashiri, G.; Antoney, J.; Jirgis, E.N.M.; Shah, M.V.; Ney, B.; Copp, J.; Stuteley, S.M.; Sreebhavan, S.; Palmer, B.; Middleditch, M.; et al. A revised biosynthetic pathway for the cofactor F420 in prokaryotes. Nat. Commun. 2019, 10, 1558. [Google Scholar] [CrossRef]
- Purwantini, E.; Daniels, L. Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J. Bacteriol. 1996, 178, 2861–2866. [Google Scholar] [CrossRef][Green Version]
- Costa, K.C.; Wong, P.M.; Wang, T.; Lie, T.J.; Dodsworth, J.A.; Swanson, I.; Burn, J.A.; Hackett, M.; Leigh, J.A. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc. Natl. Acad. Sci. USA 2010, 107, 11050–11055. [Google Scholar] [CrossRef]
- Shuber, A.P.; Orr, E.C.; Recny, M.A.; Schendel, P.F.; May, H.D.; Schauer, N.L.; Ferry, J.G. Cloning, expression, and nucleotide sequence of the formate dehydrogenase genes from Methanobacterium formicicum. J. Biol. Chem. 1986, 261, 12942–12947. [Google Scholar]
- Novotná, J.; Neužil, J.; Hoš?álek, Z. Spectrophotometric identification of 8-hydroxy-5-deazaflavin: Nadph oxidoreductase activity in Streptomycetes producing tetracyclines. FEMS Microbiol. Lett. 1989, 59, 241–245. [Google Scholar] [CrossRef]
- Kumar, H.; Nguyen, Q.T.; Binda, C.; Mattevi, A.; Fraaije, M.W. Isolation and characterization of a thermostable F420:NADPH oxidoreductase from Thermobifida fusca. J. Biol. Chem. 2017, 292, 10123–10130. [Google Scholar] [CrossRef] [PubMed]
- Kunow, J.; Schwörer, B.; Stetter, K.O.; Thauer, R.K. A F420-dependent NADP reductase in the extremely thermophilic sulfate-reducing Archaeoglobus fulgidus. Arch. Microbiol. 1993, 160, 199–205. [Google Scholar]
- Eker, A.P.M.; Hessels, J.K.C.; Meerwaldt, R. Characterization of an 8-hydroxy-5-deazaflavin: NADPH oxidoreductase from Streptomyces griseus. Biochim. Biophys. Acta 1989, 990, 80–86. [Google Scholar] [CrossRef]
- Seelbach, K.; Riebel, B.; Hummel, W.; Kula, M.R.; Tishkov, V.I.; Egorov, A.M.; Wandrey, C.; Kragl, U. A novel, efficient regenerating method of NADPH using a new formate dehydrogenase. Tetrahedron Lett. 1996, 37, 1377–1380. [Google Scholar] [CrossRef]
- Alex, L.A.; Reeve, J.N.; Ormejohnson, W.H.; Walsh, C.T. Cloning, sequence determination, and expression of the genes encoding the subunits of the nickel-containing 8-hydroxy-5-deazaflavin reducing hydrogenase from Methanobacterium thermoautotrophicum delta-H. Biochemistry 1990, 29, 7237–7244. [Google Scholar] [CrossRef]
- Tersteegen, A.; Hedderich, R. Methanobacterium thermoautotrophicum encodes two multisubunit membrane-bound NiFe hydrogenases - transcription of the operons and sequence analysis of the deduced proteins. Eur. J. Biochem. 1999, 264, 930–943. [Google Scholar] [CrossRef]
- Hocking, W.P.; Stokke, R.; Roalkvam, I.; Steen, I.H. Identification of key components in the energy metabolism of the hyperthermophilic sulfate-reducing archaeon archaeoglobus fulgidus by transcriptome analyses. Front. Microbiol. 2014, 5, 20. [Google Scholar] [CrossRef]
