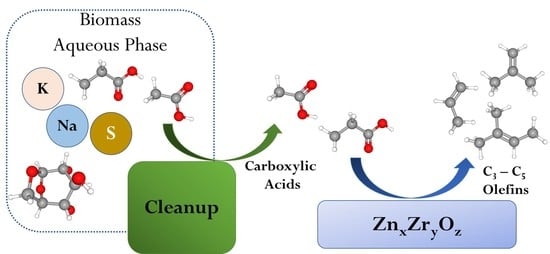
Cleanup and Conversion of Biomass Liquefaction Aqueous Phase to C3–C5 Olefins over ZnxZryOz Catalyst

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
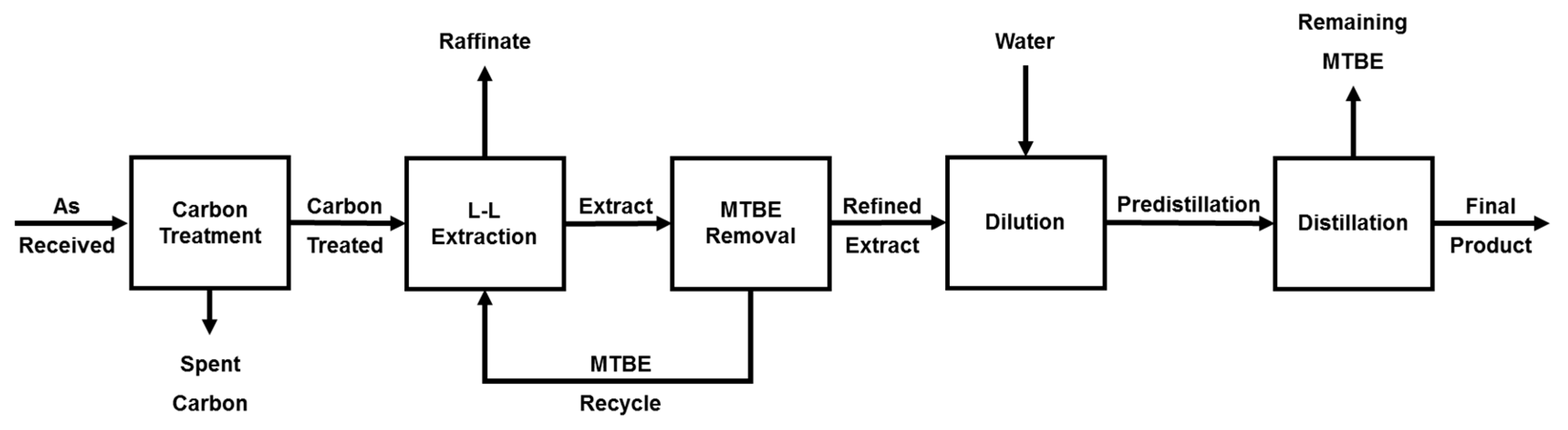
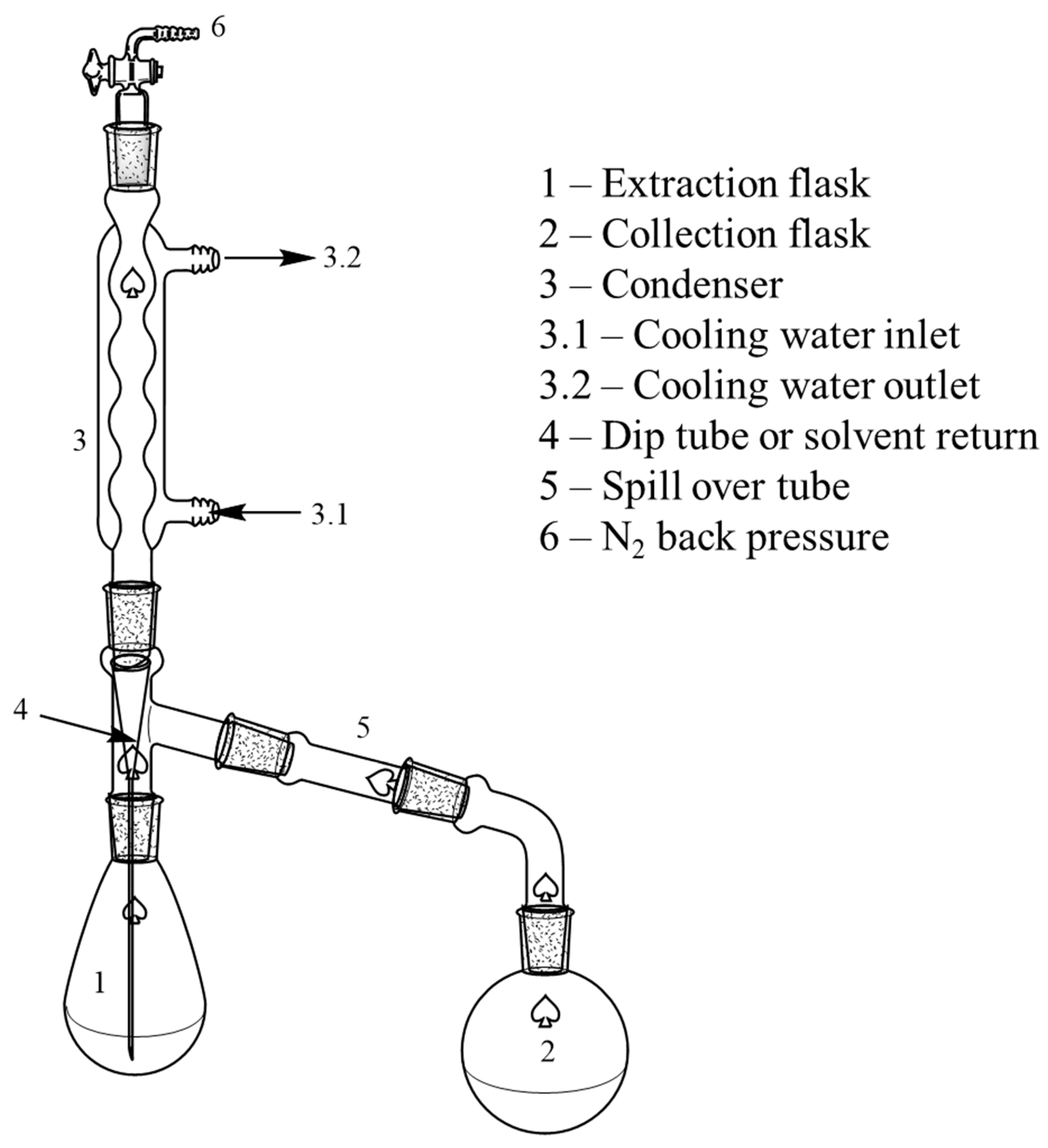
2.1. Feedstock Cleanup with Continuous Liquid–Liquid Extraction
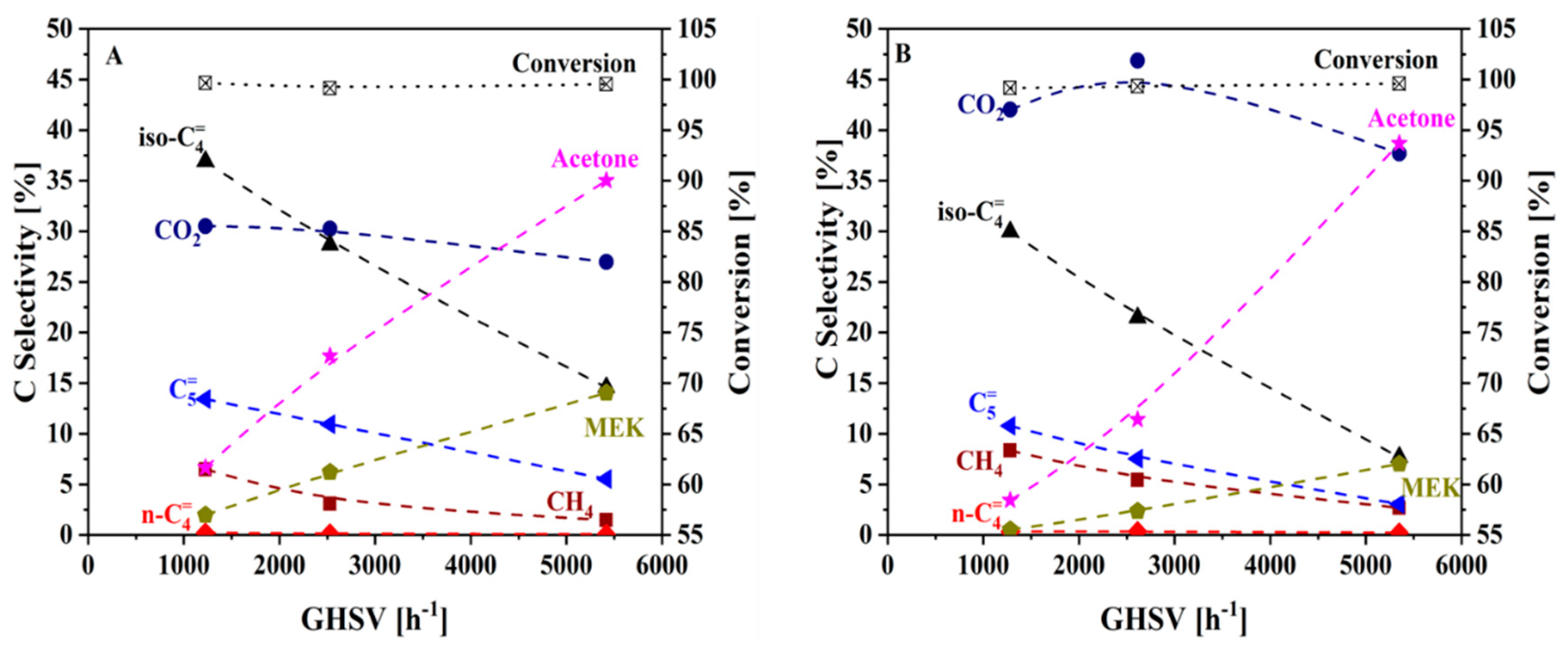
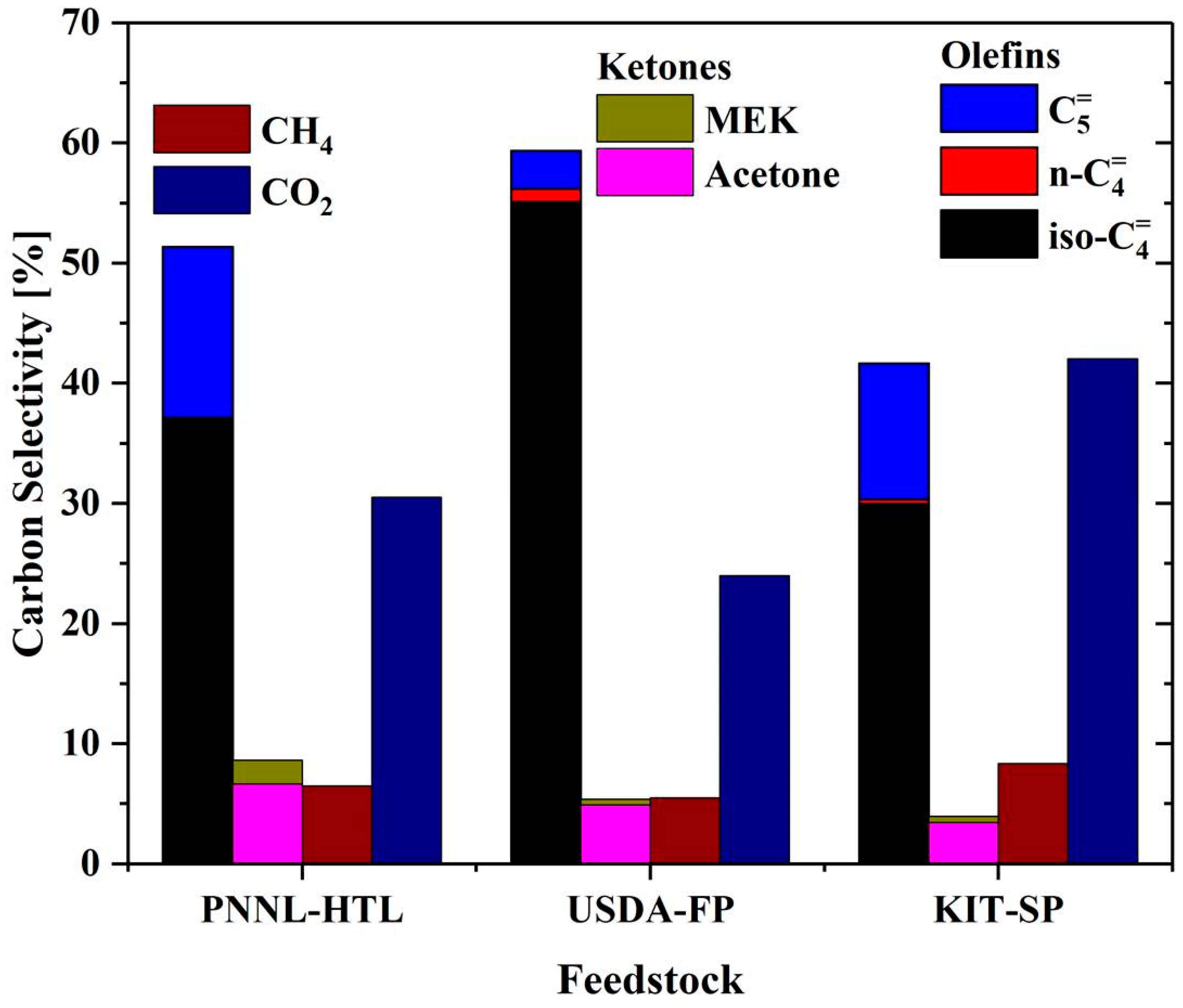
2.2. Catalytic Upgrading of Cleaned Up Aqueous Phase to Isobutene
2.3. Catalytic Upgrading of Ethanol to Propene
3. Materials and Methods
3.1. Feedstock Sources
3.2. Feedstock Clean Up
3.3. Catalyst Preparation
3.4. Reactor and Reaction Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Activated carbon |
| BDL | Below detection limit |
| DI | Deionized |
| FP | Fast pyrolysis |
| GHSV | Gas hourly space velocity |
| HTL | Hydrothermal liquefaction |
| KIT | Karlsruhe Institute of Technology |
| LLE | Liquid–liquid extraction |
| MEK | Methyl ethyl ketone (butanone) |
| MTBE | Methyl Tert-butyl ether |
| PNNL | Pacific Northwest National Lab |
| S/C | Steam-to-carbon ratio (molar ratio) |
| SP | Screw pyrolysis |
| SV | Space velocity |
| TOS | Time on stream |
| USDA | United States Department of Agriculture |
Appendix A
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.Q.; Upadhye, A.A.; Olcay, H.; Tompsett, G.A.; Jae, J.; Xing, R.; Alonso, D.M.; Wang, D.; Zhang, T.; Kumar, R.; et al. Production of renewable jet fuel range alkanes and commodity chemicals from integrated catalytic processing of biomass. Energy Environ. Sci. 2014, 7, 1500–1523. [Google Scholar] [CrossRef]