- Vitt, S.; Ma, K.; Warkentin, E.; Moll, J.; Pierik, A.J.; Shima, S.; Ermler, U. The F-420-reducing NiFe -hydrogenase complex from methanothermobacter marburgensis, the first x-ray structure of a group 3 family member. J. Mol. Biol. 2014, 426, 2813–2826. [Google Scholar] [CrossRef]
- Bashiri, G.; Rehan, A.M.; Greenwood, D.R.; Dickson, J.M.; Baker, E.N. Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLoS ONE 2010, 5, e15803. [Google Scholar] [CrossRef]
- Isabelle, D.; Simpson, D.R.; Daniels, L. Large-scale production of coenzyme F420-5,6 by using Mycobacterium smegmatis. Appl. Environ. Microbiol. 2002, 68, 5750–5755. [Google Scholar] [CrossRef] [PubMed]
- Eker, A.P.M.; Pol, A.; van der Meyden, P.; Vogels, G.D. Purification and properties of 8-hydroxy-5-deazaflavin derivatives from Streptomyces griseus. FEMS Microbiol. Lett. 1980, 8, 161–165. [Google Scholar] [CrossRef]
- Grochowski, L.L.; Xu, H.M.; White, R.H. Identification and characterization of the 2-phospho-L-lactate guanylyltransferase involved in coenzyme F-420 biosynthesis. Biochemistry 2008, 47, 3033–3037. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Lasta, D.; Hasan, M.; Guo, H.; Leichnitz, D.; Uzum, Z.; Richter, I.; Schalk, F.; Beemelmanns, C.; Hertweck, C.; et al. Metabolic pathway rerouting in Paraburkholderia rhizoxinica evolved long-overlooked derivatives of coenzyme F420. ACS Chem. Biol. 2019, 2088–2094. [Google Scholar] [CrossRef]
- Hossain, M.S.; Le, C.Q.; Joseph, E.; Nguyen, T.Q.; Johnson-Winters, K.; Foss, F.W. Convenient synthesis of deazaflavin cofactor Fo and its activity in F420-dependent NADP reductase. Organ. Biomol. Chem. 2015, 13, 5082–5085. [Google Scholar] [CrossRef]
- Alberty, R.A. Calculation of standard transformed formation properties of biochemical reactants and standard apparent reduction potentials of half reactions. Arch. Biochem. Biophys. 1998, 358, 25–39. [Google Scholar] [CrossRef]
- Alberty, R.A. Calculation of standard transformed gibbs energies and standard transformed enthalpies of biochemical reactants. Arch. Biochem. Biophys. 1998, 353, 116–130. [Google Scholar] [CrossRef]
- Benedict, M.N.; Gonnerman, M.C.; Metcalf, W.W.; Price, N.D. Genome-scale metabolic reconstruction and hypothesis testing in the methanogenic archaeon Methanosarcina acetivorans C2A. J. Bacteriol. 2012, 194, 855–865. [Google Scholar] [CrossRef]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010, 28, 977–982. [Google Scholar] [CrossRef]
- Nazem-Bokaee, H.; Gopalakrishnan, S.; Ferry, J.G.; Wood, T.K.; Maranas, C.D. Assessing methanotrophy and carbon fixation for biofuel production by Methanosarcina acetivorans. Microb. Cell Fact. 2016, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The metacyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed]







| Reaction | Family | Reference |
|---|---|---|
| Enoate reduction | ||
| Aflatoxins | FDOR | [14,18,34] |
| Coumarins | FDOR | [14,34,35] |
| Quinones | FDOR | [36] |
| Biliverdin reduction | FDOR | [12,16] |
| Nitroimidazoles | FDOR | [36] |
| Cyclohexenones | FDOR | [6,34,38] |
| Citral/Neral/Geranial | FDOR | [6] |
| Carvone | FDOR | [6] |
| Ketoisophorone | FDOR | [6] |