- Huber, G.W.; Corma, A. Synergies between Bio- and Oil Refineries for the Production of Fuels from Biomass. Angew. Chem. Int. Ed. 2007, 46, 7184–7201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Biddy, M.J.; Jones, S.B.; Elliott, D.C.; Schmidt, A.J. Techno-economic analysis of liquid fuel production from woody biomass via hydrothermal liquefaction (HTL) and upgrading. Appl. Energy 2014, 129, 384–394. [Google Scholar] [CrossRef]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Panisko, E.; Wietsma, T.; Lemmon, T.; Albrecht, K.; Howe, D. Characterization of the aqueous fractions from hydrotreatment and hydrothermal liquefaction of lignocellulosic feedstocks. Biomass Bioenergy 2015, 74, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Review of biomass pyrolysis oil properties and upgrading research. Energy Convers. Manag. 2007, 48, 87–92. [Google Scholar] [CrossRef]
- Elliott, D.C.; Beckman, D.; Bridgwater, A.V.; Diebold, J.P.; Gevert, S.B.; Solantausta, Y. Developments in direct thermochemical liquefaction of biomass: 1983–1990. Energy Fuels 1991, 5, 399–410. [Google Scholar] [CrossRef]
- Elliott, D.C.; Biller, P.; Ross, A.B.; Schmidt, A.J.; Jones, S.B. Hydrothermal liquefaction of biomass: Developments from batch to continuous process. Bioresour. Technol. 2015, 178, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Funke, A.; Tomasi Morgano, M.; Dahmen, N.; Leibold, H. Experimental comparison of two bench scale units for fast and intermediate pyrolysis. J. Anal. Appl. Pyrolysis 2017, 124, 504–514. [Google Scholar] [CrossRef]
- Boateng, A.A.; Daugaard, D.E.; Goldberg, N.M.; Hicks, K.B. Bench-Scale Fluidized-Bed Pyrolysis of Switchgrass for Bio-Oil Production. Ind. Eng. Chem. Res. 2007, 46, 1891–1897. [Google Scholar] [CrossRef]
- Huber, G.W.; Dumesic, J.A. An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery. Catal. Today 2006, 111, 119–132. [Google Scholar] [CrossRef]
- Davidson, S.D.; Lopez-Ruiz, J.A.; Zhu, Y.; Cooper, A.R.; Albrecht, K.O.; Dagle, R.A. Clean up and Catalytic Upgrading of Hydrothermal Liquefaction-Derived Aqueous Phase into Fuels and Chemicals via Ketone Intermediates. ACS Sustain. Chem. Eng. 2019. submitted. [Google Scholar]
- Saavedra, L.J.; Dagle, R.A.; Dagle, V.L.; Smith, C.; Albrecht, K.O. Oligomerization of ethanol-derived propene and isobutene mixtures to transportation fuels: Catalyst and process considerations. Catal. Sci. Technol. 2019, 9, 1117–1131. [Google Scholar] [CrossRef]
- Dagle, V.L.; Smith, C.; Flake, M.; Albrecht, K.O.; Gray, M.J.; Ramasamy, K.K.; Dagle, R.A. Integrated process for the catalytic conversion of biomass-derived syngas into transportation fuels. Green Chem. 2016, 18, 1880–1891. [Google Scholar] [CrossRef]
- Yoon, J.W.; Chang, J.-S.; Lee, H.-D.; Kim, T.-J.; Jhung, S.H. Trimerization of isobutene over a zeolite beta catalyst. J. Catal. 2007, 245, 253–256. [Google Scholar] [CrossRef]
- Marchionna, M.; Di Girolamo, M.; Patrini, R. Light olefins dimerization to high quality gasoline components. Catal. Today 2001, 65, 397–403. [Google Scholar] [CrossRef]
- Carr, A.G.; Dawson, D.M.; Bochmann, M. Zirconocenes as Initiators for Carbocationic Isobutene Homo- and Copolymerizations. Macromolecules 1998, 31, 2035–2040. [Google Scholar] [CrossRef]
- Liu, C.; Sun, J.; Smith, C.; Wang, Y. A study of ZnxZryOz mixed oxides for direct conversion of ethanol to isobutene. Appl. Catal. A Gen. 2013, 467, 91–97. [Google Scholar] [CrossRef]
- Sun, J.; Baylon, R.A.L.; Liu, C.; Mei, D.; Martin, K.J.; Venkitasubramanian, P.; Wang, Y. Key Roles of Lewis Acid–Base Pairs on ZnxZryOz in Direct Ethanol/Acetone to Isobutene Conversion. J. Am. Chem. Soc. 2015, 138, 507–517. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, K.; Gao, F.; Wang, C.; Liu, J.; Peden, C.H.F.; Wang, Y. Direct Conversion of Bio-ethanol to Isobutene on Nanosized ZnxZryOz Mixed Oxides with Balanced Acid–Base Sites. J. Am. Chem. Soc. 2011, 133, 11096–11099. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Dagle, V.; Flake, M.; Ramasamy, K.; Kovarik, L.; Bowden, M.; Onfroy, T.; Dagle, R. Conversion of Syngas-Derived C2+ Mixed Oxygenates to C3-C5 Olefins over ZnxZryOz Mixed Oxides Catalysts. Catal. Sci. Technol. 2016, 6, 2325–2336. [Google Scholar] [CrossRef]