| Alcohol oxidation/ketoreduction | ||
| Glucose-6-phosphate | LLHT | [26,50] |
| Phthiodiolone dimycocerosate | LLHT | [51] |
| Isopropanol | LLHT | [40] |
| Imine reductions | ||
| Dehydropiperidine (in thiopeptins) | FDOR | [45] |
| 4-ethylidene-3,4-dihydropyrrole-2-carboxylic acid | Flavin-dependent monooxygenase | [11] |
| Nitroreductions | ||
| Picrate | LLHT | [47,50] |
| 2,4-DNP | LLHT | [48,50] |
| Ring opening/closing | ||
| C-N bond cleavage/formation in methylenetetrahydromethanopterin | LLHT | [23,28,29,30] |
| Source | F420 Yield (μmol/g Cell Weight) | Growth Conditions | Ref |
|---|---|---|---|
| Methanobacterium thermoautotrophicum | 0.42 a,c | Grown at 60 °C using complex media in fermenter, under pressurized hydrogen | [9] |
| Methanobacterium formicium | 0.27 a,c | Grown at 37 °C using complex media in fermenters | [9] |
| Methanospirillum hungatii | 0.41 a,c | Grown at 37 °C using complex media in fermenters | [9] |
| Methanobacterium strain M.o.H | 0.53 a,c | Grown at 40 °C using complex media in fermenters | [9] |
| Methanobacterium thermoautotrophicum | 1.7 e | Grown using complex media in fermenters, under pressurized hydrogen gas | [73] |
| Streptomyces flocculus | 0.62 e | Grown using complex media in fermenters | [73] |
| Streptomyces coelicolor | 0.04 e | Grown using complex media in fermenters | [73] |
| Streptomyces griseus | 0.008 a,c | Growth conditions not mentioned in the publication | [74] |
| Rhodococcus rhodochrous | 0.11 e | Grown using complex media in fermenters | [73] |
| Mycobacterium smegmatis | 0.30 e | Grown using complex media in fermenters | [73] |
| Mycobacterium smegmatis | 3.0 d | Overexpression of F420 pathway genes, cultivation in complex media at 37 °C in shake flasks | [72] |
| Escherichia coli | 0.38 b | Overexpressing F420 pathway genes, grown in minimal media at 30 °C in shake flasks. | [59] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, M.V.; Antoney, J.; Kang, S.W.; Warden, A.C.; Hartley, C.J.; Nazem-Bokaee, H.; Jackson, C.J.; Scott, C. Cofactor F420-Dependent Enzymes: An Under-Explored Resource for Asymmetric Redox Biocatalysis. Catalysts 2019, 9, 868. https://doi.org/10.3390/catal9100868
Shah MV, Antoney J, Kang SW, Warden AC, Hartley CJ, Nazem-Bokaee H, Jackson CJ, Scott C. Cofactor F420-Dependent Enzymes: An Under-Explored Resource for Asymmetric Redox Biocatalysis. Catalysts. 2019; 9(10):868. https://doi.org/10.3390/catal9100868
Chicago/Turabian StyleShah, Mihir V., James Antoney, Suk Woo Kang, Andrew C. Warden, Carol J. Hartley, Hadi Nazem-Bokaee, Colin J. Jackson, and Colin Scott. 2019. "Cofactor F420-Dependent Enzymes: An Under-Explored Resource for Asymmetric Redox Biocatalysis" Catalysts 9, no. 10: 868. https://doi.org/10.3390/catal9100868
APA StyleShah, M. V., Antoney, J., Kang, S. W., Warden, A. C., Hartley, C. J., Nazem-Bokaee, H., Jackson, C. J., & Scott, C. (2019). Cofactor F420-Dependent Enzymes: An Under-Explored Resource for Asymmetric Redox Biocatalysis. Catalysts, 9(10), 868. https://doi.org/10.3390/catal9100868