- Dagle, V.L.; Dagle, R.A.; Kovarik, L.; Baddour, F.; Habas, S.E.; Elander, R. Single-step Conversion of Methyl Ethyl Ketone to Olefins over ZnxZryOz Catalysts in Water. ChemCatChem 2019, 11, 3393–3400. [Google Scholar] [CrossRef]
- Crisci, A.J.; Dou, H.; Prasomsri, T.; Román-Leshkov, Y. Cascade Reactions for the Continuous and Selective Production of Isobutene from Bioderived Acetic Acid Over Zinc-Zirconia Catalysts. ACS Catal. 2014, 4, 4196–4200. [Google Scholar] [CrossRef]
- Schmidt, A.J.; Lindstorm, T.; Talmadge, M.; Zhu, Y. Mid Stage 2 Report on Hydrothermal Liquefaction Strategy for the NABC Leadership Team. In PNNL-21768; Energy, U.S.D.O., Ed.; Pacific Northwest National Laboratory: Richland, WA, USA, 2012. [Google Scholar]
- Mullen, C.A.; Boateng, A.A.; Goldberg, N.M. Production of Deoxygenated Biomass Fast Pyrolysis Oils via Product Gas Recycling. Energy Fuels 2013, 27, 3867–3874. [Google Scholar] [CrossRef]
- Boateng, A.A.; Schaffer, M.A.; Mullen, C.A.; Goldberg, N.M. Mobile demonstration unit for fast- and catalytic pyrolysis: The combustion reduction integrated pyrolysis system (CRIPS). J. Anal. Appl. Pyrolysis 2019, 137, 185–194. [Google Scholar] [CrossRef]
- Tomasi Morgano, M.; Leibold, H.; Richter, F.; Seifert, H. Screw pyrolysis with integrated sequential hot gas filtration. J. Anal. Appl Pyrolysis 2015, 113, 216–224. [Google Scholar] [CrossRef]
- Tomasi Morgano, M.; Leibold, H.; Richter, F.; Stapf, D.; Seifert, H. Screw pyrolysis technology for sewage sludge treatment. Waste Manag. 2018, 73, 487–495. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PNNL-HTL | USDA-FP | KIT-SP | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial | Carbon Treated | Raffinate | Refined Extract | Final Feedstock | Initial | Carbon Treated | Raffinate | Refined Extract | Final Feedstock | Initial | Carbon Treated | Raffinate | Refined Extract | Final Feedstock | |
| %C Retained | 100 | 65.5 | 26.0 | 29.0 | 29.0 | 100 | 73.2 | 15.1 | 21.7 | 15.1 | 100 | 30.4 | 11.7 | 14.9 | 11.8 |
| %Carboxylic Acid Retained | 100 | 62.6 | 18.8 | 34.9 | 34.9 | 100 | 88.9 | 24.8 | 53.5 | 45.9 | 100 | 33.5 | 6.55 | 22.2 | 22.4 |
| Acetic Acid [wt.%] | 22.3 | 19.4 | 7.65 | 63.4 | 29.1 | 4.78 | 3.07 | 1.45 | 39.6 | 9.24 | 9.68 | 6.62 | 1.80 | 34.3 | 20.1 |
| Propionic Acid [wt.%] | 3.93 | 2.33 | 0.241 | 11.2 | 4.79 | 2.89 | 1.95 | 1.47 | 9.23 | 1.68 | 2.72 | 2.07 | 1.39 | 5.42 | 2.98 |
| Non-Participating Carbon [wt.%] | 6.40 | 6.09 | 7.89 | 5.77 | 0.16 | 5.95 | 3.00 | 5.34 | 27.33 | 1.55 | 13.8 | 8.03 | 12.2 | 41.1 | 2.57 |
| Total C [wt.%] | 32.6 | 27.8 | 15.8 | 80.4 | 34.0 | 8.35 | 8.02 | 8.26 | 76.16 | 12.5 | 26.2 | 16.7 | 15.4 | 80.8 | 25.6 |
| S/C a | 2.8 | 11.2 | 4.7 | ||||||||||||
| Na [ppm] | 7040 | 7320 | 8580 | 25.2 | 22.9 | 9.30 | 202 | 147 | 18.3 | 12.8 | 28.2 | 446 | 365 | 7.96 | 9.64 |
| K [ppm] | 746 | 1960 | 2350 | 11.6 | 3.97 | 12.3 | 1560 | 1450 | 85.2 | 17.2 | 15.7 | 4050 | 1420 | 20.5 | 12.8 |
| S [ppm] | BDLb | BDL | BDL | BDL | BDL | 10.6 | 65.1 | 44.9 | BDL | BDL | 56.5 | 68.5 | 65.8 | BDL | BDL |
| PNNL-HTL | KIT-SP | |||
|---|---|---|---|---|
| Initial | Final | Initial | Final | |
| Conversion (%) | 99.8 | 99.8 | 99.7 | 82.1 |
| Selectivity (%) | ||||
| All Olefins | 53.2 | 21.5 | 30.8 | 0.77 |
| All Ketones | 9.11 | 50.0 | 20.1 | 64.8 |
| Isobutene | 34.5 | 14.7 | 20.2 | 0.39 |
| Acetone | 6.60 | 35.3 | 16.0 | 52.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidson, S.D.; Lopez-Ruiz, J.A.; Flake, M.; Cooper, A.R.; Elkasabi, Y.; Tomasi Morgano, M.; Lebarbier Dagle, V.; Albrecht, K.O.; Dagle, R.A. Cleanup and Conversion of Biomass Liquefaction Aqueous Phase to C3–C5 Olefins over ZnxZryOz Catalyst. Catalysts 2019, 9, 923. https://doi.org/10.3390/catal9110923
Davidson SD, Lopez-Ruiz JA, Flake M, Cooper AR, Elkasabi Y, Tomasi Morgano M, Lebarbier Dagle V, Albrecht KO, Dagle RA. Cleanup and Conversion of Biomass Liquefaction Aqueous Phase to C3–C5 Olefins over ZnxZryOz Catalyst. Catalysts. 2019; 9(11):923. https://doi.org/10.3390/catal9110923
Chicago/Turabian StyleDavidson, Stephen D., Juan A. Lopez-Ruiz, Matthew Flake, Alan R. Cooper, Yaseen Elkasabi, Marco Tomasi Morgano, Vanessa Lebarbier Dagle, Karl O. Albrecht, and Robert A. Dagle. 2019. "Cleanup and Conversion of Biomass Liquefaction Aqueous Phase to C3–C5 Olefins over ZnxZryOz Catalyst" Catalysts 9, no. 11: 923. https://doi.org/10.3390/catal9110923